
Abstract
We evaluate the application of 57Fe Mössbauer spectroscopy to the determination of the composition of magnetite (Fe3O4)/maghemite (γ-Fe2O3) mixtures and the stoichiometry of magnetite-maghemite solid solutions. In particular, we consider a recently proposed model-independent method which does not rely on a priori assumptions regarding the nature of the sample, other than that it is free of other Fe-containing phases. In it a single parameter,  —the 'centre of gravity', or area weighted mean isomer shift at room temperature, T = 295 ± 5 K—is extracted by curve-fitting a sample's Mössbauer spectrum, and is correlated to the sample's composition or stoichiometry. We present data on high-purity magnetite and maghemite powders, and mixtures thereof, as well as comparison literature data from nanoparticulate mixtures and solid solutions, to show that a linear correlation exists between
—the 'centre of gravity', or area weighted mean isomer shift at room temperature, T = 295 ± 5 K—is extracted by curve-fitting a sample's Mössbauer spectrum, and is correlated to the sample's composition or stoichiometry. We present data on high-purity magnetite and maghemite powders, and mixtures thereof, as well as comparison literature data from nanoparticulate mixtures and solid solutions, to show that a linear correlation exists between  and the numerical proportion of Fe atoms in the magnetite environment:
and the numerical proportion of Fe atoms in the magnetite environment:  = Femagnetite/Fetotal
= Femagnetite/Fetotal  , where
, where  = 0.3206 ± 0.0022 mm s−1 and
= 0.3206 ± 0.0022 mm s−1 and  = 0.2135 ± 0.0076 mm s−1. We also present equations to relate
= 0.2135 ± 0.0076 mm s−1. We also present equations to relate  to the weight percentage w of magnetite in mixed phases, and the magnetite stoichiometry x = Fe2+/Fe3+ in solid solutions. The analytical method is generally applicable, but is most accurate when the absorption profiles are sharp; in some samples this may require spectra to be recorded at reduced temperatures. We consider such cases and provide equations to relate
to the weight percentage w of magnetite in mixed phases, and the magnetite stoichiometry x = Fe2+/Fe3+ in solid solutions. The analytical method is generally applicable, but is most accurate when the absorption profiles are sharp; in some samples this may require spectra to be recorded at reduced temperatures. We consider such cases and provide equations to relate  to the corresponding α value.
to the corresponding α value.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Two of the most common phases of iron oxide—magnetite (Fe3O4) and maghemite (γ-Fe2O3)—are of note in that they are ferrimagnetic at room temperature [1]. Indeed, magnetite is the most magnetic of all the naturally occurring oxides on Earth, having a room temperature saturation magnetisation by mass of Ms = 92 Am2 kg−1, while maghemite is also strongly magnetic, with Ms = 76 Am2 kg−1 at room temperature [2]. These phases play a significant role in geomagnetism and palaeomagnetism [3–6], and are also important markers in archaeomagnetism, largely due to their presence in clays and clay products such as pottery [7–9]. Maghemite is an oxidation product of magnetite, and as such the interplay and balance between the two phases is often informative, both in centuries-old samples [10] and in more contemporary materials, such as the corrosion products of carbon steels [11–13].
Both phases have found important technical applications in everyday life, from ferrofluids and magnetic oils [14, 15] and battery components [16], through to their large-scale use in water treatment plants [17, 18]. In their fine particle form they have also attracted a great deal of recent interest in biomedical applications ranging from magnetic actuation for drug delivery, magnetic hyperthermia treatments for brain and prostate cancers, and novel medical imaging agents [19, 20]. In many applications the degradation of the materials from magnetite to maghemite causes a loss of effective function due to the reduced magnetisation, while in the biomedical field there is considerable interest in the transformation between the two, given the different roles that ferrous (Fe2+) and ferric (Fe3+) ions play in the production of reactive oxygen species via the catalytic Haber–Weiss and Fenton chemistry pathways [21–23].
However, experimentally it is often difficult to distinguish between magnetite and maghemite even if they are present in their bulk, well-crystallised forms. As illustrated in figure 1, this is because crystallographically they are very closely related, with both having inverse spinel structures comprising a cubic lattice of close-packed oxygen layers in an ABCABC stacking sequence (FCC-type stacking) along the [1 1 1] direction, within which there are a selection of octahedral and tetrahedral sites at which the iron atoms may sit [1]. In magnetite there is full occupancy of these sites, as represented by the formula ![$\text{F}{{\text{e}}^{3+}}\left[\text{F}{{\text{e}}^{2+}}\text{F}{{\text{e}}^{3+}}\right]{{\text{O}}_{4}}$](https://content.cld.iop.org/journals/0022-3727/50/26/265005/revision2/daa73faieqn009.gif) , where the square brackets denote the octahedral sites. The ferrous ions only appear on the octahedral sites, and at temperatures above the Verwey transition (TV ≈ 118 K in bulk magnetite) there is fast electron hopping between neighbouring {Fe2+Fe3+} pairs, so that in some measurement techniques, including Mössbauer spectroscopy, the two octahedral Fe ions become indistinguishable, and effectively behave as two 'Fe2.5+' ions.
, where the square brackets denote the octahedral sites. The ferrous ions only appear on the octahedral sites, and at temperatures above the Verwey transition (TV ≈ 118 K in bulk magnetite) there is fast electron hopping between neighbouring {Fe2+Fe3+} pairs, so that in some measurement techniques, including Mössbauer spectroscopy, the two octahedral Fe ions become indistinguishable, and effectively behave as two 'Fe2.5+' ions.

Figure 1. Representations of the crystallographic unit cells of magnetite and of the most common maghemite phases: (a) the cubic Fd-3m space group of magnetite and of those maghemites where the octahedral site vacancies are randomly distributed through the lattice; and (b) the cubic P4132 space group of those maghemites where the octahedral site vacancies are partially ordered [30]. The oxygen anions are shown here as dark grey spheres, and the iron cations reside within mid and light grey shaded polygons that indicate, respectively, local tetrahedral and octahedral oxygen coordination.
Download figure:
Standard image High-resolution imageIn maghemite all of the Fe2+ ions are oxidised to Fe3+ ions, but given that the same fundamental structure elements are maintained, a number of vacancies (denoted here by the  symbol) occupy octahedral sites to maintain electrical neutrality [24]. The maghemite formula may then be expressed as
symbol) occupy octahedral sites to maintain electrical neutrality [24]. The maghemite formula may then be expressed as ![$\text{F}{{\text{e}}^{3+}}[\text{Fe}_{5/3}^{3+}{{\square}_{1/3}}]{{\text{O}}_{4}}$](https://content.cld.iop.org/journals/0022-3727/50/26/265005/revision2/daa73faieqn011.gif) , which emphasises its relation to the magnetite parent structure. For intermediate forms—commonly referred to as nonstoichiometric or partially oxidised magnetites, but more precisely a solid solution of magnetite and maghemite—the formula may be expressed in a form that highlights the oxidative transformation:
, which emphasises its relation to the magnetite parent structure. For intermediate forms—commonly referred to as nonstoichiometric or partially oxidised magnetites, but more precisely a solid solution of magnetite and maghemite—the formula may be expressed in a form that highlights the oxidative transformation: ![$\text{F}{{\text{e}}^{3+}}\left[\left\{\text{Fe}_{a}^{2+}\text{Fe}_{a}^{3+}\right\}\text{Fe}_{b}^{3+}{{\square}_{2-2a-b}}\right]{{\text{O}}_{4}}\,$](https://content.cld.iop.org/journals/0022-3727/50/26/265005/revision2/daa73faieqn012.gif) [25]. Here: the endpoints correspond to a = 1 and b = 0 for pure magnetite; and a = 0 and b = 5/3 for pure maghemite; and charge balance demands that for any intermediate solution 5a + 3b = 5. This form is particularly useful for Mössbauer studies of solid solutions, as it has been found that the subspectrum associated with the octahedral Fe3+ ions (the b per formula unit ions) is practically indistinguishable from the subspectrum associated with the tetrahedral Fe3+ ions [26–29].
