
Abstract
In vitro neuronal models have become an important tool to study healthy and diseased neuronal circuits. The growing interest of neuroscientists to explore the dynamics of neuronal systems and the increasing need to observe, measure and manipulate not only single neurons but populations of cells pushed for technological advancement. In this sense, micro-electrode arrays (MEAs) emerged as a promising technique, made of cell culture dishes with embedded micro-electrodes allowing non-invasive and relatively simple measurement of the activity of neuronal cultures at the network level. In the past decade, MEAs popularity has rapidly grown. MEA devices have been extensively used to measure the activity of neuronal cultures mainly derived from rodents. Rodent neuronal cultures on MEAs have been employed to investigate physiological mechanisms, study the effect of chemicals in neurotoxicity screenings, and model the electrophysiological phenotype of neuronal networks in different pathological conditions. With the advancements in human induced pluripotent stem cells (hiPSCs) technology, the differentiation of human neurons from the cells of adult donors became possible. hiPSCs-derived neuronal networks on MEAs have been employed to develop patient-specific in vitro platforms to characterize the pathophysiological phenotype and to test drugs, paving the way towards personalized medicine. In this review, we first describe MEA technology and the information that can be obtained from MEA recordings. Then, we give an overview of studies in which MEAs have been used in combination with different neuronal systems (i.e. rodent 2D and three-dimensional (3D) neuronal cultures, organotypic brain slices, hiPSCs-derived 2D and 3D neuronal cultures, and brain organoids) for biomedical research, including physiology studies, neurotoxicity screenings, disease modeling, and drug testing. We end by discussing potential, challenges and future perspectives of MEA technology, and providing some guidance for the choice of the neuronal model and MEA device, experimental design, data analysis and reporting for scientific publications.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
In vitro neuronal models represent an important tool to study the complexity of the brain and, by extension, the pathophysiology of neurological diseases. For many decades, rodents have proven to be a valuable source of mammalian neuronal cells, in the form of brain slices or cultures of dissociated neurons [1]. However, rodent neurons must be continuously isolated from fresh animals, and the inherent inter-species differences can influence the translation of results into humans [2–5]. For these reasons, in the past years, there has been a combined push in the scientific community to leave non-human models in favor of human cell-based systems, among which neuronal cultures derived from human induced pluripotent stem cells (hiPSCs) represent a promising approach. Investigating them [6–8]. Together with the shift from rodent towards human-cell based models, three-dimensional (3D) systems have been sought with the aim to replicate the 3D environmental complexity of the brain, and investigate neuronal functions in a more in vivo-like condition [9–11].
Undoubtedly, one of the key advantages of in vitro neuronal models, both rodent and hiPSCs-derived, 2D and 3D, is that they retain their electrophysiological functions and their neuronal activity can be measured by means of different techniques. Beside conventional patch-clamp, allowing the measurement of neuronal activity at single-cell level [12–14], micro electrode arrays (MEAs) (i.e. cell culture dishes with embedded micro-electrodes [15–17]) have been increasingly used to characterize neuronal activity at the network level. Nowadays, different MEA devices are available, allowing to investigate the electrophysiological activity of different neuronal systems, in both physiological and pathological conditions, in a non-invasive and relatively simple way. One of the key advantages of MEA technology is that the recorded electrophysiological activity appears to be (i) deeply shaped by the physiological characteristics of neuronal networks under investigation [18–22], and (ii) highly sensitive to the presence of any kind of compound able to influence the physiological mechanisms of neurons [23–27]. For this reasons, in vitro neuronal cultures on MEAs have been largely used for investigating physiological mechanisms, screening neurotoxic compounds, modeling neurological diseases and testing drugs.
Despite the increasing popularity of MEAs, we believe that MEA technology is not leveraged at its full potential. The reason is related to the relative novelty of this technique, but also to challenges with the interpretation of experimental results, and to the lack of guidelines for the choice of the neuronal model and MEA device, design of experiments, and analysis of data.
With the present review, we aim to provide the scientific community with an overview of in vitro neuronal cultures in combination with MEA technology for biomedical research. First, we will introduce MEA technology, including the functioning of MEA devices, the neuronal signals which are recorded by MEAs, and the information which can be obtained through the analysis of MEA recordings alone or in combination with other techniques. We will review the protocols to culture different neuronal systems (i.e. rodent 2D and 3D neuronal cultures, organotypic brain slices, hiPSCs-derived 2D and 3D neuronal cultures, and brain organoids) on MEA devices, and we will provide a description of the electrophysiological activity exhibited by these neuronal systems on MEAs. We will give an overview of the studies in which rodent and hiPSCs-derived neuronal cultures on MEAs have been used for physiology studies, neurotoxicity screenings, disease modeling and drug testing. Lastly, we will discuss potential, challenges and future perspectives for the use of MEAs in biomedical research, and we will provide some guidance for the choice of the neuronal model and MEA device, experimental design, data analysis and reporting, in order to fully harness the potential of in vitro neuronal cultures on MEAs.
2. Neuronal cell cultures: from rodent to hiPSCs-derived neurons
The brain is studied at many different levels, from the molecular and cellular physiology of the neuron to the processing of information by a whole brain region. For this purpose, several experimental models are available. Among them, in vitro neuronal cultures represent an accessible and economical system to study the complexity of the brain and, by extension, the pathophysiology of neurological diseases.
The first reported in vitro neuronal model was developed in 1910 by Harrison by isolating and growing pieces of the neural tube from the embryonic frog [28]. Later, studies on neurons and neuronal networks have been carried out on in vitro preparations from different animal models, including invertebrates with relatively simple nervous systems, such as mollusks (i.e. squids and Aplysia), and worms (i.e. leeches and C. elegans), and vertebrates with nervous systems closer to the human one, such as fish (i.e. lamprey), birds (i.e. chicken), amphibians and mammals [29]. In this context, rodents (i.e. rats and mice) have been progressively established as the most commonly used mammalian models to study the nervous system and to isolate neuronal cells for in vitro cultures, thanks to their genomic, developmental and physiological similarities with humans in combination with the relative ease of use and convenience in terms of materials, time and expertise [29].
The most common biological preparations from the rodent brain are divided into two main categories: brain slices and dissociated cell cultures. These models encompass the spectrum from ex vivo short-term preparations (i.e. acute brain slices, few hours), to in vitro medium- and long-term cultures (i.e. organotypic slices and dissociated cell cultures, few weeks up to several months) [30]. Biological preparations from other parts of the central and peripheral nervous system, such as the spinal cord [31], the retina [32, 33], and the olfactory epithelium [34, 35], are also possible. However, in this review, we will mainly focus on medium- and long-term in vitro neuronal models of the brain.
Throughout the twentieth century, techniques to isolate and culture neuronal cells from the rodent brain have been developed and progressively refined [36, 37]. Protocols to maintain medium- and long-term neuronal cultures were optimized and standardized, and media formulations were made commercially available, thereby providing access to this technique to a larger number of laboratories [38–41]. Protocols optimization led to better growth, differentiation, and long-term survival of neuronal cells under controlled conditions, allowing for better consistency and reproducibility of results.
For many decades, rodents have proven to be a valuable source of mammalian neuronal cells for most laboratories. From specific brain regions of mice and rats, wild-type or transgenic disease models, thin slices or a large number of viable dissociated neurons can be isolated and cultured, maintaining the ability to develop and mature in vitro. However, neuronal cell cultures from rodents show several shortcomings. Firstly, since neurons are not mitotically active (i.e. they are not able to go through mitosis and proliferate), primary rodent cells must be continuously isolated from fresh animals. Secondly, the inherent inter-species differences, including genomic, developmental and physiological divergences [2–5], imply that rodent models cannot fully recapitulate human brain physiology and disease. In response, in the past years, there has been a combined push in the scientific community to leave non-human models in favor of human cell-based systems.
A major obstacle for creating human in vitro models is related to obtaining an adequate amount of viable material to begin with, since accessing the nervous tissue in patients for biopsy is not possible except under very rare circumstances. In the past century, a few cell lines have been derived from human tumors, typically from surgical biopsies (e.g. SH-SY5Y line [1]). These lines can be propagated in culture indefinitely, and differentiated on demand into neuronal-like cells [1]. However, the actual usability of these cell lines for modeling the human brain is extremely limited, considering that they are derived from a pathological condition and, when differentiated, they only represent an approximation of mature neurons, with some generic neuronal properties.
Conversely, a very promising human model is represented by human stem cells-derived neurons. For many years, human embryonic stem cells (hESCs) and fetal neural stem cells (NSCs) have been studied. hESCs are cells derived from embryonic blastocysts with the properties of self-renewal and pluripotency (i.e. the potential, if subjected to the correct signals, to differentiate into any kind of somatic cell of the human body, including neurons and glial cells). Human NSCs instead are derived from the brain tissue of human fetuses, and genetically modified to obtain stable multipotent lines that can be continuously expanded in culture and differentiated in neurons and glial cells (e.g. ReNcell VM and CX lines [42]). Since hESCs and NSCs are typically collected from human donor embryos and fetuses, substantial ethical concerns and limited availability constitute two of the major limitations. Moreover, before hESCs and NSCs are differentiated into neurons, they undergo rapid and extensive proliferation, during which their genome integrity is put at high risk by a wide variety of genomic mutations [43].
The opportunity of overcoming these limitations was offered by Shinya Yamanaka's group in the past decade. In 2006, Yamanaka's group proved that it was possible to reprogram differentiated cells back into pluripotent stem cells through ectopic expression of four transcription factors [44]. The process to obtain induced pluripotent stem cells (iPSCs) was first described using mouse fibroblasts, and then successfully applied to human fibroblasts [45]. hiPSCs not only possess the same properties of self-renewal and pluripotency, and overcome the above-mentioned limitations of hESCs, but they also allow the generation of differentiated cell lines from patients with a specific genetic background, which is very promising for personalized medicine. For these reasons, in a few years, hiPSCs popularity has rapidly grown. Since obtaining human fibroblasts to produce hiPSCs requires an invasive skin biopsy [45], in the past years, there has been a push towards the use of more easily accessible cell types, such as keratinocytes [46], peripheral blood cells [47], and renal epithelial cells [48], from single hair plucks, blood and urine samples, respectively. Moreover, researchers have progressively developed and optimized protocols to differentiate hiPSCs into mix cultures of neurons and glial cells, or even into specific neuronal subtypes [6–8].
Despite the undeniable advantages, the use of hiPSCs technology is still in its early stage. One main limitation is the large amount of materials, time (up to several months) and expertise that are necessary to differentiate hiPSCs into mature and functional neurons[49]. A relevant improvement was the development of differentiation protocols based on the overexpression of single lineage-determining transcription factors, resulting in the rapid conversion into uniform populations of neurons [7, 18, 50]. The advent of hiPSCs technology and the progressive optimization of differentiation protocols into neurons, have opened the way to the development of human in vitro models of the brain and neuronal diseases, not only to study the pathophysiological phenotype and the underlying mechanisms, but also for drug discovery and testing [51–53].
The major part of in vitro neuronal models consists of 2D cultures of neurons, both dissociated from rodents or derived from hiPSCs. Even if 2D neuronal cultures have been widely used to model the brain in a simplified way, providing invaluable results, it is clear that they are inherently unable to replicate the 3D environmental complexity of the brain (such as cell–cell and cell-extracellular matrix interactions, and axons-dendrites extension in the 3D space) [9–11]. For this reason, together with the shift from rodent towards human neuronal cultures, 3D models have been sought with the aim to investigate neuronal functions in a more in vivo-like condition. By isolating brain slices from rodents, 3D structural and functional relationships between groups of cells are partially preserved [54]. However, organotypic slices cultures are difficult to maintain in vitro for a long time, and they cannot be obtained from the human brain, except under very rare circumstances. For all these reasons, long-term 3D models from both dissociated rodent neurons and hiPSCs have recently been developed [22, 55, 56].
A schematic overview of medium- and long-term in vitro neuronal models is found in figures 1(a) and (b).
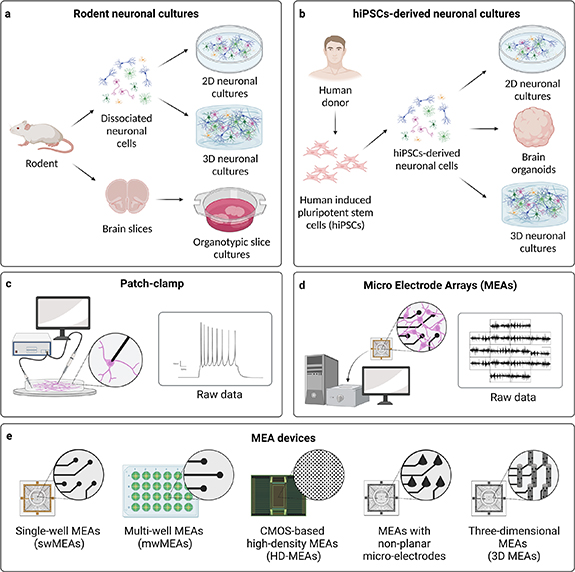
Figure 1. (a), (b) Overview of medium- and long-term in vitro neuronal models, divided into (a) rodent neuronal cultures, including 2D and 3D cultures of dissociated neuronal cells, and organotypic brain slices, and (b) hiPSCs-derived neuronal cultures, including 2D cultures, brain organoids, and 3D cultures. (c), (d) Two main electrophysiological techniques used to record neuronal activity of in vitro neuronal cultures, i.e. (c) patch-clamp, recording the electrophysiological activity of single neurons at single-cell level, and (d) MEAs, recording the electrophysiological activity of neuronal cultures at network level. (e) Different types of commercially available MEA devices for recording electrophysiological activity of in vitro neuronal cultures, i.e. single-well MEAs (swMEAs), multi-well MEAs (mwMEAs), CMOS-based high-density MEAs (HD-MEAs), MEAs with non-planar micro-electrodes, and three-dimensional MEAs (3D MEAs). Created with BioRender.com.
Download figure:
Standard image High-resolution image3. From single-cell to network level: recording the electrophysiological activity of in vitro neuronal cultures with MEAs
One of the key advantages of in vitro neuronal cultures, both isolated from rodents and derived from hiPSCs, is that they retain their electrophysiological functions. In the past century, different techniques to record and evoke the electrophysiological activity of in vitro neuronal cultures have been progressively developed.
Since its introduction in 1976, patch-clamp has been the technique of choice for electrophysiologists to investigate the activity of ion channels in electrogenic cells (i.e. central and peripheral neurons, heart cells, and muscle cells), supplying invaluable information on their electrophysiological functions [12–14]. The patch-clamp technique allows the measurement of ion channels currents flowing through the cellular membrane by performing intracellular recordings at single-cell level [12–14].
In conventional patch-clamp, the small tip (less than 1 µm in diameter) of a glass pipette is sealed to the surface of the cell membrane in order to isolate a tiny membrane area (patch) from the rest of the membrane. The glass pipette is filled with a conducting saline solution. The ground electrode, typically silver/silver chloride (Ag/AgCl), is located in the bath chamber (where cells are placed). The recording electrode, which is an Ag/AgCl wire, is in contact with the pipette solution and connects the glass pipette with the amplifier.
With patch-clamp, different experiments can be performed. According to the aim, the cell membrane is left intact (i.e. cell-attached configuration), or a patch of membrane is excised, broken by suction or perforated with antibiotics. Different configurations allow to measure the activity of a single or a small number of ion channels within the patch, or to simultaneously record the currents through all the ion channels on the entire cell membrane (called whole-cell patch-clamp) over time [57–59]. Two modes are possible: current and voltage clamp. With current clamp mode, steady currents are injected into cells, while membrane potential is recorded. Conversely, with voltage clamp mode, membrane potential is controlled, while ionic currents flowing through the cellular membrane are recorded. The possibility to control the membrane potential enables activation and manipulation of voltage-dependent channels. Moreover, the chemical composition of the pipette and of the bath solution can be supplemented with permeable and impermeable ions to suppress and/or isolate specific currents, or with drugs to affect the activity of specific channels [57, 60].
Although conventional patch-clamp has been very effective in measuring ionic currents and supplying invaluable information on ion channels, it has several limitations. First of all, conventional patch-clamp allows recording one cell at a time, and requires highly trained electrophysiologists [61]. Therefore, it represents a time-consuming technique. The volume and rigidity of conventional pipettes, along with the functions of the patch-clamp recording system which depend on the microfluidic system connected to each pipette, such as the suction application, are the major limitations preventing parallelization (i.e. recording of more than one cell in parallel) in conventional patch-clamp. Several companies have developed automated patch-clamp systems with hundreds of recording sites enabling to perform medium throughput electrophysiological experiments, in which the activity of many cells in parallel is recorded in a few hours [62–66]. Secondly, patch-clamp is an invasive technique, which inevitably implies the disruption of physiological conditions and of natural biochemical processes necessary for normal electrophysiological activity [61]. For this reason, while short-term recordings are considered reliable, long-term measurements are generally unsuccessful due to the decay of intracellular signals, and repeated recordings of the same culture are not possible. Thirdly, in patch-clamp, spatial resolution is limited by the tip size of conventional pipettes, which makes it difficult to record the electrophysiological activity of small neurons and subcellular structures, thus from more than one site in the same cell [61].
To overcome these limitations and to support the growing interest of electrophysiologists in measuring and manipulating the electrophysiological activity not only of single neurons, but of whole neuronal networks, MEAs were developed. MEA technology allows long-term recordings of the electrophysiological activity of groups of electrogenic cells simultaneously and at many sites, in a relatively simple and non-invasive way, by means of extracellular substrate-integrated micro-electrodes [15–17]. A schematic comparison between conventional patch-clamp and MEAs is found in figures 1(c) and (d) and table 1.
Table 1. Schematic comparison between conventional patch-clamp and MEAs.
| Conventional patch-clamp | MEAs | |
|---|---|---|
| Scale | Single neuron | Neuronal network |
| Type of recording | Intracellular recording of voltage (current clamp) or current (voltage clamp) | Multi-site extracellular recording of voltage |
| Single-ion channel recording | Possible | Not possible |
| Temporal resolution | <milliseconds | Milliseconds |
| Spatial resolution | Limited by tip size of pipette, cellular resolution | Dependent on dimension, number and density of micro-electrodes, up to subcellular resolution for HD-MEAs |
| Invasiveness | High | Not invasive |
| Single recording duration | Short-term recording (few hours) | Long-term recording (many hours, days) |
| Repeated recordings on the same culture | Not possible | Possible |
| Parallelization | Not possible | Medium to high throughput |
| Accessibility | Requires skilled electrophysiologists, time-consuming | Relatively easy to learn, can be conducted by a technician after a few hours of training |
| Data analysis | Relatively easy | Big amount of raw data, hard |
| Voltage control | Yes | No |
| Current control | Yes | No |
| Electrical stimulation | Possible | Possible |
| Combination with other techniques | Possible | Possible |
Nowadays, MEAs represent a promising technique for the investigation of the electrophysiological activity of in vitro neuronal networks, providing invaluable insights about the dynamics of neuronal models at the network level, in both physiology and pathology. In this chapter, the birth and evolution of MEA technology will be reviewed, along with the functioning of MEA devices, the neuronal signals which are recorded by MEAs, and the information which can be obtained through the analysis of MEA recordings alone or in combination with other techniques.
3.1. Birth and evolution of MEA technology
The beginning of in vitro neuronal network electrophysiology using MEAs can be reconducted to the pioneering studies of Thomas et al, Gross et al, and Pine [15–17]. In 1972, Thomas et al introduced the first MEA device consisting of approximately 30 platinized gold micro-electrodes integrated into a glass substrate (two rows of 15 micro-electrodes each, spaced 100 µm apart), and succeeded in recording the activity of cultured chick cardiomyocytes [15]. Five years later, Gross and his collaborators recorded the electrophysiological activity of an isolated snail ganglion [16]. Finally, in 1980, Pine was able to record the activity of a 3 weeks-long neuronal network derived from rat neurons using a MEA device with 32 gold micro-electrodes (two rows of 16 micro-electrodes each, spaced 250 µm apart) [17]. These three hallmark studies laid the foundations for and marked the beginnings of in vitro network electrophysiology using MEAs.
Since then, MEA technology has garnered interest and contributions from a very broad cross-disciplinary research community and has continuously improved during these years. Nowadays, there is great variety of MEA technology which depends on specifications such as active or passive devices, number and density of micro-electrodes, designs, shapes or materials, and number of independent wells (figure 1(e)). With all MEAs presented, spontaneous electrophysiological activity can be recorded simultaneously from the embedded micro-electrodes. In addition, electrical stimuli can be delivered to the cells from one or multiple micro-electrodes to investigate neuronal network evoked activity [67]. In the following paragraph, we will touch upon some of these properties.
3.1.1. Standard passive MEAs
The current standard passive MEAs consists of cell culture dishes with a matrix of extracellular microfabricated micro-electrodes integrated at the bottom, in a biocompatible insulation substrate (e.g. polyamide or silicon nitride/oxide) which prevents short circuits with the electrolyte bath [68–70]. The fabrication of micro-electrodes becomes an important step in developing a MEA device. It is crucial to select the material for fabrication based on its biocompatibility and electrical conductivity and to optimize the dimensions and shape of the micro-electrodes. Indeed, the main challenge is that, due to their small size, their impedance value is large, resulting in low signal-to-noise ratios (SNRs), which is not desirable [71, 72]. Micro-electrodes are typically made of Au, indium–tin oxide (ITO), titanium nitride (TiN), gold, PEDOT coated gold, or black platinum, and are biocompatible, long-term lasting, and with a low impedance (less than 500 KΩ at 1 kHz) for low thermal noise. The opposite end of each micro-electrode extends to the periphery of the chip and makes contact with an external amplifier, which passes electrical signals for further conversion, filtering, storage, and analysis of data. Neurons are directly cultured on MEAs on top of the micro-electrodes, and cell adhesion is promoted by pre-coating MEAs with components of the extracellular matrix (ECM) [69].
Traditional single-well MEAs (swMEAs) allow to record the electrophysiological activity of single neuronal networks with a wide range of resolution, depending on the number of micro-electrodes (60–256 micro-electrodes), the micro-electrode size (10–30 µm of diameter), the distance between them (100–500 µm inter-electrode spacing), the layout (i.e. single or multiple quadrant), the special organization of the micro-electrodes (micro-electrodes grid 8 × 8–6 × 10 for single quadrant, 5 × 6 for two quadrants, 4 × 4 and a center line of 1 × 8 for four quadrants), and the recording area (0.2–2 mm2).
To answer the growing need of the neuroscience community for high-throughput devices, during the past decade, multi-well MEA platforms have been developed. Multi-well MEAs (mwMEAs) provide the possibility to record, at the same time, multiple neuronal networks cultured into independent wells (6–96 independent wells). Also in this case, recordings can be performed with a wide range of resolution, depending on the number of micro-electrodes (3–64 micro-electrodes), the electrode size (50–100 µm of diameter), and the distance between them (150–700 µm inter-electrode spacing).
Companies producing swMEAs and mwMEAs are Alpha MED scientific (Osaka, Japan), Multi-Channel systems (Reutlingen, Germany), Axion Biosystems (Atlanta, USA), and 3Brain (Pfäffikon, Switzerland). Other companies, such as Ayanda-Biosystems (Lausanne, Switzerland), focus on the development of microelectrode devices only, and Plexon (Dallas, USA) developed hardware and software tools to be used together with third parties microdevices.
3.1.2. High-density MEAs (HD-MEAs)
As alluded to above, the main advantage of MEA technology is the ability to simultaneously record the neuronal network activity from different micro-electrodes. However, standard MEA devices suffer from low spatial resolution. Due to the low number of micro-electrodes, and their relatively large spatial separation and dimension, the neuronal signals recorded by MEAs result from the contribution of many neurons, rather than single ones [69]. In fact, each micro-electrode records the extracellular potentials generated by action potentials (APs) in cell bodies of neurons that are within its receptive field. With the spatial resolution of standard MEAs (inter-electrode spacing of 100–700 μm) the distance between cells and micro-electrodes typically ranges from 10 to 100 nm. As an example, a culture of 50 000 neurons coupled to 50 micro-electrodes presents an under sampling of the network activity by a factor of 103. Although this is adequate to obtain a general overview of neuronal network activity, the electrophysiological activity at the cellular and sub-cellular level is in such a way not detected [69]. In addition, the size of micro-electrodes in standard MEAs is partially constrained: on the one hand, micro-electrodes should be as small and close to the cells as possible to obtain information from localized points, on the other hand, they should have a sufficient surface to detect electrical signals with an acceptable SNR [69].
During the past decade, the growing need of neuroscientists for high-resolution investigations boosted the development of high-density MEAs (HD-MEAs). In 2009, Berdondini et al developed the first HD-MEA based on the conventional thin-film technology, whose 60 micro-electrodes size and distance were comparable to that of a neuron [73]. Four different MEA layouts (i.e. 22 and 30 μm micro-electrodes diameters, 20 and 10 μm spaced) were designed. The layout of the array was divided into 4 clusters of 15 high-density micro-electrodes each [73]. The advantage of this configuration relied on the possibility of investigating interconnected neuronal sub-populations, both on a local network basis (i.e. considering the high-density clusters) and on a whole network basis (considering the four separated high-density clusters) [73].
However, conventional thin-film technology had fabrication limitations, which created important constraints for the further development of HD-MEAs [71, 72]. In particular, limiting factors were the management of high number and density of micro-electrodes, contact pad connections, and the increasing complexity of the external amplification circuit. For this reason, complementary metal oxide semiconductor (CMOS) technology and concepts that were previously established for light imaging sensors have been used to create HD-MEAs with higher resolution (i.e. active MEAs) [30]. In this context, Berdondini and colleagues developed a CMOS technology-based solid-state active pixel sensor MEA device with 4096 pixels [74–76]. In the same period, Frey and colleagues presented a system composed of 11 011 metal micro-electrodes and 126 channels, each of which comprises recording and stimulation microelectronics [77, 78]. In another work [79], Lambacher and colleagues reported considerable progress of neuronal recording by multi-transistor array (128 × 128 sensors) chips with EOMOS transistors. More recently, Tsai and colleagues developed a CMOS-MEA device that contains 65 536 simultaneously recording and stimulating micro-electrodes [80].
Up to now, CMOS-based HD-MEA technology is produced by Maxwell Biosystems (Zurich, Switzerland, 264 000 micro-electrodes of which 1024 recording channels, 17.5 µm pitch, 3.85 × 2.1 mm2 sensor area), 3Brain (Pfäffikon, Switzerland; 4096 micro-electrodes, 42–60–80 µm pitch, 2.6 × 2.6–3.8 × 3.8–5.1 × 5.1 mm2 sensor area), and Multi-Channel systems (Reutlingen, Germany; 4225 micro-electrodes, 16–32 µm pitch, 1.04 × 1.04–2.08 × 2.08 mm2 sensor area).
In addition, since in the past years the neuroscience field asked for a combination of multi-well and high-density systems. Companies already producing HD-MEAs developed systems in a multi-well format (i.e. 3Brain: 6 wells plate with 2304 micro-electrodes; Maxwell Biosystems: 6 or 24 wells with 26 400 micro-electrodes).
The advantage of integrating active electronic components on the same substrate as actual micro-electrodes lies in the possibility of increasing the number of micro-electrodes, and their density. Furthermore, the co-integration allows to amplify the signals with an optimal quality thanks to minimal capacitance and parasitic resistances [30]. The short inter-electrode separation results in a gain of information on the micro-circuit neuronal dynamics and signal propagation, but requires the careful evaluation of the temporal resolution as well as the assessment of possible cross-talk artifacts (i.e. electrical and optical) between neighboring recording sites [73]. Even if various techniques have been utilized to minimize the effect of cross-talk (i.e. devices that constrain the generated electric fields [81]), channel interference due to cross-talk still constrains the performance of HD-MEAs, and represent a rate-limiting effect on spatial resolution of these devices [81].
3.1.3. Three dimensional MEAs (3D MEAs)
Another advancement in MEA technology is the development of MEAs with non-planar micro-electrodes and of 'true' three dimensional MEAs (3D MEAs, i.e. recording simultaneously from multiple 2D planes).
The majority of commercially available MEAs have planar micro-electrodes, since they are specifically designed for 2D neuronal cultures. However, several MEAs with 3D micro-electrodes have been developed to be used in combination with brain slices. In these devices, micro-electrodes are shaped as tips [82], pillars [83], mushrooms [84], volcanos [85] or needles [86]. The advantages of the 3D micro-electrodes MEAs for recording the activity of brain slices include: (i) tissue slice penetration that enables to avoid the superficial layer of dead or damaged cells and to reduce the distance between micro-electrodes and active neurons, (ii) increase in geometrical surface that reduces micro-electrode impedance, thus enhancing the SNR. However, in MEAs with 3D micro-electrodes, recordings take place only in a single plane as all the micro-electrodes have the same height.
A different approach is to simultaneously record the electrophysiological activity of 3D neuronal cultures from multiple 2D planes, by using a 'true' 3D MEA with micro-electrodes distributed in the entire 3D neuronal tissue volume. A few prototypes of 3D MEAs have recently been proposed, in which micro-electrodes are embedded at different heights along free-standing probes which can be inserted into neuronal networks either during or after the seeding of cultures. For instance, Soscia et al developed a polyamide-based 3D MEAs in which micro-electrodes are embedded in flexible polymer pillars that are vertically actuated [87]. Each 3D MEAs consists of 10 actuated pillars with 8 micro-electrodes along each one (for a total of 80 micro-electrodes per 3D MEA) which can non-invasively cover the 3D neuronal model for effective interaction [87]. While Soscia et al chose a 'bottom-up design' approach, retaining many of the features of traditional 2D MEAs, recently Shin et al developed a 'true' 3D MEAs with a 'top-down approach' inspired by implantable in vivo probe development [88]. Indeed, it is conceivable that in vivo 3D MEAs can also be repurposed for hiPSCs-derived organoids and other in vitro 3D models. In this sense, Shin et al reported a 3D MEA setup in which the micro-electrodes could be lowered into the sample from above using a micromanipulator. In addition to 17 individual shanks with micro-electrodes embedded at different heights (for a total of 64 micro-electrodes per 3D-MEA), one multifunctional shank integrates additional functionality, including optical fibers and microfluidic channels for drug delivery at specific sites [88]. Interestingly, 3D MEAs with a 'top-down approach' like the one of Shin et al can be applied also in vivo, allowing to compare recordings from 3D neuronal networks in vitro and in vivo.
Recently, Huang et al proposed a third type of 3D MEA, specifically designed for brain organoids. This prototype was inspired by macroscale electroencephalography (EEG) caps, and consisted of a 'shell' of self-folding polymer leaflets with embedded micro-electrodes, designed to wrap spherical organoids of different sizes [89]. In the resulting configuration, recording micro-electrodes are distributed in the 3D space on the surface of the brain organoid [89].
