

Abstract
The dynamics of a star-shaped polymer translocation pulled by a single arm through a nanochannel is investigated using three-dimensional Langevin dynamics simulations. The pulling force is applied on the terminal monomer of the leading arm in order to mimic the motion of chains subject to a combination of magnetic and optical tweezers in real experimental setups. The effect of channel dimensions and magnitude of the pulling force as well as the chain size and functionality on the chain's translocation dynamics is extensively examined. The variation of the mean translocation time 〈τ〉 with respect to channel length and diameter exhibits a non-trivial behavior characterized by an abrupt change in the translocation dynamics for chains with higher functionalities f. The dependence of 〈τ〉 upon channel aspect ratio yields also a regime change for the transport dynamics for chains with larger functionalities. Moreover, the average exit time with respect to chains total mass N and to the magnitude of the pulling force F are found to follow scaling laws in agreement with theoretical predictions.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Translocation of biomolecules through a hole of nanometric size has been an active field of research over the past two decades yielding interesting experimental [1–4], numerical [5–10], and theoretical [11–20] results. From a general point of view, the phenomenon of polymer translocation is a key process in which polymeric materials cross from the cis side to the trans side of a membrane through a nanopore. This process which can be forced or unforced exists widely in nature and can be replicated in a laboratory setup. As such, polymer translocation is very pertinent to many crucial biological mechanisms such as DNA and mRNA translocation through nuclear pores, proteins transport across membrane channels, virus injection into cells [21–23] and the likes. Furthermore, it has also significant technological potentials, including gene therapy, rapid DNA sequencing, and controlled drug delivery [24–28]. In particular, technological applications of high significance in biotechnology and medical diagnosis that employ quicker and cheaper methods for DNA sequencing are anticipated [20]. Most experimental researches have put the emphasis on forced polymer translocation. The first experimental successful forced DNA translocation through the α-homolysing channel in a lipid bilayer membrane was conducted by Kasianowicz et al in 1996 [1]. Subsequently, Li et al and Storm et al reported that such molecule translocation experiments can be conducted using a solid-state nanopore [2, 29]. In later years, translocation phenomena were used to characterize single biological entities such as single-stranded and double-stranded DNA, proteins, or even entire cells [30–34].
On the theoretical side, Sung and Park are the first to develop a polymer translocation model based on the Fokker-Planck equation to describe chain transport dynamics [35–38]. In addition, Muthukumar [12, 13], Kong and Muthukumar [19], and Slonkina and Kolomeisky [39, 40] used the nucleation theory to investigate the translocation process while Buyukdagli et al have developed an electrostatic theory of polymer translocation to show that the translocation time can be tuned via the dielectric trapping of the polymer [41]. These theoretical works have shown that chain translocation time depends on various non-trivial factors such as the polymer size, the polymer diffusion coefficient, the magnitude of the driving force, the polymer configurational entropy, the confinement geometry, and the interaction between polymers and pores.
Simulations studies on chain translocation process have been hotly pursued for quite a long time. Numerical investigations on forced and unforced translocations include (linear) polymer translocation subject to an applied external field [7, 42–45], a chemical potential gradient [18, 46], confinement [47], absorption [48], a pulling force, as well as unbiased (free) translocation [7, 44, 49–52]. One of the aspect widely investigated in the abovementioned works is the dependence of translocation time on polymer chain size. In the case of unforced translocation of linear chains, simulation results by Chuang et al [52] showed that the mean translocation time nearly equals the Rouse relaxation time and scales as 〈τ〉 ∼ N1+2ν , where ν = 1/2 for phantom chains, ν = 3/4 for two-dimensional self-avoiding chains, which deviates from the theoretical predictions based on the Fokker-Planck equation or the nucleation theory approaches, and ν = 3/5 for reals chains in three dimensions. Subsequent simulation works by Luo and collaborators corroborated the findings of Chuang et al [7, 44]. Moreover, in connection to forced translocation of linear chains in two dimensions, Luo and co-workers have found a crossover scaling from 〈τ〉 ∼ N2ν for relatively short polymers to 〈τ〉 ∼ N1+ν for longer chains in the presence of an applied external field using Monte Carlo simulations coupled with bond fluctuation model and Langevin dynamics simulations respectively [6, 44]. Huopaiemi et al [49] showed that the mean translocation time of linear chains scales as 〈τ〉 ∼ N2 for a polymer under a strong or moderate driving force regime. Moreover, in this regime, the authors have found that the dependence of the mean translocation time upon the pulling force F exhibits a scaling relation 〈τ〉 ∼ F−1.