[25]. Here: the endpoints correspond to a = 1 and b = 0 for pure magnetite; and a = 0 and b = 5/3 for pure maghemite; and charge balance demands that for any intermediate solution 5a + 3b = 5. This form is particularly useful for Mössbauer studies of solid solutions, as it has been found that the subspectrum associated with the octahedral Fe3+ ions (the b per formula unit ions) is practically indistinguishable from the subspectrum associated with the tetrahedral Fe3+ ions [26–29].
Furthermore, the manner in which the octahedral site vacancies are distributed within the lattice determines the crystal structure of the material as a whole. For example, whereas the structure of magnetite is well established and known to be that of the cubic Fd-3m space group [1], maghemite, even in its fully oxidized state, can present a variety of structures [1, 30]. The three most common forms of maghemite are: the cubic Fd-3m space group (the same as that of magnetite) wherein the vacancies are randomly distributed; the cubic P4132 space group in which the vacancies are partially ordered [30]; and the tetragonal P43212 space group in which the vacancies are fully ordered [31]. The similarity between these structures—as illustrated in figure 1 for the case of the two cubic space groups, Fd-3m and P4132—makes it problematic to crystallographically distinguish the different phases. Although this is less of an issue in well-crystallised, bulk materials, where one can seek out key distinguishing features such as lattice parameters, atomic positions and site occupancies, in poorly crystallised or fine particle samples, where size-dependent line broadening effects are frequently encountered, the x-ray analysis can become complicated and ambiguous.
An alternative is to turn to chemical analysis to determine the molar ratio of Fe2+ to Fe3+ ions in a given sample, and deduce the composition (in the case of a magnetite/maghemite mixture) or stoichiometry (in the case of a magnetite-maghemite solid solution) from the result. Various analytical methods may be used: two of the most common are a colorimetric assay based on the reaction of Fe2+ with 1,10-phenanthroline that forms a deep red solution [32, 33]; and a redox titration assay with potassium dichromate using diphenylamine as an indicator [25, 34]. However, as in all such analyses dealing with solid samples, the first step is one of acid dissolution, typically achieved using concentrated hydrochloric acid. This dissolution step may take many hours or even days to complete [35], during which time one needs to be very careful that the dissolved Fe2+ does not oxidise in situ—indeed, some researchers perform the dissolution in an anaerobic glovebox [33]. There are also other pitfalls that practitioners remark upon, such as the risk of ambient oxidation during the course of a slow-paced benchtop titration [25], or the oxidizing nitrates present in filter paper that can alter the iron-containing solution as it passes through [33].
Given the inherent ambiguity of diffractometry studies, and the intrinsic reliance on operator skills associated with chemical analyses, it is not surprising that researchers have looked for other—preferably non-destructive and non-perturbative—methods for probing the composition and/or stoichiometry of magnetites and maghemites. One of the most promising of these has been 57Fe Mössbauer spectroscopy, which has long been known to be an excellent method for discriminating between magnetite and maghemite [27, 28]—as illustrated by the room temperature spectra shown in figure 2, where there are clear differences in both the number and the positions of the absorption lines for the two phases. However, this apparent simplicity belies an underlying complexity that arises whenever non-ideal samples are measured—where, for example, the Mössbauer spectra of fine particle samples can exhibit substantial line broadening [25, 33, 36], to the extent that profile-based discrimination between phases is impossible. Even in cases where the line profiles are well resolved the process of resolving the subspectra associated with distinguishable Fe2+ and Fe3+ sites can be challenging, with numerous model-dependent constraints needing to be imposed to ensure reliable results. In the hands of experts this can be accomplished—as, for example, in the five-site deconstruction of the 4.2 K spectrum of a single crystal of pure magnetite [37], or the careful examination of the four sites apparent in the 6 K spectra of nonstoichiometric magnetite nanoparticles to identify those due to octahedral Fe2+ ions [38]—but these are exceptions rather than the rule.

Figure 2. 57Fe Mössbauer spectra, recorded at room temperature (T = 295 ± 5 K), of polycrystalline samples of magnetite, Fe3O4 (sample DTU100, α = 0.97 ± 0.02, top) and maghemite, γ-Fe2O3 (sample DTU0, α = 0.00 ± 0.01, bottom), and of mixtures of the two phases. The solid lines represent the best-fit results using the model-independent curve-fitting procedure described in section 3.
Download figure:
Standard image High-resolution imageIt is in this context that a recently proposed 57Fe Mössbauer spectroscopic method for measuring magnetite/maghemite composition or magnetite-maghemite stoichiometry is of significant interest. The method is based on that proposed by da Costa et al [25]—a team including one of the current authors—who were inspired in part by the work of Santoyo Salazar et al [39], who, in discussing a Mössbauer study of core-shell magnetite-maghemite nanoparticles, had noted that 'the mean stoichiometry can be estimated from the mean isomer shift'. The underlying physical principle here is quite straightforward, viz.: (1) that the Mössbauer absorption characteristic of every individual 57Fe nucleus in the sample is determined by the local, nearest-neighbour atomic environment of that nucleus; (2) that as the isomer shift parameter is driven by the electrostatic environment of the nucleus, it is an especially good marker for both the atomic charge state (Fe2+ or Fe3+) and for the local oxygen coordination (octahedral or tetrahedral); and (3) that as every 57Fe nucleus in the sample has an equal probability of contributing to the overall recorded absorption spectrum, the area weighted mean isomer shift—or to be less prosaic, the centre of gravity of the spectrum—should be representative of the summed probabilities of the 57Fe nuclei encountering either magnetite-like or maghemite-like local environments. The method has latterly been extended by some of the current authors to embrace a model-independent spectral fitting procedure designed to most accurately evaluate the mean isomer shift, and its associated measurement uncertainty, in a way that does not depend on a priori assumptions regarding the nature of the sample [36], other than that it is free of other Fe-containing phases.
The resulting 'centre of gravity' method establishes, in principle, a well-defined correlation between a relatively easily measured parameter and a valuable physical characteristic—the composition or stoichiometry—of magnetite and maghemite samples. However, to date the method has not been rigorously tested against independently verified standards, and has not yet entered into common usage. It is for this reason that we have undertaken to thoroughly test the method using a set of standard samples comprising mixtures of verifiably pure magnetite and verifiably pure maghemite. Our goal is to bring the application of Mössbauer spectroscopy to the magnetite versus maghemite issue to the attention of not just experienced practitioners but also to a wider audience, in the interest of highlighting in a definitive manner what we consider to be an effective but under-utilised methodology in the field. We therefore present here the results of this evaluation, and also recommend procedures for using the centre of gravity method.
2. Definitions
Before proceeding further, let us define some parameters and nomenclature relating to the composition of magnetite/maghemite mixtures and the stoichiometry of magnetite-maghemite solid solutions. We first define two parameters that relate to both mixtures and solid solutions:
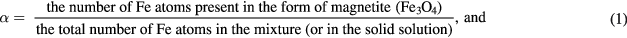
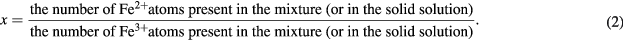
The parameter α is convenient for our purposes, as it applies to both mixtures and solid solutions, and ranges from endpoints of α = 0 for pure maghemite through to α = 1 for pure magnetite. It is a natural parameter to measure with Mössbauer spectroscopy in these samples, given that Mössbauer is an atom-based experimental technique, wherein, to a good approximation, every Fe atom contributes an equally-weighted absorption signal6.
In the case of a magnetite/maghemite mixture, when expressed as a percentage, α may be considered to be the atomic percentage (at.%) of Fe atoms present in the form of magnetite in the mixture:
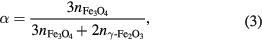
where n is number of moles present of each phase in the mixture, and where we apply the knowledge that a mole of Fe3O4 contains three moles of Fe atoms, and a mole of γ-Fe2O3 contains two moles of Fe atoms. However, we do not recommend this terminology, as it is easily confused with an alternative but different definition of 'atomic percentage', namely:
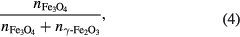
which defines it in terms of the amounts of each phase, rather than the amounts of Fe in those phases. Hence, to ensure clarity, we recommend using the α parameter defined in equation (1) to describe the number determined from the Mössbauer data. In addition, it may be appropriate in some cases to report a second, more sample-specific parameter, derived from α. In the case of solid solutions, this could be the magnetic stoichiometry, x, as defined in equation (5) below; and in the case of mixtures7, it could be the weight percentage, w, as defined in equation (9) below.