Nowadays, companies providing 3D MEAs for in vitro recordings comprise 3Brain (Pfäffikon, Switzerland), and NMI Technologietransfer GmbH (Reutlingen, Germany). For more comprehensive reviews on 3D MEAs see [90, 91].
3.2. Neuronal signals, analysis and information provided by MEAs
MEAs are used to measure the electrophysiological activity of networks of electrogenic cells. Upon the occurrence of electrical activity, ions (mostly sodium and potassium) travel across the cell membrane and generate an electric field which can be recorded by means of micro-electrodes placed outside of the cell membrane. In this way, the extracellular voltage that is produced by the cell when it undergoes an AP is recorded [30]. APs constitute the elementary unit associated with the transmission of neuronal signals in a network of functionally connected neurons, and are generated by significant variations of neuronal membrane potential [92]. For this reason, they can be easily recognized for their shape, which is characterized by a rapid rise and subsequent fall of the membrane potential of neurons from a baseline level. MEA systems are equipped with software configured hardware filters allowing to record at different frequency bands (i.e. up to 50 kHz), thus increasing the temporal resolution and enabling the detection of different events, such as extracellular APs and local-field potentials (LFPs). Moreover, depending on the resolution of the devices, the activity recorded by MEAs results from the contribution of more or less neurons. In the majority of MEA studies, characterizing the electrophysiological activity of 2D neuronal networks, extracellular APs (i.e. frequencies higher than 100 Hz) are recorded. Conversely, LFPs (i.e. frequencies lower than 100 Hz) are preferred when characterizing the electrophysiological activity of 3D neuronal structures. Indeed, LFPs are generated in neuronal networks by the summed and synchronous electrical activity of individual neurons, they have multiple sources and they are shaped by the spatial and temporal characteristics of these sources. Quantitative parameters describing LFPs (e.g. frequency, duration, amplitude, power) can be extracted [93, 94].
Extracellular APs are visualized in raw data as spikes, which are sudden changes in the extracellular voltage detected by MEA micro-electrodes, recognizable as actual peaks rising above the background noise, and exceeding a certain threshold [95, 96] (figures 2(a) and (b), in green). This threshold is usually defined relative to the background noise level, which is estimated from portions of the raw signals that do not contain spikes. The choice of this threshold is critical, since it determines which events are retained for further analysis. In MEA recordings, the presence of spikes defines the firing activity of the network under investigation [97].
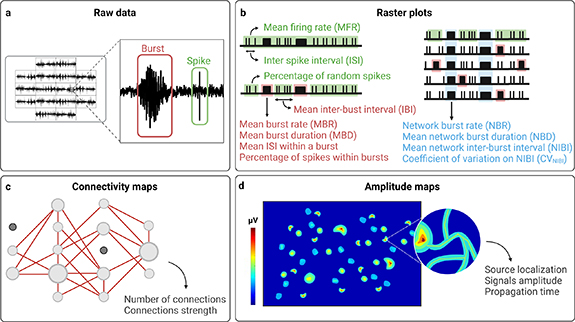
Figure 2. (a), (b) MEA raw data and schematic raster plots with a schematic overview of the most commonly used quantitative parameters which can be extracted from raw data to describe specific characteristics of spiking activity (green), bursting activity (red), and NBs activity (light blue). (c), (d) Schematic connectivity and amplitude maps with some parameters which can be extracted from raw data to describe functional connectivity and propagation of neuronal signals, respectively. Created with BioRender.com.
Download figure:
Standard image High-resolution imageSince APs are triggered only when the threshold potential of neurons is reached, and their amplitude is totally independent of the stimuli amplitude, APs are commonly defined as 'all or nothing' events. Each spike indeed can be considered indistinguishable from the others produced by the same neurons, except for the instant in time at which it occurs [98]. For this reason, variations in neuronal signals are obtained by modifying, not the amplitude, but the occurring of APs over time, and, typically, we refer to neuronal signals as the temporal sequence of APs, also called spike train [99]. The time interval between two spikes is called inter-spike interval (ISI), and the variation of this parameter indicates a change in the dynamics of neuronal signals [99].
During a spike train, periods of quiescence in which spike frequency is relatively low can be interrupted by high-frequency sequences of spikes, which take the name of bursts [23, 100, 101] (figures 2(a) and (b), in red). In MEA recordings, the presence of bursts defines the bursting activity [23, 100, 101]. To date, a common agreement about the definition of bursts has not been reached [98, 102], and a large variety of burst detection methods have been proposed. The simplest approaches involve imposing thresholds on the number of spikes and the maximum allowed ISI between them, classifying any sequence of consecutive spikes satisfying these thresholds as a burst [103, 104]. These thresholds can be chosen by the user [103, 104], or derived from raw data by adaptive burst detection algorithms [105, 106]. Other methods incorporate additional thresholds on relevant parameters (e.g. the minimum interval between two bursts, and the minimum duration of a burst) [107, 108] or take alternative approaches, including the use of statistical techniques [109, 110] (e.g. hidden Markov models [111]). From an electrophysiological point of view, the generation of bursts depends on the interaction between fast, spike-generating membrane conductances, and slower mechanisms that control when spikes occur, allowing to modulate spikes frequency more abruptly [112]. However, the electrophysiological mechanisms underlying bursting activity may vary among different types of neurons [112]. Bursting dynamics are implicated in various phenomena, including synaptic plasticity [113], selective communication between neurons [114], sensory information transmission [112], and dysfunctional states such as epileptic seizures [115].
When neurons are functionally connected in a network, synchronous sequences of spikes spatially distributed across multiple recording channels can be observed [97]. These synchronous rhythmic events, normally involving the whole network, and followed by periods where activity is relatively low, take the name of network bursts (NBs) [105, 116] (figure 2(b), in light blue). NBs are characterized by a phase of increasing activity, which reaches a widespread and intense peak, followed by a relative long-lasting phase of activity, which decreases and finally ends in a refractory silent phase [117, 118]. Due to their complex dynamics, many algorithms have been proposed to identify and detect NBs in MEA recordings, and can be divided in two approaches: (i) those setting a rate-threshold to detect NBs whenever the activity rate (i.e. the number of spikes or active micro-electrodes) exceeds a specific value [97, 117, 119, 120], (ii) those setting an ISI-threshold to detect bursts whenever the ISI between consecutive spikes is less than a specific value, thereby restricting the detection of NBs only when the high activity originates from bursts in different recording channels [98, 105, 116]. Rate-threshold detectors simply bin together the spike times from all recording channels within a specified time window in order to create a firing rate histogram. In its most basic implementation, two parameters need to be set: the time window and the activity rate threshold [97, 117, 119, 120]. Conversely, ISI-threshold detectors consider that periods of low and high ISIs correspond to spikes occurring within and outside of bursts, respectively. At its most basic implementation, the ISI threshold is the only parameter required [98, 105, 116].
Compared to the activity of the adult brain, network bursting activity recorded in in vitro neuronal cultures resembles the spindles observed in the electroencephalogram (EEG) of sleeping brains, as well as epileptic activity [121]. How NBs originate in in vitro neuronal cultures is not completely clear. Experimental evidence suggests that isolated populations of oscillatory neurons within the network spontaneously synchronize and generate periodic bursts involving the whole network [122]. This particular behavior, as compared to the wide repertoire of electrical activity patterns found in wake conditions in vivo, has been partly justified by the absence of afferent inputs [123–125]. In this sense, network bursting activity would represent an exploring dynamic of close in vitro systems that, missing the natural input-output pathways of the in vivo brain, have to find a stable state with properly formed synapses [121]. As it is known from the literature, indeed, network bursting activity has an important role in establishing appropriate connections in the developing brain [126, 127]. In this sense, while spikes and bursts are already visible in the early stages of neuronal development in vitro, NBs are normally observed only later, in mature (i.e. functionally connected) neuronal networks [18, 97, 107, 117, 118, 128, 129].
The signals detected by MEAs can be graphically represented in raster plots, whose visual observation allows to qualitatively appreciate the electrophysiological activity patterns of the neuronal networks under investigation (figure 2(b)). Moreover, quantitative parameters can be extracted to describe specific characteristics of firing activity, bursting activity, and network bursting activity. Some of these parameters are easily obtainable [130], and, for this reason, they are most commonly used (figure 2(b), table 2). These include, for instance, the number of active micro-electrodes (i.e. number of micro-electrodes exhibiting a number of spikes in time exceeding a specific value), and the mean firing rate (MFR, i.e. number of spikes in time per electrode, averaged among all active micro-electrodes in a well), both describing the firing activity of the network. Similarly, the number of actively bursting micro-electrodes (i.e. number of micro-electrodes exhibiting a number of bursts in time exceeding a specific value), and the mean burst rate (MBR, i.e. number of bursts in time per electrode, averaged among all actively bursting micro-electrodes in a well), can be extracted to describe the bursting activity. Bursts can be further characterized by calculating the mean ISI within bursts (i.e. mean time interval between two consecutives spikes within bursts), the mean burst duration (MBD, i.e. average duration of all bursts detected), the mean inter-burst interval (IBI, i.e. mean time interval between two consecutive bursts), and the percentage of spikes within bursts or of random spikes (i.e. not incorporated in bursts). Also NBs, after detection, can be described by quantitative parameters extracted from raw data, including the network burst rate (NBR, i.e. number of NBs detected in time in a well), and the network burst duration (NBD, i.e. average duration of all NBs detected).
Table 2. Examples of the most commonly used parameters that can be extracted from MEA recordings to describe spiking activity, bursting activity, and network activity.
| Type of measure | Parameter | Description |
|---|---|---|
| Spiking activity | Mean firing rate (MFR) | Number of spikes in time per electrode, averaged among all active micro-electrodes in a well. Typically reported in spikes s−1. |
| Number of active micro-electrodes | Number of micro-electrodes exhibiting a number of spikes in time exceeding a specific value | |
| Inter-spike interval (ISI) | Time interval between two consecutive spikes. | |
| Bursting activity | Mean burst rate (MBR) | Number of bursts in time per electrode, averaged among all actively bursting micro-electrodes in a well. Typically reported in burst min−1. |
| Number of actively bursting micro-electrodes | Number of micro-electrodes exhibiting a number of bursts in time exceeding a specific value. | |
| Mean ISI within a burst | Mean time interval between two consecutives spikes within bursts. | |
| Mean burst duration (MBD) | Average duration of all bursts detected. | |
| Mean inter-bust interval (IBI) | Mean time interval between two consecutive bursts. | |
| Percentage of spikes within bursts | Percentage of spikes that occur within bursts. | |
| Percentage of random spikes | Percentage of spikes that are not incorporated in bursts. | |
| Network activity | Network burst rate (NBR) | Number of network bursts detected in time in a well. Typically reported in network bursts min−1. |
| Mean network burst duration (NBD) | Average duration of all network bursts detected | |
| Mean network inter-burst interval (NIBI) | Mean time interval between two consecutive network bursts. | |
| Coefficient of variation on NIBI (CVNIBI) | Calculated by diving the standard deviation of all NIBI values to the mean, the value ranges between 0 (very regular network bursts) to 1 (very irregular network bursts). |
Besides these most common parameters, also others can be extracted ad hoc by using specific algorithms in order to describe specific characteristics of network activity that may arise, for instance, by the visual observation of raster plots. As an example, network bursting activity can be further characterized by extracting the coefficient of variation of the network burst interval (CVNIBI), which allow to better describe the patterns of synchronous rhythmic activity that can be easily appreciated in raster plots [19]. Data from MEA recordings can also be analyzed to estimate functional connectivity [131]. Correlation-based methods (including independent components analysis and various measures of synchrony [132], cross-correlation [133], correlation coefficient [134], and partial correlation [135]) are the most commonly used, and enable to evaluate not only the interactions among the elements of a neuronal network, but also strength of connections, represented in connectivity maps (figure 2(c)). Other algorithms allow to analyze the propagation of neuronal signals (i.e. to identify one or more sources of APs, and to characterize their propagation in the neuronal network) [22]. With HD-MEAs, neuronal signals propagation can be characterized even at sub-cellular level, by tracking APs along neurons axons [136, 137] (figure 2(d)).
The electrophysiological activity of in vitro neuronal cultures is deeply shaped by the underlying physiological characteristics of the neuronal networks under investigation. As it will be largely discussed in the following paragraphs, MEA recordings appear to be markedly different according to (i) the species of origin of neuronal cultures, (ii) the genetic background, (iii) the developmental stage, (iv) the culturing conditions, (v) the type of neurons, and (vi) the region of the brain to which they belong. For instance, by comparing rodent and hiPSCs-derived neuronal networks, differences in spikes rate, network synchronicity and bursts features can be observed [20]. The same applies when comparing the raster plots and the parameters extracted from the same cultures at different days during maturation or differentiation [19, 97] or in different culturing conditions [138, 139], from hiPSCs-derived neuronal networks containing different ratios of excitatory and inhibitory neurons [18, 21], or from neuronal cultures composed of cortical or hippocampal neurons [140]. For this reason, neuronal cultures on MEAs have been identified as optimal platforms for physiology studies and disease models, since by analyzing MEA recordings, precious information about the cellular and molecular mechanisms occurring in the neuronal networks under investigation can be easily obtained.
For the same principle, the electrophysiological activity of neuronal cultures recorded by MEAs appears to be highly sensitive to small changes to the chemical environment, in particular to the presence of any kind of compound able to influence the physiological mechanisms of neurons. A simple example is set by the two main excitatory and inhibitory neurotransmitters (i.e. glutamate and gamma-aminobutyric acid (GABA), respectively) (figure 3). It was observed that the application of rising concentrations of glutamate, or of agonists of glutamate receptors such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA), finely modulated the excitatory synaptic transmission in a concentration-dependent manner, resulting in the electrophysiological activation of networks (i.e. increase of firing and bursting activity) at low concentrations, and in the loss of electrophysiological activity, due to excitotoxic effects, at higher concentrations [23, 140] (figure 3). Conversely, the addition of high concentrations of GABA in hiPSCs-derived neuronal networks including both excitatory glutamatergic and inhibitory GABAergic neurons, completely abolished firing and bursting activity immediately, reflecting the upregulation of inhibitory synaptic transmission over excitatory one [18] (figure 3).
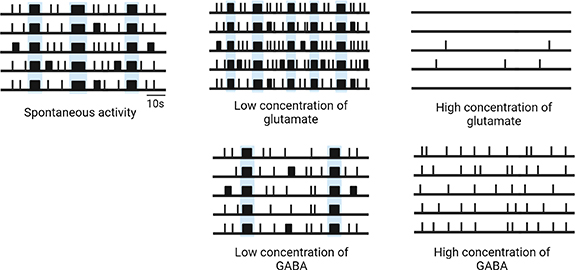
Figure 3. Schematic raster plots describing the effect of low and high concentrations of glutamate and GABA on the spontaneous activity of neuronal networks on MEAs. NBs are highlighted in light blue. Created with BioRender.com.
Download figure:
Standard image High-resolution imagePioneering works from Gross et al [24], and many others following them [23, 25–27], saw the potential to utilize in vitro neuronal networks coupled to MEAs as biosensors. Their studies aimed to identify the most suitable parameters for characterizing their unique substance-specific profiles in response to neurotransmitters, blockers, signaling molecules, drugs, and other neuroactive compounds [23–27], paving the way for today's high-throughput neurotoxicity screenings on MEAs.
3.3. Combination with other techniques
MEA recordings are frequently combined with other electrophysiology techniques, including patch-clamp and calcium imaging, biological methods, such as immunostainings and transcriptome analysis, pharmacological treatment and genetic manipulation, with the aim to obtain more comprehensive information about the characteristics of the neuronal cultures under investigation.
Electrophysiology techniques, such as patch-clamp and MEAs, measuring the electrophysiological activity at different levels and with different resolution, can be combined in order to use the advantages and overcome the limitations of each. MEAs offer the opportunity of recording the electrophysiological activity of neuronal networks, simultaneously and at many sites, allowing a detailed investigation of electrophysiological activity and dynamics at the network level. Conversely, patch-clamp enables to detect those subthreshold signals which could not be recorded by MEAs (e.g. excitatory and inhibitory post-synaptic potentials), and to investigate the contribution of specific ion channel currents to the phenotype observed at the network level, for instance, in disease models [60, 141, 142]. By fine-tuning the setup settings to avoid electrical interference, patch-clamp and MEAs can be combined in the same experiment: while MEA micro-electrodes are recording or stimulating (e.g. to induce synaptic plasticity), single neurons are patched, and intracellular activity is measured [143]. Alternatively, data from MEAs and patch-clamp experiments can be integrated, allowing to obtain a more comprehensive overview of the electrophysiological activity of the neuronal networks under investigation [142].
MEAs can also be combined with calcium imaging to investigate the contribution of intracellular calcium transients to neuronal activity measured at the network level. For this purpose, ad hoc MEA devices and setups have been developed to combine MEAs with optical microscopy [144], allowing to simultaneously record neuronal network activity and measure intracellular calcium transients [145]. Alternatively, data from independent MEAs and calcium imaging experiments can be integrated. This allows, for instance, to study the specific effect of drugs on neuronal network activity and calcium signaling [146], or to evaluate the involvement of calcium-dependent pathways in both physiological (e.g. synaptic plasticity [147]), and pathophysiological mechanisms [148, 149].
Electrophysiological recordings from MEA experiments are frequently integrated also with data obtained through other biological methods, such as immunostainings and transcriptome analysis. Immunostainings techniques can be used in combination with MEA recordings to evaluate, for example, neuronal morphology [20, 150, 151], cell viability [140, 148, 152, 153], and structural connectivity (i.e. anatomical synapses) [131, 154]. Transcriptome analysis, conversely, help researchers shed light on the molecular mechanisms underlying the electrophysiological phenotype observed at the network level, by revealing, for instance, the expression profile of receptors, ionic channels, and other determinants of neuronal electrophysiology [155–157]. Often, neuronal networks on MEAs are pharmacologically treated to block the activity of specific receptors, ionic channels and enzymes, and investigate their contribution to the observed electrophysiological phenotype, hence their involvement in the physiological or pathological mechanisms under investigation [18, 155, 158, 159].
Neuronal cultures on MEAs can also be genetically modified with different purposes. Commonly used techniques of genetic manipulation include genome editing (e.g. CRISPR-Cas9) which allow to insert, replace, or delete DNA sequences, and technologies aiming to induce the expression of heterologous genes (i.e. genes which are not normally expressed in neurons), or to control the expression of receptors, ionic channels, enzymes and other proteins which are normally expressed in neurons but at different levels. These techniques can be used, for instance, to investigate how mutations, genetic variants, or the level of expression of certain proteins affect the electrophysiological phenotype observed at the network level [148, 158, 160–162], but also to label or target specific neuronal populations (e.g. for optogenetic stimulation which allows to activate or inhibit specific neuronal populations [88, 131, 137, 163]).
Numerous examples of studies in which MEA recordings were combined with electrophysiology techniques, biological methods, pharmacological treatment, and genetic manipulation, will be provided in the following chapters.
4. Rodent neuronal cultures on MEAs
To date, dissociated neuronal cultures from rodents have been widely used in neurophysiology as an accessible and economical system to model the complexity of brain physiology and pathology. In this chapter we will first describe the activity exhibited by 2D rodent neuronal cultures on MEAs, and we will look at some relevant studies in which they were combined to MEA technology with different experimental aims (i.e. neurotoxicity screening, disease modeling and drug studies). Afterwards, we will review some of the few studies in which protocols to obtain 3D neuronal networks from dissociated rodent cells on MEAs were developed. Lastly, we will briefly discuss organotypic slice cultures, which provide a compromise between the longevity of dissociated cell cultures and the preservation of the 3D organization of the brain.
4.1. 2D rodent neuronal cultures on MEAs
Rodent neuronal cultures are generally obtained from the embryonic or newborn brain of rats or mice. Neurons and glial cells from the desired brain region (e.g. cortex, hippocampus, thalamus) are first dissociated, and then plated on MEAs on top of the micro-electrodes. After plating, neurons grow out dendrites and axons, and form new synapses including glutamatergic excitatory synapses and GABAergic inhibitory synapses. After a maturation period of 3 weeks, neuronal networks reach a steady state of activity and are considered mature. From that moment they can remain viable for up to several months [164]. In most of the studies with MEAs, glial cells are maintained in culture with neurons to sustain neuronal networks maturation and function. However, their overgrowth can be limited by controlling the composition of the culture medium [159]. In other techniques, such as patch-clamp, it is preferred to deplete them by using anti-glial cell agents (e.g. cytosine arabinoside).
Methods to isolate neurons and glial cells from the brain of adult rats and mice have also been developed. However, protocols are more challenging to perform, and the resulting neuronal networks are more susceptible to stressors and less suited for long-term studies. For this reasons, in this review we will mainly focus on rodent neuronal cultures dissociated from embryonic or newborn rodents, which are the most used in combination with MEAs [165].
4.1.1. Activity exhibited by 2D rodent neuronal networks on MEAs
Regardless of the brain region from which neurons and glia are dissociated, spontaneous activity emerges in rodent neuronal cultures on MEAs toward the end of the first week in vitro, when a few random spikes and almost no bursts can be observed on a small portion of the recording micro-electrodes [97, 118, 128, 166]. At the end of the second week, neuronal networks exhibit a rich and stable activity behavior characterized by an evident increase in firing and bursting activity, both temporally (i.e. firing and bursting rate) and spatially (i.e. number of involved micro-electrodes) [97, 118, 128, 166]. Bursts progressively synchronize on a large number of recording micro-electrodes into long and periodic NBs, and the percentage of spikes within a burst reaches its maximum, indicating that the bursting activity dominates this stage [97, 118, 128, 166]. However, network synchronization is restricted to a few sites and does not involve all the active micro-electrodes. After three weeks in vitro, the network dynamics change dramatically, and long NBs start to be substituted by shorter NBs, with a reduced IBI and increased CVNIBI, denoting a higher variability in the burst generation process. At this age, the network displays frequent bursting patterns and high random firing rate (i.e. spikes not incorporated in bursts), which starts to strongly decrease in the weeks thereafter [97, 118, 128, 166]. During the fourth and the fifth week in culture, the network reaches a stable condition of maturation, exhibiting rich and elaborated temporal patterns of bursting activity, characterized by NBs involving most of the recording micro-electrodes, with nearly similar values for the burst features, and almost no random spikes [97, 118, 128, 166] (figure 4(a), table 3).
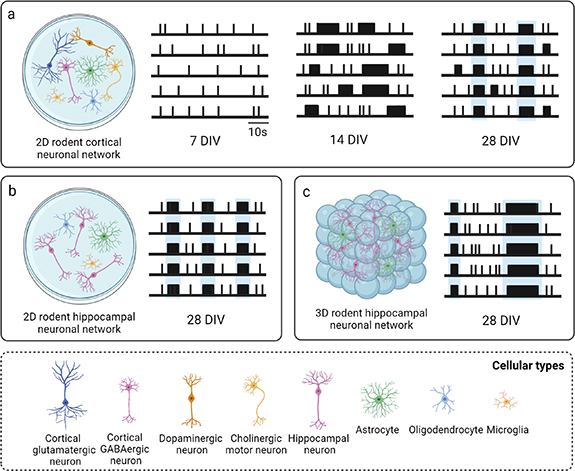
Figure 4. Schematic raster plots describing the spontaneous activity of rodent neuronal networks during development and at maturity, including (a) 2D cortical neuronal networks, (b) 2D hippocampal neuronal networks, and (c) 3D hippocampal neuronal networks. NBs are highlighted in light blue. The most relevant parameters characterizing their mature activity with the respective values are reported in table 3. Created with BioRender.com.
Download figure:
Standard image High-resolution imageTable 3. Representative MEA studies providing a characterization of the spontaneous activity of cortical and hippocampal neuronal networks during development and at maturity, with the most relevant parameters and respective values describing mature activity. DIV = days in vitro, MFR = mean firing rate, MBR = mean burst rate, MBD = mean burst duration, IBI = mean inter-burst interval, PRS = percentage of random spikes, NBR = network burst rate, NBD = network burst duration.
| Neuronal type | Reference | Days in vitro for recording | System and plate format | Micro-electrodes per well | Relevant parameters and values characterizing mature activity | ||||
|---|---|---|---|---|---|---|---|---|---|
| Random spikes | Bursts | Network bursts | Mature activity | ||||||
| 2D | Cortical neurons | Chiappalone et al [103] | 7 DIV | 14 DIV | 14 DIV | 28 DIV | Multichannel System swMEA | 60 | MFR ∼ 1,5 spikes s−1 |
| MBR ∼ 3 bursts min−1 | |||||||||
| MBD ∼ 500 ms | |||||||||
| IBI ∼ 35 s | |||||||||
| Cortical neurons | Charlesworth et al [166] | 7 DIV | 14 DIV | 14 DIV | 28 DIV | Multichannel System swMEA | 60 | MFR ∼ 1 spikes s−1 | |
| MBR ∼ 6 bursts min−1 | |||||||||
| MBD ∼ 1000 ms | |||||||||
| PRS ∼ 25% | |||||||||
| Cortical neurons | Martens et al [142] | 10 DIV | 13 DIV | 13 DIV | 17 DIV | Multichannel System swMEA | 60 | MFR ∼ 1,5 spikes s−1 | |
| PRS ∼ 25% | |||||||||
| NBR ∼ 12 NB min−1 | |||||||||
| Hippocampal neurons | Frega et al [22] | — | — | — | 28 DIV | Multichannel System swMEA | 60 | MBR ∼ 20 bursts min−1 | |
| MBD ∼ 250 ms | |||||||||
| PRS ∼ 15% | |||||||||
| NBR ∼ 20 NB min−1 | |||||||||
| NBD ∼ 600 ms | |||||||||
| Hippocampal neurons | Charlesworth et al [166] | 7 DIV | 14 DIV | 14 DIV | 28 DIV | Multichannel System swMEA | 60 | MFR ∼ 1,2 spikes s−1 | |
| MBR ∼ 5 bursts min−1 | |||||||||
| MBD ∼ 1000 ms | |||||||||
| PRS ∼ 15% | |||||||||
| 3D | Cortical neurons | Smith et al [93] | — | — | — | 21 DIV | Multichannel System swMEA | 60 | MFR ∼ 0,2 spikes s−1 |
| Mean local field potential power ∼ 1 × 10−6 dB mV−2 | |||||||||
| Hippocampal neurons | Frega et al [22] | — | — | — | 28 DIV | Multichannel System swMEA | 60 | MBR ∼ 5 bursts min−1 | |
| MBD ∼ 300 ms | |||||||||
| PRS ∼ 35% | |||||||||
| NBR ∼ 5 NB min−1 | |||||||||
| NBD ∼ 900 ms | |||||||||
The developmental profile of spontaneous activity is similar in rodent neuronal cultures dissociated from the cortex and the hippocampus. Nevertheless, slight differences have been observed both during development and at maturity, and have been reported in the comparative study of Charlesworth et al [166]. Qualitative differences in activity patterns and in the burst shape were already appreciated from a preliminary observation of raw data and raster plots, moreover quantitative differences were found by comparing the parameters extracted from MEA recordings of cortical and hippocampal neuronal networks (figures 4(a) and (b), table 3). In particular, statistically significant differences in firing rate and in the fraction of spikes occurring within bursts (both higher in hippocampal networks), were observed during development, although these differences were no longer significant by the fourth week in vitro [166]. At maturity, three features showed to be critical to differentiate cortical from hippocampal cultures: CVIBI (i.e. hippocampal spike trains tended to fire in bursts that were more regularly spaced than spike trains from cortical neurons), the percentage of theta bursting (i.e. 4–10 Hz oscillations), and the mean correlation between pairs of micro-electrodes, which were both higher in hippocampal networks [166]. However, overall, the spontaneous activity of rodent neuronal networks, dissociated either from the cortex or from the hippocampus, can be considered comparable, except for a few slight differences.
In table 3, we reported some representative MEA studies providing a characterization of the spontaneous activity of cortical and hippocampal neuronal networks during development and at maturity, with the most relevant parameters and their respective values describing mature activity.
According to the research aim, MEA recordings can be performed during network development (e.g. to investigate physiological and pathophysiological mechanisms occurring during development), or later, when neuronal networks are fully mature. Moreover, rodent neuronal cultures dissociated from the cortex or from the hippocampus can be preferred, on the basis of the relevance of the brain region in the physiological mechanism, or disease, under investigation. For instance, cortical neuronal networks represent a good system for physiology studies, and for disease modeling of most neurological disorders. Conversely, neuronal cultures isolated from the hippocampus might be preferred to investigate synaptic plasticity phenomena occurring in the hippocampal circuits, and to model neurodegenerative disorders in which the hippocampus is involved.
4.1.2. Neuronal network physiology studies
Rodent neuronal networks cultured on MEAs are widely used to investigate the physiological mechanisms occurring in the brain.
Neuronal cultures on MEAs are a valuable experimental model for observing changes in the neuronal dynamics at different stages of development [97, 107, 117, 118, 128]. Indeed, the evolution of electrophysiological activity as recorded by MEAs, has been related to the structural and functional changes occurring in neuronal network during the in vitro development, in line with morphological studies. During the first week in vitro, synapses density is not significant, which is reflected by the presence of random spikes recorded by a few micro-electrodes. During the second week, a rapid chemical synaptogenesis occurs, with the exploration of new connections, which is translated into a global excitation, an increased firing and bursting rate, and longer NBs. From the third week, neuronal networks reach a stable state of maturation. The number of synapses undergoes a transient decline, as indicated by the decrease of firing activity, and the structure of synaptic connectivity of the network is shaped [97, 167].
Moreover, the electrophysiological activity as recorded by MEAs at a certain stage of development reflects the presence and activity of the receptors and ion channels which are expressed and functional in that specific moment. In this regard, for instance, a study from Edwards et al compared the spontaneous network activity of embryonic and adult rat hippocampal neurons highlighting marked differences in firing and bursting activity parameters [168]. By exposing the cultures to synaptic transmission antagonists against the excitatory NMDA and AMPA receptors (NMDARs and AMPARs) and comparing the resulting changes in activity, they deduced a significantly different expression profiles of these receptors in embryonic and adult networks [165]. More recently, it was demonstrated that the same excitatory receptors directly influence the duration of bursts and NBs. Specifically, studies using receptor-type specific antagonists showed that AMPARs-driven bursts have short durations, while NMDARs-driven bursts have comparatively longer durations [168].