The above works have put the emphasis on the translocation of linear chains, a topic reasonably well understood by now. However, little attention was given to the effect of polymer architecture with regards to translocation dynamics until recently. Star polymers represent an important class of branched polymers with a single branching point which are widely utilized across numerous fields, with applications in the pharmaceutical and petrochemical industries [53]. The prospect of employing star polymers in the biomedical field as means for drug and genes deliveries is also widely shared among researchers. When it comes to specific applications, star polymer solutions or melts with a narrow distribution of arm numbers and arm molecular weight of are often preferable as their physical properties can be monitored with more accuracy. In this view, star polymer translocation through a nanopore has been investigated as a possible approach for characterizing and separating such polymers [54–57]. It is noteworthy that the phenomenon of star polymer transport across a nano-scaled pore has attracted increasing attention over the last decade [58–60].
One of the first works that studied the dependence of star polymer translocation time upon chain functionality was performed by Liu et al [60]. They simulated flow-induced star polymer translocation through a nanopore by employing dissipative particle dynamics. They reported that the mean exit time exhibits a power-law dependence upon the total number of monomers while the translocation process is strongly affected by star arm arrangement. In addition, Katkar and Muthukumar investigated the effect of arms number and nanopore dimension upon the mean exit time for the electrophoresis of star polyelectrolytes through a nanopore [58]. They found that, for a fixed value of chain size, the variation of 〈τ〉 with respect to chain functionality f is non-monotonic with a minimum for a particular value of f. The non-monotonic behavior of 〈τ〉 with chain functionality for chains of constant molecular weight is also observed for flow induced translocation, a result reported by Nagarajan and Chen [59]. Moreover, in our recent study on star polymer translocation out of a confining cylindrical cavity subject to a pulling force, we have found that 〈τ〉 exhibits a minimum for a critical functionality which defines a boundary between two translocation regimes [51]. In that study, we have shown that the translocation dynamics is impacted by a change in the dimensions of the confining tube with the presence of a specific tube aspect ratio for which the fastest translocation is recorded.
In the present paper, we intend to investigate the transport dynamics of star polymers across a cylindrical nanochannel subject to a pulling force by carrying out three dimensional Langevin dynamics simulations. We focus our attention on the effect of chain functionality as well as nanochannel dimensions, in terms of channel length and diameter, upon the mean translocation time. In addition, the existence of scaling laws relating the mean translocation time to chain total mass and to the magnitude of the pulling force is also investigated. The novelty of the present work lies on the fact that we investigate the translocation of star polymers through a nanochannel subject to a pulling force. Such investigation has not been undertaken before to the best of our knowledge.
The rest of the paper is organized as follows: in section 2, the simulation model and the method used are described in detail, in section 3, simulations results of star polymer translocation obtained under various conditions are discussed, and finally, key points of this paper are summarized in section 4.
2. Model and method
2.1. Simulation model
In this paper, the translocation of a self-avoiding star polymer through a 3-dimensional nanochannel is performed using a Langevin dynamics simulation approach. The chain enters the nanochannel through a circular nano-scaled pore of diameter D. The chain then propagates across the nanochannel subject to a pulling force applied on the end monomer of the leading arm. The origin of the coordinate system inside the simulation box is set at the center of the pore. The nanochannel has a cylindrical shape with a radius R and length L and parallel to the x axis. The simulation box is divided into a cis side and a trans side by a membrane located at the nanochannel entrance and perpendicular to the x-axis as depicted in figure 1.

Figure 1. Schematic representation of 4 − arms star polymer with identical arm length translocating across a nanochannel subject to a constant pulling force F applied at the end monomer of the leading arm. The wall located at the channel entrance and the nanochannel length L and diameter D are made of immobile beads shown in red.
Download figure:
Standard image High-resolution imageStar polymers considered in the present study belong to a special class of branched polymers with a single branching point called core connected to f-flexible linear arms We have limited our investigation to homogeneous star polymers with equal arm length. Such polymers consist of N identical monomers including the core unit. N represents then the total mass of the polymer and is given by N = fNa + 1, where f is the number of arms or the chain functionality and Na is the number of monomers per arm. In addition, polymers used for the purpose of the present study are electrically neutral. A coarse-grained bead-spring model is used to represent star polymers. According to this model, each monomer is represented by a spherical bead of diameter σ. Figure 1 depicts such chain with beads belonging to arms colored in black while the central core is in green.
2.2. Interactions
As mentioned previously, polymer chains are modeled as bead-spring chains, that is, a collection of particles or beads interacting via the Lennard-Jones (LJ) potential while connected particles interact through the finitely extensible non-linear elastic (FENE) potential
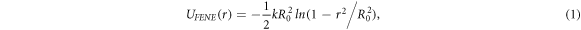
where r is the distance between two connected beads, k is the spring constant, and R0 is the maximum allowed separation between connected beads. The excluded volume interactions of beads is realised by the short range repulsive LJ potential given by
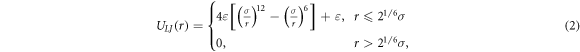
where σ is the diameter of a bead, ε is the depth of the potential, and r is the distance between two beads.