In the case of a magnetite-maghemite solid solution, the concept of 'an Fe atom present in the form of magnetite' requires some further explanation. Namely: that this is a construct specific to Mössbauer spectroscopy, and one that is based on the local nature of the 57Fe nucleus' sampling of its electrostatic environment. In essence we are positing here that, even in a solid solution, a given 57Fe nucleus will experience a local environment that can be uniquely identified as that of magnetite, as opposed to that of maghemite. This is a rather subtle point, but it is important in that it establishes that the centre of gravity method may be applied to the study of both mixtures and solid solutions, and even to samples of an indeterminate nature where it is not known whether they are a mixture of phases or a single-phase solid solution.
In mixtures x is referred to as the molar ratio Fe2+/Fe3+, or else 2x is reported as a percentage and is referred to as the molar percentage of magnetite in the mixture. (Note that the 'molar percentage' here is the same as the 'atomic percentage' in equation (4).) In solid solutions x is referred to as the magnetite stoichiometry [33]. In any case, algebraically it can be shown (see the supplementary information sections S1(a) and S1(b) (stacks.iop.org/JPhysD/50/265005/mmedia)) that in both mixtures and solid solutions:
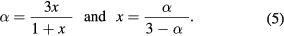
In solid solutions it may be preferable to report the stoichiometry in terms of the formula unit of the material. In the case that the formula unit is written as ![$\text{F}{{\text{e}}^{3+}}\left[\left\{\text{Fe}_{a}^{2+}\text{Fe}_{a}^{3+}\right\}\text{Fe}_{b}^{3+}{{\square}_{2-2a-b}}\right]{{\text{O}}_{4}}$](https://content.cld.iop.org/journals/0022-3727/50/26/265005/revision2/daa73faieqn013.gif) the a and b occupancy parameters may be derived from α (see supplementary information S1(c)) using:
the a and b occupancy parameters may be derived from α (see supplementary information S1(c)) using:
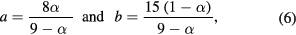
and in the case that the formula unit is written as  the
the  parameter may be related to α (see supplementary information S1(d)) using:
parameter may be related to α (see supplementary information S1(d)) using:
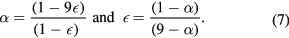
Lastly, we define a parameter that applies only to mixtures:
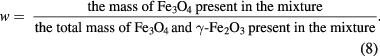
This is a number that ranges from 0 to 1. When expressed as a percentage, w is the weight percentage (wt.%) of magnetite in the mixture. It too can be expressed in terms of α (see supplementary information S1(e)):
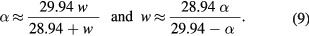
Henceforth we will describe our experimental results in terms of α, on the understanding that α can at any time be transformed into either x, ε or w using equations (5), (7) or (9).
3. 'Centre of gravity' method
The 'centre of gravity' method allows for the area weighted mean isomer shift,  , of a magnetically split 57Fe Mössbauer spectrum to be determined without the need to assume any specific underlying model for the microenvironment of the Fe atoms. There are, however, certain requirements on the data that must be met for the method to be applied. These are (i) that the data collected should obey the fundamental physical properties of thin, texture free absorbers, and (ii) that the (usually folded) spectrum should possess a flat, well defined background. The former is usually a simple matter of preparing suitably thin and homogeneously dispersed polycrystalline absorbers, in keeping with best practice [40], to ensure that the absorber is not so thick that self-absorption prevents each of the 57Fe atoms in the sample experiencing the same flux of γ-rays [41]. The latter is a matter of ensuring that the mechanical aspects of the Mössbauer spectrometer are well-behaved, e.g. by carefully setting up the instruments and regularly recording α-Fe foil calibration spectra, and by operating the spectrometer at a large enough velocity range to comfortably accommodate all of the absorption lines present in the spectrum—a range of ca. ±15 mm s−1 usually being sufficient for this purpose.
, of a magnetically split 57Fe Mössbauer spectrum to be determined without the need to assume any specific underlying model for the microenvironment of the Fe atoms. There are, however, certain requirements on the data that must be met for the method to be applied. These are (i) that the data collected should obey the fundamental physical properties of thin, texture free absorbers, and (ii) that the (usually folded) spectrum should possess a flat, well defined background. The former is usually a simple matter of preparing suitably thin and homogeneously dispersed polycrystalline absorbers, in keeping with best practice [40], to ensure that the absorber is not so thick that self-absorption prevents each of the 57Fe atoms in the sample experiencing the same flux of γ-rays [41]. The latter is a matter of ensuring that the mechanical aspects of the Mössbauer spectrometer are well-behaved, e.g. by carefully setting up the instruments and regularly recording α-Fe foil calibration spectra, and by operating the spectrometer at a large enough velocity range to comfortably accommodate all of the absorption lines present in the spectrum—a range of ca. ±15 mm s−1 usually being sufficient for this purpose.
The centre of gravity method is then based on the principle of modelling a given spectrum as the sum of component subspectra, wherein the isomer shift of every subspectrum is invariant with respect to changes in the quadrupole shift and/or hyperfine field used to define that subspectrum. According to the method, the fitted spectrum Stotal is constructed from a superposition of subspectra, Si:
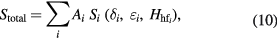
where each Si subspectrum is either: (i) a singlet, with isomer shift  and area
and area  ; or (ii) a doublet, with isomer shift
; or (ii) a doublet, with isomer shift  and quadrupole splitting 2
and quadrupole splitting 2 , wherein both absorption lines are constrained to have the same area,
, wherein both absorption lines are constrained to have the same area,  ; or (iii) a sextet with isomer shift
; or (iii) a sextet with isomer shift  , quadrupole shift
, quadrupole shift  , and hyperfine field
, and hyperfine field  , wherein the area ratios of the outermost: middle: innermost pairs of lines are constrained to be 3 : 2 : 1, so that in order from most-negative to most-positive velocity the areas of the six lines are
, wherein the area ratios of the outermost: middle: innermost pairs of lines are constrained to be 3 : 2 : 1, so that in order from most-negative to most-positive velocity the areas of the six lines are  respectively. The centre of gravity is then:
respectively. The centre of gravity is then:
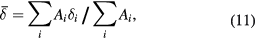
where  is the area of the ith subspectrum, be that a singlet, doublet, or sextet.
is the area of the ith subspectrum, be that a singlet, doublet, or sextet.
The need to constrain the relative areas of the doublet and sextet subcomponent lines is a rather subtle point, which is perhaps best illustrated by an example. Consider a spectrum with an absorption line of area 1 at 0 mm s−1 and a second absorption line of area 2 at 0.6 mm s−1. If we allow the line areas of a doublet fit to vary, we would obtain an asymmetric doublet fit with quadrupole splitting 2 = 0.6 mm s−1 and isomer shift
= 0.6 mm s−1 and isomer shift  = 0.3 mm s−1—which is clearly not the centre of gravity of the spectrum. If instead we require that any doublet subspectra must have equal area lines, we are led to model the spectrum as the superposition of a doublet of area A1 = 2 with
= 0.3 mm s−1—which is clearly not the centre of gravity of the spectrum. If instead we require that any doublet subspectra must have equal area lines, we are led to model the spectrum as the superposition of a doublet of area A1 = 2 with  = 0.3 mm s−1 and 2
= 0.3 mm s−1 and 2 = 0.6 mm s−1 and a singlet of area A2 = 1 with
= 0.6 mm s−1 and a singlet of area A2 = 1 with  = 0.6 mm s−1. The mean isomer shift for the spectrum is then
= 0.6 mm s−1. The mean isomer shift for the spectrum is then  = 0.4 mm s−1, which is its centre of gravity.