In addition to developmental studies, rodent neuronal cultures, dissociated from both the cortex and the hippocampus, in combination with MEAs are used to study synaptic plasticity and its involvement in learning and memory consolidation. In the majority of studies, synaptic plasticity is induced through electrical stimulation protocols in which high-frequency (i.e. tetanic stimulation) and/or low-frequency (more physiological) stimuli are delivered by one or more micro-electrodes of the array. Synaptic plasticity phenomena such as long-term potentiation (LTP) and long-term depression (LTD) are reflected by changes (i.e. increase or decrease, in both spontaneous and evoked response to later electrical stimuli) [143, 169–176]. In the work of Jäckel et al, HD-MEA technology was combined with patch-clamp enabling to deliver electrical stimuli at a microsecond resolution to specific neurons, and to identify the contributions of individual presynaptic synapses to postsynaptic potentials [143]. This allowed to induce, for instance, short- and long-term synaptic plasticity through the manipulation of multiple synaptic inputs to specific single neurons [143]. In another recent study, Dias et al investigated the physiological basis of memory consolidation [175]. In particular, they focused on the involvement of cholinergic modulation and synchronized activity. By electrically stimulating cortical neurons in presence of high or low cholinergic tone, they demonstrated that high cholinergic activity, the absence of synchronized patterns, and low network excitability prevented memory consolidation [175] (for more comprehensive reviews about in vitro studies of synaptic plasticity using MEAs see [177]).
MEA devices also give the opportunity to investigate network functional connectivity and signal propagation. For this purpose, MEAs can be incorporated with microfluidic devices in which microchambers are connected by microchannels, narrow enough to prevent the passage of somas, and long enough to allow the passage of axons but not dendrites [178–180]. Unidirectional connections are achieved by adding a 'barbed' design to the microtunnels to hinder axon growth in the opposite direction, or by plating one chamber before the others, allowing axons from only one chamber to fill the microtunnels [181, 182]. By using this approach, Kanagasabapathi et al built a dual compartment system for co-culturing and study the propagation of electrical activities between distinct neuronal sub-populations, specifically between cortical and thalamic networks [183]. Microtunnels have also been to measure signal propagation speed along the axons [184]. In this regard, HD-MEAs are particularly well-suited for the investigation of axonal conduction, since they enable to detect and track APs propagating along neurons axons at high temporal and spatial resolution [136, 137, 185, 186]. For instance, a recent study of Shimba et al used this approach to investigate the spatial characteristics of saltatory conduction along the myelinated axons of peripheral sensory neurons [137].
Neuronal cultures on MEAs can also be used to study the effect of different neuromodulators, such as magnesium [187], endocannabinoids [188], acetylcholine [175, 189] and norepinephrine [190], on neuronal network activity. Some of these modulators have been investigated for their involvement in the sleep-wake rhythms. Indeed, the activity of neuronal networks on MEAs, characterized by synchronized, low frequency firing patterns resulting from the absence of excitatory inputs, is very similar to in vivo slow wave oscillations, which are a key feature of sleep-like state. By modulating the spontaneous firing pattern through the administration of specific neurotransmitters, such as acetylcholine, it was possible to mimic the characteristic high-frequency waves of wakefulness, thereby obtaining a simplified in vitro model of the sleep-wake cycle [191–193].
4.1.3. Neurotoxicity screening
One of the research fields in which MEA technology has evidently proved its potential is neurotoxicology. Indeed, when in 2007 the Nation Academy of Sciences report on 'Toxicity testing in the 21st century' highlighted the need for efficient in vitro methods to screen potentially neurotoxic chemical compounds [194], MEA devices were immediately identified as an ideal platform to perform neurotoxicity screening for a wide number of reasons.
Firstly, the impairment of ion channels, receptors and other determinants of neuronal electrophysiology is a key event in the toxicity pathways of many known neurotoxins, and often precedes or occurs in the absence of other biochemical or morphological changes. Thereby, to perform screenings of neurotoxic compounds, physiological assessment is crucial.
Secondly, as previously pointed out, the activity of neuronal networks as recorded by MEAs appears to be highly sensitive to the presence of any kind of compound able to influence the molecular and cellular mechanisms of neurons [23–27, 140], including neurotoxic ones. Since the electrophysiological activity patterns recorded by MEAs are shaped by the physiological characteristics and mechanisms occurring in the neuronal networks, MEAs can not only be used to detect and identify neurotoxic compounds, but also to classify them on the basis of their mechanism of action and potency [108]. In addition, since neuronal networks cultured on MEAs are viable for up to several months and MEAs are a non-invasive technique, both acute and chronic toxicity can be investigated [195, 196], along with developmental neurotoxicity [108, 197–199].
Lastly, the advantage of using mwMEAs for neurotoxicity screenings is the high throughput. By using mwMEAs, indeed, large numbers of chemicals and drugs are easily and rapidly screened for their neurotoxic potential [200]. To further increase the throughput, single higher concentrations of the compounds under investigation can be screened to identify 'hits' (i.e. compounds which alter specific parameters of the network activity beyond a predefined threshold) [201]. Hits are then followed up with a concentration-response characterization, and a deeper investigation of which parameters are affected, which can give insight about the mechanism of action of the identified neurotoxic compounds. As an example, in 2006 Gramowski et al observed that the administration of three sedative and mild antidepressant herbal extracts to neuronal cultures on MEAs caused a substance-specific decrease in the firing and bursting activity, revealing a common mode of action, but significant differences in potency [202]. By using specific blockers of receptors involved in the inhibitory response they demonstrated that the herbal extracts acted on GABA and serotonin receptors, which are well-recognized targets of pharmacological antidepressant treatments [202].
Beyond the advantage of testing larger numbers of chemicals in short periods of time, mwMEAs also enable parallel screening of neurotoxic compounds in neuronal cultures dissociated from different brain regions, thereby providing information about the potential regional specificity of certain neurotoxic compounds [195].
Several studies indicated the reliability of neurotoxicity screenings on MEAs [203–205] and the comparability of results to in vivo models [206, 207]. For instance, Xia and Gross found that ethanol began to induce a decrease of firing rate in neuronal cultures from the frontal cortex at 20 mM, with EC50 at 48 mM, which are in line with the estimated blood-alcohol concentration (i.e. 20–50 mM) that cause typical behavioral effects in rodents and humans [207]. Similar results were observed in the study of Croom et al who investigated the neurotoxicity kinetics of lindane, a GABAA receptor antagonist which causes seizures in vivo due to increased neuronal excitability. In their study, Croom et al characterized the time- and concentration-dependent neurotoxic effect of lindane (i.e. increase of firing rate) and demonstrated the predictivity of results as regards doses and timing reported in the literature for human and rodents [206].
For all these reasons, MEA devices are considered a promising platform to perform high throughput and rapid screenings for neurotoxicity testing, and they have already been widely used to characterize the neurotoxic effect of a large number of compounds, such as chemicals commonly contained in insecticides, bactericides and fungicides [196, 208–211], metals [199, 212–214], bacterial toxins [215], drugs [195, 202, 212, 216, 217], and psychoactive substances [218, 219] (for more comprehensive reviews on the use of MEAs for neurotoxicity screenings see [220, 221]).
4.1.4. Disease modeling
Another research field in which MEA technology shows its potential is in vitro modeling of neurological disorders. Similarly to what observed in neurotoxicity, in neuropathology the impairment of ion channels and receptors involved in neuronal electrophysiology and synaptic communication quickly occurs, resulting in visible alterations of neuronal activity. In this sense, MEAs represent an ideal platform to perform functional phenotyping assays. By comparing the electrophysiological activity of affected neuronal networks with healthy controls, it is possible to define a phenotype for the disease under investigation. Once established, a well-defined disease model can give insights about underlying molecular and cellular pathological mechanisms and potential therapeutical targets, or it can be used to test candidate treatment strategies.
Several neurological disorders have been modeled by using rodent neuronal networks cultured on MEAs, including Alzheimer's disease (AD), dementia with Lewi's bodies (DLB), epilepsy, autism spectrum disorders (ASD), psychiatric disorders, such as schizophrenia (SCZ) and bipolar disorder (BP), Kleefstra syndrome (KS), Fragile X syndrome (FXS) and other brain genetic diseases, encephalitis, ischemic stroke, and traumatic brain injury (TBI). An overview of MEA studies using 2D rodent cultures in disease modeling is found in table 4.
Table 4. Overview of MEA studies using 2D rodent cultures in disease modeling. AD = Alzheimer's disease, DLB = dementia with Lewi's bodies, ASD = autism spectrum disorder, TSC = tuberous sclerosis complex, FXS = fragile X syndrome, TBI = traumatic brain injury, AE = autoimmune encephalopathies. MFR = mean firing rate, ISI = inter-spike interval, MBR = mean burst rate, MBD = mean burst duration, IBI = mean inter-burst interval, PRS = percentage of random spikes, PSB = percentage of spikes within bursts, NBR = network burst rate, NBD = network burst duration, NIBI = mean network inter-burst interval, CV = coefficient of variation.
| Reference | Disease model | Neuronal cultures | How the model was obtained | System and plate format | Micro-electrodes per well | Extracted quantitative parameters and phenotype on MEA |
|---|---|---|---|---|---|---|
| Görtz et al [222] | AD | Cortical cultures | Treatment with Aβ peptides | Multichannel System swMEA | 60 | MFR ↓, MBR ↓ |
| Charkhkar et al [159] | AD | Cortical cultures | Treatment with Aβ oligomers | ALA Scientific Instruments swMEA | 60 | MFR ↓ |
| Varghese et al [141] | AD | Hippocampal cultures | Treatment with Aβ oligomers | Multichannel System swMEA | 60 | MFR ↓ |
| Gao et al [223] | AD | Hippocampal cultures | Treatment with Aβ oligomers | Multichannel System swMEA | 60 | MFR ↓, spike duration, peak to peak value |
| Mitroshina et al [148] | AD | Hippocampal cultures | Treatment with Aβ oligomers | Multichannel System swMEA | 60 | MFR ↓, NBR ↓, PSB, number of key network elements (hubs) and connections |
| Theiss et al [224] | DLB | Hippocampal cultures | Application of CSF from DLB and PD patients | Multichannel System swMEA | 60 | MFR ↓, NBR ↓, peak firing rate |
| Hales et al [225] | Epilepsy | Cortical and hippocampal cultures | Electrical stimulation | Multichannel System swMEA | 60 | Visual observation of average power spectra |
| Vedunova et al [226] | Epilepsy | Hippocampal cultures | Treatment with hyaluronidase | Alpha MED Scientific swMEA | 64 | MFR ↑, MBR ↓, number of superbursts ↑, IBI ↓, PSB ↓ |
| Colombi et al [227] | Epilepsy | Hippocampal cultures | Treatment with bicuculline (BIC) | Multichannel System swMEA | 60 | MFR↑, MBR ↑, MBD ↑, PSB ↑ |
| Gullo et al [228] | Epilepsy | Cortical cultures | Neurons dissociated from FVB-Tg(tTA:Chrnb2V287L) transgenic rodents | Multichannel System swMEA | 60 | Autocorrelation function, MBD ↑, MFR ↑, PSB, IBI, Fano factor |
| Jewett et al [158] | Epilepsy | Cortical cultures | Treatment with picrotoxin (PTX) | Axion Biosystems swMEA | 64 | MFR ↑, synchrony index ↑, PSB ↑ |
| McSweeney et al [229] | Epilepsy | Cortical cultures | miRNA-128 knockdown | Axion Biosystems 12wMEA | 64 | MFR ↑, MBR ↑, PSB ↑ |
| Kreir et al [230] | Epilepsy | Cortical cultures | Treatment with 8 epileptogenic compounds | Axion Biosystems 48wMEA | 16 | MFR ↑, MBR, MBD ↑, PSB, IBI, CVIBI, NBR ↑, NBD, CVNIBI, half width at half height of normalized cross correlation |
| Bradley et al [231] | Epilepsy | Cortical cultures | Treatment with 15 epileptogenic compounds | Axion Biosystems 48wMEA | 16 | MFR ↑, MBR ↑, PSB ↑, PRS, CVISI, MBD ↑, IBI |
| Fan et al [232] | Epilepsy | Hippocampal cultures | Treatment with 6 epileptogenic compounds | Multichannel System swMEA | 60 | MFR ↑ |
| Axion Biosystems | 16 | MFR ↑ | ||||
| 48wMEA | ||||||
| Ahtiainen et al [233] | Epilepsy | Cortical cultures | Treatment with 4-aminopyridine (4-AP) and gabazine | Multichannel System swMEA | 60 | MFR ↑, MBR ↑, number of active electrodes, PSB, IBI, fold change of MFR and MBR |
| Mincheva-Tasheva et al [234] | PCDH19-Clustering Epilepsy (PCDH19-CE) | Hippocampal cultures | Neurons dissociated from Pcdh19 knockout rodents and genome editing of Pcdh19 | Multichannel System 24wMEA | 12 | MFR, MBD, PSB |
| Erata et al [235] | Epilepsy Aphasia Syndrome (EAS) | Cortical cultures | Neurons dissociated from Cnksr2 knockout rodents | Axion Biosystems | 16 | MFR ↑, MBR ↑ |
| 48wMEA | ||||||
| Martens et al [142] | Kleefstra syndrome | Cortical cultures | Downregulation of Ehmt1 gene | Multichannel System swMEA | 60 | MFR, MBR, MBD, IBI, CVISI, Fano Factor, CVIBI ↑, independent measure for irregularity, autocovariance |
| Frega et al [236] | Kleefstra syndrome | Cortical cultures | Downregulation of Ehmt1, Smarcb1, Mll3, or Mbd5 genes | Multichannel System swMEA | 60 | MFR ↑, number of active and bursting electrodes, MBR, MBD, IBI, NBR ↑, NBD, NIBI, PRS ↑ |
| Lu et al [237] | ASD | Cortical cultures | Neurons dissociated from Shank3 knockout rodents | Multichannel System 6wMEA | 9 | MFR ↓, array-wide spike detection rate |
| Bateup et al [238] | TSC | Hippocampal cultures | Tsc1 gene knockout | Alpha MED Scientific swMEA | 32 | MFR ↑ |
| MacLaren et al [239] | Schizophrenia and bipolar disorder | Hippocampal cultures | Knockdown of mental disorder susceptibility genes | Multichannel System swMEA | 64 | MFR, PSB ↑, MBR, MBD, burst pattern ↓, network size, correlation index |
| Moskalyuk et al [240] | FXS | Cortical cultures | Neurons dissociated from Fmr1 knockout rodents | Multichannel System swMEA | 60 | Number of active electrodes, MFR ↑, MBR ↑, MBD ↑, IBI ↓, PSB ↑ |
| Cao et al [241] | Fragile X-associated tremor/ataxia syndrome (FXTAS) | Hippocampal cultures | Neurons dissociated from preCGG knock-in rodents | Alpha MED Scientific swMEA | 64 | MFR ↑, MBR, MBD ↑ |
| Feng et al [242] | Niemann-Pick Type C1 (NPC1) disease | Cortical cultures | Neurons dissociated from NPC1 mutant rodents | Axion Biosystems | 16 | MFR ↓, MBR ↓, MBD, burst period ↑, IBI, PSB, event rate ↓, event period ↑ |
| 48wMEA | ||||||
| Le Feber et al [243–245] | Ischemic stroke | Cortical cultures | Exposition to hypoxia | Multichannel System swMEA | 60 | Array-wide firing rate ↓, post-stimulus time histograms, functional connectivity ↓ |
| Muzzi et al [246] | Ischemic stroke | Cortical cultures | Exposition to hypoxia | Multichannel System swMEA | 60 | Array-wide firing rate ↓, number of active electrodes |
| Prado et al [247] | TBI | Cortical cultures | Mechanical insult | In-house fabrication swMEA | 64 | MFR ↓, IBI ↑, number of spikes within bursts ↓ |
| Rogers et al [248] | TBI | Cortical cultures | Mechanical insult | In-house fabrication swMEA | 64 | MFR ↓ |
| Otto et al [249] | TBI | Cortical cultures | Application of CSF from TBI patients | Multichannel System swMEA | 60 | MFR ↑, spike amplitude, MBR ↓, IBI, ISI, MBD, intra-burst inter-spike interval ↑, PSB ↓, agreement coefficient Cohen's kappa |
| Schwarz et al [250] | Hepatic encephalopathy (HE) | Cortical cultures | Treatment with ammonium chloride (NH4Cl) | Multichannel System swMEA | 60 | MFR ↑, MBR ↑, agreement coefficient Cohen's kappa (i.e. degree of synchrony) ↓ |
| Jantzen et al [251] | AE | Cortical cultures | Application of CSF from AE patients | Multichannel System swMEA | 60 | MFR ↓, MBR ↓, agreement coefficient Cohen's kappa (i.e. degree of synchrony) |
| Koch et al [252] | AE | Hippocampal cultures | Application of CSF from AE patients | Multichannel System swMEA | 60 | MFR ↑, NBR ↑ |
MEA-based models of neurological diseases can be obtained in different ways: (i) treatment of neuronal cultures dissociated from healthy rodents (e.g. treatment with drugs, neurotoxic compounds, electrical stimulation, mechanical insults), (ii) genetic manipulation of neuronal cultures dissociated from healthy rodents, and (iii) dissociation of neuronal cultures from rodents carrying pathogenic mutations or genetic variants linked to specific neurological disorders.
Neuronal cultures can be dissociated from specific brain regions of healthy mice or rats, cultured on MEAs and then treated in order to mimic the desired pathological condition. In this regard, a typical example is epilepsy, which is defined by a wide spectrum of excitability disorders and disposes of several in vitro models, among which many on MEAs. An epileptic phenotype, in this case, is easily achieved by treating healthy neuronal cultures with the application of drugs. Some recent studies took advantage of the high throughput potential of mwMEAs to perform a screening of well-known and candidate epileptogenic compounds for their ability to increase neuronal network activity [230–232]. The resulting phenotype was characterized by increased spike, burst, and network burst rates as well as enhanced synchronicity, which are common features of epileptic phenotypes. Commonly used drugs to induce epileptic phenotypes in neuronal cultures on MEAs include the GABA antagonists pentylenetetrazol, picrotoxin (PTX), bicuculline (BIC), gabazine, and endosulfan, along with several other compounds, including the potassium channel blocker 4-aminopyridine (4-AP), and the excitatory neurotransmitters glutamate and NMDA [108, 158, 196, 200, 227, 230–233, 253].
Alternatively, in vitro models of epilepsy on MEAs can be obtained by electrically stimulating neuronal networks cultured on MEAs. In this case, the ability of MEAs to deliver electrical stimuli through the recording micro-electrodes in a predefined fashion represents a convenient advantage. As an example, Hales et al used electrical stimulation on both hippocampal and cortical neuronal cultures in order to obtain and investigate the pathophysiology of evoked high frequency oscillations, which are a common feature of in vivo epilepsy models [225].
Once established, a MEA-based model can give insight about the underpinning molecular and cellular mechanisms contributing to the disease, thereby revealing new candidates for treatment strategies. As an example, Jewett et al investigated the role of Mdm2-p53-Nedd4-2 signaling in a PTX-induced epilepsy model. They observed that (i) the elevation of neuronal activity induced by PTX triggered Mdm2-dependent degradation of the tumor suppressor p53, (ii) blocking p53 degradation further enhanced neuronal network synchrony, (iii) genetically reducing the expression of Nedd4-2, a direct target gene of p53, elevated neuronal network activity, occluding the effect of a subsequent administration of PTX [158]. All together these results suggested a role for Mdm2-p53-Nedd4-2 signaling in the regulation of neuronal network synchrony, thus in seizure susceptibility, revealing a new potential therapeutic target for hyperexcitability-associated disorders [158].
Also neurodegenerative disorders, such as AD, can be modeled in vitro on MEAs by treating healthy neuronal cultures to mimic the pathological condition. The most commonly used AD models are obtained through the application of amyloid-β (Aβ) peptides or oligomers, which play a well-recognized key-role in early AD pathophysiology, long before amyloid plaque formation and neurodegeneration occurring [141, 148, 159, 222, 223]. The use of a MEA-based model, by allowing to record the electrophysiological activity on the same population of cells over an extended period of time, enables to investigate both the acute and chronic neurotoxic effects of Aβ oligomers on neuronal network activity. The results reveal a progressive decrease in both firing and bursting activity which was related to the degeneration of neuronal network functionality and connectivity [141, 159, 222, 223]. Moreover, by using one of these models, Charkhkar et al not only characterized the concentration-dependent neurotoxic effect of Aβ peptides and oligomers on hippocampal neuronal networks, but also demonstrated the involvement of ionotropic glutamate receptors in the neurodegenerative pathways induced by Aβ oligomers, by using a specific NMDARs antagonist [159].
Another approach to model neurological disorders on MEAs consists in treating healthy rodent neuronal cultures with the cerebrospinal fluid (CSF) from patients suffering from a specific disease, such as DLB, Parkinson's disease (PD), TBI, and autoimmune encephalopathies (AE) associated with auto-antibodies against different proteins of the nervous system [224, 251, 252, 254]. Neurological diseases, indeed, frequently induce pathological changes of CSF. As the CSF-brain barrier is partially permeable, particularly under pathological conditions, this might secondarily influence brain activity. In all the reported studies, the application of CSF from patients induced disease-specific alterations in the neuronal network activity recorded by MEAs as compared to the application of CSF from control healthy subjects. For instance, the exposition to CSF from DLB patients led to a reduced spike and burst rate compared to CSF from PD patients and controls [224]. Investigating the disease-specific changes in neuronal networks activity not only have a diagnostic potential, but can give insights about the pathological mechanisms caused or aggravated by factors present in the CSF from patients. For example, Otto et al reported that CSF samples from patients with TBI suppresses synchronous activity of in vitro neuronal networks partially driven by increased NMDA receptors activity [254]. Similarly, Jantzen et al investigated the effects induced by the application of CSF from patients with anti-NMDARs encephalitis on the activity of neuronal networks, and found that it suppressed global spike and burst rate, but left unchanged synchronicity, thereby suggesting that auto-antibodies against NMDARs selectively regulate distinct parameters of the network activity while sparing their functional connectivity [251].
Pathological conditions such as cerebral ischemic stroke and TBI were mimicked on MEAs by exposing healthy rodent neuronal cultures to low levels of oxygen (i.e. hypoxia) or by simulating a concussion with a mechanical insult, respectively [243–245,246–248]. To model cerebral ischemic stroke, which is associated with failure of brain circulation, the group of Le Feber exposed cortical neuronal networks cultured on MEAs to controlled hypoxic conditions of varying depth and duration [243–245]. By using a MEA-based model, they had the opportunity to investigate the progression of damage during time (observed as a decrease of general electrophysiological activity), and the recovery or further deterioration, often decisive, occurring at longer timescales [243–245]. In such a way, they found that synaptic failure occurs rapidly after the induction of hypoxia, while neurons remain viable and able to generate APs for hours. Moreover, they observed that low activity induced by hypoxia triggered compensatory mechanisms aiming to maintain the total network activity within a certain range. Interestingly, the increased excitability of networks exposed to hypoxia only became apparent upon reoxygenation, when higher stimulus responses than during baseline were observed [243–245].
A different approach to model disease with neuronal cultures dissociated from healthy rodents and cultured on MEAs is by genetically modifying them. Naturally, this approach fully expresses its potential in disease modeling of neurological disorders with a more or less relevant genetic component. Different techniques of genetic manipulation, such as RNA interference and Cre-Lox recombination system, are used in order to perform a knockdown (i.e. reduce expression) or a knockout (i.e. disrupt expression) of one or more genes involved in the disease under investigation [142, 229, 236, 238, 239]. For instance, McSweeney et al downregulated the expression of the microRNA-128 in cortical neuronal networks cultured on MEAs, in order to mimic a pathological condition called mature microRNA-128 deficiency and characterized by lethal seizures in mice [229]. In line with clinical evidence, they observed that the reduced expression of microRNA-128 in neuronal networks induced a significant increase of firing rate, burst rate, and burst duration, reflecting a global increase of excitability [229].
Another example has been set by Martens et al and Frega et al which used genetic manipulation to alter the expression of genes involved in KS, a neurodevelopmental disorder characterized by autistic-like features and severe intellectual disability [142, 236]. The knockout of well-known genes causative for KS was achieved through RNA interference during the development of cortical neuronal networks on MEAs, thereby giving the opportunity to investigate the pathophysiology of KS during neurodevelopment. By combining MEAs and patch clamp recordings, they observed that the loss of function of genes causative for KS impaired the neural network activity during the transition from random spiking to synchronized network bursting, resulting in less regular network bursting patterns later in development, and, ultimately, in hyperactive networks with altered network organization and excitatory-inhibitory balance [142, 236]. Similarly, MacLaren and colleagues used RNA interference in order to downregulate the expression of four susceptibility genes for SCZ and BP, and investigate the resulting effects on the network activity of hippocampal networks cultured on MEAs [239]. The knockdown of genes associated with psychiatric disorders resulted in abnormal hyperactivity, as reflected by an increase in either bursting rate, duration and pattern, and disrupted development of neural networks in vitro [239].
The MEA-based disease models described up to this point have a common feature: they are all based on neuronal cultures dissociated from healthy rodents, cultured on MEA devices and treated with drugs, electrical stimulation, genetic manipulation, neurotoxic compounds or other conditions, in order to mimic the neurological disorder under investigation. Another approach is to directly dissociate neuronal cultures from the brain of rodents carrying pathogenic mutations or genetic variants linked to specific neurological disorders, which can be obtained by genetically modifying embryos and via selective breeding.
One of the advantages of this approach is that nowadays countless strains of mice and rats with well-defined genotypes and phenotypes, and the purpose to be used as reproducible disease models, are commercially available. Besides in vivo experiments, they can be utilized for the isolation of neuronal cultures for in vitro experiments, including electrophysiology studies with MEAs. Similarly to disease models based on genetic manipulation of healthy neuronal cultures, this approach is clearly more suitable for modeling genetic disorders. Examples of neurological disorders which have been modeled on MEAs by isolating and culturing neuronal cultures from rodents carrying specific pathogenic mutations include Autosomal Dominant Sleep-related Hypermotor Epilepsy (ADSHE) [228, 255] (formerly known as Autosomal Dominant Nocturnal Frontal Lobe Epilepsy, ADNFLE), Epilepsy Aphasia Syndromes (EAS) [235], PCDH19-Clustering Epilepsy (PCDH19-CE) [234], ASD [237], FXS [240, 241], and Niemann-Pick Type C1 (NPC1) disease [242].
As an example, Gullo et al isolated cortical neuronal networks from a transgenic line of mice expressing mutant neuronal nicotinic acetylcholine receptors (nAChR) which cause a partial sleep-related epilepsy called ADSHE [228]. Once cultured on MEAs, ADSHE neuronal networks showed spontaneous hyperexcitability (i.e. considerably higher burst duration, and systematically generated prolonged synchronized bursts, with durations of 20–30 s). Moreover, in patch-clamp recordings, no distortion of the AP waveform was observed, suggesting that the mutation mainly affected the excitatory/inhibitory synaptic dynamics at the network level [228]. Interestingly, the spontaneous nature of the long bursts observed in ADSHE neuronal networks, and the similarity between their duration and the average duration of seizures in transgenic mice suggested that the abnormal excitability of the cortical cultures in vitro might reproduce some features of in vivo seizures [228].
In vitro models derived from genetically modified rodents allow to compare and integrate the results from experiments performed in vitro (e.g. electrophysiological recordings, cellular and molecular analysis) and in vivo (e.g. EEG data, behavioral and cognitive functions testing) [142, 236, 256]. As an example, Erata et al generated a line of transgenic mice carrying a knock-out mutation of CNKSR2, which in vivo causes a highly disruptive disorder (i.e. EAS), during infancy and childhood [235]. In their study, they investigated the behavioral, electrophysiological, and molecular changes resulting from Cnksr2 loss both in vivo and in vitro, by combining the results obtained from MEA recordings, EEG data, behavioral and cognitive functions tests, and molecular analysis [235]. The epileptiform phenotype was observed in MEAs and EEG recordings, and confirmed by spontaneous seizures in vivo. Moreover, Cnksr2 KO mice displayed significantly increased anxiety, impaired learning and memory, and a progressive and dramatic loss of ultrasonic vocalizations, which all are phenotypic traits strikingly similar to those of human patients [235]. Lastly, proteomics analysis reveals that Cnksr2 loss resulted in significant alterations of the synaptic proteome, including proteins implicated in epilepsy disorders. Altogether their results validated that loss of CNKSR2 leads to EAS, and highlighted its role in synaptic organization and neuronal network activity [235].
4.1.5. Drug studies
Once established, MEA-based disease models can be used as a point of reference for phenotypic rescue experiments using genetic interventions or other pharmacological and non-pharmacological treatments. Indeed, in the majority of the studies cited in the previous paragraph, researchers took advantage of their newly established disease models on MEAs to test both well-known and candidate treatments strategies aiming to improve, or ideally reverse, the pathophysiological phenotype as defined by the analysis of MEA recordings [148, 158, 223, 225, 228, 241, 242].
Well-known drugs can be used to provide a further proof of validity of the newly established MEA-based disease models [159, 227, 228]. As an example, Colombi et al studied the acute pharmacological effects of two commonly used antiepileptic drugs, carbamazepine and valproate, in an in vitro model of epilepsy, based on hippocampal neuronal networks cultured on MEAs and treated with the pro-convulsant BIC [227]. Both of them showed to reduce the BIC-induced abnormal neuronal network activity in terms of firing and burst rate, at concentrations close to those clinically relevant, indicating a relationship between the drug sensitivity of neuronal networks cultured on MEAs and the expected effects in vivo [227].
Moreover, MEA devices represent an ideal platform to perform rapid high-throughput screenings. Healthy neuronal networks on MEAs are used to study, for instance, well-known drugs and non-pharmacological treatments, such as hypothermia and neural stimulation techniques, which are already commercially available and widely used in clinic, with the aim to investigate their therapeutic and potential side-effects effect on neuronal network activity and connectivity [119, 216, 217, 257–259]. Screenings can be also performed on neuronal cultures dissociated from different brain regions to identify potential tissue-specificity [119, 217, 260].
Alternatively, candidate treatment strategies can be tested, and, from the results, dose-response curves and kinetics of the pharmacological effects and chemical dissociation are easily defined [119, 216, 260, 261]. In this regard MEA devices have the undeniable advantage of being a non-invasive technique, thus allowing the measurement of electrophysiological activity of the same neuronal network for a long period of time, which is more like to what occurs in patients. For instance, Varghese et al took advantage of this aspect to test the effect of curcumin, an inhibitor of Aβ oligomerization, in their in vitro model of AD [141]. As stated by the authors, the use of MEAs, compared to patch-clamp experiments (which were performed in the same study), allowed them to follow the time course of the positive action of curcumin on the Aβ-modified activity of the same populations of cells over a longer period of time, thereby closer to the clinical scenario [136].