2.3. Pulling force
The pulling force is a contact force acting on the end monomer of the leading arm of the polymer. This driving force is expressed as
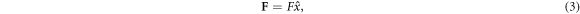
where F is the magnitude of the force. Note that this force acts inside the channel and extends to the trans side along the x axis and is directed towards the trans side of the channel.
2.4. Langevin dynamics simulations
The Langevin dynamics simulation consists in integrating the Langevin equation which includes the abovementioned two potentials (equation (1) and equation (2)) and the pulling force defined in equation (3) as well as other forces. A given bead i is thus subject to conservative, frictional, and random forces respectively, while only the end monomer of the leading arm (i = 1) is in addition subject to the pulling force
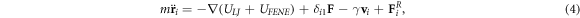
where m is the mass of a bead, γ is the friction coefficient, vi
is the beads velocity and  is the random force satisfying the fluctuation-dissipation theorem [61, 62]. The integration of the Langevin equation is performed by the method proposed by Ermak and Buckholz [63]. The trajectory integration is implemented by using the molecular dynamics package ESPResSo, an extensible simulation package for research on soft matter systems [64].
is the random force satisfying the fluctuation-dissipation theorem [61, 62]. The integration of the Langevin equation is performed by the method proposed by Ermak and Buckholz [63]. The trajectory integration is implemented by using the molecular dynamics package ESPResSo, an extensible simulation package for research on soft matter systems [64].
2.5. Parameters
LJ parameters ε and σ together with the bead mass m are used as units for measuring the energy, length and mass respectively, while a time  and a force ε/σ units in the order of ps and pN respectively are defined by combining these parameters. The bead diameter σ is equal to 1.00 while the potential depth ε is also set to 1.00. A short-range repulsive LJ parameter with a cut-off distance rc
= 21/6
σ = 1.12246σ is used to model the interaction between non-bonded beads as well as the interaction between beads and the channel wall. The interaction between bonded beads is modeled via the FENE spring potential with the values R0 = 2σ and k = 7ε/σ. In addition, κB
T = 1.0ε while the friction coefficient is γ = 1.0. The integration time step used in all simulations is dt = 0.001tLJ
.
and a force ε/σ units in the order of ps and pN respectively are defined by combining these parameters. The bead diameter σ is equal to 1.00 while the potential depth ε is also set to 1.00. A short-range repulsive LJ parameter with a cut-off distance rc
= 21/6
σ = 1.12246σ is used to model the interaction between non-bonded beads as well as the interaction between beads and the channel wall. The interaction between bonded beads is modeled via the FENE spring potential with the values R0 = 2σ and k = 7ε/σ. In addition, κB
T = 1.0ε while the friction coefficient is γ = 1.0. The integration time step used in all simulations is dt = 0.001tLJ
.
2.6. Simulation procedure
The translocation through the nanochannel is conducted by applying a contact force on the end monomer of the leading arm as to mimic the action of optical tweezers in actual experiments [65–68]. We start by placing the chain in such a way that the terminal monomer of the leading arm is located at the entrance of the channel. Next, the chain undergoes thermal relaxation while the end monomer of the leading arm is kept immobilized. The translocation starts at t = 0 by releasing the constrained monomer and by applying a pulling force on it. A successful translocation is recorded when all the monomers cross the channel on the trans side. For each set of parameters, the mean translocation time is computed over 1000 independent runs for which successful translocations are recorded.