= 0.4 mm s−1, which is its centre of gravity.
In the case of a sextet,  is independent of both
is independent of both  and
and  so long as the relative areas of the outermost: middle: innermost pairs of lines are constrained to be 3 : 2 : 1. An intuitive way to see this by considering the effect of the quadrupole shift on the line positions in a hyperfine sextet, as illustrated schematically in figure 3. Alternatively, one can consider the counter-example in which the pairwise relative areas in the component sextets are different, e.g. 3 : x : y. A quadrupole shift
so long as the relative areas of the outermost: middle: innermost pairs of lines are constrained to be 3 : 2 : 1. An intuitive way to see this by considering the effect of the quadrupole shift on the line positions in a hyperfine sextet, as illustrated schematically in figure 3. Alternatively, one can consider the counter-example in which the pairwise relative areas in the component sextets are different, e.g. 3 : x : y. A quadrupole shift  would shift the outermost pair of lines by +
would shift the outermost pair of lines by + towards a more positive velocity, whereas the other four lines would be shifted by −
towards a more positive velocity, whereas the other four lines would be shifted by − towards a more negative velocity. As such, the isomer shift of the sextet as a whole would no longer be
towards a more negative velocity. As such, the isomer shift of the sextet as a whole would no longer be  , but will be
, but will be  , and the centre of gravity methodology would have fallen down. (See supplementary information S2(a) for more details.) To avoid this we require that x + y = 3, in which case
, and the centre of gravity methodology would have fallen down. (See supplementary information S2(a) for more details.) To avoid this we require that x + y = 3, in which case  is invariant, irrespective of the value of
is invariant, irrespective of the value of  . For additional simplicity and ease of implementation, we recommend that the 3 : 2 : 1 relative area constraint is applied, for which equation (11) holds and remains valid.
. For additional simplicity and ease of implementation, we recommend that the 3 : 2 : 1 relative area constraint is applied, for which equation (11) holds and remains valid.

Figure 3. Schematic of the 'centre of gravity' method for analysing magnetically split 57Fe Mössbauer spectra: (a), (b) illustrating that provided the area ratios of the outermost: middle: innermost pairs of lines in a component sextet are 3 : 2 : 1, the isomer shift δi is unaffected by changes in either (a) the quadrupole shift εi, or (b) the hyperfine field Hhfi; (c) noting the optional use of Voigtian line profiles, alongside or in preference to the more standard Lorentzian line profiles; and (d) illustrating that the fitted spectrum is a superposition of subcomponent spectra—in this case of subcomponent sextets, but more generally also with singlet or doublet subcomponents.
Download figure:
Standard image High-resolution imageThe invariance of δi to changes in εi and Hhfi means that a truly model-independent approach can be taken to finding the centre of gravity of the spectrum, where one simply adds as many subspectra Si as is necessary to obtain an acceptable fit. This is a simple and direct protocol, albeit that some care needs to be taken to avoid adding superfluous components [36]. In practice, it is rarely necessary to add more than three or four subspectra to achieve a good quality fit. That said, it is sometimes beneficial to employ Voigtian line profiles rather than the more standard Lorentzian line profiles, as Voigtians are themselves a convenient superposition of Lorentzian lines [42]. (See supplementary information S2(b) for more details.) Originally used to model the effect of a distribution of hyperfine fields, Voigt profiles are a convolution of a Lorentzian profile with a Gaussian distribution—see figure 3—but in the centre of gravity method they are a simple way to allow greater flexibility in the model-independent fitting procedure.
It should also be noted that the centre of gravity is temperature dependent:  . This is because
. This is because  embodies the combined effects of the local s electron charge density at the nucleus and the mean square of the atom velocity due to lattice vibrations—respectively the isomer shift and the second order Doppler shift—both of which are temperature dependent [40]. Although at first sight this is a complicating factor, in practice the
embodies the combined effects of the local s electron charge density at the nucleus and the mean square of the atom velocity due to lattice vibrations—respectively the isomer shift and the second order Doppler shift—both of which are temperature dependent [40]. Although at first sight this is a complicating factor, in practice the  variations are predictable (the isomer shift temperature dependence is usually small, and the second order Doppler shift follows a Debye law), and there may be benefits to changing the temperature at which the spectrum is recorded—for instance, making it possible to select a temperature at which a spectrum is well resolved (e.g. with sharper absorption lines) so that the centre of gravity may be more readily measured [36].
variations are predictable (the isomer shift temperature dependence is usually small, and the second order Doppler shift follows a Debye law), and there may be benefits to changing the temperature at which the spectrum is recorded—for instance, making it possible to select a temperature at which a spectrum is well resolved (e.g. with sharper absorption lines) so that the centre of gravity may be more readily measured [36].
4. Experimental methods
4.1. Standard samples
Magnetite and maghemite standard samples, denoted DTU100 and DTU0 respectively, were supplied by the Department of Physics at the Technical University of Denmark. Although these were of unknown provenance, they were known to be decades old, and were said to be 'pure' samples of magnetite and maghemite. Both were free flowing powders, with the magnetite being black in appearance, while the maghemite was a dark brown colour (see supplementary information S3(a)). Both materials were available in sufficient quantity to allow a series of standard magnetite/maghemite mixtures to be prepared.
4.2. Chemical analysis
The ratio of the ferrous iron content to the total iron content was determined for the DTU100 and DTU0 samples by the redox titration of potassium dichromate against Fe2+, using sodium diphenylamine-4-sulfonate as the indicator [25, 34]. To determine the Fe2+ content, the powder sample was dissolved in concentrated hydrochloric acid in a capped Erlenmeyer flask, a process intended to maintain the Fe2+ to Fe3+ ratio. Prior to titration H3PO4 was added to decolourise the solution by the formation of colourless Fe3+ complexes, and then the indicator was added. During both dissolution and titration, argon gas was flushed over the solution to avoid atmospheric oxidation of Fe2+ to Fe3+. Total iron content was determined using the same titration method, but with samples in which all of the Fe3+ ions were first reduced to Fe2+ ions by treatment with tin(II)chloride in hot hydrochloric acid, with excess Sn2+ removed prior to titration by reaction with mercury(II)chloride to produce insoluble mercury(I)chloride.
4.3. X-ray diffraction
Powder x-ray diffraction was performed at room temperature with a D8 Advance Bruker diffractometer, with Bragg–Brentano geometry and using Cu Kα radiation, to confirm the crystal structure and chemical composition of the two standard samples. The XRD patterns were analysed by the Rietveld method using the FullProf software suite [43]. The samples were mounted on a Si single-crystal low background sample holder, which was rotated during data collection to reduce possible preferred orientation effects in the samples.
4.4. SQUID magnetometry
Magnetometry measurements were performed using a SQUID-VSM instrument (Quantum Design, USA). Powder samples of ca. 5–15 mg in mass were mounted in a polycarbonate holder and mounted on a brass sample rod. The masses were measured to ±0.001 mg accuracy using a Sartorius Cubis microbalance (Sartorius AG, Germany). Prior to measurement, the samples were demagnetised at 300 K using alternating field steps from H = ±5.6 MA m−1 (µ0H = ±7.0 T) to 0.0 A m−1.
Magnetisation M versus field H measurements were recorded step-wise at room temperature up to H = ±5.6 MA m−1. The magnetic moment was measured five times at each field using automatic sample tracking, automatic lock-in amplifier gain and a VSM oscillation amplitude of 5 mm. Zero field cooled (ZFC) and field cooled (FC) magnetisation curves were measured between 5 K and 300 K using an applied field of H = 8 kA m−1 and a sweep rate of 5 K min−1 with continuous measurement of magnetic moment.
4.5. Thermogravitometry
The thermomagnetic properties of the pure magnetite and maghemite samples were monitored using a method based on thermogravimetric analysis (TGA). The technique, known as 'magneto-TGA', involves using an externally applied inhomogeneous magnetic field in an otherwise standard TGA measurement to provide an additional downwards magnetic force on a sample that then allows for the Curie temperature (TC) of the sample to be established [44]. If there are changes in the magnetic state of the sample during heating there will be a change in the force exerted on it and thus a change in the apparent mass, in addition to any changes in the physical mass of the sample.