The characterization of disease phenotype offered by MEA-based models can give precious insights about the underlying molecular and cellular pathophysiological mechanisms, thereby suggesting new potential therapeutical targets. Similarly, by observing the effects of specific treatments on the neuronal network activity as recorded by MEAs, the mechanisms of action of these treatments can be further investigated, and new ones can be uncovered. As an example, Hales et al, by using their model of epilepsy characterized by electrical stimulation-evoked high frequency oscillations, found that the addition of carbenoxolone (i.e. a putative gap junction blocker) either reduced the amplitude and duration of the oscillations or completely abolished them, while the pharmacological blockade of voltage-dependent sodium channels and direct synaptic blockade were ineffective [225]. This was in line with previous studies suggesting that pathological high-frequency oscillations may be due to direct electrical coupling. Moreover, clinical trials utilizing carbenoxolone demonstrated improved cognitive scores in elderly man. Although the effect was mainly attributed to other mechanisms (i.e. reductions in glucocorticoid concentrations), an alternative hypothesis based on this work suggested that a decreasing in pathological high frequency oscillations may also be responsible for the improved cognition [225].
It is worth underlying that, besides traditional pharmacological and non-pharmacological treatments, disease models based on MEAs also allow genetic interventions. In this case, genetic techniques can be used to manipulate the DNA in order, for instance, to reverse a pathogenic mutation in a genetic neurological disorder model [148, 158]. The aim might be to link or to quantify the contribution of a specific mutation to the observed pathological phenotype, but also to test new treatment strategies based on genetic manipulation, such as gene therapy.
4.2. 3D rodent neuronal cultures on MEAs
2D neuronal cultures are relatively simple to obtain and show clear advantages related to controllability and observability. However, they also have major limitations, as they are inherently unable to exhibit certain characteristics of in vivo systems. Indeed, it has become clear that morphological and electrophysiological properties of neuronal cells are substantially influenced by their immediate extracellular surroundings, yet the features of this environment are difficult to mimic in vitro [9–11].
In 2D neuronal models the structure is constrained by the presence of a rigid planar substrate, and this implies that the morphology of neurons is unrealistically flat, the interactions with glial cells and components of the ECM are limited to 2D, and axons-dendrites outgrowth cannot occur in all directions [9–11]. Moreover, various criticisms have been introduced on the validity of 2D models in order to investigate neuronal network activity. One of the major issues is related to the poor dynamics exhibited by 2D neuronal networks, often dominated by bursting activity encompassing most of the neurons in the network [121, 128], which is in contrast to the wide repertoire of electrical activity patterns found in in vivo studies [123, 124].
From this perspective, it is clear that 2D models for studying the characteristic of in vivo systems are extremely reduced, and that there is a tremendous need to develop culture systems that more closely model the complexity of the brain. These reasons justified the combined push in the neurophysiology community to introduce in vitro models based on 3D neuronal networks, which would allow the investigation of cellular behavior and network activity in a more physiologically relevant environment. A growing body of evidence, indeed, suggests that neurons grown in 3D cultures better represent in vivo cellular behavior than cells cultured in monolayers, as regards gene expression, differentiation, morphology, cellular signaling, and viability [9–11]. 3D neuronal cultures may also be more appropriate for electrophysiological studies than 2D counterparts. For instance, in 3D culture, Na+/H+ exchangers have been shown to have polarized expression to the apical membrane, which is difficult to maintain in monolayer cultures [262]. Moreover, neurons in a 2D environment show exaggerated Ca2+ dynamics in comparison to 3D cultures [263]. Lastly, it is worth underlying that 3D neuronal cultures preserve the primary advantages of traditional in vitro systems, such as control of cellular environment, and accessibility for repeated imaging.
To obtain 3D neuronal cultures from dissociated rodent neurons, biocompatible scaffolds mimicking the ECM are employed. This allows neurons and glial cells to grown within it, reproducing the 3D cytoarchitecture of the neuronal tissue in the brain. In the past years, two main approaches have been developed: (i) ECM-based scaffolds, and (ii) microbeads-based scaffolds.
For the design of ECM-based scaffolds, numerous substrates have been successfully developed, including biopolymers [93, 264], agarose [265], collagen [88, 266], chitosan [267, 268], silk proteins [269] and gel-like substances [267]. The majority of them are biocompatible polymer gels or solid porous matrices, that can be further coated with specific ECM components to support cell development, and guide axons-dendrites outgrowth [270]. It has been shown that the use of these materials induces the spontaneous formation of 3D neuronal networks with arborizations in the 3D space, closely representing the in vivo conditions [93, 265, 271]. Although this approach is very versatile, only a few studies used MEAs to characterize the electrophysiological activity of 3D neuronal networks grown within ECM-based scaffolds [88, 93, 266, 268], and some of them pointed out some limitations.
As an example, Smith et al utilized a 3D cell culture scaffold made of Alvetex, a polystyrene-based material, comprised of voids of variable sizes with interconnecting pores in which rodent mixed neuronal-glial cultures were seeded [93]. Interestingly, astrocytes morphology was notably different (i.e. extensive, intricate processes), more consistent with the one observed in vivo in comparison to 2D cultures. Moreover, by using planar MEAs, the authors demonstrated that primary neuronal-glial cultures could successfully grow in Alvetex 3D scaffolds, producing 3D neuronal networks that exhibited a spontaneous electrophysiological activity with features closer to the ones of brain slices, rather than 2D cultures of the same cells [93]. In particular, the firing rate observed in 3D cultures was significantly lower than that seen in 2D cultures, although immunostainings showed that this lower rate was not a result of a loss of viability, and LFPs occurred more frequently outside burst events. In the discussion, they suggested that the lower firing rate may arise from technical limitations related to the use planar MEAs to record the activity of 3D neuronal networks (i.e. signal attenuation due to a greater distance between cells and MEA micro-electrodes) and the possibility that some viable neurons within the 3D scaffolds lay outside of the MEA micro-electrodes receptive fields [93].
Another approach to obtain 3D neuronal cultures from dissociated rodent neurons is based on the use of microbeads. This innovative technique was proposed for the first time in 2008 by Patout et al, and involves the use of microbeads to build a modular 3D scaffold for the growth of 3D neuronal networks [266]. In Patout et al work, silica microbeads coated with ECM components were designed to be large enough to provide an adhesion surface for neuronal cell bodies, and to support growth and differentiation of axons and dendrites in the 3D space [272]. Microbeads were added in succession and spontaneously assembled into ordered 3D hexagonal arrays. Layers containing distinct subsets of neurons were formed, and neuronal processes could grow creating synaptic connections between neurons on different beads and from different layers. Layers of beads coated with chemical attractants were included to promote axonal growth and orient functional neuronal connections between different layers [272]. In the resulting 3D neuronal cultures, neuronal processes grew between the beads over the course of 3 weeks in culture to form highly interconnected networks. Despite the small number of glial cells on each bead, they were sufficient to maintain neurons health and support uniform and robust neuronal processes outgrowth throughout the bead layers. Moreover, the void spaces between the beads permitted the necessary medium exchange to maintain healthy growing conditions. Both the number of cells per bead and the number of neuronal processes were similar in all layers of the array, sign of uniform neuronal cultures [272].
The main advantage of using microbeads-based scaffolds, as compared to other methods (such as ECM-based scaffolds), is that they allow to have a high control over the final structure, organization and cell density of 3D neuronal networks, depending on the density of cells during plating and on the dimension of beads. Moreover, the final density of cells achieved in 3D neuronal cultures within microbeads-based scaffolds (i.e. 75 000 cells mm−3 in Patout et al [272].) is close to the 91 000 cells mm−3 observed in vivo [273], and definitely higher than the density for optimal viability in biocompatible polymer gels or solid porous matrices (i.e. ∼4000 cells mm−3).
Regarding electrophysiological recording on MEAs, microbeads-based scaffolds demonstrated to be well-suited to record the electrophysiological activity of 3D neuronal networks [22, 274, 275]. In a study of Frega et al, the electrophysiological activity of 3D cortical networks grown on beads was recorded and compared with the one exhibited by 2D cultures [22]. Rodent hippocampal neurons were cultured in 5–8 interconnected layers of glass microbeads coated with adhesion molecules on planar MEAs. MEA recordings showed that the quasi-synchronous network bursting observed in 2D neuronal cultures was maintained also in 3D networks, while global frequency was decreased and global synchrony was lost [22] (figure 4(c), table 3). In the discussion, they speculated that global asynchronous activity patterns may result from the higher complexity of the network. In particular, the wider and longer interactions may contribute to desynchronize and temporally differentiate the network activity, producing bursting activities confined to sub-populations in the network [22]. More recently, chitosan microbeads were used as 3D scaffold for rodent neuronal cultures on MEAs [274]. Chitosan is a biocompatible polymer with a structure similar to that of glycosaminoglycans, one of the components of ECM. Recently, it has been found that chitosan itself (i.e. without pre-treatment with adhesion molecules) is able to sustain primary neurons growth, allowing the formation of functional neuronal cultures [275]. Moreover, chitosan microbeads show to have internal micro-porosities that support not only the exchange of nutrients and other molecules, but also the outgrowth of neuronal arborizations with characteristics more similar to in vivo conditions [274]. Similarly to what observed by Frega et al, MEA recordings of 3D neuronal networks grown on chitosan microbeads showed a rich repertoire of complex and asynchronous activity patterns, which seems to better recapitulate activity dynamics of in vivo brain regions, when compared to the traditional networks' dynamics from 2D cultures [274, 275].
It is worth underlying that in all these reported studies, researchers employed traditional planar MEAs to characterize the electrophysiological activity of 3D neuronal networks. This is probably due to the fact that even though 3D models have become more common, almost all commercially available MEAs are designed for 2D cultures. Although this approach is valid and can provide a general view of the activity of 3D neuronal networks, the full benefit of having a 3D model is not achieved, as planar MEAs can only provide data from a single 2D plane. As discussed at the end of 3.1.3, several prototypes of MEAs with non-planar micro-electrodes and of 'true' 3D MEAs (i.e. recording simultaneously from multiple 2D planes) have recently been developed. To our knowledge, to date only Shin et al used their 'true' 3D MEA device with a top-down approach to record the electrophysiological activity of 3D rodent neuronal networks grown in ECM-based scaffolds [88]. Certainly, 3D MEAs represent a promising approach to provide the high level of spatial and temporal resolution necessary to fully harness the potential of 3D models.
4.3. Organotypic slice cultures on MEAs
In addition to dissociated neurons, which can be cultured in 2D or 3D networks, thin slices can be isolated from the whole brain or from different brain regions (e.g. cerebellum and hippocampus) of rodents. While in dissociated neuronal cultures, the in vivo organization of the neuronal tissue (i.e. the relative in vivo positions and connections between cells) is no longer preserved, brain slices maintain some of the 3D structural and functional synaptic connections within local brain regions, since, after isolation, there is no opportunity for cells to alter their relative organization from the initial in vivo state. Brain slices are cut at a thickness ranging from approximately 100–400 μm, and are normally prepared from the brain of embryonic and newborn mice and rats up to postnatal day 12 [54]. At this age, indeed, the cytoarchitecture of the neuronal tissue is established, the brain is larger and easier to manipulate, and neurons are likely to survive explantation, and the mechanical trauma caused when neuronal processes are cut, thanks to the high levels of plasticity [276].
While acute slice preparations allow to maintain neuronal viability and functionality up to a few hours, critically limiting the time-window for experiments, organotypic brain slices can be kept in culture for longer times, even for several weeks [54]. To achieve this goal, a perfusion system is necessary for the delivery of gases and nutrients, which are required to sustain cell viability and physiology [277]. After a few days, organotypic slice cultures stabilize with high cell viability, and can be maintained in culture up to several weeks, during which experiments are performed [94]. In this sense, organotypic slice cultures represent a compromise between the longevity of dissociated cell cultures, and the preservation of the cytoarchitecture of the brain. Organotypic slice cultures, indeed, preserve the 3D organization of the neuronal tissue, at least partially, and appear to be representative of the brain region from which they are derived. Both neuronal and non-neuronal cells are the same that are found in vivo [278], glial cells are present in similar proportions to those observed in vivo [279], and vascular cells are maintained [280]. A further advantage is that the development of cells and synapses in brain slices from embryonic and postnatal rodents mimics the development of the brain in vivo [278, 281]. For instance, developmental changes in spine density and shape, and increased connectivity recapitulate the in vivo phenotype observed in acute slices from age-matched time points [281].
4.3.1. Activity exhibited by organotypic slices on MEAs
Organotypic slice cultures have been used to study the electrophysiological activity of neurons, synapses, and neural circuits under controlled conditions, in isolation from the rest of the brain and the body, in both physiological and pathophysiological scenarios. Indeed, organotypic slices maintain electrical properties comparable to those observed in acute brain slices from age-matched time points [281, 282]. In the past years, several groups have published methods for culturing organotypic slice cultures on MEAs [283–286], aiming to record and characterize the electrophysiological activity of organotypic slices isolated from different regions of rodents' brains [287–292].
In 1998, Egert et al was the first to record the electrophysiological activity from organotypic cultures of hippocampal slices living for up to 4 weeks by means of MEAs [94]. The principal cytoarchitecture of the hippocampus (i.e. with defined subregions, such as CA1 and CA3) was preserved, and both spontaneous APs and electrically-evoked LFPs were recorded. Interestingly, spontaneous spike activity was detected in a different percentage by the micro-electrodes beneath different subregions (e.g. 30% was found at the micro-electrodes beneath the stratum granulosum of the dentate gyrus) [94]. Moreover, the temporal characteristics of spiking activity differed between the cultures with spontaneous LFPs and those in which LFPs occurred only when evoked by stimulation. In the latter case, distributed, non-bursting activity prevailed, while in cultures with spontaneous LFPs, periodic burst patterns resembled epileptic activity [94]. Stimulation of the Schaffer collaterals in CA3 through the micro-electrodes elicited spikes, or LFP responses, or both, which were recorded in the CA1 region, with onset-latencies of 4–10 ms after stimulation. This allowed to estimate an approximate conduction velocity of 30 cm s−1 in the hippocampal circuits [94]. As for spiking activity, LFPs amplitude varied among different subregions, depending on the relative number of neurons involved in synchronized activity in each area [94].
In another study, Kessler et al investigated the electrophysiological activity of Purkinje cells in organotypic cerebellar slice cultures [281]. The first aim of the study was to characterize the spontaneous activity of single Purkinje cells, which could be clearly identified throughout the slices by their size and dendrite structure, using immunohistochemical staining. Only channels which showed large spikes, that clearly originated from single neurons, were analyzed, whereas channels with contributions from more cells were excluded from analysis [287]. The number of cells which exhibited spontaneous activity varied considerably between cultures. However, many cultures exhibited an intermediate level of activity that made it possible to identify the activity of individual neurons at several micro-electrodes, and to carry out quantitative analyses (e.g. of spike waveform and frequency, ISI, and burst patterns) [287]. The second goal of the study was to investigate how spontaneous activity of Purkinje cells was modulated by agents that affect ion channels, excitatory and inhibitory transmission, modulatory transmitter systems, and intracellular signaling mechanisms. As expected, spontaneous activity was highly sensitive to the presence of these agents, and several parameters obtained from the above-mentioned quantitative analysis were modulated in a substance-specific way [287].
Both these studies were performed by using conventional MEAs. More recently, Ito et al used a 512-channel HD-MEA device to measure the activity of organotypic cultures of cortical and hippocampal brain slices [288]. This allowed to record the spontaneous activity of hundreds of neurons simultaneously with high spatial and temporal resolution (i.e. 60 mm and 50 ms, respectively), enabling the analysis of functional connectivity between neurons at different frequency ranges, and the comparison between the neuronal network structures of cortex and hippocampus [288]. In particular, they investigated three frequency ranges (i.e. gamma (30–80 Hz), beta (12–30 Hz), and high frequency (100–1000 Hz) ranges). While the lower two frequency ranges showed similar network structure between cortex and hippocampus, many significant differences were observed in the high frequency range [288]. For the first time, this study provided a method to characterize frequency dependent differences of network architecture from different brain regions, underlying the importance of high temporal resolution recordings for the understanding of functional networks in neuronal systems [288].
4.3.2. Physiology studies and disease modeling
Thanks to the partial preservation of structural and functional synaptic connections within local brain regions, organotypic slice cultures on MEAs are particularly well-suited for studies on synaptic plasticity. Indeed, by analyzing MEA data, changes in electrophysiological activity can be investigated in both time and space. This allows to characterize both spatial and temporal properties of synaptic plasticity mechanisms, to map the structural and functional connectivity remodeling in different microcircuits and layers within a single slice, and to reveal alterations under pathological conditions [293]. Moreover, unlike acute brain slices, organotypic slice cultures allow the investigation of long-term synaptic plasticity mechanisms, such as LTP and LTD [294].
In 2002, Shimono et al investigated LTP in organotypic hippocampal slices cultured on MEAs [294]. LTP was elicited by delivering trains of high frequency stimulations (i.e. tetanus stimulation) to one set of pyramidal cells in the CA1 subregion. Afterwards, field excitatory post-synaptic potentials (fEPSPs) were evoked, at different timepoints after delivering tetanus stimulation, and recorded. Potentiated responses (i.e. with higher amplitude) were observed after 1 h in 75% of slices, moreover, a significant number of slices exhibited a non-decaying LTP that lasted more than 48 h. Moreover, LTP induction was completely and reversibly blocked by APV, an antagonist of NMDARs [294]. The results indicated that trains of high-frequency stimulation in organotypic hippocampal slices were able to elicit a NMDARs-dependent form of LTP that, in a significant number of slices, lasted for several days. The authors underlined that theirs was the first report in which LTP was observed in an in vitro preparation with a duration similar to that found in vivo, indicating that organotypic slice cultures might be ideally suited for the study of mechanisms underlying long-term synaptic plasticity [294].
In addition to physiology studies, organotypic slice cultures have been widely used for modeling neurological diseases. In particular, organotypic hippocampal slices have been increasingly used as an in vitro model of post-traumatic epilepsy. Indeed, some studies reported that principal neurons in organotypic slice cultures tended to form aberrant excitatory connections with other principal cells in response to slicing-induced deafferentation, in a similar way to the mechanisms underlying epileptogenesis in post-traumatic epilepsy [295–299]. It was observed that these cultures spontaneously developed seizures about one week after isolation [295–297]. Epileptogenesis in organotypic hippocampal slices, thereby, had a compressed timescale, and could be monitored with chronic imaging, electrical recording, or by detecting biochemical markers of seizures, providing an easy-to-access in vitro model for investigation of epileptogenesis mechanisms and antiepileptic drug discovery [295–297]. Recently, Liu et al developed a novel hybrid microfluidic-MEA device enabling to monitor the electrophysiological activity of up to 6 organotypic hippocampal cultures on a single chip [299]. This device provided a high-throughput platform for screening and characterizing anti-epileptic drugs. Continuous electrical recordings of LFPs were conducted over 2 weeks after isolation, and false color maps based on spike frequency were constructed from micro-electrodes data with ictal (i.e. seizures) and interictal (i.e. between seizures) activities [299]. In this study, results allowed to characterize epileptogenesis in organotypic hippocampal slices: a latent period after isolation, then spontaneous appearance of interictal and ictal events which continued in the later period, and progressively increased with the age of cultures [299]. The effect of phenytoin, a commonly used anticonvulsant, was tested in order to validate the model. As expected, seizure-like activity was transiently abolished when phenytoin was applied, and rebounded after the termination of drug application. Afterwards, 12 receptor tyrosine kinases inhibitors were screened, and two of them were identified as novel antiepileptic compounds [299].
Organotypic slice cultures can also be treated in order to mimic specific neurological disorders. For instance, mechanical traumas and oxygen-glucose deprivation can be performed in order to model traumatic brain injury [300], and ischemic stroke [301], respectively. Alternatively, organotypic slices can be isolated from transgenic rodents carrying pathogenic mutations modeling neurological diseases, such as AD [302–305], Rett syndrome [306], and neurodevelopmental disorders associated to GRIN2B mutations [307]. Despite the large availability of organotypic slice cultures-based models of disease, to our knowledge, almost no one has been characterized from an electrophysiological perspective by means of MEAs.
As discussed at the end of the previous paragraph, the use of traditional planar MEAs to measure the electrophysiological activity of 3D neuronal cultures, such as organotypic slices, can provide a valid characterization of 3D neuronal networks activity. However, it does not allow to fully benefit of having a 3D model. Moreover, the use of planar MEAs in brain slices may yield fewer stable recordings due to a layer of dead cells (∼50 μm in depth) at the surface of the slice caused by cutting. This forms an electrically passive layer producing a shunt between the planar MEAs and active cells inside the slice, resulting in a low SNR and small signal amplitudes [41]. Since reducing the distance between the recording micro-electrodes and active cells is a good way to obtain high-amplitude signals, MEAs with 3D protruding micro-electrodes, able to penetrate the 'dead cell layer' and make contact with active cells, were developed [82, 83]. Although several prototypes of MEAs with non-planar micro-electrodes are nowadays available (reviewed at the end of paragraph 3.1.3), only a few of them have actually been used to record and characterize the electrophysiological activity of brain slices, but only of acutely-prepared brain slices [82, 83]. These results showed that larger signal amplitudes and stronger signals with higher power content could be detected by using 3D micro-electrodes devices, as compared to traditional planar MEAs [83]. It would be really interesting to apply these 3D micro-electrodes devices to the characterization of the electrophysiological activity in organotypic cultures, in order to improve the signals amplitude in long-term slice cultures.
Organotypic slice cultures show several advantages, including the preservation of some of the 3D structural and functional connections between groups of cells, and the possibility to be kept in culture for long times (i.e. even for several weeks). Nevertheless, they also have many limitations. In general, the approach to isolate and culture organotypic slices from rodent brains is technically more challenging than acute slices preparation, since it generally requires thinner slices and sterility must be maintained throughout their life in vitro. Moreover, some studies reported the formation of aberrant connections [297], and high variability between cultures, that might affect reliability and reproducibility of results [287]. Lastly, organotypic slices are normally obtained from young donors, and even if they undergo further development during their life in culture, in vitro development may differ from in vivo one [287]. This limits their usability to model disease. On one hand, many neural circuits relevant for neurological disorders may have not yet been fully developed in embryonic and newborn animals [287]. On the other hand, organotypic slices from so young animals might be inappropriate for studies on brain ageing, and to model many age-related disorders [302].
5. hiPSCs-derived neuronal cultures on MEAs
As described in the previous chapter, rodent neuronal cultures have been widely utilized in neurophysiology, for investigating brain physiology, screening neurotoxic compounds, modeling neurological disorders and performing drug studies, providing invaluable results. Nevertheless, they show several limitations. Firstly, since neurons do not proliferate in vitro, primary rodent cells must be continuously isolated, involving frequent euthanasia of live animals and arising substantial ethical concerns. Secondly, transability into humans appears to be critically influenced by inter-species differences, including genomic, developmental and physiological divergences, between rodent and human neurons [2–5]. For instance, in large-scale gene profiling of rodent and human cortical neurons, differences were found in both the expression pattern (i.e. neuronal populations) and expression level of a large percentage of genes related to neuronal function, connectivity and signaling [3, 5]. Moreover, differences in proportions of inhibitory neuronal populations, likely to influence microcircuits functions, and in the morphological features of glial cells have been observed [2, 5]. Thirdly, human behavioral and cognitive functions can hardly be evaluated in rodent models. Similarly, human neurological disorders, which normally are highly complex, with a more or less relevant genetic component, or with unclear causes, are frequently correlated to behavioral and cognitive impairments, and to a high level of heterogeneity among patients, in both phenotype and response to treatments [53, 308]. All these aspects can poorly be reproduced in rodent models. Therefore, in the past years, it has become more and more clear than rodent neuronal cultures cannot fully recapitulate human brain physiology and disease in the hope they might provide more predictive results.
In response, the biomedical community have gradually attempted to leave non-human models in favor of human cell-based systems. In this sense, human pluripotent stem cells are believed to be capable to fulfill this attempt. While the use of ESCs is limited by ethical concerns and scarce availability, hiPSCs represent a promising opportunity to overcome these limitations and open new perspectives in the research field of neurophysiology and neuropathology. Moreover, since they enable to generate differentiated cell lines from patients with a specific genetic background, they pave the way towards personalized medicine.
For this reason, in this review we will mainly focus on human neuronal cultures derived from hiPSCs.
5.1. 2D hiPSCs-derived neuronal cultures on MEAs
As soon as hiPSCs technology was introduced in 2006 by Yamanaka et al, several research groups focused their efforts on developing methods to differentiate hiPSCs into the cell types of the human body, including neurons and glial cells. In this chapter, we will firstly describe the most commonly used methods of differentiation of hiPSCs into neuronal cultures, giving particular attention to the ones optimized for experiments on MEAs. Afterwards, we will look at some relevant studies in which hiPSCs-derived neuronal networks on MEAs were used to study the brain physiology, model neurological disorders, investigate disease pathophysiology, and test new therapeutic treatments.
5.1.1. Protocols of neuronal differentiation of hiPSCs on MEAs
The majority of differentiation protocols for generating functional neurons from hiPSCs are based on two approaches: (i) the 'dual-SMAD inhibition' principle, and (ii) the overexpression of lineage-determining transcription factors.
In protocols based on dual-SMAD inhibition, various mitogenic and morphogenic molecules are added to culture media over a period of multiple weeks, to mimic the extracellular signaling cues involved in embryonic development [6]. In particular, compounds such as SB431542 and dorsomorphin are used to inhibit TGF and BMG signaling pathways, that in vivo channel differentiation toward endodermal and mesodermal derivates. As a consequence, hiPSCs are pushed to differentiate into neuronal progenitor cells (NPCs) of the neuroectodermal lineage, that represent the first step in the development of the nervous system [6]. Afterwards, different mitogenic and morphogenic molecules are added to culture media to promote differentiation of NPCs into more specific neuronal populations, such as cortical glutamatergic and GABAergic neurons [49, 309], dopaminergic neurons [310], and motor neurons [311]. In any case, the final result of differentiation protocols based on dual-SMAD inhibition is a mature and functional neuronal culture, including a more or less heterogenous population of post-mitotic neurons, as well as glial cells.
Based on this principle, Shi et al optimized a multistep protocol for neuronal differentiation of hiPSCs into cortical neuronal cultures, consisting in the differentiation of hiPSCs into NPCs, followed by an extended period of cortical neurogenesis, neuronal terminal differentiation to acquire mature electrophysiological properties, and functional excitatory synaptic network formation [309]. In 2019, Hyvärinen et al used this protocol in combination with MEAs and showed that such differentiation could produce human cortical-like neurons displaying firing and bursting activity, and functional connectivity after two months of differentiation [20]. Immunocytochemistry and pharmacological analysis showed the presence of functional glutamatergic and GABAergic neurons, from both deep and upper cortical layers, as well as astrocytes [20]. Interestingly, the differentiation of neuronal cultures with this protocol mimics the timescale of human cortical development in vivo (i.e. 2–3 months). This aspect suggests that hiPSCs-derived cortical neurons obtained with dual-SMAD inhibition may represent a trustful model to study cortical development. However, the length of this protocol also constitutes a major limitation in terms of materials, time and expertise.
Conversely, other neuronal differentiation protocols are based on the overexpression of lineage-determining transcription factors. For instance, Pang et al and Zhang et al proposed a one-step approach for the differentiation of hiPSCs into upper layer cortical neurons by using lentiviral overexpression of the mouse transcription factor Neurogenin 2 (Ngn2) [7, 50]. Later, Frega et al optimized the protocol from Zang et al to differentiate hiPSCs lines with stable Ngn2 overexpression into functional neuronal networks with a reduced variability over the final neuronal density, which is crucial for electrophysiological recording with MEAs [129]. The neuronal networks in such a way obtained, co-cultured with rodent astrocytes to sustain neuronal maturation, showed mature neuronal morphology and functional activity within 3 weeks of differentiation. The protocol from Frega et al also showed to be extremely efficient, yielding the differentiation of nearly 100% of cells into mitogen activated protein-2 (MAP2) positive neurons that received only excitatory post-synaptic potentials in patch-clamp, suggesting them to be almost entirely glutamatergic excitatory neurons [129]. Similar protocols utilizing overexpression of other lineage-determining transcription factors (e.g. mouse distal-less homeobox 2 (Dlx2) and achaete-scute family bHLH transcription factor 1 (Ascl1)) have been developed for differentiation into homogeneous GABAergic inhibitory populations [18, 312, 313]. By using this approach, Mossink et al optimized a protocol to generate hiPSCs-derived GABAergic neurons that could be co-cultured with hiPSCs-derived glutamatergic neurons on MEAs at predefined ratios [18]. By evaluating the neuronal network activity as recorded by MEAs over time and under treatment with PTX (i.e. a GABA antagonist), they found that GABAergic neurons were functionally mature after 6–7 weeks [18]. In the past years, several groups and companies optimized protocols of neuronal differentiation based on the overexpression of lineage-determining transcription factors, as a rapid and efficient approach to obtain neuronal cultures with controlled composition of neuronal types and glial cells [18, 129, 314, 315]. Over the years, several approaches for differentiating hiPSCs into different cells of the nervous system, including astrocytes, microglia, and specific neuronal populations, have been proposed [316–325], allowing researchers to choose and combine the most relevant cell types and neuronal populations to model the physiological mechanism or the neurological disorder under investigation.
Moreover, biotechnology companies such as Axol Bioscience (Cambridge, UK) and Cellular Dynamics (Tokio, Japan) developed proprietary protocols and made commercially available kits to differentiate hiPSCs into different neuronal populations [326], or already differentiated hiPSCs-derived neurons [153, 314, 327–329].
5.1.2. Activity exhibited by 2D hiPSCs-derived neuronal networks on MEAs
Similarly to that described for rodent neuronal cultures, the electrophysiological activity of hiPSCs-derived neuronal networks at maturity is generally characterized by rich and elaborated temporal patterns of bursting activity, which may vary depending on the neuronal populations included in the network under investigation. Also the development of electrophysiological activity retraces the steps described for rodent neuronal networks, but with different timescales according to the neuronal differentiation protocol which is adopted.