3. Results and discussion
3.1. Effect of channel length L on 〈τ〉
Our first investigation focuses on how the nanochannel length affects the mean translocation time for polymer chains of various functionalities but having the same mass N = 121. The channel length L is varied from 2 to 17, while two distinct values of channel diameter and pulling force are considered in this first study. Chains' functionalities are varied from 2 to 12. Figure 2 depicts the average exit time as a function of channel length for a relatively narrow channel with diameter D = 4.6 and, for a moderate F = 10, figure 2(a) and strong F = 100, figure 2(b) pulling forces. In both pulling force regimes, and for star polymers of a given functionality f, the exit time increases almost linearly with the channel length, which is an expected result. A close inspection of figure 2(a) reveals that chains with the largest functionalities, i.e., f ≥ 10, exhibit the largest exit times. Moreover, in the moderate pulling force regime, our simulations show that no successful translocation is recorded for chains with larger functionalities, i.e., f ≥ 8, when the channel length is greater than a given value. Indeed, these chains get indefinitely stuck inside channels whose length is beyond a certain channel length. Interactions opposing chain movement inside the channel are obviously responsible for the stalling of the translocation process. Clearly, the arm-arm and arm-wall interactions which depend upon chains' functionality are the ones hindering chains motion inside the channel. The interplay between these two forces when chain functionality varies can be better understood by considering the mean radius of gyration. The mean radius of gyration of a star polymer when viewed as an ideal chain is given by [69]:
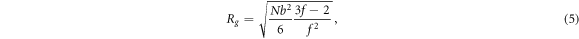
where b is the size of a monomer. Numerical values of the mean radius of gyration of chains under study are presented in table 1 together with the ratio between the channel diameter D and the chain diameter 2Rg /D. According to table 1, the ratio which is close to 2 for the linear chains and decreases and becomes close to 1 for f ≥ 8, implying that the interaction between chain arms and channel wall is significantly reduced when f is increased. One can then safely conclude that the observed translocation slow-down or stalling is not primarily due to wall-chain arms interaction since although chains with small functionality exhibit larger ratios, which translates into greater arm-wall interaction, they translocate relatively easily through the channel. In fact the explanation lies in the change of chain topology resulting from the increase of chain functionality. Indeed, as f increases, the star polymer inside the channel changes its shape from a linear to a quasi-spherical geometry and behaves like a colloidal particle subject to an increased arm-arm interactions. This in turn leads to an increase in arm-wall interactions, greatly hindering chain translocation through the nanochannel. It is then reasonable to assume that while arm-arm interactions have an indirect role, it is the arm-wall interactions that are responsible for the observed stalling of these chains inside the channel. To further understand why chains with higher functionality can't cross the channel, one needs to use arguments based on chain geometry. Indeed, chains should be able to significantly modify their shape in order to cross a narrow channel. But for chains with higher functionality, steric hindrance emanating from arm-arm interaction prevents them to sufficiently modify their shape inside a channel whose diameter is close to or even less than their mean diameter. From table 1, one can note that the geometric size of chains with f > 10 is close to 4 which happens to be the upper limit of channel length that these chains can successful cross. Their translocation is made possible up to that limit because some of their arms remain outside the channel at any time, either on the cis or trans side of the channel, during the translocation process, which in turn helps them maintain a greater entropy (compared to chains fully inside the channel) large enough to cross the entropic barrier constituted by the cavity. However, when the channel length is greater than their mean diameter, these chains would be completely inside the channel at some point during their translocation, which explains why they get stuck.

Figure 2. Plot of 〈τ〉 versus channel length L for D = 4.6, N = 121, and in the case of a moderate pulling force, F = 10 (panel (a)), and strong pulling force, F = 100 (panel (b)). Functionalities are varied from f = 2 to 12.
Download figure:
Standard image High-resolution imageTable 1. Radius of gyration of star polymers of mass of N = 121 with various functionalities. The ratio between chain size, 2Rg, and channel diameter D with D = 4.6 and 6.4 is also given.
| f | Rg | 2Rg /D | 2Rg /D |
|---|---|---|---|
| D = 4.6 | D = 6.4 | ||
| 2 | 4.49 | 1.95 | 1.40 |
| 3 | 3.96 | 1.72 | 1.24 |
| 4 | 3.55 | 1.54 | 1.11 |
| 5 | 3.24 | 1.41 | 1.01 |
| 6 | 2.99 | 1.30 | 0.93 |
| 8 | 2.63 | 1.14 | 0.82 |
| 10 | 2.38 | 1.04 | 0.74 |
| 12 | 2.18 | 0.95 | 0.68 |
Moreover, one can note a non-monotonic relation between 〈τ〉 and f insofar as the minimum exit time is registered for f = 5. This particular value of f constitutes an optimal functionality for which the two forces opposing chain's motion are minimized. Note that the dependence of 〈τ〉 upon f will be further investigated in subsequent sections. As for the case of the large pulling force regime, panel (b) of figure 2, one can note a fairly different picture whereby all chains irrespective of their functionality experience successful translocations as well as a steadily decreasing 〈τ〉 with increasing f. Obviously, for the latter case, the pulling force which has overcome all interactions hindering chain motion inside the channel such as arm-wall, arm-arm and monomer-solvent interactions is the sole dominant factor controlling the translocation dynamics.
Next, we investigate how the translocation dynamics is affected when a larger channel diameter, D = 6.4, is considered using the same set of values for f and F as in the previous study. For the case of a weak driving force, i.e., F = 10, (figure 3(a)) the obtained result is quite different from the previous one with chains with higher functionality now recording successful translocations irrespective of channel length. This is because channel diameter is large enough to accommodate the whole chain without the need for the chain to be subject to a severe shape deformation.

Figure 3. 〈τ〉 as a function of L for different functionalities, varying between 2 and 12, and magnitude of a pulling force: (a) F = 10, (b) F = 100. The values of D and N are 6.4 and 121, respectively.