TGA and magneto-TGA data were collected using a Discovery TGA 7 instrument (TA Instruments, USA) at a heating rate of 20 °C min−1. Samples comprised ca. 3–4 mg of powder placed in open platinum crucibles in an atmosphere of flowing nitrogen (to minimise oxidation), and the mass (or apparent mass) m was monitored as a function of temperature.
4.6. Mössbauer spectroscopy
Mössbauer spectroscopy was carried out independently, at the Technical University of Denmark (DTU) and at University College London (UCL), on identical magnetite/maghemite mixture samples. In both laboratories measurements were performed using conventional Mössbauer spectrometers, either built in-house (DTU), or manufactured by Wissel GmbH, Germany (DTU), or by SeeCo Inc., USA (UCL). In all cases the spectrometers operated in the constant acceleration mode, in transmission geometry, with a 57Co in Rh foil as the source of the 14.4 keV γ-rays. Velocity (and therefore Doppler-shifted γ-ray energy) calibration was performed by recording a reference spectrum from a 10 µm thick foil of α-Fe at room temperature8. At UCL the spectra were then folded and baseline corrected using cubic spline parameters derived from fitting the α-Fe calibration spectrum, following a protocol implemented in the Recoil analysis program [45]. At DTU the spectra were folded and calibrated using a previously described in-house protocol [36]. Low temperature spectra were recorded at DTU using either a bespoke liquid nitrogen cryostat to reach from room temperature to 78 K, or a closed cycle helium refrigerator (APD Cryogenics Inc., USA) to cover the range from room temperature to 17 K.
At DTU the spectra were fitted using an in-house analysis program in MATLAB (MathWorks Inc., USA) that was designed to automatically assign and fit three or four sextets to the data. Both Lorentzian and Voigtian line shapes were used, the latter comprising up to two Gaussian distributions of Lorentzian lines for each component to describe the distribution in hyperfine fields. Components with a total area below 1% or a maximum intensity below 0.5% of the baseline transmission count were automatically removed from the model, as were components with unphysically high centre shifts (viz. >1 mm s−1) or unfeasibly broad lines (viz. full width at half maximum >10 mm s−1). At UCL the fittings were more conventionally user-controlled, using the Windows-based Recoil curve-fitting program [45], albeit applying the same constraints with regard to the 3 : 2 : 1 area ratios of the subcomponent sextets. At both DTU and UCL the uncertainty of the centre of gravity was calculated by incorporating the uncertainty in the calibration of the Mössbauer instrument and the uncertainty of the fitted parameters [36]. It is reported as one standard deviation, corresponding to the 66% confidence interval.
5. Results and discussion
5.1. Verification of the quality and purity of the standard samples
Experiments were performed to test the quality and purity of the DTU100 and DTU0 powder samples. In the first of these, redox titration was used to measure the Fe2+ and total Fe contents of both samples. This chemical analysis was repeated five times for the DTU100 sample, yielding Fe2+/Fetotal values with a mean and 95% confidence interval of 0.30 ± 0.10. The accuracy of this result must be regarded as poor, and is in our opinion a reflection of the inherent difficulty in applying the redox titration method to an inherently highly oxidative material such as magnetite. Nevertheless, we note that the value obtained is consistent with the 0.33 value of pure magnetite. The analysis was performed four times on the DTU0 sample. One aliquot was very difficult to dissolve, taking more than a day to do so, and gave an Fe2+/Fetotal value of ca. 0.12. However, the other three results were more uniform at 0.03 ± 0.03, implying that the first result was anomalous, and that the DTU0 sample was consistent with being pure maghemite with zero Fe2+ content.
Figure 4 shows the XRD patterns for both at room temperature, with the corresponding set of refined parameters listed in table 1. Initial refinements including variable site occupancies were determined to be unreliable given the possible confounding effects of texture in the samples, so that phase-specific model site occupancies were used thereafter. Rietveld refinement of the DTU100 sample was consistent with the cubic Fd-3m crystal structure for magnetite [46], with no other phases being evident within the estimated ±2 vol.% uncertainty of the measurement9. In contrast the refinement of the DTU0 sample was not possible using the Fd-3m structure—as seen for example in the presence of extra reflection peaks near 2θ = 25°—and instead the pattern was consistent with the cubic P4132 space group. This space group has previously been used to explain the crystallographic structure of maghemite [30]. Analysis of the line profiles provided estimates of the crystallite sizes, viz. greater than or equal to ca. 150 nm in DTU100, and of order ca. 34 nm in DTU0.

Figure 4. Rietveld refinements of the x-ray diffraction pattern of (a) the DTU100 magnetite and (b) the DTU0 maghemite powder samples at room temperature. The calculated profile, as well as the residual, is represented with solid lines. The vertical tick marks indicate the positions of the allowed diffraction peaks: for the cubic Fd-3m phase in the case of magnetite, and for the cubic P4132 phase in the case of maghemite. The insets highlight some of the peaks present in the maghemite data that cannot be indexed with the cubic Fd-3m phase.
Download figure:
Standard image High-resolution imageTable 1. Crystal structure data for magnetite [46] and maghemite [30] (top panel) and results from Rietveld analysis of the room temperature diffraction patterns in figure 5 using those structures (bottom panel), from which it is concluded that the DTU100 and DTU0 samples are consistent with 98+ vol.% pure magnetite and maghemite respectively. Note that although the lattice parameter a of the two materials are similar, they are distinguishable. Crystallite sizes D and strain ε (measured in parts-per-ten-thousand) were estimated from the broadening line profile using a Thompson-Cox-Hastings function to assure a good peak profile description in the FullProf refinements. Also listed are the Bragg R-factor and χ2 figures of merit for the goodness of the refinement.
| Atom | Wyckoff position | x | y | z | Occupancy |
|---|---|---|---|---|---|
| Magnetite—sample DTU100—space group Fd-3m in the origin choice 2 | |||||
| O | 32 e | 1/4 | 1/4 | 1/4 | 1 |
| Fe octahedral | 16 d | 1/2 | 1/2 | 1/2 | 1 |
| Fe tetrahedral | 8 a | 1/8 | 1/8 | 1/8 | 1 |
| Maghemite—sample DTU0—space group P4132 | |||||
| O1 | 24 e | 1/8 | 1/8 | 5/8 | 1 |
| O2 | 8 c | 5/8 | 5/8 | 5/8 | 1 |
| Fe octahedral 1 | 4 b | 7/8 | 7/8 | 7/8 | 1/3 |
| Fe octahedral 2 | 12 d | 1/8 | 7/8 | 1/8 | 1 |
| Fe tetrahedral | 8 c | 1/2 | 1/2 | 1/2 | 1 |
| Variable | Rietveld DTU100 | Rietveld DTU0 |
|---|---|---|
| a (Å) | 8.3952 (1) | 8.3464 (2) |
| ρ (g/cm3) | 5.198 | 4.864 |
| D (nm) | >150 | 34 (1) |
| ε (0/000) | <5 | 15 (1) |
| Bragg R-factor (%) | 11.9 | 7.73 |
| χ2 | 3.64 | 1.16 |
Room temperature magnetisation curves were recorded for both samples (figure 5, top panel), with saturation values at H = ±4.0 MA m−1 of Ms = 93.1 ± 0.1 Am2 kg−1 for DTU100 and 75.9 ± 0.1 Am2 kg−1 for DTU0. These compare reasonably well with textbook 'rule of thumb' values of ca. 92 Am2 kg−1 for magnetite and ca. 76 Am2 kg−1 for maghemite [2]. Both samples exhibited hysteresis, with the coercivity in DTU100 magnetite (Hc = 2.5 ± 0.2 kA m−1) being significantly less than that in DTU0 maghemite (Hc = 29.2 ± 0.2 kA m−1), which is likely to be an indication of a multidomain (as opposed to single domain) magnetic structure in the larger DTU100 particles. In contrast, the 'knee' of the magnetisation curve, i.e. the field at which M(H) exhibits a maximum in the curvature function10:
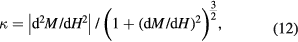
was significantly higher in DTU100 (ca. 230 kA m−1) than in DTU0 (ca. 120 kA m−1). This indicates that the DTU100 magnetite has a higher magnetic anisotropy than does the DTU0 maghemite, which is not surprising given that the magnetocrystalline anisotropy of magnetite, at ca. 13 kJ m−3, is significantly higher than that of maghemite, at ca. 5 kJ m−3 [48].