Differentiation protocols based on dual-SMAD inhibition result in the formation of mature and functional neuronal cultures whose electrophysiological activity can be recorded by means of MEAs. So far, the vast majority of studies characterized long-term development of network activity and connectivity of cortical neuronal networks [326, 330, 331], in some cases, with the aim of performing a direct comparison with rodent neuronal cultures, held as the 'gold standard' in the MEAs field [4, 20]. In Hyvärinen et al, for instance, hiPSCs-derived cortical networks were plated on MEAs at day 32 of differentiation, and regularly recorded over the span of 4 months [20]. Widespread activity was expressed from the first day on MEAs onwards. In particular, during the first two weeks on MEAs (i.e. days 32–46 of differentiation), spike rate increased, and uncorrelated spikes started to organize into NBs. Both the number of bursts and the percentage of spikes participating in bursts progressively increased. Moreover, while, initially, NBs were short and at high frequency, once networks matured, bursts became longer and less frequent [20]. After 66 d of differentiation, hiPSCs-derived cortical networks reached a mature and stable state of activity, characterized by a rich repertoire of bursting activity which was still apparent after 4 months [20] (figure 5(a), table 5). As compared to rodent counterparts, hiPSCs-derived cortical networks developed slowly (i.e. 66 d of differentiation versus 28), and at maturity were characterized by different dimension and morphology, bursts with longer duration and lower spike frequencies, and by a higher number of random spikes [20]. Other studies report even longer developmental timescales. For instance, Odawara et al investigated the development of spontaneous electrophysiological activity of hiPSCs-derived cortical networks obtained with the differentiation protocol from Axol Bioscience. Neuronal network activity was recorded for over 1 year, and the complete maturation of spontaneous firing, evoked responses, and pharmacological modulation by glutamatergic and GABAergic receptors antagonists and agonists was observed after 5–7 months [326] (table 5).
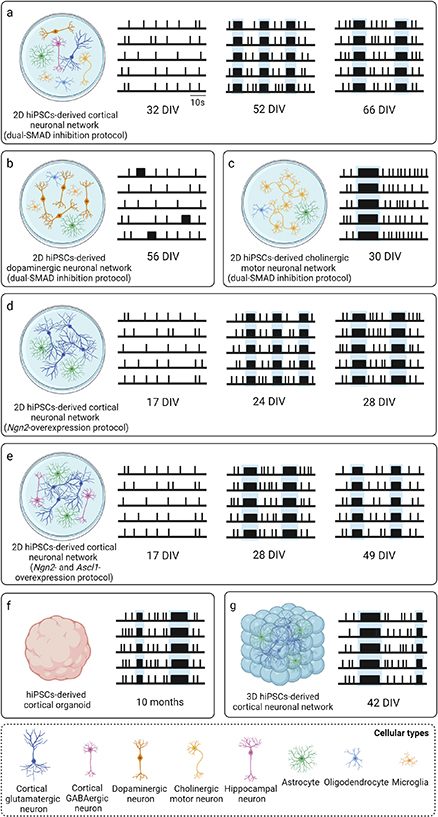
Figure 5. Schematic raster plots describing the spontaneous activity of hiPSCs-derived neuronal networks during development and at maturity, including (a) 2D cortical neuronal networks, (b) dopaminergic neuronal networks, and (c) motor neuronal networks obtained with dual-SMAD inhibition; (d), (e) 2D cortical neuronal networks including glutamatergic neurons only, or glutamatergic and GABAergic neurons, obtained with Ngn2- and Ascl1-overexpression; (f) cortical organoid; (g) 3D cortical neuronal networks. NBs are highlighted in light blue. The most relevant parameters characterizing their mature activity with the respective values are reported in table 5. Created with BioRender.com.
Download figure:
Standard image High-resolution imageTable 5. Representative MEA studies providing a characterization of the spontaneous activity of hiPSCs-derived neuronal networks obtained by using different protocols of neuronal differentiation during differentiation and at maturity, with the most relevant parameters and respective values describing mature activity. DIV = days in vitro, WIV = weeks in vitro, MFR = mean firing rate, MBR = mean burst rate, MBD = mean burst duration, IBI = mean inter-burst interval, PRS = percentage of random spikes, NBR = network burst rate, NBD = network burst duration.
| Neuronal differentiation protocol | Neuronal type | Reference | Days in vitro for recording | System and plate format | Micro-electrodes per well | Relevant parameters and values characterizing mature activity | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Random spikes | Bursts | Network bursts | Mature activity | |||||||
| 2D | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons) | Hyvärinen et al [20] | 32 DIV | 46 DIV | 52 DIV | 66 DIV | Axion Biosystems 12wMEA | 64 | MFR ∼ 2 spikes s−1MBR ∼ 5 bursts min−1 MBD ∼ 800 ms PRS ∼ 40% |
| Axol Bioscience (proprietary) | Mixed (cortical glutamatergic neurons, GABAergic neurons) | Odawara et al [326] | 2 WIV | 7 WIV | 10–13 WIV | 20–30 WIV | Alpha Med Scientific swMEA | 64 | MFR ∼ 6 spikes s−1NBR ∼ 3 bursts min−1 NBD ∼ 350 ms | |
| Dual-SMAD inhibition | Dopaminergic neurons | Sundberg et al [332] | — | — | — | 56 DIV | Axion Biosystems48wMEA | 16 | MFR ∼ 0,5 spikes s−1MBR ∼ 0 bursts min−1 MBD ∼ 300 ms | |
| Dual-SMAD inhibition | Motor neurons | Kim et al [150] | — | — | — | 30 DIV | Axion Biosystems48wMEA | 16 | MFR ∼ 7 spikes s−1NBR ∼ 1 bursts min−1 NBD ∼ 5 s | |
| Overexpression of Ngn2 | Cortical glutamatergic neurons | Frega et al [155] | 17 DIV | 17 DIV | 24 DIV | 28 DIV | Multichannel System 24wMEA | 12 | MFR ∼ 5 spikes s−1MBR ∼ 4 bursts min−1 MBD ∼ 800 ms PRS ∼ 30% | |
| Overexpression of Ngn2 and Ascl1 | Cortical glutamatergic and GABAergic neurons | Mossink et al [18] | — | — | — | 49 DIV | Multichannel System 24wMEA | 12 | MFR ∼ 3 spikes s−1NBR ∼ 3 NB min−1 NBD ∼ 750 ms PRS ∼ 50% | |
| Brain organoids | Guided method | Cortical organoid | Trujillo et al [333] | — | — | 2 months | 10 months | Axion Biosystems12wMEA | 64 | MFR ∼ 15 spikes s−1MBR ∼ 0,3 bursts s−1 Synchrony index ∼ 0,8 |
| Other 3D structures | Overexpression of Ngn2 | Cortical glutamatergic neurons | Muzzi et al [55] | 17 DIV | 28 DIV | 38 DIV | 42 DIV | Multichannel System swMEA | 60 | MFR ∼ 0,5 spikes s−1PRS ∼ 95% NBR ∼ 0,5 bursts min−1 NBD ∼ 600 ms |
Interestingly, the electrophysiological activity of hiPSCs-derived neuronal cultures obtained with dual-SMAD inhibition is critically different according to the neuronal type which is included in the network under investigation. While cortical glutamatergic and GABAergic neuronal cultures are characterized by elaborated temporal patterns of bursting activity, dopaminergic and motor ones show a completely different electrophysiological behavior. Differences can be clearly appreciated by visual observation of raster plots, and by comparing the quantitative parameters extracted from raw data: dopaminergic neuronal cultures are characterized by a low and sporadic firing activity, and almost no bursting activity [162, 332], while motor neurons are characterized by high firing activity, and few but very long bursts [150] (figures 5(b) and (c), table 5).
As mentioned in the previous paragraph, other neuronal differentiation protocols are based on the overexpression of lineage-determining transcription factors, such as Ngn2 and Ascl1 for differentiation into cortical glutamatergic and GABAergic neurons, respectively. In this case the developmental profile of network activity is similar, but with a different, generally shorter, timescale [18, 155]. In Frega et al [155], for instance, spontaneous activity emerged in homogeneous glutamatergic neuronal networks on MEAs after only a few weeks of differentiation (i.e. days in vitro, DIV 17). At this stage, only random spikes and almost no bursts were observed, indicating that neurons were electrophysiologically active, but still immature and not yet integrated in a functionally connected network [155]. Afterwards, electrical activity increased, reaching its highest level during the fourth week (i.e. DIV 24). By this timepoint, neuronal networks exhibited a rich bursting activity, and rhythmic synchronous events occurring across all the micro-electrodes (i.e. NBs), meaning that neurons have self-organized into a synaptically connected network [155]. Similarly to neuronal cultures obtained with dual-SMAD inhibition, initially, networks bursts appeared short and at a high frequency, while once network were fully mature by the end of the fourth week (i.e. DIV 28), less frequently occurring and longer-duration NBs became the more dominant form of activity [155] (figure 5(d), table 5).
Interestingly, when cortical glutamatergic and GABAergic neurons are co-cultured, GABA signaling shows to deeply shape network activity [18]. In this regard, Mossink et al performed a comprehensive analysis comparing different network compositions of either glutamatergic neurons alone, or in co-culture with GABAergic neurons on MEAs [18]. Over development, they detected a shortening of NBD, as well as reduced NBR and MFR, in contrast to increased percentage of random spikes, in neuronal networks in which glutamatergic and GABAergic neurons were co-cultured. All these parameters only became significantly different after 6 weeks of differentiation (i.e. DIV 42), when GABAergic neurons were fully mature, indicating that these differences in network activity were dependent on GABA signaling [18] (figure 5(e), table 5). Moreover, by recording spontaneous activity of mature networks with different ratios of glutamatergic and GABAergic neurons, they found that the NBD was negatively correlated to the percentage of GABAergic neurons, meaning that the effect of GABA signaling on network activity was scalable to the amount of inhibition in the network [18].
In table 5, we reported some representative MEA studies providing a characterization of the spontaneous activity of hiPSCs-derived neuronal networks obtained with different protocols during development and at maturity, with the most relevant parameters and their respective values describing mature activity.
To conclude, it is worth underlying that, besides the protocol of neuronal differentiation, and the neuronal populations included in the network (i.e. neuronal type and ratio), several other factors influence the functional development, and deeply shape the activity of neuronal networks cultured on MEAs, including the culturing conditions [139, 328] (e.g. the ECM substrates used for coating) and the presence of glial cells [334, 335]. For instance, a study from Tukker et al demonstrated that co-cultures of cortical glutamatergic and GABAergic neurons containing hiPSCs-derived astrocytes, developed spontaneous neuronal activity earlier, with increased bursting behavior later during development, as compared to neuronal cultures without glial cells [334]. All these factors should be always taken into account when developing a hiPSCs-based model on MEAs.
5.1.3. Neuronal network physiology studies
hiPSCs-derived neuronal cultures on MEAs represents a valuable in vitro system for modeling and understanding the physiological mechanisms occurring in human neuronal networks.
Firstly, they can be used to investigate how neuronal network activity develops during differentiation. For instance, Mäkinen et al used hiPSCs-derived neuronal networks on MEAs to study the contribution of GABA signaling and gap junctions communication to the development of synchronous activity [336]. By using GABA agonist and antagonist and by blocking gap junctions, they demonstrated that the earliest form of synchronous activity during development is dependent on gap junctions, and that its occurrence corresponds to the excitatory-to-inhibitory GABA switch [336]. Many factors, including coating [139], the ratio of excitatory and neurons [18], the presence of astrocytes [328, 329, 334, 335], and electrical stimulation [337], influence the development and mature activity of neuronal networks on MEAs. Their investigation can give precious insights for understanding the pathophysiological mechanisms occurring in neurodevelopmental disorders.
Secondly, hiPSCs-derived neuronal cultures in combination with MEAs can be used to study synaptic plasticity, as one of the main mechanisms involved in learning and memory consolidation. In 2016, Odawara et al demonstrated that high-frequency stimulation can produce LTP and LTD in human neuronal networks on MEAs [338]. In particular, LTP and LTD resulted in changes in the firing pattern during the first hour after stimulation, and late-phase LTP, which requires slower mechanisms such as gene expression and protein synthesis, was observed after 24 h since high-frequency stimulation [338].
In another recent study, hiPSCs-derived dopaminergic, glutamatergic and GABAergic neurons on MEAs were used to mimic the sleep-wake states and to investigate the response to neuromodulator known to be involved in sleep–wake regulation [339]. An awake-like state was achieved by adding serotonin, which increased the number of synchronized bursts, to the medium of neuronal cultures. Two experimental conditions (culture medium and culture medium with serotonin) were alternately repeated three times in a 12 h cycle for a total of 72 h in order to mimic the sleep-wake cycle [339]. Conversely, a sleep-like state was modeled by applying a protocol of low-frequency electrical stimulation in the range of the non-REM sleep slow waves. The stimulation reduced firing and synchronized bursts repeatedly, mimicking the sleep-induced brain activity changes in vivo [339].
5.1.4. Disease modeling
Undoubtedly, the research field in which hiPSCs-derived neuronal cultures have mainly been used in combination with MEA technology, is in vitro modeling of disease.
To date, several neurological disorders have been modeled by using hiPSCs-derived neuronal networks cultured on MEAs, including ASD, tuberous sclerosis complex (TSC), FXS, psychiatric disorders (e.g. SCZ and BP), epilepsy, neurodegenerative diseases (e.g. AD and ALS) and ischemic stroke. An overview of MEA studies using 2D hiPSCs-derived cultures in disease modeling is found in table 6.
Table 6. Overview of MEA studies using 2D hiPSCs-derived cultures in disease modeling. ASD = autism spectrum disorder, ADHD = attention-deficit hyperactivity disorder, TSC = tuberous sclerosis complex, FXS = fragile X syndrome, AD = Alzheimer's disease, PD = Parkinson disease, ALS = amyotrophic lateral sclerosis. MFR = mean firing rate, wMFR = weighted mean firing rate, ISI = inter-spike interval, MBR = mean burst rate, MBD = mean burst duration, IBI = mean inter-burst interval, PRS = percentage of random spikes, PSB = percentage of spikes within bursts, NBR = network burst rate, NBD = network burst duration, NIBI = mean network inter-burst interval, CV = coefficient of variation.
| References | Disease model | Neuronal differentiation protocol | Neuronal cultures | How the model was obtained | System and plate format | Micro-electrodes per well | Extracted quantitative parameters and phenotype on MEA |
|---|---|---|---|---|---|---|---|
| Marchetto et al [152] | ASD | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from ASD patients | Axion Biosystems 12wMEA | 64 | MFR ↓, NBR ↓ |
| DeRosa et al [340] | ASD | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from ASD patients | Axion Biosystems 12wMEA | 64 | MFR ↓ |
| Russo et al [341] | ASD | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from ASD patients | Axion Biosystems 12wMEA | 64 | MFR ↓ |
| Deneault et al [160] | ASD | Overexpression of Ngn2 | Cortical glutamatergic neurons | CRISPR/Cas9-mediated genome editing of ASD-relevant genes | Axion Biosystems 48wMEA | 16 | MFR ↓, MBR ↓, NBR ↓ |
| Deneault et al [342] | ASD | Overexpression of Ngn2 | Cortical glutamatergic neurons and rodent astrocytes | hiPSCs from ASD patients | Axion Biosystems 48wMEA | 16 | wMFR ↑, NBR ↑ |
| Amatya et al [343] | ASD | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from ASD patients | Axion Biosystems 96wMEA | 8 | Minimum embedding dimension, CVISI ↓, number of bursting electrodes ↓ |
| Mossink et al [18] | ASD and ADHD | Overexpression of Ngn2 and Ascl1 | Cortical glutamatergic and GABAergic neurons, rodent astrocytes | Downregulation of CDH13 gene | Multichannel System 24wMEA | 12 | NBD ↓, network burst shape |
| Frega et al [155] | Kleefstra syndrome | Overexpression of Ngn2 | Cortical glutamatergic and rodent astrocytes | hiPSCs from Kleefstra syndrome patients | Multichannel System 24wMEA | 12 | MFR ↓, MBR ↓, MBD ↑, IBI ↑, CVIBI ↑, PRS |
| Sharma et al [344] | Rett syndrome | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from Rett syndrome patients | Axion Biosystems 12wMEA | 64 | NBR |
| Mok et al [345] | Rett syndrome | Overexpression of Ngn2 | Cortical glutamatergic and rodent astrocytes | CRISPR/Cas9-mediated genome editing of MECP2 | Axion Biosystems 12wMEA | 64 | wMFR, NBR ↓, NBD ↑ |
| Nadadhur et al [151] | TSC | Dual-SMAD inhibition | Cortical glutamatergic and GABAergic neurons, human oligodendrocytes | hiPSCs from TSC patients | Multichannel System swMEA | 60 | MFR ↑ |
| Winden et al [315] | TSC | Overexpression of Ngn2 | Cortical glutamatergic neurons and human astrocytes | hiPSCs from TSC patients | Axion Biosystems 48wMEA | 16 | wMFR ↑ |
| Alsaqati et al [346] | TSC | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from TSC patients | Axion Biosystems 24wMEA | 16 | MFR ↑, MBR ↑, NBR ↑, PRS ↑, NBD ↑, NIBI ↑ |
| Liu et al [347] | FXS | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from FXS patients | Axion Biosystems 12wMEA | 64 | MFR ↑ |
| Utami et al [156] | FXS | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | CRISPR/Cas9-mediated genome editing of FMR1 | Axion Biosystems 12wMEA | 64 | MFR ↓, maximum spike frequency ↓, number of unresponsive channels ↑ |
| Graef et al [348] | FXS | Overexpression of Ngn2 | Cortical glutamatergic neurons | hiPSCs from FXS patients | Axion Biosystems 48wMEA | 16 | wMFR ↑ |
| Snow et al [349] | Alternating hemiplegia of childhood (AHC) | Overexpression of Ngn2 | Cortical glutamatergic neurons | hiPSCs from AHC patients | Axion Biosystems 48wMEA | 16 | MFR |
| van Rhijn et al [162] | Brunner syndrome | Dual-SMAD inhibition | Dopaminergic neurons and rodent astrocytes | hiPSCs from Brunner syndrome patients | Multichannel System 24wMEA | 12 | MFR ↑, MBR ↑, NBR ↑ |
| Multichannel System 6wMEA | 9 | ||||||
| Linda et al [350] | Koolen-de Vries Syndrome (KdVS) | Overexpression of Ngn2 | Cortical glutamatergic neurons and rodent astrocytes | hiPSCs from KdVS patients and genome editing of KANSL1 | Multichannel System 24wMEA | 12 | MFR, NBR ↓¸ PRS ↑, CVNIBI↑ |
| Sundberg et al [332] | 16p11.2 CNV | Dual-SMAD inhibition | Dopaminergic neurons and human astrocytes | CRISPR/Cas9-mediated genome editing of 16p11.2 | MaxWell Biosystems HD-MEA | 1024 | MBR ↑, PSB, MBD, IBI |
| Axion Biosystems 48wMEA | 16 | MFR ↑, MBR ↑, MBD | |||||
| Nageshappa et al [351] | MECP2 duplication syndrome | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | hiPSCs from MECP2 duplication syndrome patients | MED64 swMEA | 64 | MFR ↑, NBR ↑ |
| Sarkar et al [357] | Schizophrenia | Dual-SMAD inhibition | Hippocampal CA3 pyramidal neurons | hiPSCs from schizophrenia patients | Axion Biosystems 96wMEA | 8 | MFR ↓, number of active electrodes, MBR ↓, NBR ↓, synchronicity index |
| Kathuria et al [154] | Schizophrenia | Dual-SMAD inhibition | Cortical GABAergic neurons | hiPSCs from schizophrenia patients | MED64 12wMEA | 16 | MFR ↓ |
| Ishii et al [352] | Schizophrenia and bipolar disorder | Overexpression of Ngn2 and Ascl1 + Dlx2 | Cortical glutamatergic and GABAergic neurons | hiPSCs from schizophrenia and bipolar disorder patients | Axion Biosystems 48wMEA | 16 | wMFR |
| Wang et al [161] | Schizophrenia | Overexpression of Ngn2 and Ascl1 | Cortical glutamatergic and GABAergic neurons, rodent astrocytes | CRISPR/Cas9-mediated genome editing of SETD1A gene | Multichannel System 24wMEA | 12 | MFR, NBR ↑, NBD ↑ |
| Odawara et al [330] | Epilepsy | Dual-SMAD inhibition | Cortical glutamatergic and GABAergic neurons, human astrocytes | Treatment with pentylenetetrazol and 4-aminopryridine (4-AP) | Alpha Med Scientific 24wMEA | 16 | MFR ↑, number of active channels, NBR ↑ |
| Alpha Med Scientific swMEA | 64 | Peak time of spikes during synchronized burst firing | |||||
| Quraishi et al [327] | Epilepsy | Cellular Dynamics (proprietary) | Cortical glutamatergic and GABAergic neurons | Nuclease-mediated genome editing of KCNT1 gene | Axion Biosystems 48wMEA | 16 | MFR ↑, MBR ↑, synchronicity index ↑, MBD ↓, burst intensity |
| Que et al [353] | Epilepsy | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | CRISPR/Cas9-mediated genome editing of SCN2A gene | Axion Biosystems48wMEA | 16 | MFR ↑, MBR ↑, MBD ↓, bursting intensity, synchrony index ↑ |
| Mzezewa et al [354] | Epilepsy | Dual-SMAD inhibition | Mixed (cortical glutamatergic neurons, GABAergic neurons and glia cells) | Treatment with kainic acid | Axion Biosystems48wMEA | 16 | MFR ↓, MBR ↑, MBD ↓, IBI ↓, NBR ↓ |
| Van Hugte et al [355] | Dravet syndrome (DS)-related epilepsy | Overexpression of Ngn2 | Cortical glutamatergic neurons and rodent astrocytes | hiPSCs from DS patients and CRISPR/Cas9-mediated genome editing of SCN1A gene | Multichannel System 24wMEA | 12 | MBR ↑, PRS ↑, NBR ↑ |
| Caneus et al [153] | AD | Cellular Dynamics (proprietary) | Cortical glutamatergic and GABAergic neurons, human astrocytes | Treatment with Aβ and tau oligomers, or brain extracts from AD transgenic mice | Multichannel SystemswMEA | 60 | MFR ↓ |
| Ronchi et al [314] | PD | Cellular Dynamics (proprietary) | Dopaminergic neurons and human astrocytes | Nuclease-mediated genome editing of SNCA gene | MaxWell BiosystemsHD-MEA | 26400 | MFR ↓, mean spike amplitude, CVISI, percentage of active electrodes, MBD, IBI ↓, CVIBI ↓, MBR ↓, network burst shape, spike propagation in space and time |
| Kim et al [150] | ALS | Dual-SMAD inhibition | Motor neurons and human astrocytes | CRISPR/Cas9-mediated genome editing of SOD1 gene | Axion Biosystems48wMEA | 16 | MFR ↓, NBR ↓, NBD ↑, PSB |
| Ronchi et al [314] | ALS | Cellular Dynamics (proprietary) | Motor neurons and human astrocytes | Nuclease-mediated genome editing of TDP-43 gene | MaxWell BiosystemsHD-MEA | 26 400 | MFR ↑, mean spike amplitude, CVISI, percentage of active electrodes, MBD, IBI ↑, CVIBI ↑, MBR ↑, network burst shape, spike propagation in space and time |
| Klein Gunnewiek et al [356] | Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) | Overexpression of Ngn2 | Cortical glutamatergic neurons and rodent astrocytes | hiPSCs from MELAS patients | Multichannel System24wMEA | 12 | MFR ↓, MBR ↓, PRS ↑, NBR ↓, NIBI, CVNIBI |
| Pires Monteiro et al [131] | Ischemic stroke | Overexpression of Ngn2 and Ascl1 | Cortical glutamatergic and GABAergic neurons, rodent astrocytes | Exposition to hypoxia | Multichannel System 24wMEA | 12 | MFR ↓, NBR ↓, PSB ↓, NBD ↓ |
In vitro models of neurological diseases based on hiPSCs-derived neuronal networks are obtained in two main ways: (i) by directly deriving hiPSCs from patients, or (ii) by genetically modifying or treating (e.g. with drugs and neurotoxic compounds) hiPSCs derived from healthy subjects.
In the following paragraphs, some relevant studies in which human neuronal networks on MEAs were used to model neurological disorders will be presented, showing the potential of hiPSCs-derived MEA-based in vitro models not only to characterize the phenotype of the disease under investigation, but also to uncover the underlying pathophysiological mechanisms, and to perform phenotypic rescue experiments.
5.1.4.1. Use of MEAs to characterize the patient-specific disease phenotype
hiPSCs-derived neuronal networks on MEAs are a powerful tool for performing phenotyping assays with the aim to investigate how the electrophysiological activity of human neuronal networks is altered in specific neurological disorders.
In most of the studies, hiPSCs are directly derived from the cells of a clinically characterized cohort of patients. This is particularly advantageous when it comes to model highly complex disorders with a more or less relevant genetic component, or with an unclear cause (i.e. idiopathic disorders), usually correlated to a high level of heterogeneity in the patient population. A typical example is ASD [152, 340–343]. By using this approach, Marchetto et al reprogrammed fibroblasts to generate hiPSCs, NPCs and neurons, from a clinically characterized group of ASD patients who had a common anatomical phenotypic trait that occur in about 20%–30% of idiopathic ASD and is frequently associated with poor prognosis: an early developmental enlargement of brain volume [152]. ASD-derived NPCs displayed increased cell proliferation, whereas ASD-derived neurons showed abnormal neurogenesis and reduced synaptogenesis leading to functional defects in neuronal networks (i.e. lower number of spikes and NBs) [152]. The use of MEAs allowed to investigate how differences in the neuronal network activity between ASD and non-ASD derived neurons emerged and increased during differentiation. Moreover, the choice of a long protocol of differentiation, including differentiation into NPCs and an extended period of cortical neurogenesis, and mimicking the timescale of human cortical development in vivo, is particularly well suited to gain insights about the pathophysiological mechanisms occurring during development in neurodevelopmental diseases.
Different studies used hiPSCs-derived neuronal cultures from ASD patients to investigate how potentially pathogenic alterations of key neurodevelopmental genes interacted with each other and impacted on the development of neuronal networks. The results allowed to characterize different aspects of the pathophysiological phenotypes observed in ASD-derived neuronal networks, and to link them to specific genetic variants [340, 342].
Similarly, the electrophysiological phenotype of hiPSCs-derived neuronal networks derived from TSC patients was characterized [151, 315, 346]. In a study led by Winden et al, hiPSCs were obtained from a female patient with a functional loss of the TCS2 gene [315]. By using the differentiation protocol based on the overexpression of Ngn2, they cultured neuronal networks, consisting of glutamatergic neurons only, on MEAs, and they observed that loss of TSC2 caused morphological and physiological changes in neurons, resulting in hyperexcitability [315]. In another study of Alsaqati et al, hiPSCs derived from TSC patient with a functional loss of TCS2, were differentiated into mixed cultures of glutamatergic and GABAergic neurons, by using the dual-SMAD inhibition approach [346]. hiPSCs developed a dysfunctional neuronal network characterized by hyperactivity, reduced synchronization of neuronal bursting, and lower spatial connectivity [346]. These deficits of network function were associated with elevated expression of genes for inhibitory GABA and glutamate signaling, indicating a potential abnormality of synaptic inhibitory-excitatory signaling. In the discussion, Alsaqati et al compared their results with the ones obtained by Winden et al, suggesting that their model might better represent the pathophysiological phenotype of TSC, since it included both glutamatergic and GABAergic neurons [346].
Also, psychiatric disorders, such as SCZ and BP, have been modeled on MEAs by using hiPSCs-derived neuronal networks from patients [154, 352, 357]. In the study of Sarkar et al, four patients were originally selected on the basis of the high likelihood of a genetic component to disease, and subject to psychiatric characterization [357]. The derived neuronal networks, consisting of hippocampal CA3 pyramidal neurons, displayed significant reductions in the number of spikes, and in network properties such as number of NBs, spike per NB, and synchronicity [357]. In a study of Ishii et al neuronal networks were differentiated from hiPSCs derived from two BP patients with PCDH15 deletion and one SCZ patient with RELN deletion [352]. Although BP and SCZ are distinct neuropsychiatric diseases, they share a subset of similar symptoms. Thereby, the aim of the study was to characterize the phenotype of BP and SCZ neuronal cultures in vitro, to underlie potential cell biological basis for shared clinical features between these two different disorders [352]. Both type of neurons differentiated from patient-derived from hiPSCs exhibited shorter dendrites and reduced formation of excitatory and inhibitory synapses. While spontaneous neuronal activity was comparable between patient-derived neurons and control neurons, BP and SCZ neurons showed higher sensitivities in AMPA receptor and GABA receptor stimulation, which might represent a compensatory mechanism to maintain spontaneous activity of neurons [352].
To date, several neurological disorders have been modeled on MEAs by using hiPSCs-derived neuronal networks derived from patients, including KS [155], FXS [347, 348], Rett syndrome [344, 345], Koolen-de Vries Syndrome [350], Dravet syndrome-related epilepsy [355] and Brunner syndrome [162], allowing a detailed characterization of the electrophysiological phenotype of patients at the network level.
Another way to model neurological disease with hiPSCs-derived neuronal cultures is by using genome editing techniques, such as CRISPR-Cas9, which allow to insert, replace, or delete DNA sequences, in order to reproduce the desired pathogenic mutation. Moreover, the modification of DNA provides the possibility to easily obtain isogenic controls, which can be used in comparison to affected neuronal networks in order to better characterize the pathophysiological phenotype under investigation. Genome editing techniques are used to obtain models of monogenic diseases, such as FXS [156] and monogenic epilepsies [327, 353], but also to investigate the contribution of specific genetic alterations to the pathophysiological phenotype of multigenic disorders, such as ASD [18, 160], SCZ [161], PD [314], and ALS [150, 314]. By using this approach, for instance, Que et al investigated the role of a genetic variant of SCN2A (i.e. gene encoding encodes the voltage-gated sodium channel Nav1.2) which was identified in multiple patients with epileptic encephalopathy and intractable seizures [353]. By introducing the L1342P variant into hiPSCs, they found that cortical neurons derived from hiPSCs carrying L1342P variant had a significantly increased intrinsic excitability, and enhanced bursting and synchronous network firing, suggesting an hyperexcitability phenotype [353]. In another study, Wang et al used CRISPR-Cas9 to generate cortical neuronal networks from hiPSCs with a mutation of SETD1A (i.e. gene encoding a subunit of histone H3 lysine 4 methyltransferase), which is related to a neurodevelopmental syndrome and increased risk of SCZ [161]. Neuronal cultures derived from hiPSCs with the mutation showed an altered neuronal network organization with an aberrant synchronized activity at different developmental stages. Interestingly, abnormalities in neural synchronization in vivo are suggested as one of the core pathophysiological mechanisms in SCZ, supporting the validity of the in vitro model [161].