Download figure:
Standard image High-resolution imageMoreover, the mean translocation time exhibits two distinct regimes: 〈τ〉 increases moderately until a given point inside the channel followed by a more marked increase beyond that point. In addition, the change in the slope of 〈τ〉 is farther inside the channel and becomes more abrupt as chain functionality increases. In the first regime, the translocation time decreases with increasing functionality with the fastest translocation corresponding to the largest functionality, i.e., f = 12. In the second regime, the fastest translocation takes place for f = 6, while the chain with the highest functionality experiences the slowest translocation as it is also the case of the linear chain. This change in slope indicates a slowing down of the translocating chain mobility. The observed deceleration is obviously due to frictional forces emanating from solvent-monomer and chain-wall interactions and whose action become increasingly dominant as the chain advances further inside the channel. The case of strong pulling force depicted in figure 3(b) is quite similar to the one registered for a narrower channel confirming our previous conclusion, that is, the strong pulling force is by far greater than the forces opposing the chain motion and, as such, it solely dominates the translocation dynamics.
3.2. Channel diameter versus mean translocation time
As seen in the previous section, the channel diameter can dramatically alter the translocation dynamics of star polymers. Indeed forces arising from the chain state of confinement impede chain movement inside the channel and may become so dominant that they can even prevent the chain from crossing the channel. Figure 4 depicts the mean translocation time as a function of channel diameter for a channel of length L = 2 and for the case of small (Figure 4(a)) and large (figure 4(b)) driving forces. As a matter of fact, the channel is so short that it can actually be viewed as a nanopore. In the case of small driving force and low functionalities, f ≤ 4, the mean translocation time is almost not affected by the change in diameter of the channel, implying that arm-wall interactions are not significantly affecting the translocation dynamics. However, when f > 4, one can note a dependence of 〈τ〉 on D which becomes more marked for larger functionalities. In particular, for star polymers with f ≥ 8, 〈τ〉 exhibits such an abrupt decrease with increasing D that these polymers undergo a dramatic change in velocity from being the slowest to becoming the fastest. This substantial acceleration is due to the decrease of arm-wall interaction, as the channel diameter becomes large, combined with the reduction of chains radius of gyration which in turn decreases the monomer-solvent interaction. In the case of a large pulling force, figure 4(b), the obtained result is fairly different from the previous one in so far as now the mean translocation time does not exhibit a strong dependence upon D irrespective of polymer functionalities. This confirms our previous conclusion, that is, when the pulling force is sufficiently large, it can overcome all other forces opposed to chain motion inside the channel and, as such, it becomes the only force controlling the translocation dynamics.

Figure 4. Plot of 〈τ〉 versus channel diameter D for N = 121, in the case of a short channel, L = 2, and two distinct pulling forces: (a) F = 10 and (b) F = 100. The functionality is varied from f = 2 to 12.
Download figure:
Standard image High-resolution image3.3. Isovolume
In order to investigate the impact of nanochannel dimensions upon the translocation dynamics, we have computed the mean translocation time by varying the channel aspect ratio a = L/D while keeping the channel volume constant, V = 50. Figure 5 depicts the mean exit time as a function of the aspect ratio for chains of various functionalities, 2 ≤ f ≤ 12, but with the same mass N = 121. A strong pulling force F = 100 is applied while the values of channel diameter decreases from D = 7.96 to 2.82 as the channel length is increased from L = 1 to 8, leading to a variation of the aspect ratio from a = 0.13 and 2.84. For small values of the functionality, i.e. 2 ≤ f ≤ 4, the mean translocation time is almost constant over the range of aspect ratio values into consideration. One can therefore infer that for such values of f, the translocation dynamics does not depend upon the aspect ratio, in other words, on the channel shape, but rather on the magnitude of the pulling force. On the other hand, for functionalities greater or equal than 5, the mean exit time increases with increasing aspect ratio. Moreover, a close inspection of the curves reveals the existence of a translocation regime change for f > 5 marked by a slight increase of 〈τ〉 around a particular value of a. This regime change occurs around the same value of the aspect ratio equal to ac = 1.4 for all functionalities. In addition, the regime change is more pronounced for chains with higher functionalities since they are the ones experiencing the largest increase in 〈τ〉. Indeed, chains with higher functionality record the smallest mean exit time in the first regime, i.e., for a < ac . However, they exhibit the largest 〈τ〉 in the second regime (a > ac ). This clearly indicates that the shape of the channel in the first regime seems to favour a fast translocation for chains with high functionality whereas the opposite takes place in the second regime, that is, a faster translocation for chains with low functionality. In order to understand this result, it is necessary to draw a correlation between the channel shape and the most probable conformation for a chain of a given f inside the channel that minimizes monomer-wall interaction. As the aspect ratio increases, the shape of the channel goes from a flat disk (L = 1.00 and D = 7.96) to an elongated narrow tube (L = 8.00 and D = 2.82). A chain with high functionality tends to adopt a spherical conformation due to its numerous short arms extending outward from the central monomer in order to minimize arm-arm interactions. For such chain, a channel shaped like a flat disk will obviously reduce its interaction with the wall resulting thereby in a faster translocation. A chain with a lower functionality has few but lengthy arms Such a star polymer inside a channel with a small aspect ratio, a < ac , will be subject to a stronger arms-wall interaction which will result in a relatively slow translocation. On the other hand, chains with lower functionality are less affected by the channel narrowing that occurs above ac insofar as they can adopt with a relative ease an elongated (tubular) conformation in opposition to chains with high functionality for which steric hindrance arising from arm-arm interaction prevents them to easily fit inside such channel, thereby severely affecting their motion inside the channel.