Figure 5. Magnetic and magneto-thermogravimetric data for the DTU100 magnetite and DTU0 maghemite samples. Top panel: room temperature magnetisation curves, with values indicated for the saturation magnetisation Ms and maximum curvature field Hknee. Middle panel: temperature dependent zero field cooled and field cooled magnetisation curves, showing the distinctive Verwey transition in the DTU100 magnetite sample. Bottom panel: thermogravimetric data showing an apparent increase of mass as the samples are subjected to an externally applied magnetic field and field gradient, Hext, followed by an apparent loss in mass on heating beyond the respective Curie temperatures, TC.
Download figure:
Standard image High-resolution imageZero field cooled and field cooled magnetisation curves were recorded for both samples (figure 5, middle panel). A sharp drop was observed in the ZFC curve for the DTU100 magnetite at ca. 107 K, with a corresponding peak in the FC curve at the same temperature. No equivalent feature was observed in the DTU0 maghemite sample. A sharp fall in the M(T) curve is a classic indicator of the Verwey transition in magnetite, albeit in pure magnetites the Verwey temperature TV generally lies in the range from 113 K to 125 K [49], implying that it is most likely that the DTU100 sample is not strictly 100% pure. Indeed, according to the trends identified by Shepherd et al [50], a Verwey temperature of 107 K corresponds to α ≈ 0.97 rather than 1.00 (see supplementary information S3(b) for details).
Magneto-TGA data (figure 5, bottom panel) provide clear indications of the Curie temperatures for both samples, viz. TC ≈ 840 K in DTU100 magnetite and TC ≈ 905 K in DTU0 maghemite, although inspection of the derivatives of the mass change with temperature curves in the vicinity of TC shows that the magnetite transition is significantly sharper than the maghemite one. These figures compare favourably with literature reports of TC ≈ 850 K in magnetite and TC ≈ 820–986 K in maghemite [1]. Smaller features in the DTU100 data, viz. the 'hump' at ca. 650 K and the apparent increase in mass above TC, were found to be repeatable. We speculate that these features may be associated with an impurity and/or an underlying oxidative transition from Fe3O4 to the heavier α-Fe2O3 or γ-Fe2O3 phases, driven by the presence of trace levels of oxygen in the flowing nitrogen gas in the TGA chamber. However, we considered it to be beyond the scope of the current work to pursue this further.
Lastly, the room temperature 57Fe Mössbauer data for the two samples (figure 2, top and bottom traces) are informative with regard to quality and purity, as Mössbauer spectroscopy is an efficient method for identifying impurities or mixed phases. The DTU100 spectrum shows the characteristic two-sextet pattern of magnetite, with one sextet from the Fe3+ ions at tetrahedral sites (Bhf ≈ 48.9 T, δ ≈ 0.27 mm s−1, ε ≈ 0.00 mm s−1) and a second, more intense sextet representing the 'Fe2.5+' ions at octahedral sites (Bhf ≈ 45.8 T, δ ≈ 0.67 mm s−1, ε ≈ 0.00 mm s−1). The area ratio r of the two sextets is ca. 1:1.8 which is slightly lower than the value expected for pure magnetite. Following the method described in da Costa et al [25], this area ratio corresponds to that of a slightly oxidised magnetite with α ≈ 0.97 (see supplementary information S3(c) for details), which is in good agreement with the value estimated from the Verwey transition temperature. The DTU0 spectrum shows a sextet with hyperfine parameters typical of maghemite (Bhf ≈ 49.6 T, δ ≈ 0.32 mm s−1, ε ≈ 0.01 mm s−1), and also features the subtle but characteristic sextet asymmetry known to maghemite spectra (most clearly seen from line 6 being broader and less intense than line 1) due to small variations in the hyperfine parameters for Fe3+ at the different sites in maghemite [25]. (For further information on both fits, see supplementary information S5.) None of the spectra in figure 2 show any absorption lines not attributed to magnetite or maghemite.
Taken together, these data indicate that there are no significant impurities or extraneous phases in either sample, within the limits of detection of the measurement methods employed. That said, for DTU100 both the Verwey temperature TV and the Mössbauer area ratio r indicate that it is a slightly oxidised magnetite; we estimate its α value to be 0.97 ± 0.02. For DTU0, no features were found in either the Mössbauer or XRD data that did not correspond to those expected for maghemite; we therefore estimate its α value to be 0.00 ± 0.01.
5.2. Room temperature Mössbauer spectroscopy of magnetite/maghemite mixtures
Although in principle it is to be expected that the Mössbauer spectrum of a mixture of two substances will be a simple resolvable superposition of the two component subspectra, in practice the overlapping of absorption lines makes the analysis nontrivial, and the results somewhat unpredictable. For this reason we undertook a comparative analysis of a series of mixtures formed from the pure magnetite and maghemite powders. Room temperature Mössbauer spectra were recorded on the DTU100 magnetite and DTU0 maghemite standards, and on nine intermediate magnetite/maghemite mixtures, at two independent laboratories (DTU and UCL)—meaning that each sample was measured twice. Representative spectra from this series are shown in figure 2, along with best fits obtained using the model independent curve fitting method described in section 3. As each of the mixtures was prepared with a known mass of the two powders, the α parameter as defined in equation (1) was an a priori known quantity for each of the mixtures (see supplementary information S6). Every spectrum obtained was independently analysed, at both DTU and UCL, to determine the centre of gravity parameter,  . (Representative details of these fits are given in supplementary information S7.)
. (Representative details of these fits are given in supplementary information S7.)
The results of all four sets of analysis are plotted in figure 6, where it is evident that a well-defined linear correlation exists between the measured  values and the known α values. There is a small but significant scatter in the datapoints, which is indicative of the absolute experimental error that may be attributed to the measurements due to both the process of data collection and manipulation (including calibration and folding), and the computational analysis procedure applied (including the choice of fitting programme). The linear correlation was fitted as:
values and the known α values. There is a small but significant scatter in the datapoints, which is indicative of the absolute experimental error that may be attributed to the measurements due to both the process of data collection and manipulation (including calibration and folding), and the computational analysis procedure applied (including the choice of fitting programme). The linear correlation was fitted as:

with slope m = 0.2135 ± 0.0076 mm s−1, and intercept  = 0.3206 ± 0.0022 mm s−1, where the calculated uncertainties correspond to the 66% confidence limit of the linear regression taking into account the uncertainties in the α values of the DTU100 and DTU0 samples (see supplementary information S8 for details). The endpoint values of
= 0.3206 ± 0.0022 mm s−1, where the calculated uncertainties correspond to the 66% confidence limit of the linear regression taking into account the uncertainties in the α values of the DTU100 and DTU0 samples (see supplementary information S8 for details). The endpoint values of  (α = 0) = 0.321 ± 0.002 mm s−1 and
(α = 0) = 0.321 ± 0.002 mm s−1 and  (α = 1) = 0.534 ± 0.005 mm s−1 are both in agreement with the literature values, 0.32 mm s−1 and 0.53 mm s−1, of the room temperature isomer shift of pure maghemite and pure magnetite respectively [29].
(α = 1) = 0.534 ± 0.005 mm s−1 are both in agreement with the literature values, 0.32 mm s−1 and 0.53 mm s−1, of the room temperature isomer shift of pure maghemite and pure magnetite respectively [29].