Depending on the neurological disease under investigation, other approaches can be adopted. For instance, epilepsy models can be obtained by treating hiPSCs-derived neuronal networks with epileptogenic compounds [330, 354], AD models were realized by applying amyloid-β and tau oligomers [153], ischemic stroke can be mimicked by exposing the human cultures to controlled hypoxic conditions [131].
A large part of the above-mentioned models consists of neuronal networks in which cortical glutamatergic and GABAergic neurons, and glial cells, are present, in order to mimic the cerebral cortex. Conversely, in other studies, specific neuronal populations have been chosen because of their relevance in the neurological disorder under investigation. As an example, Ronchi et al modeled PD and ALS by using dopaminergic and motor neurons neuronal network, respectively [314]. In this study, a CMOS-based HD-MEAs with 26 400 micro-electrodes was used to characterize the pathophysiological phenotype of PD dopaminergic and ALS motor neurons [314]. This allowed not only to extract parameters describing the firing and bursting activity, and synchronicity of the affected neuronal networks, but also to investigate other aspects, such as AP propagation along axons. For instance, by comparing the AP propagation velocity between controls and diseased neuronal networks, they found an increased AP conduction velocity in ALS motor neurons, which could be correlated with altered axonal excitability reported in previous ALS studies [314].
5.1.4.2. Use of MEAs to investigate cellular and molecular mechanisms underlying the disease phenotype
MEAs represent a powerful platform to perform phenotyping assays of affected neuronal networks. However, their potential does not stop there. Once established, a well-defined human disease model on MEAs can give precious insights about the cellular and molecular mechanisms underpinning the pathophysiological phenotype. By observing how the electrophysiological activity is altered in affected neuronal networks, hints about candidate disease mechanisms can be collected, and successively investigated by means of MEAs combined with other (i) electrophysiology techniques, such as patch-clamp and calcium imaging, (ii) biological methods, including immunostainings and transcriptome analysis, (iii) pharmacological treatment (i.e. the use of inhibitors of specific receptors, ionic channels, and enzymes), and (iv) genetic manipulation.
Several of the studies reviewed in table 6 combined MEAs with other techniques to investigate the cellular and molecular mechanisms underlying the pathophysiological phenotype previously observed at the network level.
For instance, Quraishi et al [327] and Que et al [353] combined MEA recordings with patch-clamp experiments to characterize how mutations in KCNT1 (encoding KNa1.1 channel) and the L1342P variant in SCN2A (encoding Nav1.2), respectively, resulted in a significant increase of intrinsic excitability of neurons, explaining the hyperactive phenotype observed at the network level. Similarly, Alsaqati et al investigated the mechanisms underlying the pathophysiological phenotype observed in hiPSCs-derived neuronal networks from TSC patients with a functional loss of TSC2 gene, characterized by neuronal hyperactivity, reduced synchronization and lower spatial connectivity [346]. They found that these deficits were associated with an elevated expression of target genes of the rapamycin complex 1 (mTORC1) pathway for inhibitory GABA signaling and excitatory glutamate signaling. By using the specific inhibitor or inducers of the protein kinases involved in mTORC1 activity, they managed to suppress neuronal hyperactivity, and to restore other aspects of network activity, thereby demonstrating the involvement mTORC1 pathway in the pathophysiological phenotype resulting from TSC2 functional loss [346]. In another study, hiPSCs-derived dopaminergic neurons from Brunner syndrome patients were characterized at both single cell and neuronal network level, showing reduced synaptic density, but hyperactive network activity [162]. While intrinsic functional properties and AMPARs-mediated synaptic transmission were not affected, they found that hyperactivity was mediated by upregulation of two subunits of NMDARs, resulting in increased NMDARs-mediated currents [162]. By correcting a pathogenic mutation with CRISPR/Cas9-mediated genome editing, they restored the expression of NMDARs subunits, NMDARs function and neuronal network activity to control levels [162].
Also the observation of burst shape can suggest the involvement of specific cellular and molecular mechanisms. Mossink et al observed that the knockdown of cadherin-13 (CDH13), which is associated with ASD, resulted in no changes in the number of inhibitory presynapses (i.e. where CDH13 is normally localized), but in increased inhibitory synaptic strength, as demonstrated by reduced NBD together with an altered average burst shape in MEA recordings, and confirmed by following patch-clamp experiments [18]. The mechanisms by which CDH13 regulates inhibitory synaptic strength were further investigated by biological methods, including transcriptome analysis, immunostainings and immunoprecipitation assays [18].
In a similar way, the electrophysiological phenotype of hiPSCs-derived neuronal networks from KS patients carrying a mutation in the EHMT1 gene (i.e. reduced number of NBs with a longer duration and increased temporal irregularity) suggested the involvement of NMDARs-mediated currents, which are known to directly influence the duration of bursts [155]. Patch-clamp and transcriptome analysis showed increased NMDARs-mediated currents, whereas the pharmacological inhibition of NMDARs resulted in the rescue of the pathophysiological phenotype. In the discussion, they postulated that those changes were mediated by the abnormal upregulation of NMDARs subunit 1, due to the reduced activity of a repressive mark, catalytic product of EHMT1 [155]. hiPSCs-derived MEA-based models have also been used to investigate how the disease phenotype is influenced by cellular and molecular mechanisms other than those strictly related to the intrinsic functional properties of neurons and neuronal networks. These includes, for instance, the interactions between affected neurons and glial cells [151, 341], the intercellular signaling mediated by exosomes [344], or the release of inflammatory cytokines [354]. As an example, Sharma et al observed that treating hiPSCs-derived neuronal cultures lacking MECP2 (i.e. a model of the neurodevelopmental disorder Rett syndrome) with exosomes released by isogenic control neuronal cultures rescued deficits in neuronal proliferation, differentiation, synaptogenesis, and synchronized firing, whereas exosomes from MECP2-deficient neuronal cultures lacked this capability [344]. This indicated that the protein cargo and signaling bioactivity of exosomes play a critical role in the regulation of neuronal networks development which can contribute to disease pathogenesis [344]. Moreover, MEA-based models have been used also to investigate how physiological mechanisms, such as LTP, are affected in pathological conditions. For instance, Caneus et al investigated how the treatment with Aβ oligomers, tau oligomers, or brain extracts from AD transgenic mice, could cause deficits in LTP in hiPSCs-derived neuronal networks [153]. By means of MEAs, LTP was easily induced by delivering high frequency stimulation through the micro-electrodes, and the deleterious effects of Aβ and tau oligomers on the cellular and molecular mechanisms underpinning synaptic plasticity, including the recruitment of receptors at synapses and gene expression, were evaluated, demonstrating the impairment of LTP without loss of viability [153].
5.1.4.3. Use of MEAs in phenotypic rescue experiments
In vitro models on MEAs represents a valuable tool for performing phenotypic rescue experiments using genetic interventions or other pharmacological and non-pharmacological treatments.
In several studies cited in the previous paragraphs, well-known drugs were tested. For instance, Marchetto et al observed that IGF-1, a drug that is currently in clinical trials for ASD, rescued the defects observed in ASD-patient neuronal networks [152]. Conversely, Que et al observed a degree of resistance of L1342P neuronal culture to the anti-convulsant drug phenytoin, which recapitulated aspects of clinical observation of patients carrying the same variant [353]. In a recent study, van Hugte et al tested four different anti-seizure drugs in hiPSCs-derived neuronal networks from patients with two types of epilepsy arising from mutations in the SCN1A gene, i.e. Dravet syndrome (DS)-related epilepsy and generalized epilepsy with febrile seizures plus (GEFS+) [355]. While no differences were observed in the electrophysiological phenotype on MEA, characterized by hyperactivity in both cases, anti-seizure drugs only affected GEFS+, but not DS neuronal networks. Interestingly, in vitro response (i.e. responsiveness or not) to the administration of anti-seizure drugs in DS and GEFS+ neuronal networks exactly corresponded to those observed in the respective patients [355].
In other cases, the administration of commonly used drugs was not beneficial, or even deleterious [330, 349]. These results do not necessarily invalidate the disease model, but can give insights about the cellular targets and the mechanisms of actions of these drugs. For instance, Snow et al suggested that the inefficacy of treatment with flunarizine in improving the pathophysiological phenotype of hemiplegia of childhood (AHC) neuronal networks, might indicate that this drug, commonly used to prevent AHC episode, might exert an influence on other neuronal populations, such as GABAergic neurons, which were not present in their model [349].
In other studies, researchers used the acquired information about the cellular and molecular mechanisms underlying the phenotype of their newly established disease models to test new treatment strategies. For instance, Nageshappa et al characterized the phenotype of neuronal networks carrying a duplication in MECP2, which in vivo results in a dramatic neurodevelopmental phenotype [351]. Knowing that MECP2 acts at the epigenetic level, they screened a library of compounds with a defined activity on epigenetic pathways for their ability to reverse the pathophysiological phenotype. With this approach, one histone deacetylase inhibitor, NCH-51, which had never been considered before as a therapeutic alternative for neurological disorders, was validated as a potential clinical candidate [351].
Besides pharmacological treatments, genetic techniques can be easily used in hiPSCs-derived models to test innovative treatment strategies such as gene therapy. Several studies used CRISPR-Cas9 technique to reverse the pathogenic mutation responsible for the disease, demonstrating the possibility of fully rescuing the pathophysiological phenotype [162, 347, 348]. In another interesting study, Graef et al used a combination of CRISPR-Cas9, antisense and expression technologies to generate a mixed and mosaic neuronal culture system composed of FMRP-negative (i.e. diseased) and FMRP-positive (i.e. healthy) cells [348]. The aim was to determine the level of FMRP protein and the percentage of FMRP-positive cells necessary to correct the phenotype of FXS patients-derived neuronal networks. The results defined a threshold for the overall FMRP expression levels and for the percentage of FMRP-expressing neurons in a mosaic population, that can be used for evaluating potential therapeutic interventions focused on reactivating FMRP in hiPSCs-derived neurons from FXS patients [348].
5.2. 3D hiPSCs-derived neuronal cultures on MEAs
The most commonly used methods of neuronal differentiation enable to differentiate hiPSCs into 2D neuronal cultures consisting of different neuronal populations, and glial cells, depending on the adopted protocol. Nevertheless, as discussed for rodent cultures in 4.2, it has become clear that 2D neuronal models have major limitations, since cultures are constrained into monolayers, thereby they are inherently unable to reproduce the 3D environment and cytoarchitecture of the human brain [9–11]. For this reason, researchers have combined their efforts to develop protocols to differentiate hiPSCs into 3D neuronal cultures, which would allow the investigation of cellular behavior and network activity in a more physiologically relevant environment. Interestingly, unlike dissociated neuronal cultures from rodents, hiPSCs offer the opportunity to obtain brain organoids, which are self-assembled 3D aggregates of neuronal cells, with cell types (i.e. neurons and glial cells) and cytoarchitecture that resemble the embryonic human brain [56]. For this reason, brain organoids represent a promising system for modeling neurogenesis and neurodevelopmental disorders.
In this section, we will review some relevant protocols to differentiate hiPSCs into brain organoids and other 3D structures on MEAs, afterwards we will look at some first studies in which brain organoids in combination with MEAs were used to model neurological disorders.
5.2.1. Brain organoids and other 3D structures
Unlike dissociated neuronal cultures from rodents, hiPSCs are able to self-assemble without the use of 3D scaffolds into brain organoids, which are 3D spheroidal aggregates with an organized cytoarchitecture, composed of NPCs, neurons and glial cells, resembling the embryonic human brain [56, 358–360].
To generate brain organoids two different approaches are used: (i) unguided methods, and (ii) guided methods. Unguided methods fully rely on the intrinsic potential of hiPSCs aggregates of spontaneously acquiring various neuronal cell identities, and hence to establish multiple brain regions within a single organoid [56]. Conversely, guided methods require supplementation of external patterning factors to induce hiPSCs to differentiate with a certain regional specificity [56] (e.g. cerebral cortex [360, 361], hippocampus [362], and midbrain [358]). In both cases, the resulting organoids contain various cell types (i.e. neurons, glia, and microglia) that normally constitute the brain tissue [56]. hiPSCs-derived brain organoids recapitulate the human brain not only at the cellular level, but also in terms of cytoarchitecture and developmental trajectory, that resemble those of the actual embryonic brain [56]. While unguided organoids are suitable for exploring cell-type diversity during whole-brain development, guided organoids better recapitulate the cytoarchitecture of specific brain regions with less heterogeneity, allowing the investigation of region-specific neurodevelopmental mechanisms.
Several approaches have been proposed for the differentiation of brain organoids in vitro, including the use of spinning bioreactors [359, 363], matrices of microwells [364–366], cell aggregates on adherent substrates protocols [367, 368], and hanging drop methods [369–371] (for a more comprehensive review on hiPSCs-derived brain organoid methods see [56]). Each approach has advantages and disadvantages depending on the experimental aim. For instance, the protocol to obtain brain organoids through hiPSCs-derived cell aggregates is relatively easy, but it lacks of control over the final position and size of the organoids [367, 368]. Conversely, the use of matrices of microwells, or hanging drop methods allow to obtain many uniform spheroidal aggregates from the same or different hiPSCs lines can be cultured in parallel in the same plate, thereby reducing the variability and allowing high-throughput screenings [364–366, 369–371]. Lastly, the use of spinning bioreactors enables to develop brain organoids in a way that is more similar to in vivo development. Indeed, thanks to the continuous perfusion of medium containing oxygen and nutrients, organoids are grown in suspension, and tend to preserve a 3D structure. Nevertheless, commercial bioreactors are bulky and consume large volumes of culture medium, limiting the efficiency and throughput of organoid cultures [359, 363].
So far, organoids mimicking the cerebral cortex have been better characterized, and more frequently used than other brain organoids. The reason is probably related to the fact that the cerebral cortex is the most unique and evolutionarily expanded region of the human brain, as compared to that of other animals, and it is often affected in neurological disorders. However, like all in vitro models, cortical organoids are not identical replicas of their in vivo counterparts, and show several limitations. Firstly, they are much smaller in size compared with the human cerebral cortex (i.e. they can at most expand to ∼4 mm in diameter) [372, 373]. Secondly, whereas cortical organoids may develop a rudimentary cytoarchitecture resembling that of the actual embryonic brain, they still lack the fine cytoarchitecture of all six layers of the human brain cortex [56]. In this regard, the small size of current organoids represents one of the main limiting factors preventing brain organoids to fully recapitulate both the early and late stages of human brain development. Lastly, due to a lack of vascularization, and to the fact that diffusion allows oxygen and nutrients to penetrate less than 1 mm into tissues, a necrotic core (i.e. a core of not viable cells) inevitably builds up in the organoid's interior, and the actual viable thickness is further limited [372, 373]. A solution could be to cut the brain organoids into slices, which, similarly to classic organotypic slice cultures, can be maintained in cultures by using a perfusion system to deliver gases and nutrients and sustain cell viability [149, 374, 375]. Afterwards, slices can be transferred, for instance, on MEAs to record their electrophysiological activity [149, 374]. Although this approach is valid to perform electrophysiological recordings of neuronal networks which have formed within brain organoids, it does not fully take advantage of having a 3D model, since, similarly to classic organotypic slice cultures, the 3D cytoarchitecture and neuronal connections are only partially preserved in slices. Alternatively, some recent studies proposed the development of vascularized brain organoids, in which epithelial cells were added to form complex vascular-like networks with blood–brain barrier characteristics, able to sustain cell viability and physiology even in the deep core of organoids [376–378].
Undoubtedly, one of the main limitations of current brain organoids is that protocols to obtain them generally have an extremely long timescale (i.e. up to several months [56]). Indeed, it has been seen that the development of in vitro brain organoids dynamically mimics the temporal progression of human brain development [333, 379], which is both an advantage and a disadvantage for researchers. On the one hand, brain organoids of different ages recapitulate their corresponding in vivo counterparts, offering researchers a versatile platform to investigate different developmental stages [333, 379]. On the other hand, from a practical point of view, brain organoids take an extremely long time to grow and mature, raising the cost and hindering the efficiency of experiments. To overcome this limitation, a recent study proposed an approach to adapt a well-established protocol of differentiation of hiPSCs into neuronal cultures, to the development of 3D neural aggregates within just 3 weeks [331], representing a relevant advance in 3D neuronal cultures differentiation methods.
Despite the huge success of brain organoids, in the past years, only a few studies focused on investigating how the electrophysiological activity of brain organoids spontaneously develops over time [333, 374, 379, 380]. Among them, the study of Trujillo et al provided a detailed description of the electrophysiological properties of cortical organoids grown on MEAs, with the aim to make a comparison with the development of the human brain in vivo [333]. Over the course of 10 months, cortical organoids exhibited consistent increases in electrophysiological activity, as indicated by increases in firing rate, burst frequency, and synchrony (figure 5(f), table 5). Cultures displayed a robust pattern of activity, switching between long periods of quiescence and short bursts of spontaneous network-synchronized spiking. These synchronous events were periodic (∼0.05 Hz) but infrequent early in development (i.e. first 2 months), occurring roughly every 20 s and decayed monotonically after the initial onset. From 4 months onward, a secondary peak emerged 300–500 ms after the initial network activation, leading to the presence of a faster oscillatory (∼2–3 Hz) pattern up to 6 months in culture. Afterwards, the oscillatory activity of cortical organoids became more variable spatiotemporally [333]. To quantify this network complexity, the regularity, expressed as coefficient of variation (CV) of inter-event intervals, was tracked. The inter-event interval CV consistently increased over 10 months of differentiation, from extremely regular latencies (CV ≅ 0) at 2 months to irregular (CV ≅ 1) at 10 months. In addition, spatial and temporal irregularity on a shorter timescale (i.e. within event) also increased with development [333]. In Trujillo's et al also LFPs were recorded, revealing the development of cortical organoids across different network states: from sparse activity with extreme rigidity and regularity to one acquiring repetitive and regular oscillatory patterns, until it finally reaches a stage of higher spatiotemporal complexity and variability that is reminiscent of self-organized networks [333]. In order to make a comparison with the development of the human brain in vivo, Trujillo et al used an EEG dataset from human preterm infants. Interestingly, they observed similarities in the developmental trajectory of some electrophysiological features between the two datasets, and a regression model based on EEG characteristics of preterm infants was able to predict the developmental trajectory of LFPs activity in organoids with a very strong correlation from week 25 onwards [333]. This provided strong evidence that hiPSCs-derived brain organoids can model the development of functional activity in neuronal networks during early neurodevelopment.
Similarly to hiPSCs-derived 2D neuronal cultures on MEAs, the electrophysiological activity of brain organoids was expected to be highly sensitive to the presence of any kind of compound able to influence the molecular and cellular mechanisms responsible for neuronal activity. Yokoi et al tested this hypothesis by evaluating the response of cortical organoids on MEAs to commonly used convulsants and antiepileptic drugs [381]. As expected, the administration of convulsants, such as PTZ, induced a sudden and persistent seizure-like firing (i.e. increase in the frequency of oscillations and amplitude) in a concentration-dependent manner. Conversely, the administration of antiepileptic drugs caused a concentration-dependent decrease in the frequency of oscillations [381]. This study supported hiPSCs-derived brain organoids on MEAs as a useful tool for predicting the seizure liability of drugs and evaluating the effect of anticonvulsant dugs on 3D human neuronal cultures.
Other studies used MEAs to investigate the development of electrophysiological activity of non-cortical brain organoids, including 'whole-brain' [379], and midbrain organoids [380]. For instance, Fair et al recorded the electrophysiological activity of whole-brain organoids over the span of 5 months, during which they observed the transition from immature electrical activities during the first 2 months of culture (as evidenced by random, low-amplitude, spike events), to more robust electrophysiological properties, which gave rise to complex network bursting events, emerging by the fourth month, and increasing throughout the fifth month of culture [379]. This evolution of electrophysiological activity reflected the development of highly connected neural networks, and was complemented with immunohistochemical and transcriptomic analyses to characterize the cellular and molecular development underlying the functional development [379]. Monzel et al, conversely, recorded the electrophysiological activity of midbrain organoids, containing spatially organized groups of dopaminergic neurons, astrocytes and oligodendrocytes [380]. By treating the organoids with a dopaminergic receptor agonist, they confirmed the presence of functional networks of dopaminergic neurons, characterized by functional synaptic connections and spontaneous neuronal activity [380]. In this sense, the midbrain organoid from Monzel et al appeared as a promising system to model PD pathophysiology.
As previously mentioned, brain organoids show several limitations, including high variability due to limited control over the final size, cellular composition (i.e. cell type and ratio), and density of neural aggregates [56], extremely long protocols [333, 379], lack of vascularization affecting the diffusion of oxygen and nutrients and resulting in a necrotic core [372, 373]. For these reasons, recently, Muzzi et al took inspiration from the work of Frega et al [22] and Tedesco et al [274] (described in 4.2), to generate 3D hiPSCs-derived neuronal networks grown within modular microbeads-based scaffold [55]. To obtain controllability over cell type, density, and structure, and to ensure rapid functional investigations, the hiPSCs differentiation protocol based on Ngn2 overexpression was adapted [129], and combined with chitosan microbeads as scaffold. 3D human neuronal networks were cultured on MEAs, and their electrophysiological activity was recorded over the span of 2 months, characterized and compared with the one exhibited by 2D neuronal cultures grown on MEAs coated with chitosan. 2D cultures showed spontaneous activity from DIV 17, which was mainly composed by isolated spikes and bursts [55]. During development, the general level of activity of the network as well as the number of micro-electrodes involved increased, and organized itself in NBs by DIV 24. From DIV 32–35 onwards, all these parameters tended to stabilize. The dynamics exhibited by 3D cultures during development was similar. Firing activity was detected starting from DIV 17 while single channel bursting activity appeared around DIV 28. NBs, conversely, were not detected in all samples until DIV 38 [55] (figure 5(g), table 5). However, when compared at a late and stable developmental stage, 2D and 3D cultures exhibited some differences. 3D networks showed lower spiking and bursting activities as compared to 2D. The synchronous network bursting activity characterizing 2D neuronal networks dynamic in absence of external inputs was not maintained in 3D cultures. The absence of NBs in 3D neuronal networks might indirectly indicate that neurons on the electrode plane received inputs from neurons of the upper layers of the structure, thus giving rise to a more heterogeneous network dynamic [55].
Undoubtedly, 3D neuronal cultures grown within a scaffold do not allow to mimic in vivo development and cytoarchitecture, as brain organoids might do. Nevertheless, they represent an interesting approach from an engineering point of view. Indeed, they allow to have a good reproducibility, and full control over the final size, structure, cellular composition (i.e. cell type and ratio) and density, depending on the dimension of beads and the differentiation protocol used. Moreover, single beads can be coated with chemical attractants to orient functional neuronal connections between different layers [272]. Lastly, as pointed out by the authors, this approach is well-suited to be combined with 3D MEAs, since the inter-porosity of microbeads-based scaffolds could guarantee the insertion of the 3D micro-electrodes afterwards, without forcing the construction of the culture around the micro-electrodes. In this regard, it is worth underlying that although several prototypes of MEAs with non-planar micro-electrodes and of 'true' 3D MEAs (reviewed at the end of paragraph 3.2.1) have recently been developed, to our knowledge, only a few have actually been tested in combination with brain organoids [89, 149], or 3D hiPSCs-derived neuronal networks [87]. Nevertheless, they certainly represent a promising approach for the future to fully harness the potential of 3D models, which is partially lost whether the electrophysiological activity of 3D neuronal networks is investigated with traditional planar MEAs.
5.2.2. Disease modeling
In the last years, several groups have seen the advantage of using brain organoids to establish 3D in vitro models of neurological disorders (reviewed in [382–384]). Similarly to 2D hiPSCs-derived cultures, organoids-based models of specific neurological diseases can be obtained with different approaches, including the isolation of hiPSCs directly from patients, and the genetic manipulation of healthy hiPSCs line to reproduce the desired pathogenic mutations [382–384]. The vast majority of the studies focused on investigating how pathophysiology altered some aspects of neuronal physiology, such as proteome and transcriptome profiles, cell viability, and structural connectivity, neuronal morphology and cytoarchitecture. Conversely, quite little has been done to evaluate how electrophysiological activity is affected in brain organoids under specific pathophysiological conditions.
To our knowledge, only in a few studies brain organoids have been used in combination with MEA technology to study neurological diseases, such as genetic syndromes [385], psychiatric disorders [157, 386] and neurodegenerative diseases [149, 387] (table 7).
Table 7. Overview of MEA studies using brain organoids in disease modeling. AD = Alzheimer's disease. MFR = mean firing rate, NBR = network burst rate.
| Reference | Disease model | Brain organoid | How the model was obtained | System and plate format | Micro-electrodes per well | Extracted quantitative parameters and phenotype on MEA |
|---|---|---|---|---|---|---|
| Kathuria et al [386] | Schizophrenia | Cortical organoid | hiPSCs from schizophrenia patients | Axion Biosystems 24wMEA | 16 | MFR |
| Kathuria et al [157]) | Bipolar disorder | Cortical organoid | hiPSCs from bipolar disorder patients | Axion Biosystems 24wMEA | 16 | MFR |
| Szebényi et al [149] | Amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD) | Cortical organoid | hiPSCs from ALS/FTD patients | Multichannel Systems 3D MEA | 60 | MFR, spike-timing tiling coefficient (STTC) |
| Ghatak et al [387] | AD | Cortical organoid | hiPSCs from AD patients | Axion Biosystems 12wMEA | 64 | MFR ↑, NBR ↑, synchrony index ↑ |
| Trujillo et al [385] | Rett syndrome | Cortical organoid | CRISPR/Cas9-mediated genome editing of MECP2 | MED64 swMEA | 64 | MFR ↓ |
Among them, the group of Kathuria et al generated cortical organoids from hiPSCs of patients with SCZ and BP [157, 386], and compared their transcriptomic profiles and functional characteristics with organoids from hiPSCs of healthy subjects. In both cases, they found a different expression of genes involved in synapses, neurodevelopment and immune response signaling. MEA recordings showed no differences in the baseline electrical activity (i.e. spontaneous firing rate) between control and SCZ organoids. Nevertheless, when firing activity was evaluated with electrical stimulation, or in in setting of neuronal depolarization, firing rate was significantly increased in control organoids but not in affected organoids. As pointed out by the authors, these preliminary results might give precious insights about the molecular and cellular mechanisms underlying the pathophysiology of SCZ and BP, in this case the involvement of NMDARs [157, 386].
In another study, Ghatak et al used hiPSCs carrying familial AD mutations to differentiate cortical organoids, and characterize their electrophysiological phenotype by using patch-clamp, calcium imaging and MEA recordings [387]. As compared to isogenic control organoids, AD organoids showed increased spontaneous APs, slow oscillatory events (∼1 Hz), and hypersynchronous network activity. Interestingly, this was in line with in vivo evidence of hyperexcitability, which has been suggested to contribute to the loss of synapses, which in turn is related to cognitive dysfunction in early stages human AD brains [387]. In addition, two drugs were tested for their ability to abrogate hyperactivity, suggesting hiPSCs-derived organoids as a useful tool for screening drugs, in this case, directed at the treatment of hyperexcitability and related synaptic damage in AD [387]. Similarly, Trujillo et al genetically modified hiPSCs from healthy subjects to obtain brain organoids with a functional knock-out of MECP2 gene, which in vivo causes Rett syndrome [385]. As compared to control organoids, MECP2-KO organoids exhibited decreased spiking activity. Two currently available lead compounds were tested, and one of them was able to rescue the pathophysiological phenotype observed on MEAs, suggesting it as a potential candidate for clinical trials [385].
Overall, in literature there are only few studies in which brain organoids combined with MEAs have been used to characterize the electrophysiological phenotype of 3D neuronal networks in pathological conditions. Nevertheless, we believe that they could be more widely applied as outstanding human 3D models of neurological diseases.
6. Potential and challenges of MEA technology for biomedical research
6.1. The experimental aim guides the choice of the in vitro neuronal model
In MEA studies, a priori considerations are required to obtain in vitro neuronal models which can provide the most valuable and accurate information on the physiological mechanisms or the diseases under investigation, in compromise with the availability of materials, time, and expertise. In this sense, the experimental aim is critical to guide the decisions to be taken, in particular, regarding the choice of (i) the neuronal source (i.e. rodent or hiPSCs), (ii) the neuronal network composition (i.e. heterogeneous or homogeneous cultures, which cell types and neuronal populations, from which brain region), and (iii) the structural complexity (i.e. 2D or 3D neuronal networks). For each of these decisions, advantages and disadvantages should be considered.
The first decision to be taken regards the neuronal source (i.e. whether to isolate neurons from rodent brains or to differentiate them from hiPSCs lines). Compared to hiPSCs, rodents are an accessible source of mammalian neurons beyond comparison for many laboratories. Since their introduction (more than a century ago), protocols to isolate and culture rodent neurons have been highly optimized and standardized, and countless lines of transgenic mice and rats, with well-defined genotypes and phenotypes, have been made commercially available. This provided neuronal cultures with reduced variability, and more reproducible and reliable results [148, 228, 234, 235, 237, 240–242]. For this reason, 2D rodent neuronal networks represent the 'gold standard' in the MEAs field, and studies using MEAs to characterize the neuronal activity of healthy rodent cultures, both during development and at maturity, are largely available in the literature [97, 118, 128, 166].
Several studies have demonstrated the validity of results provided by rodent models when it comes to investigate physiological and pathological mechanisms that occur in similar ways in both rodent and human brains. Frequently, indeed, results obtained from rodent neuronal networks on MEAs have been confirmed by following works based on hiPSCs-derived neuronal networks, or by in vivo observations. This is the case, for instance, of studies investigating synaptic plasticity phenomena [143, 169–176] and neuromodulation of neuronal activity [187–193], screening neurotoxic compounds [203–207], and characterizing the phenotype of certain neurological disorders [225, 227, 243–245]. In all these cases, rodent neuronal cultures still represent the most rapid, economical, and accessible way to gain valuable insights about the human brain, in both physiology and pathology.
Nevertheless, the use of rodent neurons for biomedical research shows several limitations. Firstly, the use of rodent cells requires frequent euthanasia of live animals and arises substantial ethical concerns. Secondly, besides the genomic, developmental, and physiological inter-species differences [2–5], problems of translatability are also related to the fact that behavioral and cognitive functions, typical of the human brain, can poorly be reproduced in rodent models. By extension, many neurological disorders, particularly those correlated to complex behavioral and cognitive impairments such as psychiatric and neurodevelopmental disorders, can hardly be modeled by using rodent models. In this sense, hiPSCs-derived neurons are believed to represent a promising attempt to reduce the use of animals in biomedical research, and to overcome problems of translatability into human patients.