Figure 5. Effect of channel aspect ratio upon mean translocation time for chains of fixed total mass N = 121 but varying functionality. The volume of the channel is kept constant at V = 50 while a strong pulling force, F = 100, is applied.
Download figure:
Standard image High-resolution image3.4. Influence of star polymer functionality upon exit time
Another interesting aspect of star polymers translocation through nanochannels is the impact of polymer functionality upon the translocation dynamics. In this section, we study how the translocation time is impacted as the number of arms of the chain are varied while keeping the polymer total mass constant. Simulations of chain translocation are performed by applying a pulling force of magnitude F = 10 for a channel of diameter D = 4.6 while two distinct values of channel length are considered: L = 2 and 10. Figure 6 represents normalised exit time distributions of star polymers with varying functionality but whose total mass is kept at N = 121. The distributions are symmetric irrespective of channel length and chain functionality. A close inspection of the curves reveals that the distributions become broader with increasing f for both channel lengths. However, the broadening of the distributions does not follow a monotonic dependence on f insofar as the highly-peaked narrowest distribution is not for the smallest value of f but rather for f = 4, this being more obvious for the longer channel (figure 6(b)). Moreover, for small functionalities, i.e., f ≤ 4, the most probable exit time identified by the location of the distributions peak tends to coincide with the mean translocation time whereas it becomes smaller than 〈τ〉 for f > 5.

Figure 6. Mean exit time distributions of star polymers with different functionalities and for a channel length (a) L = 2 and (b) L = 10. The nanochannel diameter is D = 4.6, while the total polymer mass is N = 121, and the magnitude of the pulling force F = 10.
Download figure:
Standard image High-resolution imageFigure 7 depicts the variation of the mean translocation time in terms of chain functionality for smaller (figure 7(a)) and larger (7(b)) channel diameters. The chains mass and the magnitude of the pulling force are kept constant while two channel lengths are considered. A significant difference based on the channel diameter can be observed. For a small channel diameter, the translocation time has a non-monotonic dependence upon chain functionality with a minimum around f = 5 for both channel lengths. Chains with small functionality tend to have a larger entropy which undergoes a drastic reduction when they are placed inside the nanochannel. The nanochannel thus behaves like an entropic barrier for these chains as their motion is reduced inside the channel. As chains functionality is gradually incremented up to f = 5, their entropy decreases as it is also the case of the entropic barrier resulting in chains faster translocation. This can be viewed as the first regime of the translocation process for which a decrease of 〈τ〉 is observed. However, as f increases further (beyond f = 5), excluded volume interactions arising from the polymer arm-arm interactions become dominant offsetting the gain made from the entropic barrier reduction and leading thereby to an increased arm-wall interactions that hinders the translocation process. Obviously, f = 5 constitutes an optimal functionality at which forces hampering chain motion inside the channel are minimized. This non-monotonic behavior of the mean exit time with respect to f has been previously observed for star polymer translocation through a nanopore [51, 58]. In contrast to the previous result, the mean exit time decreases continuously as f increases for a larger channel diameter. In the latter case, the large channel diameter leads to a reduction of the arm-wall interaction, especially for chains with higher functionality whose radius of gyration is clearly smaller than the channel diameter which explains the monotonic decrease of 〈τ〉 with increasing f.
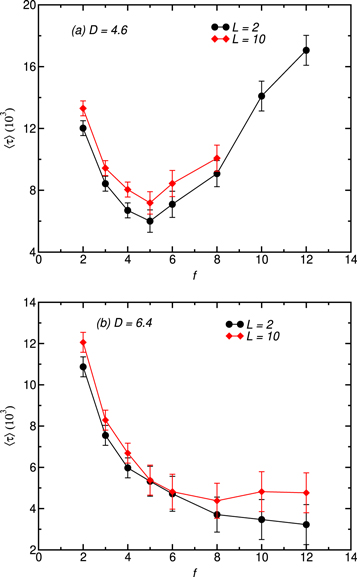
Figure 7. Plot of 〈τ〉 against f for N = 121, F = 10, two distinct values of channel length and channel diameter (a) D = 4.6 and (b)D = 6.4.