Figure 6. Correlation between the centre of gravity (a.k.a. the area weighted mean isomer shift,  ) of the room temperature (T = 295 ± 5 K) 57Fe Mössbauer spectra of mixtures of pure magnetite, Fe3O4 (α = 1.0) and pure maghemite, γ-Fe2O3 (α = 0.0), and the composition of the mixture as given by the α parameter defined in equation (1). To estimate the experimental errors associated with environment and practice, identical samples were measured and analysed independently in two laboratories, DTU and UCL. The displayed equation is the best linear fit to the accumulated data. Τhe 95% confidence limit of the result is indicated by the grey shaded region either side of the fitted line.
) of the room temperature (T = 295 ± 5 K) 57Fe Mössbauer spectra of mixtures of pure magnetite, Fe3O4 (α = 1.0) and pure maghemite, γ-Fe2O3 (α = 0.0), and the composition of the mixture as given by the α parameter defined in equation (1). To estimate the experimental errors associated with environment and practice, identical samples were measured and analysed independently in two laboratories, DTU and UCL. The displayed equation is the best linear fit to the accumulated data. Τhe 95% confidence limit of the result is indicated by the grey shaded region either side of the fitted line.
Download figure:
Standard image High-resolution imageEquation (13) may be rearranged to yield an expression for α as a function of the measured  :
:

and also a useful approximate expression for the uncertainly dα associated with the derived α as a function of the uncertainty  in the measured
in the measured  :
:
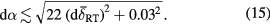
The derivation of the expression for dα is based on consideration of the covariance matrix of the uncertainty, details of which are provided in supplementary information S9.
5.3. Variable temperature Mössbauer spectroscopy of magnetite and maghemite
Variable temperature Mössbauer spectra were recorded for the DTU100 magnetite and DTU0 maghemite standard samples. Fits with the centre of gravity method yielded the  values shown in figure 7. As an approximation to the theoretically predicted Debye law dependence, on inspection it was found that the
values shown in figure 7. As an approximation to the theoretically predicted Debye law dependence, on inspection it was found that the  values for the magnetite and the maghemite could be phenomenologically described by a simple parabolic model:
values for the magnetite and the maghemite could be phenomenologically described by a simple parabolic model:
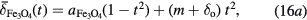
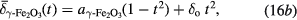
where t = T/To is the reduced temperature, with To = 295 K;  = 0.647 ± 0.002 mm s−1 and
= 0.647 ± 0.002 mm s−1 and  = 0.435 ± 0.002 mm s−1 are the extrapolated values, at t = 0, of the centres of gravity of the Mössbauer spectra of magnetite and maghemite respectively; and m and
= 0.435 ± 0.002 mm s−1 are the extrapolated values, at t = 0, of the centres of gravity of the Mössbauer spectra of magnetite and maghemite respectively; and m and  are the slope and offset of the room temperature correlation defined in equation (13). Note here that equation (16a) describes the temperature dependence for 100% pure magnetite (α = 1.00), which was estimated from the measured temperature dependence of the DTU100 sample (α = 0.97), as described in the supplementary information S9(a). These fitted parabolic curves are shown as the solid lines in figure 7.
are the slope and offset of the room temperature correlation defined in equation (13). Note here that equation (16a) describes the temperature dependence for 100% pure magnetite (α = 1.00), which was estimated from the measured temperature dependence of the DTU100 sample (α = 0.97), as described in the supplementary information S9(a). These fitted parabolic curves are shown as the solid lines in figure 7.

Figure 7. Variation with temperature of the centre of gravity  of the 57Fe Mössbauer spectra of almost-pure magnetite (DTU100, α = 0.97 ± 0.02) and pure maghemite (DTU0, α = 0.00 ± 0.01), and of a commercial magnetite/maghemite nanoparticle system with an intermediate α value of 0.61 ± 0.02. Solid lines are fits using a phenomenological parabolic model to approximate the Debye law behaviour. The grey shaded regions either side of the fitted lines represent the 95% confidence level of the fits. The dashed line is a predicted curve based on the derived relationship between
of the 57Fe Mössbauer spectra of almost-pure magnetite (DTU100, α = 0.97 ± 0.02) and pure maghemite (DTU0, α = 0.00 ± 0.01), and of a commercial magnetite/maghemite nanoparticle system with an intermediate α value of 0.61 ± 0.02. Solid lines are fits using a phenomenological parabolic model to approximate the Debye law behaviour. The grey shaded regions either side of the fitted lines represent the 95% confidence level of the fits. The dashed line is a predicted curve based on the derived relationship between  and α, as described in equation (17).
and α, as described in equation (17).
Download figure:
Standard image High-resolution imageAn advantage of applying a simple parabolic model in this way is that it is then straightforward to rearrange the equations (16a) and (16b) to be able to determine  from any measured
from any measured  , using:
, using:
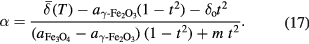
It is also possible to estimate the upper limit to the uncertainly dα associated with the derived α as a function of the uncertainty  in the measured
in the measured  :
:
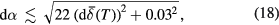
which is an expression that holds so long as the uncertainty in the measurement temperature is  3 K. Further details of this derivation are provided in supplementary information S9(b), and a MATLAB function for the calculation of equation (17) and
3 K. Further details of this derivation are provided in supplementary information S9(b), and a MATLAB function for the calculation of equation (17) and  valid for all
valid for all  is provided in the supplementary materials.
is provided in the supplementary materials.
The formulation was tested by recording a temperature-dependent series of Mössbauer spectra on a commercially produced magnetite/maghemite nanoparticle material, 'MM03', supplied by Micromod GmbH, Rostock, Germany11. The spectra were fitted to extract the  values that are plotted as the open circles in figure 7. (Further details are given in supplementary information S10.) The dashed grey line in figure 7 shows the predicted
values that are plotted as the open circles in figure 7. (Further details are given in supplementary information S10.) The dashed grey line in figure 7 shows the predicted  curve derived from equation (17) with an α value of 0.61 ± 0.02. It is clear on inspection that the observed
curve derived from equation (17) with an α value of 0.61 ± 0.02. It is clear on inspection that the observed  values fall close to the predicted line, providing evidence that the model is indeed valid.
values fall close to the predicted line, providing evidence that the model is indeed valid.
5.4. Comparison with literature data on mixtures and solid solutions
One of the underlying principles of the centre of gravity method is that the 57Fe Mössbauer spectrum of a given sample will yield a valid estimate of the α parameter—and hence also the magnetite/maghemite composition or magnetite-maghemite stoichiometry—irrespective of whether the sample is a mixture or a solid solution. In the current work we have tested the method on a mixture of standard magnetite and maghemite powders, leading to the correlation shown in figure 6. It is possible, however, to apply the method to previously published data on both mixtures and solid solutions, as illustrated in figure 8, in cases where the authors have obtained independent confirmation of the composition or stoichiometry by chemical analysis.

Figure 8. Comparison of the centre of gravity method correlation between  or
or  and α with literature data on magnetite/maghemite mixtures from Santoyo Salazar et al [39] and da Costa et al [25], and with literature data on magnetite-maghemite solid solutions from Yang et al [51] and Gorski et al [33]. Note that the Gorski data in panel (d) was measured at 140 K, and that the solid line
and α with literature data on magnetite/maghemite mixtures from Santoyo Salazar et al [39] and da Costa et al [25], and with literature data on magnetite-maghemite solid solutions from Yang et al [51] and Gorski et al [33]. Note that the Gorski data in panel (d) was measured at 140 K, and that the solid line  has been derived from equation (17). Panels (e) and (f) show the results of re-fitting the original data using the model independent analysis described in the text.
has been derived from equation (17). Panels (e) and (f) show the results of re-fitting the original data using the model independent analysis described in the text.
Download figure:
Standard image High-resolution imageFour datasets are displayed in figure 8, viz.: a series of 10–40 nm core-shell magnetite/maghemite nanoparticles [39]; a series of 10–30 nm magnetite/maghemite nanoparticles, in which the individual nanoparticles were either magnetite or maghemite [25]; a series of 150–200 nm stoichiometric and cation-deficient magnetites prepared by chemical synthesis followed by heat treatment [51]; and a series of ca. 20 nm non-stoichiometric magnetites prepared by H2O2 oxidation of stoichiometric magnetite nanoparticles [33]. It is clear on inspection that all four datasets show linear correlations that are near to, and in some cases overlapping with, the predicted line. This is a strong point in favour of the centre of gravity method, as it has not previously been shown that the correlation holds for both mixtures and solid solutions, and also because the literature data had been obtained using a variety of different model-dependent fitting procedures.