A key advantage of the use of hiPSCs-derived neurons on MEAs for biomedical research is that hiPSCs technology enables to derive neuronal cultures directly from patients with a well-defined genetic background and clinical phenotype [152, 155, 162, 342, 343, 346, 357]. On one side, this gives the opportunity to develop patient-specific in vitro platforms to test which drugs and therapeutic treatments are more likely to be effective in the specific patient, paving the way toward personalized medicine. On the other side, it enables to investigate the pathophysiology of complex disorders more deeply, in particular those with a relevant genetic component or with unclear causes, by linking specific alterations of the electrophysiological activity of neuronal networks recorded on MEAs, to specific pathogenic mutations, and phenotypic traits [152, 161, 340, 342, 352, 353]. Unluckily, the complexity of neurological disorders, which are usually correlated to an elevated level of heterogeneity in patients' population, makes it difficult to establish this link. For this reason, it is preferred to have access to a cohort of patients sharing, for instance, pathogenic mutations in the same gene [155, 162], or a common phenotypic trait [152, 352], rather than a single patient, to avoid the risk to model the patient-specific phenotype, hence to gain insights about pathophysiological mechanisms and candidate therapeutic targets which cannot be generalized to other patients.
Another advantage of the use of hiPSCs-derived neurons is that hiPSCs lines can be easily genetically manipulated by means of genome editing techniques. This allows to differentiate affected neuronal networks carrying specific pathogenic mutations and genetic variants from healthy hiPSCs lines [18, 150, 156, 160, 314, 327, 353], and isogenic controls from patients' hiPSCs lines [155, 161, 346–348], which simplifies the characterization of the pathophysiological phenotype and enable to more clearly isolate and study the contribution of specific mutations and variants to the disease.
To date, the main limitations of hiPSCs-derived neurons are related to the fact that hiPSCs technology is still in its early stage. Although several protocols of neuronal differentiation are available nowadays, most of them have not been fully optimized and standardized yet. For this reason, often, a large amount of materials, time, and expertise is required, and it is not easy to compare neuronal cultures obtained through different protocols.
Once the neuronal source is decided, a choice should be made regarding the composition of neuronal networks. In the case of rodent neuronal cultures, dissociated neurons or organotypic slices can be isolated from different brain regions according to their relevance to the physiological or pathological mechanism under investigation. In most of MEA-based studies for biomedical research, rodent neurons are isolated from the cerebral cortex, which is involved in complex brain functions, such as memory and learning, and is frequently affected in neurological disorders. Therefore, cortical neuronal networks represent a good system for physiology studies, such as the investigation of synaptic plasticity phenomena [143, 169–175], signal propagation [185, 186], and neuromodulation [187–193], and for disease modeling of neurological disorders, including AD [159, 222], epilepsy [158, 228–231, 233], neurodevelopmental disorders [142, 236, 237], TBI [247, 248, 254], and encephalopathies [250–252]. In other cases, neuronal cultures isolated from the hippocampus are preferred. For instance, thanks to the partial preservation of structural and functional synaptic connections, organotypic slices from the hippocampus are particularly well suited to investigate synaptic plasticity phenomena occurring in the hippocampal circuits [294], whereas dissociated hippocampal neurons can be used to model neurological disorders in which the hippocampus might be involved, such as AD [141, 148, 223], DLB [224], epilepsy [226, 227, 232] and psychiatric disorders [239].
The large availability of protocols to differentiate hiPSCs into different cells of the nervous system represents a great advantage for both physiology studies and disease modeling. Researchers can choose the cell types and neuronal populations which are the most relevant to the physiological mechanisms or the pathophysiology of the disease under investigation, including glial cells (i.e. astrocytes, oligodendrocytes, and microglia) and specific neuronal populations (i.e. cortical glutamatergic and GABAergic neurons, dopaminergic neurons, motor neurons) [316–325].
Different cell types and neuronal populations can be combined into heterogeneous cultures, aiming to mimic the complexity of the human brain and to model the pathophysiological phenotype resulting from the interplay of different neuronal populations. This is the case of models based on highly heterogeneous cortical cultures, including neurons and glial cells obtained with dual-SMAD inhibition protocols [8, 309]. Other models, conversely, are based on the generation of homogeneous populations of cells (i.e. neurons obtained with Ngn2 or Ascl1 induction, glial cells) that can be cultured alone or together in co-cultures. The aim, in this case, is precisely to reduce the complexity of the model, in order to characterize the contribution of specific neuronal populations [18, 314], or of the interactions between specific neuronal populations and glial cells [151, 341] to the pathophysiological phenotype.
Similarly to rodent models, hiPSCs-derived cortical cultures are the most used for modeling neurological disorders, including ASD [18, 152, 160, 340–342], TSC [151, 315, 346], FXS [156, 347, 348], psychiatric disorders [154, 161, 352, 357], epilepsy [327, 330, 353, 354], and AD [153]. Other disorders, such as PD [314] and amyotrophic lateral sclerosis (ALS) [150, 314], conversely, have been modeled by using homogenous cultures of dopaminergic and motor neurons, respectively, since they represent the neuronal populations which are specifically affected in these neurodegenerative diseases.
Another choice that should be based on the characteristic of the disease under investigation and on the experimental aim is the differentiation protocol. Several protocols of neuronal differentiation are nowadays available, and are based on two approaches: (i) the dual-SMAD inhibition principle, and (ii) the overexpression of lineage-determining transcription factors. Differentiation protocols based on dual-SMAD inhibition allow to obtain heterogenous cultures of neurons and glial cells in about 2 months [8, 309–311]. Since they mimic the timescale of human cortical development in vivo [8, 309], they are particularly well-suited to investigate pathophysiological mechanisms occurring in the cerebral cortex during development [152]. However, they are long protocols, requiring a large amount of materials and time, and providing a limited control over the final density and composition of the networks. Conversely, protocols based on the overexpression of transcription factors enable to obtain homogeneous cultures of glutamatergic neurons [7, 50], or co-cultures of glutamatergic and GABAergic neurons [18, 312, 313], in a shorter time (i.e. few weeks), with a reduced variability over the final density and composition of the network (e.g. neuronal networks with controlled percentage of glutamatergic and GABAergic neurons can be obtained [18]). Since, in many cases, it is not necessary to use long protocols based on dual-SMAD inhibition to obtain the desired pathophysiological phenotype, protocols based on the overexpression of transcription factors often represent the most rapid and convenient way to obtain human in vitro models of neurological disorders [18, 155, 161, 356].
Finally, besides the composition of neuronal networks in terms of cell types, a decision should be taken regarding the structural complexity of the model (i.e. whether culturing neurons and glial cells into 2D or 3D neuronal networks). 2D neuronal networks are the most widely used, since they are simpler to culture, they make changes in morphology easy to monitor with various imaging techniques, and they provide valuable results in both physiological studies and disease modeling. Nevertheless, the lack of 3D cytoarchitecture makes them inherently unable to model certain aspects of the brain, such as the interactions cell-cell and cell-ECM in the 3D space. As a consequence, also the electrophysiological behavior of 2D neuronal cultures is less complex, and farther than what observed in vivo [22]. On the contrary, 3D models allow to study neuronal networks in a more physiologically relevant environment. These include (i) 3D neuronal cultures, from both rodents and hiPSCs, (ii) organotypic brain slices isolated from rodent brains, and (iii) hiPSCs-derived brain organoids. 3D neuronal cultures are obtained by culturing neurons, dissociated from rodents or differentiated from hiPSCs, within a 3D scaffold, consisting of biocompatible polymer gels, solid porous matrices, or microbeads, to mimic the ECM [55, 88, 93, 266, 268, 274, 388]. This allows neurons and glial cells to grow arborizations in the 3D space, partially reproducing the cytoarchitecture of the neuronal tissue. The electrophysiological activity of 3D neuronal cultures recorded by MEAs showed to be deeply shaped by the 3D cytoarchitecture, less stereotypical and similar to what observed in vivo [22, 55, 274]. Undoubtedly, neuronal cultures grown within a 3D scaffold do not fully mimic in vivo conditions, but they allow to have a good reproducibility, and full control over the final size, structure, cellular composition (i.e. cell type and ratio) and density. Another 3D model which can be combined with MEAs is represented by organotypic slice cultures isolated from rodents. Organotypic slices partially preserve the 3D organization of the neuronal tissue, and the local structural and functional connections of the brain region from which they are derived [54, 278–281], however they cannot be maintained in culture for long periods of time (i.e. few weeks). In this sense, they represent a compromise between the longevity of dissociated cell cultures, and the preservation of the 3D cytoarchitecture of the brain. Undoubtedly, hiPSCs-derived brain organoids represent the most intriguing option to model the complexity of the human brain in both physiology and pathophysiology [56, 358–360]. hiPSCs, indeed, are able to self-assemble without the use of a 3D scaffold, into spheroidal aggregates with an organized cytoarchitecture, composed of NPCs, neurons and glial cells, resembling the embryonic human brain at different developmental stages [56, 333, 358–360, 379]. However, they are a relatively new technique with critical limitations related to protocols lack of standardization, high variability, and inaccessibility in terms of time and materials (i.e. extremely long protocols, up to several months) [329]. Despite the undeniable advantage of having a 3D model which so well mimic the cytoarchitecture, development, and electrophysiology of the in vivo brain, the above-mentioned limitations fully explain why hiPSCs-derived brain organoids in combination with MEA technology are not widely used for biomedical research.
To conclude, when it comes to developing a neuronal model on MEAs, advantages and disadvantages of the different possibilities should be critically evaluated according to the experimental aim, with the purpose of obtaining a simplest model providing the most valuable and accurate information in compromise with the availability of materials, time, and expertise.
6.2. The neuronal model and required information guide the choice of MEA device
Nowadays, several MEAs are available, including (i) traditional low-density swMEAs and mwMEAs, (ii) HD-MEAs, (iii) MEAs with non-planar micro-electrodes, and 'true' 3D MEAs. Right after the choice of a neuronal model, according to the characteristics of the chosen model and to the information required for the experimental aim, a MEA device should be selected from the available ones, which is not always trivial. In this paragraph, we provide some guidelines for the choice of MEA devices. In this sense, strengths and weaknesses of each device should be evaluated, with the aim to choose the MEAs providing the most valuable and accurate information, in compromise with the availability of materials, time, and expertise.
Traditional swMEAs and mwMEAs are classified as low-density devices. swMEAs are available in different formats, with a number of micro-electrodes ranging from 60 to 256, organized in different layouts, whereas mwMEAs are available in plate formats with up to 96 independent wells, with a limited number of micro-electrodes. mwMEAs represent an outstanding high-throughput platform, enabling to measure the electrophysiological activity of several neuronal networks at the same time. This is particularly advantageous in neurotoxicity and drug screenings, when it comes to screen a large number of neurotoxic chemicals and drugs [200], to characterize their dose-response curve [201], or to test the same compound in neuronal networks with different characteristics, for instance, representative of different brain regions [195]. Similarly, mwMEAs can be used in disease modeling to characterize the electrophysiological phenotype of neuronal networks derived from the hiPSCs of different patients, which are often characterized by high variability. In all these cases, mwMEAs represent the most convenient and rapid way to obtain a general, but valuable, overview of the network activity of neuronal cultures under different conditions. However, the downside of mwMEAs is that the number of micro-electrodes per well decreases with the increase in the number of wells (i.e. Multi-Channel Systems: 12 micro-electrodes for 24-well plate and 3 micro-electrodes for 96-well plate; Axiol Biosystems: 16 micro-electrodes for 24-well plate and 8 micro-electrodes for 96-well plate). Although the spatial resolution is adequate to observe the overall neuronal network activity, it is not good enough to obtain other types of information (i.e. functional connectivity, signal propagation). In this case, devices with higher number of micro-electrodes should be chosen (swMEAs or HD-MEAs).
In particular, the recently developed HD-MEAs present a much higher number and density of micro-electrodes (up to thousands of micro-electrodes per mm2), allowing to record the activity of neuronal networks with spatial-temporal resolution and signal quality once unimaginable, and to integrate the extracellular recordings at subcellular, cellular, and network levels from the same culture [74–80]. For these reasons, HD-MEAs are particularly well-suited to investigate particular physiological and pathological mechanisms with high spatial-temporal resolution, including the contributions of individual presynaptic synapses to postsynaptic potentials in short- and long-term synaptic plasticity [143], or how AP propagation along axons is affected in neurodegenerative diseases such as PD and ALS [314]. Recently, Ito et al used a 512-channel HD-MEAs to measure the activity of organotypic cultures of cortical and hippocampal brain slices [288]. In this case, the use of HD-MEAs enabled the analysis and characterization of functional connectivity between neurons at different frequency ranges, pointing out differences of network architecture in different brain regions, made evident thanks to the high spatial-temporal resolution provided by HD-MEAs [288]. HD-MEAs should be also preferred to record the electrophysiological activity of hiPSCs-derived brain organoids, given their small size [372, 373]. However, the increased spatial and temporal resolution reached with HD-MEAs results in a huge amount of raw data (e.g. a 10 min recording with a 4096 electrode MEA device and sampling rate of 20 kHz is approximately 90 GB in size [30]), which on one hand are easily obtainable, but on the other hand are difficult to handle as regards to data storage and analysis. In addition, the use of HD-MEAs requires the careful evaluation of possible cross-talk artifacts, which can affect the electrophysiological recordings. Channel interference due to cross-talk is can be partially addressed, via continuous interleaved sampling or post-data acquisition spike sorting [81]. For these reasons, it is preferred to use HD-MEAs only when high spatial and temporal resolution is needed to investigate particular physiological and pathological mechanisms [143, 314].
Lastly, MEAs with non-planar micro-electrodes and 'true' 3D MEAs (i.e. recording simultaneously from multiple 2D planes) are nowadays available. These devices are particularly well-suited for recording the electrophysiological activity of 3D neuronal cultures, including 3D neuronal networks from both rodents and hiPSCs grown within a biocompatible scaffold, organotypic brain slices isolated from rodent brains, and hiPSCs-derived brain organoids. In these cases, electrophysiological recordings acquired by non-planar micro-electrodes, or by micro-electrodes distributed on multiple 2D planes, allow to better characterize the complex dynamics of 3D neuronal networks, similar to those observed in vivo [22, 55, 274]. However, to date, only a few studies used 3D MEA devices in combination with 3D neuronal cultures [87, 88, 149], while the vast majority of studies were limited to a general, even if valuable, overview of 3D neuronal networks activity provided by traditional planar MEAs. This is probably due to the fact that even though 3D models have become more and more common, 3D MEA technology is still in its infancy, and almost all commercially available MEAs are designed for 2D cultures. However, without any doubt, 3D MEA devices should be preferred to characterize the network activity of 3D neuronal cultures, to fully benefit of having a 3D model.
6.3. Guidelines to fully harness the potential of MEA technology are urgently needed
Despite the increasing popularity of MEAs, there is little insight into how MEA-based neuronal models should be used (i.e. which recommendations should be followed in regards of control neuronal networks, experimental design, data analysis and reporting) to fully harness the potential of MEA technology in the field of biomedical research.
In this sense, a recent work from Mossink et al provided a set of guidelines for the evaluation of control networks and the design of experiments for functional phenotyping on MEAs [19]. In particular, they used hiPSCs derived from 10 healthy subjects which were differentiated into excitatory neuronal networks through Ngn2 overexpression, and cultured on MEAs by different researchers over a period of several years. They showed that different control networks were highly comparable when used in the time window between DIV 27 and 35, as networks generated by Ngn2 overexpression presented stable activity at this stage [19]. However, many factors can influence the timing of this stable network activity, including the differentiation protocol and coating. Therefore, one must define the stable developmental period depending on each protocol, before pooling and comparing data [19]. They found that astrocytes and MEAs batch introduced variability, and advised to use at least 12 wells per condition, divided over two MEA batches with the same astrocytic batch [19]. In addition, they suggested to exclude from analysis wells with low or uneven cell density, or with low activity, indicating a set of values of MFR, NBR and active channels, indicative of the level of activity of healthy neuronal networks [19]. In regard of disease modeling, they advised to use hiPSCs lines derived from multiple healthy subjects and patients, or isogenic sets, to reliably characterize the disease phenotype, since differences in genetic background between hiPSCs donors dominate the variability at the transcriptional level [19].
Although these guidelines are valid only for homogeneous neuronal networks of excitatory neurons obtained through a very specific protocol of differentiation (i.e. Ngn2 overexpression), we believe that the work of Mossink et al is extremely inspiring. To our knowledge, clear indications about how the electrophysiological activity of healthy neuronal networks on MEAs should look like are largely missing. Even if the electrophysiological activity of rodent neuronal networks on MEAs, both during development and at maturity, has been characterized in the past 50 years, we noticed a great variability in the values of MFR, MBD and NBR reported for control neuronal networks (table 3) [22, 103, 142, 166]. This issue is even more critical with regard to 3D neuronal networks [22, 55, 93], hiPSCs-derived networks of specific neuronal populations (such as dopaminergic and motor neurons) [150, 332], and brain organoids [333] (tables 3 and 5), since studies characterizing the electrophysiological phenotype of these neuronal networks on MEAs are scarce or none. Although this can be partially explainable with the relative newness of these techniques, which can be in themselves related to protocols lack of standardization, high variability, and inaccessibility in terms of time and materials, this scenario can be extremely confusing to those who want to approach MEA technology for the first time. Undoubtedly, the present review represents a step in the right direction. However, we believe that more studies like the one of Mossink et al, providing a set of guidelines for the evaluation of healthy neuronal networks, and for the experimental design of functional phenotyping assays, should be carried out and made available to the scientific community in order to fully harness the potential of MEA technology.
The huge amount of data that can easily be obtained from MEA recordings shifts the challenges from the experimental to the analysis side. Improvements in data analysis have greatly simplified the extraction of quantitative parameters from raw data. However, data analysis itself can remain a hurdle, especially for researchers with no background in programming. For example, it is possible that the observed phenotype cannot be described with any of the most commonly used parameters, and new parameters need be introduced to capture these signatures and reveal previously unseen phenotypes [19]. In this case, new ad hoc algorithms should be written, in order to describe specific characteristics of network activity which may arise, for instance, by the visual observation of raster plots. Then, special attention should be given for the choice of different analysis settings and algorithms for the detection of spikes and bursts, or for the identification of NBs, since they largely influence the results. These settings need to be fine-tuned, depending on the visual observation of raster plots to accurately detect the phenotypic signatures of the network under investigation. For instance, NBs exhibited by affected neuronal networks might be incorrectly detected with commonly used settings, since these were conventionally chosen based on NBs in control networks [19]. Finally, the complex spatiotemporal organization of the electrophysiological activity generated by neuronal networks on MEAs represents a relevant challenge when it comes to selecting meaningful phenotyping metrics. In literature, MEA-based studies often report one single parameter (i.e. the MFR) as it is easily extracted from raw data. However, in the study from Mossink et al investigating the variability of MEA parameters in hiPSCs-derived neuronal networks, the MFR was found to be one of the most variable parameters, thereby not trustworthy to describe, for instance, the phenotype of disease models [19]. In addition, it only describes the general level of activity and is largely dependent on cell density. Describing the electrophysiological phenotype by any single parameter in isolation is likely to omit crucial information about how neuronal network activity is affected in pathological conditions, which could provide important clues about the underlying disease mechanisms. Thus, to adequately capture the disease phenotype of neuronal networks different quantitative parameters are needed (i.e. multiparametric approach). In particular, to use MEA parameters with high variability, such as the MFR, to describe the disease phenotype, one should include multiple supporting MEA parameters to describe the network activity characteristics [19].
Another important consideration for researchers involves standardization in MEA data reporting. In the present review, we found considerable variation, not only in the choice of the algorithms and analysis settings do the detection of spikes, bursts, and NBs, but also in the name, definition, and unit of measurement of the quantitative parameters extracted from MEA recordings. We believe that researchers should be sure to report a full explanation of these parameters and of the procedure to extract them. Moreover, raster plots of representative network activity should be included as a supplement to any quantitative metrics used to characterize the electrophysiological phenotype of neuronal networks under investigation. This would provide some more intuitive understanding of neuronal network activity, but also would allow other researchers to quickly assess the quality of raw data, ensuring there are no technical artifacts, inactive micro-electrodes or excessively noisy channels. The inclusion of representative raster plots would be particularly important in studies including phenotypic rescue assays. Indeed, the effectiveness of certain pharmacological interventions can easily be misrepresented by selectively reporting only those parameters which see substantial improvement with drug treatment, even if that improvement does little to change the qualitative electrophysiological phenotype observed in affected neuronal networks [389]. We believe that these recommendations are a step in the right direction towards ensuring high standards of scientific integrity and transparency in data reporting, but also accessibility of data for those who want to approach the field.
6.4. Neuronal models on MEAs give insights about cellular and molecular mechanisms
The electrophysiological activity as recorded by MEAs (i.e. network activity patterns, the features of spikes and bursts, the quantitative parameters extracted from raw data) is deeply shaped by the underlying physiology of neuronal networks. Thereby, MEA-based models can provide precious information about the cellular and molecular mechanisms occurring in neuronal networks in both physiological and pathophysiological conditions.
Neuronal cultures on MEAs, both isolated from rodents or differentiated from hiPSCs lines, represents a valid experimental model for investigating events occurring during the development of neuronal networks [97, 107, 117, 118, 128, 336]. During the development of neuronal tissue, the evolution of the electrophysiological activity can be related to neurodevelopmental processes, including synaptogenesis of both excitatory and inhibitory synapses, shaping of structural and functional connectivity, and changes in the expression profiles of receptors and ion channels during development. For instance, the emergence of network burst activity reflects the formation of synapses to form a functional connected network [97, 167]. Or else, the shortening of NBs duration in late differentiation stages of hiPSCs-derived neurons indicates the full maturation of GABAergic neurons [18].
In the same way, the electrophysiological activity of mature neuronal cultures on MEAs reflects the underlying characteristics and physiological mechanisms, such as the excitatory-inhibitory balance [18], structural and functional connectivity [97], synaptic plasticity phenomena (such as LTP and LTD) [170, 172–174, 338], neuromodulation of neuronal network activity [175, 187–190], and the expression of receptors and ion channels [168]. MEAs represent a valid experimental for investigating these physiological characteristics and mechanisms occurring in healthy neuronal networks, but also in affected ones, since all these aspects are frequently impaired in pathophysiological conditions.
In the context of disease modeling, MEAs are a powerful tool to perform phenotyping assays, aiming to characterize how the electrophysiological activity of neuronal networks is altered in specific pathophysiological conditions. However, their potential does not stop there. Once established, a well-defined disease model can give insights about the underpinning molecular and cellular mechanisms contributing to the pathophysiological phenotype, and, in some cases, revealing new candidates for treatment strategies. Indeed, by observing how the activity patterns, and the parameters describing the features of spikes and bursts, are altered in affected neuronal networks, information about the underlying disease mechanisms can be deducted. In figure 6, we provided some relevant examples taken from the literature, of neuronal network activity phenotypes observed on MEAs suggesting hypothesis about the molecular and cellular mechanisms underlying pathophysiology. For instance, high firing and bursting rates, or abnormal synchronous activity, characterize a hyperactive phenotype which can be related to different causes, such as the imbalance of excitatory and inhibitory synapses [238], the overactivation of signaling pathways involved in the regulation of synaptic function [346, 354] and neuronal network synchrony [158], or intrinsic characteristics of neurons [228, 230, 327, 353]. Conversely, a decrease in firing and bursting activity can be due to the degeneration of neuronal network functionality and/or connectivity [131, 148, 153, 162, 222, 224, 243]. Also, a change of the burst shape, in particular of the NBD can give precious hints about the underlying disease mechanisms. For instance, Mossink et al observed that the knockdown of CDH13, which have been associated with ASD, caused a significant reduction of the NBD, suggesting the involvement of CDH13 in maintaining the excitatory/inhibitory balance in neuronal networks [18]. Similarly, Frega et al, based on the observation of longer NBs in hiPSCs-derived neuronal networks differentiated from KS patients, postulated the involvement of NMDARs-mediated currents, which are known to directly influence the duration of bursts [155].