Download figure:
Standard image High-resolution imageNext we investigate how the chain total mass N affects the dependence of the mean translocation time upon chain functionality. Figure 8 shows the variation of the mean translocation time as a function of f for four different values of the chain mass and for L = 2 (Figure 8(a)) and L = 10 (figure 8(b)). The mean translocation time exhibits a non-monotonic dependence on f for large chain sizes irrespective of channel length. However, for small chain sizes and a shorter channel, L = 2, 〈τ〉 decreases monotonically as f increases. Obviously for smaller star polymers, the increase in chain functionality which leads to a reduction of the chain radius of gyration allows a faster translocation. In this case, the benefit gained in monomer-wall interaction reduction outweighs the increase in arm-arm interaction. On the contrary, for a longer channel length and smaller polymer mass, the increase of f results only in a shift of the minimum of 〈τ〉 towards larger f since the arm-arm interaction becomes greater than monomer-wall interaction resulting in a slower translocation beyond that point.

Figure 8. Mean translocation time versus functionality for D = 4.6, F = 10, and cavity length (a) L = 2 and (b) L = 10. The functionality is varied between f = 2 and 12.
Download figure:
Standard image High-resolution imageThe last investigation aims at understanding how the pulling force affects the dependance of the mean translocation time upon chain functionality. The parameters that are kept constant for this study are the channel diameter which is equal to D = 4.6 and the chain total mass equal to N = 121. Simulations are run for two distinct channel lengths, that are L = 2 and L = 10. Figure 9 depicts the dependence of 〈τ〉 upon f for five different values of the pulling force magnitude. For both channel lengths 〈τ〉 exhibits two distinct behaviors according to the magnitude of the pulling force. For F ≤ 20, the mean translocation time assumes a non-monotonic variation with respect to f, with the minimum being shifted towards greater f as the magnitude of the pulling force increases. For larger values of the pulling force, i.e F > 20, 〈τ〉 decreases monotonically as f increases.

Figure 9. Plots of 〈τ〉 versus f for D = 4.6, N = 121, and a channel length L = 2 (a) and L = 10 (b). The functionality ranges between 2 and 12. The inset depicts the variation of the mean exit time with respect to functionality for F ≥ 20.
Download figure:
Standard image High-resolution image3.5. Polymer mass N versus mean exit time 〈τ〉
The dimension of star polymers with f-linear arms is expressed in equation (5) as  . Moreover, the diffusion coefficient of such chains based on Rouse scaling model is D ∼ N−1 while the diffusion time over a length equal to their size can be approximated by τ ∼ R2/D [70]. By combining these three relations, one can derive an expression of star polymers exit time confined inside a tube
. Moreover, the diffusion coefficient of such chains based on Rouse scaling model is D ∼ N−1 while the diffusion time over a length equal to their size can be approximated by τ ∼ R2/D [70]. By combining these three relations, one can derive an expression of star polymers exit time confined inside a tube
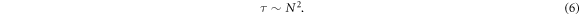
According to the above relation, the translocation time of star polymers through a confining tube should follow a power-law relation with respect to their mass with an exponent close to 2.
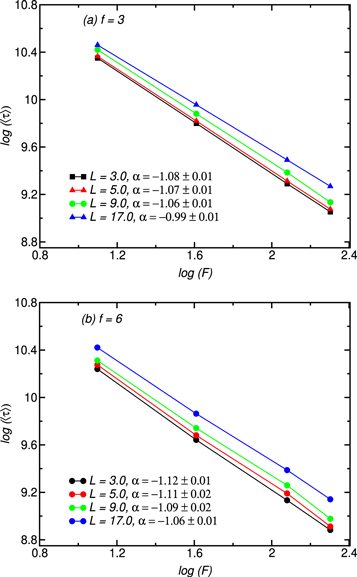
In order to verify the validity of this assertion, we have investigated the influence of the chain total mass N upon translocation time for two star polymers with functionalities f = 3 and f = 6. For both studies, a pulling force of magnitude F = 10 and a channel of diameter D = 4.6 are considered while simulations are carried out for various channel lengths ranging from L = 3 up to L = 17. The results depicted in figure 10 show the existence of a scaling relation between the translocation and the chain mass, τ ∼ Nα , for both functionalities with α in the range [1.721.96] for f = 3 (Figure 10(a)) and in the range [1.942.12] for f = 6 (figure 10(b)). Note that the exponent α decreases with increasing channel length L. However, despite its slight variation, α can be considered close to the theoretical prediction of 2. Moreover, a close inspection of the curves reveals that for a given chain size and channel length, the exit time of chains with f = 3 is systematically higher than of chains with f = 6 in favour of equation (6) which predicts a scaling τ ∼ N2.

Figure 10. log-log plot of 〈τ〉 as a function of N for f = 3 (a) and f = 6 (b). The channel lengths are given in the plots. The nanopore diameter and magnitude of a driving force are D = 4.6 and F = 10, respectively.