To test whether the fitting procedure was a significant factor in the determinations of the mean isomer shifts in these cases, we applied the model-independent fitting procedure described in section 3 to two sets of raw data kindly provided to us by da Costa et al [25] and by Gorski et al [33]. In both cases the model-independent fits produced largely similar results (see figure 8; further details are given in supplementary information S11), although it is clear that at least some of the model-independent da Costa datapoints lie closer to the predicted line than was the case for the original fit data.
6. Conclusions
The composition or stoichiometry of magnetite and maghemite mixtures or solid solutions is an important parameter in the physical and geological sciences, and materials science. It is also significant in biomedical science, where magnetic nanoparticles are used both in vitro and clinically, and where both ferrous and ferric iron ions are known to play active roles in the production of reactive oxygen species. However, the accurate determination of the composition/stoichiometry can be problematic, as it requires either well-crystallised samples suitable for x-ray diffraction studies, or it relies on destructive testing methods involving chemical dissolution that, depending on the nature of the sample, are often either unfeasible or inappropriate.
In this context the 57Fe Mössbauer spectroscopic centre of gravity method is a useful non-destructive and potentially in situ method whereby—as we have demonstrated in this work—the composition or stoichiometry can be unambiguously determined. The method is relatively new, and has not yet achieved what might be regarded as mainstream recognition. Nevertheless, it is simple and straightforward, and so long as appropriate measures and protocols are observed—such as those set out in table 2—we expect that users will find little difficulty in its implementation.
Table 2. Recommended 57Fe Mössbauer spectroscopic procedure for determining the composition of magnetite/maghemite mixtures, or the stoichiometry of magnetite-maghemite solid solutions.
| 1. | Prepare the sample | Ensure that the sample is not too thick: a rule of thumb is to not exceed ca. 3 mg of magnetite and/or maghemite per absorption line per cm2 of absorber area, or to not exceed ca. 6–8% in spectral absorption in any given line. Avoid preferred orientations or uneven distributions of sample material, e.g. by mixing with an inert and γ-ray-transparent matrix such as finely ground sugar or boron nitride. |
| 2. | Record the spectrum | It is preferable that the spectrum should have well defined, sharp absorption lines. If necessary, and possible, it may be preferable to cool the sample and record data at lower temperatures. Ensure good measurement statistics: in typical systems this corresponds to accumulated γ-ray counts in excess of one million per channel, in spectra where the count for the most intense absorption peak is at least 2–3% lower than the baseline value. |
| 3. | Evaluate the centre of gravity 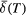 |
Fit the spectrum to estimate its centre of gravity, 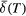 . A model-independent procedure, such as that used in this paper, is recommended. If doublets are used, the lines should be constrained to have equal area; if sextets are used, the area ratios of the outermost: middle: innermost pairs of lines should be 3: 2: 1. Estimate the uncertainty . A model-independent procedure, such as that used in this paper, is recommended. If doublets are used, the lines should be constrained to have equal area; if sextets are used, the area ratios of the outermost: middle: innermost pairs of lines should be 3: 2: 1. Estimate the uncertainty 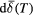 , noting that there may be many sources of error, not just the computational fit uncertainty [36]. , noting that there may be many sources of error, not just the computational fit uncertainty [36]. |
| 4. | Calculate α and dα | If the spectrum was recorded at room temperature (T = 295 ± 5 K), calculate α and its uncertainty dα using equations (14) and (15). If the spectrum was recorded at a lower temperature, then use equations (17) and (18). |
| 5. | Report α, w, or x | If it is known that the sample in question is a magnetite/maghemite mixture, then you may choose to convert the measured α ± dα into a weight percentage w ± dw using equation (9). If it is a magnetite-maghemite solid solution you may prefer to report the stoichiometry x ± dx using equation (5). |
Lastly, although we have focused in this work on the application of the method to materials for which the more conventional techniques are not easily applied—such as in nanoparticles or poorly crystallised particles—it is somewhat salutary to note that the Mössbauer method may find application even in more routine assays. A case in point is shown in figure 9, where the centre of gravity method has been used to assess the purity of purchased samples of magnetite and maghemite powders purchased from two reputable multinational suppliers12. In both cases the supplied maghemite samples were indeed maghemite, but also in both cases, the supplied magnetite samples were not the 95–97% purity that the manufacturers claimed, but much less than that, viz.: having Mössbauer α parameters of ca. 0.58 and 0.63. Assuming that the samples were mixtures of magnetite and maghemite, this would place the weight percentages of magnetite in the supplied samples at w = 57 wt.% and 62 wt.% respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Centre of gravity Mössbauer analysis (at room temperature) of commercially available magnetite and maghemite powders. The Alfa Aesar and Sigma Aldrich magnetite powders were advertised as being 95% and 97% pure (α = 0.95 and 0.97) respectively, but the spectra indicate significantly lower values. In contrast, both maghemite powders are almost 100% pure (α = 0.00).
Download figure:
Standard image High-resolution image{kind=link}
Acknowledgments
We are grateful to Geraldo da Costa (Federal University of Ouro Preto, Brazil) and to Christopher Gorski and Michelle Scherer (University of Iowa, USA) for providing their raw Mössbauer data for analysis using our model-independent protocol. We thank Luis Fernández Barquín (University of Cantabria, Spain) and Simon Hattersley (Bickley, UK) for their helpful advice regarding the magnetic measurements. This work was supported by the European Union Seventh Framework Programme through the NanoMag project 'Nanometrology standardisation methods for magnetic nanoparticles', under grant agreement no. 604448.
Footnotes
- 6
Fundamentally, the equivalence depends on the degree to which the 'recoil-free fraction' or 'f-factor' varies from site to site—see chapter 1.4 in [40]. In magnetite and maghemite this is small enough to be neglected. Experimentally, the equivalence depends on every Fe atom experiencing the same flux of incoming γ-rays. This equates to making a sufficiently thin absorber to avoid 'thickness' effects—a practice that most practitioners are familiar with.
- 7
Note that we use the term 'mixture' to cover a variety of mixed phase systems, such as one comprising separate populations of magnetite and maghemite particles, or alternatively one comprising a single population of two-phase particles with a magnetite core and a maghemite shell.
- 8
Room temperature was measured to be T = 295 ± 3 K at DTU, and T = 295 ± 5 K at UCL.
- 9
XRD uncertainty estimated from both the measurement error, evaluated as the concentration equivalent of 2 times the standard deviation of the observed background level, as described in chapter 4 of [47], and by modelling the influence that other phases would have if present. For example, re the latter, the strongest diffraction peak for a 2 vol.% hematite impurity, if it was present, would appear at 2θ ≈ 33° with a line intensity of ca. 5% of that of the magnetite peak at 2θ ≈ 30°.
- 10
In evaluating κ it is advisable to use the volumetric magnetisation rather than the mass magnetisation so that both M and H are expressed in the same units, and to focus on the anhysteretic 'backbone' of the first quadrant curve. See supplementary information S4 for further details.
- 11
The MM03 sample is derived from Micromod GmbH's BNF-Dextran series (catalogue no. 10-00-701), the only difference being that it has a smaller mean hydrodynamic diameter than the commercially available 80 nm and 100 nm particles.
- 12
Samples purchased (a) from Alfa Aesar, Thermo Fisher Scientific, UK: magnetite, 97% purity, cat. no. 47141, CAS no. 1317-61-9; and maghemite, cat. no. 44896, CAS no. 1309-37-1; and (b) from Sigma-Aldrich Co. Ltd., UK: magnetite, 95% purity, cat. no. 310069, CAS no. 1317-61-9; and maghemite, cat. no. 544884, CAS no. 1309-37-1.