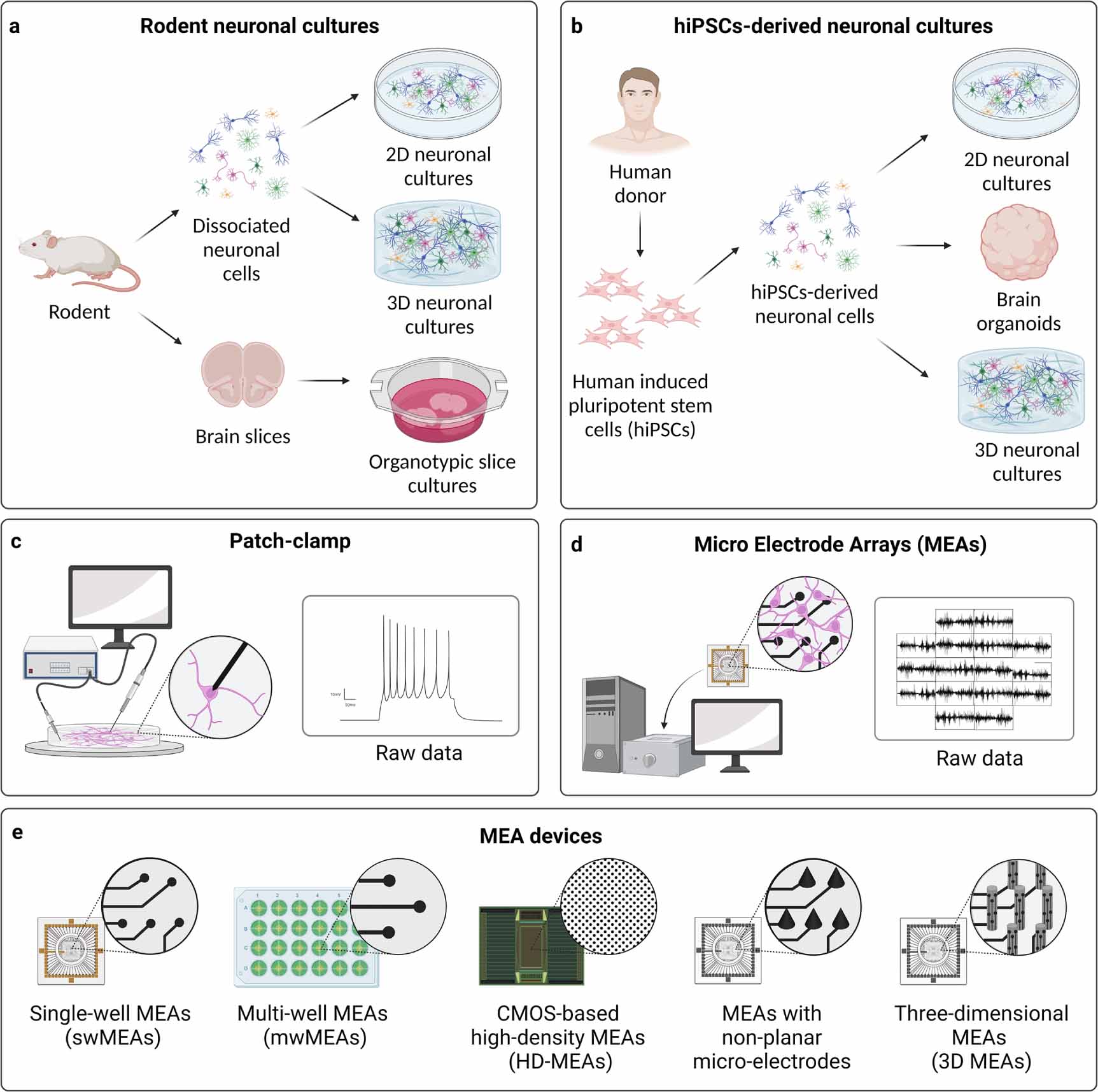{kind=link}
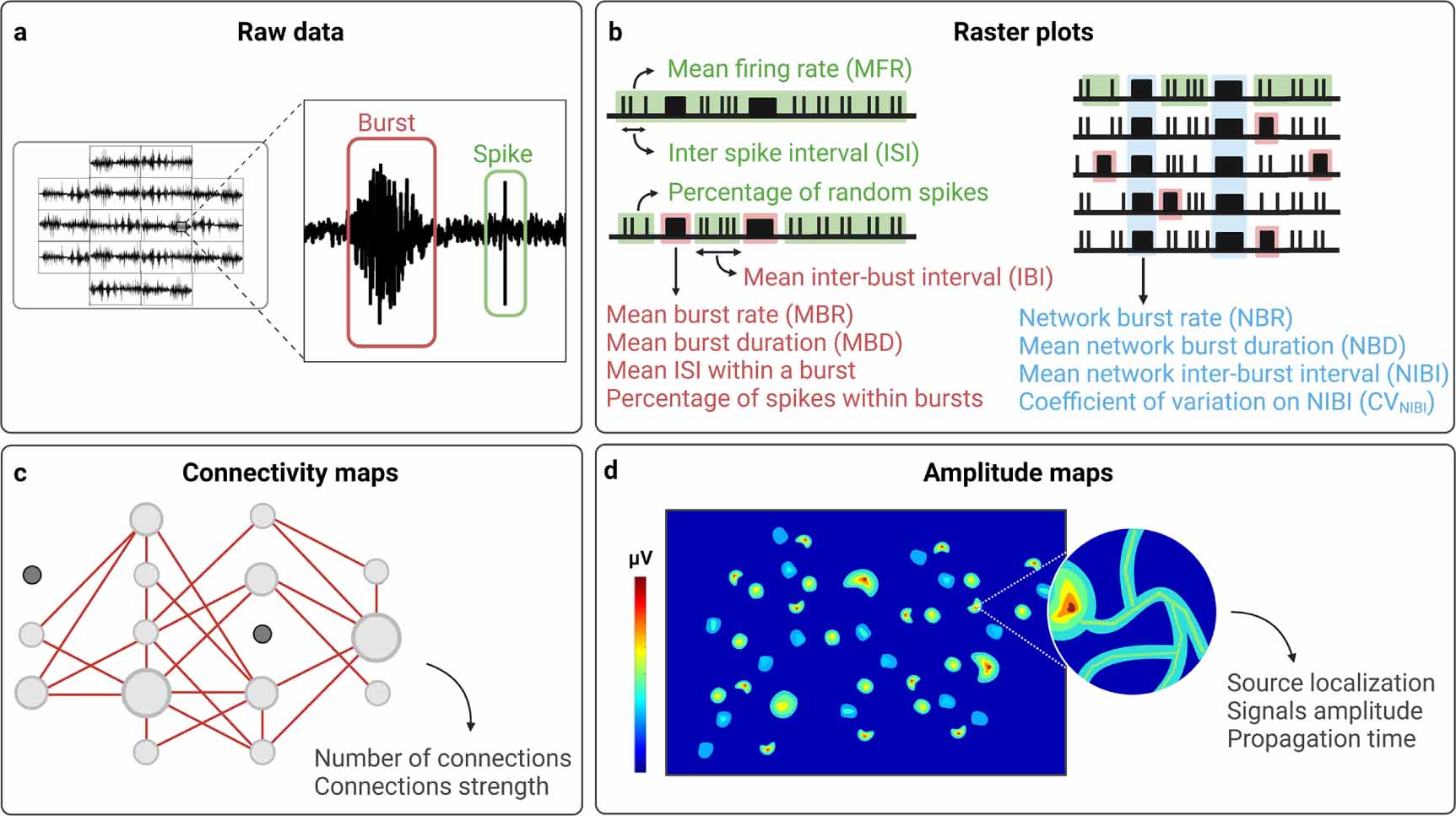{kind=link}
{kind=link}
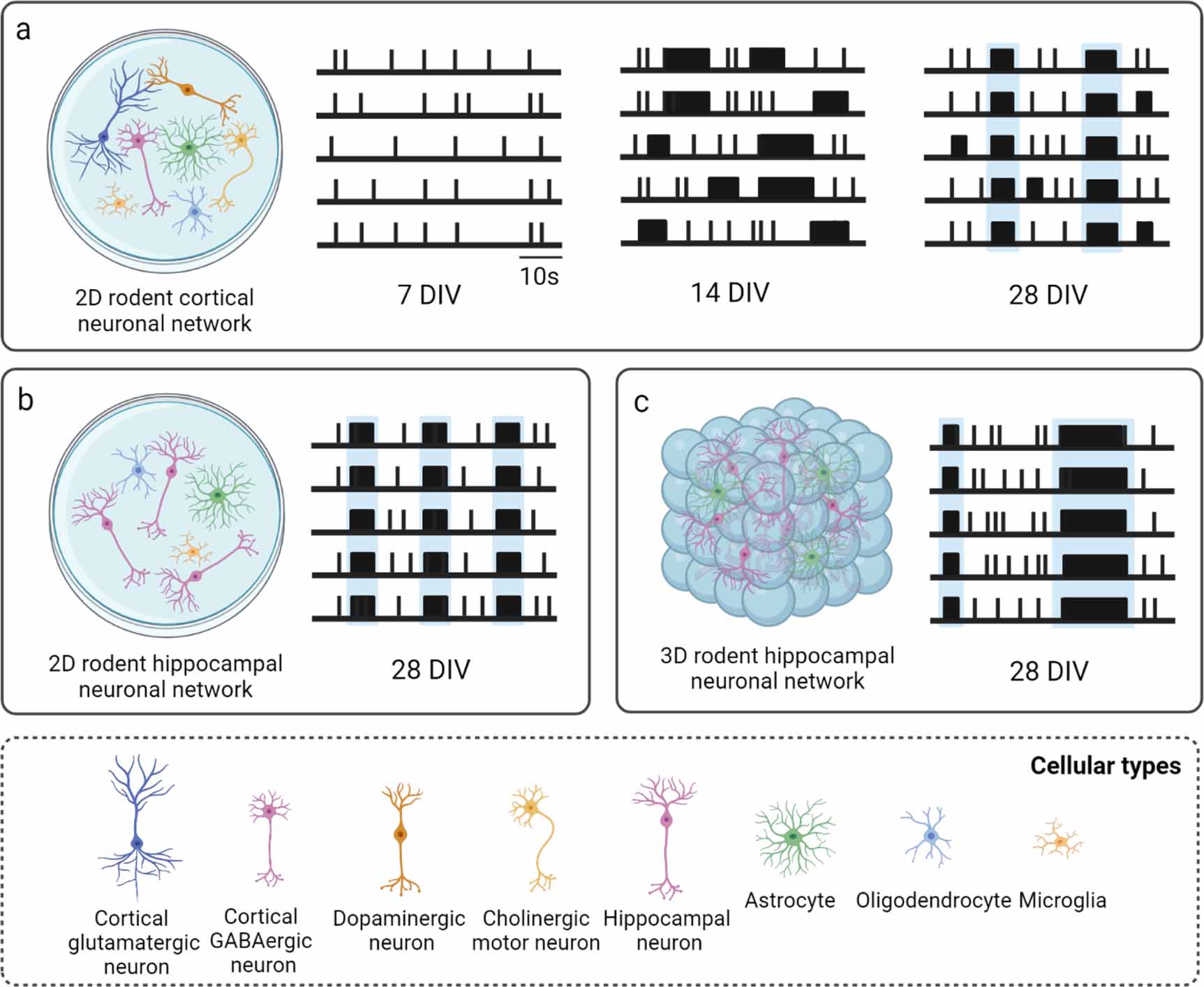{kind=link}
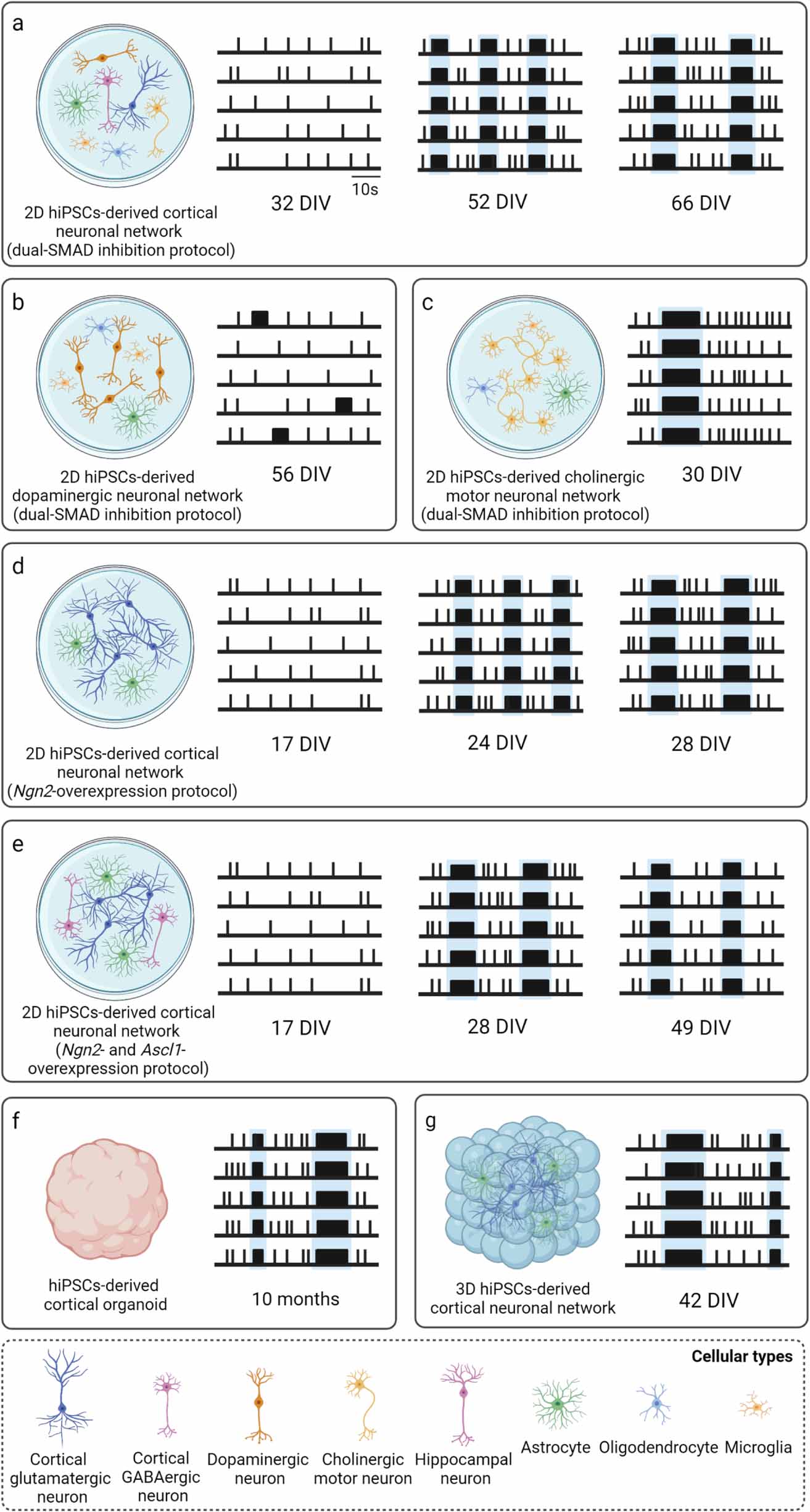{kind=link}
Figure 6. Schematic raster plots describing the phenotype on MEAs showed by affected neuronal networks in comparison with healthy neuronal networks, suggesting hypothesis about the molecular and cellular mechanisms underlying pathophysiology. NBs are highlighted in light blue. Created with BioRender.com.
Download figure:
Standard image High-resolution image{kind=link}
These 'hints' can be easily obtained by characterizing the electrophysiological activity of neuronal networks in specific pathophysiological conditions, and can guide researchers in the further investigation of the underlying disease mechanisms by means of MEAs themselves and (i) other electrophysiology techniques, such as patch-clamp and calcium imaging, (ii) biological methods, including immunostainings and transcriptome analysis, (iii) pharmacological treatment (i.e. the use of inhibitors of specific receptors, ionic channels, and enzymes) and (iv) genetic manipulation. For instance, in the above-mentioned study from Frega et al, researchers combined recordings from MEAs with patch-clamp experiments, transcriptome analysis and pharmacological inhibition of NMDARs-mediated currents, in order to test their hypothesis [155]. Patch-clamp and transcriptome analysis showed increased NMDARs-mediated currents, whereas the pharmacological inhibition of NMDARs resulted in the rescue of the pathophysiological phenotype of KS patients-derived neuronal networks. This work not only demonstrated the validity of their hypothesis, but also suggested a new candidate target for treatment strategies (i.e. NMDARs-mediated currents) [155]. Besides this clear example of MEAs potential in the investigation of disease mechanisms, several other studies in which MEA devices were used to investigate the cellular and molecular mechanisms underpinning the pathophysiological phenotype of affected neuronal networks, can be found in 4.1.4 and 5.1.4.2. As previously mentioned, the complexity of neurological disorders, which are usually correlated to a high level of heterogeneity in the patient population, makes it difficult to clarify the pathophysiological mechanisms underlying the electrophysiological phenotype observed in affected neuronal networks on MEAs. However, we believe that MEA technology may represent a valuable, convenient and relatively easy tool to start with in the investigation of pathophysiology of complex neurological disorders.
6.5. Findings from in vitro neuronal models are relevant for biomedical research
In vitro neuronal models are simplified systems with the aim to provide insights about complex neurological mechanisms and diseases. Therefore, a key aspect is the possibility to translate findings obtained in vitro into in vivo animal models, and, finally, humans. In this regard several studies suggested the comparability of the results found in in vitro MEA models to observations in in vivo models and in humans.
Several in vitro models of neurological disorders on MEAs recapitulate the dysfunctional electrophysiological activity observed in vivo. For instance, models of epilepsy on MEAs, obtained through the treatment with pro-convulsant drugs or electrical stimulation, are characterized by hyperexcitability, increased activity and synchronicity, which are common features of epileptic phenotypes in vivo [158, 227, 230–233]. In one of these models, Hales et al also observed evoked high frequency oscillations, which have been described in both animal models and humans in association with epilepsy [225]. Similarly, MEA-based models of neurodegenerative diseases, such as AD and DLB, showed a progressive decrease in electrophysiological activity which has been related to the degeneration of neuronal network functionality and connectivity, and to the impairment of cognitive processing observed in vivo [141, 159, 222–224]. In an in vitro model of ischemic stroke, Pires Monteiro et al observed electrophysiological responses to transient hypoxia (i.e. suppression and subsequent restoration of network activity) on timescales similar to patients with cerebral ischemia [131]. The same was found in regard to the therapeutical time window: similarly to what observed in patients, the functionality of neuronal networks was restored only if normoxia was re-established within 24 h [131].
Neurotoxicity screenings on MEAs showed that neurotoxic compounds not only affected the electrophysiological activity of neuronal networks in a way that is qualitatively similar to what observed in vivo, but often showed a time- and concentration-dependent effect which was in line to reported studies in rodent models and humans [206, 207]. As an example, Xia and Gross found that ethanol induced a decrease of firing activity in neuronal cultures isolated from the frontal cortex of rodents at concentrations comparable to those estimated in blood to cause the typical behavioral effects in vivo [207]. Similarly, Croom et al characterized the neurotoxicity kinetics of lindane (i.e. a GABAA receptor antagonist which causes seizures in vivo) in rodent neuronal cultures on MEAs, and demonstrated the predictivity of results in regard to doses and timing reported in the literature for human and rodents [206].
In addition, several drug studies supported the relationship between the sensitivity of neuronal networks cultured on MEAs to drugs and the expected effects in vivo. For instance, Colombi et al and Que et al investigated the pharmacological effect of commonly used antiepileptic drugs in two different in vitro models of epilepsy on MEAs [227, 353]. Colombi et al studied the effect of carbamazepine and valproate in rodent neuronal networks treated with the pro-convulsant BIC, and found that both were effective in ameliorating the epileptic phenotype at concentrations close to those clinically relevant [227]. Que et al used hiPSCs-derived neuronal networks carrying a genetic variant that in vivo causes seizures. Besides increased excitability, they observed a degree of resistance to the anti-convulsant drug phenytoin, which recapitulated aspects of clinical observation of patients carrying the same genetic variant [353]. Similarly, van Hugte et al tested four different anti-seizure drugs in hiPSCs-derived neuronal networks from patient with two different types of epilepsy, and found that in vitro responses to different drugs exactly corresponded to those observed in patients [355]. All these studies suggest that hiPSCs-derived neuronal networks from patients cultured on MEAs might be used to develop patient-specific in vitro platform to test which drugs are more likely to be effective in the specific patient, paving the way toward personalized medicine.
Several studies demonstrated that 3D neuronal cultures on MEAs could better represent in vivo cellular behavior than cells cultured in monolayers. For instance, 3D rodent neuronal networks showed a repertoire of complex and asynchronous global activity patterns, resulting from the higher complexity of the network, which seemed to better recapitulate activity dynamics of in vivo brain regions [22, 274, 388]. In this regard, a particularly relevant example is set by Trujillo et al which developed a human cortical organoid on MEAs in order to investigate the development of neuronal network activity over the span of several months [333]. Interestingly, they observed similarities in the developmental trajectory of some electrophysiological features between in vitro organoids and human preterm infants, supporting the validity of hiPSCs-derived brain organoids on MEAs as a model of the human brain [333].
7. Future perspectives of MEA technology for biomedical research
Neuronal cultures grown on MEAs provide a unique opportunity to investigate neuronal networks dynamics in vitro. For this reason, they have become a key in vitro system to study the development of neuronal networks and physiological mechanisms, to model neurological disorders, and to investigate the effects of neurotoxic compounds and drugs. With the advent of hiPSCs technology and the possibility to differentiate neurons maintaining the genetic background of the donor, MEAs has become a robust tool for phenotyping human neuronal networks and conduct patient-specific investigations. Indeed, neuronal networks derived from healthy subjects or patients show a robust and replicable in vitro functional phenotype, and various genotype/phenotype correlations have been established [19]. Furthermore, since the electrophysiological activity as recorded by MEAs is deeply shaped by the underlying characteristics of neuronal networks, it can give insights about underpinning molecular and cellular mechanisms contributing to the observed phenotype of affected neuronal networks, and, in some cases, revealing new candidates for treatment strategies.
With the advancements in cell cultures and MEA technology, new challenges have arisen. Thus, we believe that new tools should be exploited to boost even more the use of MEA technology for biomedical research. In the following paragraphs we will illustrate challenges and future perspectives related to data analysis, identification of molecular and cellular mechanisms from neuronal network activity, and translation of in vitro findings into humans.
7.1. Statistical and computational approaches help the identification of phenotypic signatures
With the advent of hiPSCs and the possibility to derive human neuronal networks from patients, a highly heterogeneous variety of novel signatures have been observed in MEA recordings. This leads to new challenges in the MEAs field, in particular related to the identification of the most relevant parameters to describe the observed neuronal network phenotype.
As previously mentioned, multiparametric approaches should be used to describe the phenotype of affected neuronal networks, in order to avoid omission of crucial information and misinterpretation of results. However, they can present their own issues when it comes to draw insights about the underlying pathophysiological mechanisms, or to evaluate and compare the effect of different drugs and treatments strategies in phenotypic rescue experiments. In these cases, it is crucial to understand which quantitative parameters are the most physiologically relevant to the neurological disorders under investigation, which is not always trivial. Nowadays, in most studies the choice of the meaningful parameters to be extracted from MEA raw data is dependent on the visual observation of raster plots by researchers performing the analysis. Also, depending on the disease of investigation, the set of meaningful parameters can change. Undoubtedly, this represents a time-consuming procedure, inevitably affected by human error, subjectivity, intra and inter-observer variability. For this reason, we believe that advanced statistical and computational methods might provide a promising approach for an in-depth and unbiased analysis of MEA recordings.
A clear example is set by Mossink and colleagues [19]. In their study, they extracted 17 parameters in total to describe the neuronal network activity and connectivity of hiPSCs-derived neuronal networks from healthy subjects, KS patients and MELAS patients [19]. All the parameters were combined in a principal-component analysis, a statistical technique for the analysis of large datasets containing a high number of dimensions per observation, enabling the visualization of multi-dimensional data in intuitive plots. By performing this analysis, they observed that neuronal networks from KS and MELAs patients clustered separately from controls, and were able to identify the parameters that explained the differences in the electrophysiological behavior of control and affected cultures, and to calculate the percentage of variance explained by each of these parameters [19].
Another approach which might be adapted to the analysis of MEA recordings is artificial intelligence (AI), including traditional machine learning and deep learning. These methods are widely used in image recognition. Briefly, a typical machine learning workflow for image recognition consists in the preparation of a training dataset of images associated to categories. The training dataset is presented to the model, which identify and extract the key features that can be used to differentiate the images of different categories. In this way, the model 'learns' to analyze and classify new images without being explicitly programmed to do so. Machine learning approaches have been used, for instance, for the recognition of interictal epileptiform discharges (i.e. epileptiform transients indicating an increased likelihood of seizures) in EEG recordings, providing highly accurate and robust results [390, 391]. Similarly, machine learning might be adopted for the analysis of MEA recordings, for instance, for the classification of raster plots from healthy or affected neuronal networks, or for the classification of drugs based on their effectiveness in rescuing the phenotype of affected neuronal networks. In these cases, machine learning approaches could be used to analyze and classify raster plots obtained from MEA raw data, as they were images. Raster plots, indeed, contain all the information regarding firing, bursting and network bursting activity of neuronal networks. Conventional analysis of MEA recordings is based on pre-established methods to detect spikes, burst and NBs, whose settings need to be fine-tuned to properly describe the phenotype of the neuronal network under investigation. Similarly, the quantitative parameters to be extracted from raw data are decided by researchers conducting the study on the basis of visual observation of raster plots. In addition to variability resulting from human error and subjectivity, this approach inevitably introduces a limitation in terms of number of parameters which can be extracted and further analyzed, for instance, to identify differences between healthy and affected neuronal networks. For all these reasons, analysis of MEA recordings and conventional analysis methods and parameters might fail in the characterization of the electrophysiological phenotype observed in raster plots, especially for newly established disease models. Therefore, AI might assist researchers in the extraction of meaningful features from raster plots, giving the possibility to capture changes in neuronal network activity without choosing settings to detect spikes, bursts and NBs, or pre-established parameters.
Recently, Zhao et al developed a deep learning model able to split long MEA recordings into smaller slices and classify them according to the genotype of neuronal cultures (i.e. rodent wild-type versus delta-catenin knock-out neuronal networks, and hiPSCs-derived control versus Williams syndrome neuronal networks) [392]. Zhao et al reported that their model succeeded in classifying MEA recordings with a good accuracy, and could be easily adapted to the analysis of raw data from MEAs with a larger number of probes, such as HD-MEAs [392]. Similarly, Matsuda et al proposed an analysis method using deep learning to extract 4096-dimensional features from raster plot images [393]. By using MEA recordings from hiPSCs-derived neuronal networks grown on MEAs, they showed that seizure-causing compounds and seizure-free compounds were distinguished by using machine learning algorithms trained on raster plots. With this model, features that were not detected using single-parameters analysis were identified, and detailed differences between compounds were detected even when their main effect was the same [393].
We believe that approaches as the ones presented here hold promise for the future of in vitro MEA-based model for biomedical research, helping researchers to identify the phenotypic signatures of neuronal networks under investigation, regardless of visual observation of raster plots and a priori choice of analysis settings and parameters.
7.2. In silico models are a powerful tool to uncover cellular and molecular mechanisms
The possibility to obtain 'hints' from MEA recordings that can guide researchers in the investigation of the pathophysiology of affected neuronal networks is impressive. However, the identification of cellular and molecular mechanisms underlying the neuronal network phenotype observed on MEAs remains challenging. Conventional approaches are defined as hypothesis-driven, since they aim to test the contribution of candidate mechanisms to the neuronal network phenotype. They include, for instance, the external perturbation of the network with electrical stimulation, drug administration or genetic manipulation, or the integration of MEA recordings with results from other techniques, such as patch clamp, calcium imaging and biological methods. However, these approaches can be very time-consuming and expensive, and need a priori hypothesis to test.
In silico models of neuronal networks can complement and assist in vitro experiments and observations of MEA recordings, representing a powerful tool to facilitate the identification of the cellular and molecular causes of the observed phenotype. With in silico models, the experimental setup can be recreated and simulations can be run to validate or reject a large number of hypotheses. In addition, in silico models allow to extend experimental setups and to include a virtually unlimited array of parameters, which might not be easy to setup experimentally [394].
In silico models of neuronal networks consist of a system of equations able to explain biological, chemical and physical phenomena occurring at the network level. The study of computational models started in 1952 with the pioneering work of Hodgkin and Huxley, who published five papers describing a series of experiments and an empirical model of AP in a squid giant axon [92, 395]. The so-called Hodgkin-Huxley model consists of a system of four coupled nonlinear ordinary differential equations describing the generation of APs, and it represents the starting point of many models developed in neuroscience [396]. An important result of the Hodgkin–Huxley studies is that neurons are dynamical systems, and, as such, they consist of a set of variables that describe their state and a law that describes the evolution of the state variables with time [396]. Furthermore, in silico models that are based on artificial neural networks have been produced via simulation of real neural networks according to Hebbian theory [397].
Researchers developing and testing computational models often face the challenge of finding the sensitive trade-off between computational time and model accuracy. One example is given by the model introduced by Izhikevich in 2003 [398]. Izhikevich explained that the model for a single neuron must be both computationally simple, yet capable of producing rich firing patterns exhibited by real biological neurons [398]. In this sense, using biophysically accurate Hodgkin–Huxley type models is computationally prohibitive, since only a handful of neurons can be simulated in real time. Conversely, using an integrate-and-fire model is computationally effective, but it is unrealistically simple and incapable of producing rich spiking and bursting dynamics exhibited by cortical neurons. The model presented by Izhikevich is the fair compromise within the two bounds, it depends on four parameters, and reproduces spiking and bursting behavior of known types of cortical neurons [398].
The explosive increase in computational power and resources of recent times has pushed the implementation of in silico models of neuronal network activity of cultured neurons in several applications [101, 399–407], but not much literature has been produced yet on in silico models of neuronal networks grown on MEAs. As an example, Dazza et al used in silico models to describe how periodic bursts are generated and propagated in a spatial network in various neuronal systems with a novel phase-based analysis [408]. Even if presented with simulations, they showed that this methodology can be applied to other forms of neuronal spatiotemporal data (e.g. MEA recordings with sufficiently high spatiotemporal resolution). Simulations were carried out with the adaptive Exponential Integrate and Fire model [409], via the NNGT python library [410] and NEST simulator [411]. In silico models were also used in combination with in vitro MEA recordings to study the neuronal network phenotype of hiPSCs-derived excitatory neurons carrying Rett syndrome-associated MECP2 mutations [345]. Mok et al recreated the electrophysiological phenotype in a computational neuronal network model and showed that the intrinsic adaptation currents in individual neurons explained the aberrant activity [345]. Within the same scenario, in silico neuronal networks predicting the changes in neuronal spiking activity expected to result from isolated rescue of synaptic structure have been generated [385].
Most of the studies using in silico models are based on hiPSCs-derived cardiomyocytes cultured on MEAs [412–416]. Even if not directly related with our field, they can teach us new methods and approaches adaptable to neuroscience research. In Shi et al, for instance, the applicability of an in vitro/in silico approach was investigated to predict human cardiotoxicity of candidate anti-addiction drugs [412]. In particular, they combined in vitro parameters extracted from MEA recordings from hiPSCs-derived cardiomyocytes with physiologically based kinetic in silico models [412]. In another study related to safety pharmacology in heart research, a strategy to analyze the signals acquired from hiPSCs-derived cardiomyocytes on MEAs was proposed with the aim to automatically deduce the channels affected by tested drugs [413].
To conclude, we believe that one way to optimize the identification the mechanisms underlying the neuronal network phenotype observed in MEA recordings is to integrate in vitro MEA recordings and experiments with in silico models. In silico models, indeed, allow to test a large number of hypotheses suggested by the analysis of MEA recordings, and to predict candidate mechanisms that can be further investigated with in vitro experiments. This approach holds the potential to make research less time-consuming and expensive.
7.3. Multi-scale models represent the ultimate translational approach
To date, there is a big gap between research and clinical practice. It is indeed very challenging to relate the signals recorded in vitro to the ones recorded from the brain, and this contribute to make challenging translation of results from in vitro experiments into humans.
Electro- and magnetoencephalography (EMEG) recordings are non-invasive methods to study electrical neuronal activity from the brain with fine temporal resolution [417]. While EMEG are intensively used in the clinic to identify biomarkers of normal and abnormal brain dynamics, these so-called 'macro-scale' techniques suffer from difficulty in interpretability in terms of the underlying cellular and molecular level events [417]. Vice versa, in vitro neuronal models, such as neuronal networks on MEAs, are used in research to investigate the pathophysiology of neurological disorders at cellular and molecular levels, and to test candidate drugs and treatment strategies. Little is known about the correlation between the neuronal signals recorded by MEAs in vitro and by EMEG in vivo, since micro-scale phenotypes observed in MEA recordings are hard to relate to macro-scale events detected with EMEG. Thus, there is a huge need of bridging macro- and micro-scales in research of brain physiology and disease.
To address this, several attempts have been proposed in the literature. Hagen et al, for example, developed an open-source software (i.e. LFPy2.0, a more recent upgrade of LFPy [418]) able to build multimodal modeling of neuronal network activity [419]. LFPy2.0 allows for modeling networks of multicompartment neurons with concurrent calculations of extracellular potentials and current dipole moments. The current dipole moments are then, in combination with suitable volume-conductor head models, used to compute EMEG-like signals [419]. In LFPy and LFPy2.0, micro- and macro-scale models are combined together, making use of different toolboxes: NEURON [420], to compute transmembrane currents of multicompartment neurons, and electrostatic forward models. For the latter, volume conduction models with four compartments with different electrical conductivities (i.e. scalp, skull, CSF and brain) are considered. The level of complexity of these anatomical models can be potentially increased and the accuracy of such simulations improved both for EEG and MEG [421–424].
Similarly, the virtual brain is a neuroinformatic platform for full brain network simulations using biologically realistic connectivity [425]. The micro- and meso-scale model consists of neuronal mass models or assembles of neuronal mass models, generating the so-called mass field. This simulation environment enables the model-based inference of neurophysiological mechanisms across different brain scales that underlie the generation of macroscopic neuroimaging signals including EEG and MEG [426–429]. The Virtual Brain has been used for many applications to investigate different diseases, such as neurodegenerative disorders [430], PD [431], dementia [432], TBI [433], AD [434–436], epilepsy [437–440], brain tumor [441], chronic stroke [442], or physiological brain dynamics [431, 443, 444]. Nevertheless, to our knowledge, there is no literature about the explicit integration of MEAs models in the computation.
Another example of multi-scale model platform was presented in the work of Neymotin et al [417]. In their study, they developed the human neocortical neurosolver (HNN), a modeling tool designed to provide researchers and clinicians an easy-to-use software platform to develop and test hypothesis regarding the neuronal origin of their data [417]. At the microscopic level, a neocortical model explaining the underlying cellular- and network-level activity was considered as the source that generates the macroscopic signal recorded on the scalp (i.e. EEG), or far away from it (i.e. MEG) [417]. Also in this scenario, realistic models of MEAs were not considered.
From our perspective, relating in vivo EMEG measurements of the brain to in vitro recordings from neuronal cultures on MEAs would boost not only pharmacological investigations and personalized treatment, but also basic research in neuroscience as a whole. To the best of our knowledge, such models have not yet been developed, but represent a valid future perspective for improve translation of in vitro findings into clinic.
8. Conclusion
With the present review, we aimed to provide an overview of in vitro neuronal cultures in combination with MEA technology for biomedical research. We introduced MEA technology, and described in vitro neuronal systems (i.e. rodent 2D and 3D neuronal cultures, organotypic brain slices, hiPSCs-derived 2D and 3D neuronal cultures, and brain organoids), and the electrophysiological activity exhibited by different neuronal systems in MEA recordings. We reviewed studies in which rodent neuronal cultures on MEAs have been used for physiology studies, neurotoxicity screenings, disease modeling and drug testing. Moreover, we showed how hiPSCs-derived neuronal networks on MEAs can be used to develop patient-specific in vitro platforms to characterize the pathophysiological phenotype, investigate the underlying disease mechanisms, and test which drugs and therapeutic treatments are more likely to be effective in vivo, paving the way toward personalized medicine. Lastly, we discussed potential, challenges and future perspectives for the use of MEAs in biomedical research. We provided some guidance for the choice of the best neuronal model and MEA device according to the research aim, for experimental design, data analysis and reporting in scientific publications. We showed how neuronal cultures on MEAs can be used not only to characterize the electrophysiological phenotype of disease models, but also to uncover the underlying cellular and molecular disease mechanisms, and to reveal new candidates for treatment strategies. We gave an overview of studies demonstrating the comparability of results found in in vitro models on MEAs to observations in in vivo models and in humans. Lastly, we suggested the importance of in silico models and other new statistical and computational approaches, as complement and support to MEA experiments.
We believe that the present review will help the scientific community understand the potential of in vitro neuronal cultures on MEAs for biomedical research, and will provide some precious guidelines to both researchers working in the field, and those who want to approach MEA technology for the first time.
Acknowledgments
Figures were created with BioRender.com.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).