Download figure:
Standard image High-resolution image3.6. Influence of the magnitude of the pulling force F on 〈τ〉
Lastly, an investigation with the purpose of understanding the impact of the pulling force strength upon the mean translocation time was undertaken. Figure 11 depicts the mean translocation time for chains with f = 3 (Figure 11(a)) and f = 6 (figure 11(b)) as a function of F where the magnitude of the pulling force is varied from F = 3.0 to F = 10.0. Various tube length ranging from L = 3 to L = 17 are investigated while the tube diameter and the chain mass are fixed at D = 4.6 and N = 121 respectively. First, we note that for a given value of F, 〈τ〉 increases with the channel length for both functionalities, which is an expected result. Next, chains translocation becomes faster as the strength of the applied force increases resulting in a power law decay of the mean exit time 〈τ〉 ∼ F−α , with the scaling exponent α close to 1 for both functionalities. Indeed, this is not a surprising result insofar as an increase of the pulling force produces a faster translocation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 11. log-log plot of 〈τ〉 versus F for N = 121, D = 4.6, and functionality (a) f = 3 and (b) f = 6. The channel length is varied from L = 3 to 17.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
Using three-dimensional Langevin dynamics simulations, we report the dynamics of a star-shaped polymer translocation through a nanochannel under a localized pulling force, by focusing on the dependence of the mean exit time on the channel length and diameter, polymer functionality, polymer mass, and magnitude of the driving force.
According to the obtained results, for longer channel lengths, no successful translocation is observed for star-shaped polymers with relatively larger functionalities when they are subject to a pulling force of small magnitude. However, in the case of a large pulling force, all chains experience successful translocation while the mean translocation time exhibits a monotonic dependence on the chain functionality. On the other hand, successful translocations are recorded for a wider channel irrespective of the magnitude of the applied force. While the dependence of the exit time upon channel length is almost linear for most of the cases under investigation, the case of the wider channel and weak applied force presents unique features: we note a dramatic change in the translocation dynamics around a particular channel length, L = 12, whereby chains with larger functionalities go from being the fastest to becoming the slowest at this particular point.
Next, the impact of channel dimensions upon the translocation dynamics is examined by varying the channel aspect ratio a while keeping its volume constant. The mean exit time 〈τ〉 is not significantly affected by the change in the aspect ratio for smaller functionalities, i.e., up to f = 4. However, for functionalities beyond that value, the translocation dynamics exhibits a regime change marked by an increase of 〈τ〉. The regime change takes place at a particular value of the aspect ratio, ac = 1.4, while the change is more pronounced for chains with higher functionalities. This result can be understood by relating the chain conformation, which depends on its functionality, to the channel shape. Indeed, as the aspect ratio increases, the channel shape evolves from a flat disk cavity suitable for chains with larger functionalities to a long narrow tube in which the same chains need to deform in order to adopt an elongated conformation which results in an increase in arm-wall interaction, leading to a slower translocation.
On the other hand, the distribution profiles of the exit time are symmetric irrespective of channel length and polymer functionality. They become broader as the chain functionality increases. Interestingly, the dependence of the mean escape time upon the star's functionality exhibits distinct behaviors based on the size of the channel diameter and magnitude of the pulling force. Indeed, when a moderate pulling force is applied, the escape time is a monotonically decreasing function of f for a channel with a large diameter, whereas a non-monotonic dependence is found for a narrow channel, with the presence of a minimum exit time  at a particular value of f. In the latter case, the minimum exit time is shifted towards larger values of chain functionality when the chain size N decreases. Moreover in the case of a strong pulling force, 〈τ〉 exhibits a monotonic decrease with respect to f irrespective of the channel diameter.
at a particular value of f. In the latter case, the minimum exit time is shifted towards larger values of chain functionality when the chain size N decreases. Moreover in the case of a strong pulling force, 〈τ〉 exhibits a monotonic decrease with respect to f irrespective of the channel diameter.
Finally, the dependences of the mean exit time with respect to polymer total mass N and magnitude of the driving force F, respectively are best described by scaling relations. The power-law relationship relating 〈τ〉 and N can be approximated by 〈τ〉 ∼ Nα where the exponent alpha is close to 2 but with a weak dependence upon channel length and chain functionality. As for the pulling force, we found an exponent close to −1 which also exhibits a slight dependence upon channel length and chain functionality. These results are in agreement with theoretical predictions based on the Rouse model.
A possible avenue for further investigation is to study the translocation dynamics through nanochannels of star polymers with more than a one arm subject to a pulling force.
Acknowledgments
We, the authors, would like to thank the International Science Program, Uppsala University, Sweden and Addis Ababa University for provided computational resources.
Data availability statement
The data generated and/or analysed during the current study are not publicly available for legal/ethical reasons but are available from the corresponding author on reasonable request.
Conflict of interest
The authors declare no conflict of interest.