Abstract
Non-aqueous electrolytes play a prominent role in the redox reactions of the oxygen electrode in the non-aqueous Li-air battery. In all electrolytes the initial O2 reduction reaction (ORR) product is superoxide, O2−, whose stability is determined by the Lewis acidity of the ion-pairing cations present in the medium as governed by the Hard Soft Acid Base (HSAB) theory. Our results suggest that depending on the basicity of the solvent as measured by its Donor Number (DN), the superoxide will be stabilized to varying lengths of time before transforming to O22− via a chemical or an electrochemical reaction. Organic solvents decrease the Lewis acidity of Li+ through the formation of solvates, Li+(solvent)n. As a result, high DN solvents such as dimethyl sulfoxide (DMSO), which decrease Li+ acidity more than low DN solvents, provide longer life-time for the soft base O2− by forming the ion pair, Li+(DMSO)n–O2−. In low DN solvents such as 1, 2-dimethoxy ethane (DME) and tetraethyleneglycol dimethyl ether (TEGDME), the LiO2 rapidly decomposes to the peroxide, Li2O2. As the ionic bond strength between the O2 reduction product and the conducting salt cation becomes stronger, its rechargeability becomes poorer. Solvent electron donor property affects ORR catalysis also.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
There is intense world-wide research and development of a rechargeable lithium battery based on the Li/O2 chemical couple, popularly known as the Li-air battery, encouraged by its very high theoretical specific energy (5200 Wh/kg) and low cost of the materials from which it can be fabricated.1,2 The theoretical specific energy of the Li-air battery is surpassed only by a battery based on the Li/F2 couple.3 However, F2, because of its high reactivity, is an impractical electrode material to construct batteries for powering consumer products such as cellphones, laptop computers, tablets, digital cameras, and electric vehicles. This leaves Li-air as the candidate of choice for an ultra-high energy density, low cost (air is free) rechargeable lithium battery. The Li-air battery is environmentally friendly as non-toxic electrolytes and other materials, besides Li and O2, needed for fabricating it can be formulated. It is a safe battery despite having a Li metal anode because the cathode active material is not stored inside the battery cell.
The Li-air battery is one of many metal-air batteries that could conceivably be fabricated (Table I).
Table I. Voltage and Specific Energies of Metal-air Batteries.1
| Theoretical specific energy, Wh/kg | |||
|---|---|---|---|
| Metal/air battery | Calculated OCV, V | Including oxygen | Excluding oxygen |
| Li/O2 | 2.91 | 5200 | 11140 |
| Na/O2 | 1.94 | 1677 | 2260 |
| Ca/O2 | 3.12 | 2990 | 4180 |
| Mg/O2 | 2.93 | 2789 | 6462 |
| Zn/O2 | 1.65 | 1090 | 1350 |
Among the various metal-O2 couples, the Li-air is the most attractive battery since the cell discharge reaction involving metallic Li and oxygen to yield Li2O, according to the reaction 4Li + O2 → 2Li2O, has an open circuit voltage of 2.91 V and a theoretical specific energy of 5200 Wh/kg (Table I). In practice, oxygen does not have to be stored in the battery which makes the theoretical specific energy excluding oxygen to be 11140 Wh/kg when the battery is initially fabricated. The battery weight will increase and its specific energy will decrease as it is discharged due to the accumulation of the discharge product in the pores of the carbon cathode.
We demonstrated the first non-aqueous Li-air battery with the use of a Li metal anode, a porous carbon cathode and a gel polymer electrolyte membrane which served as both the separator and ion-transporting medium in it.1,2 The gel-polymer electrolyte in our first cells was based on either a polyacrylonitrile (PAN)3 or a polyvinylidene fluoride (PVdF) polymer4 host. The discharge/charge voltage versus capacity curves of such a Li-air battery we demonstrated in 1996 are depicted in Figure 1.
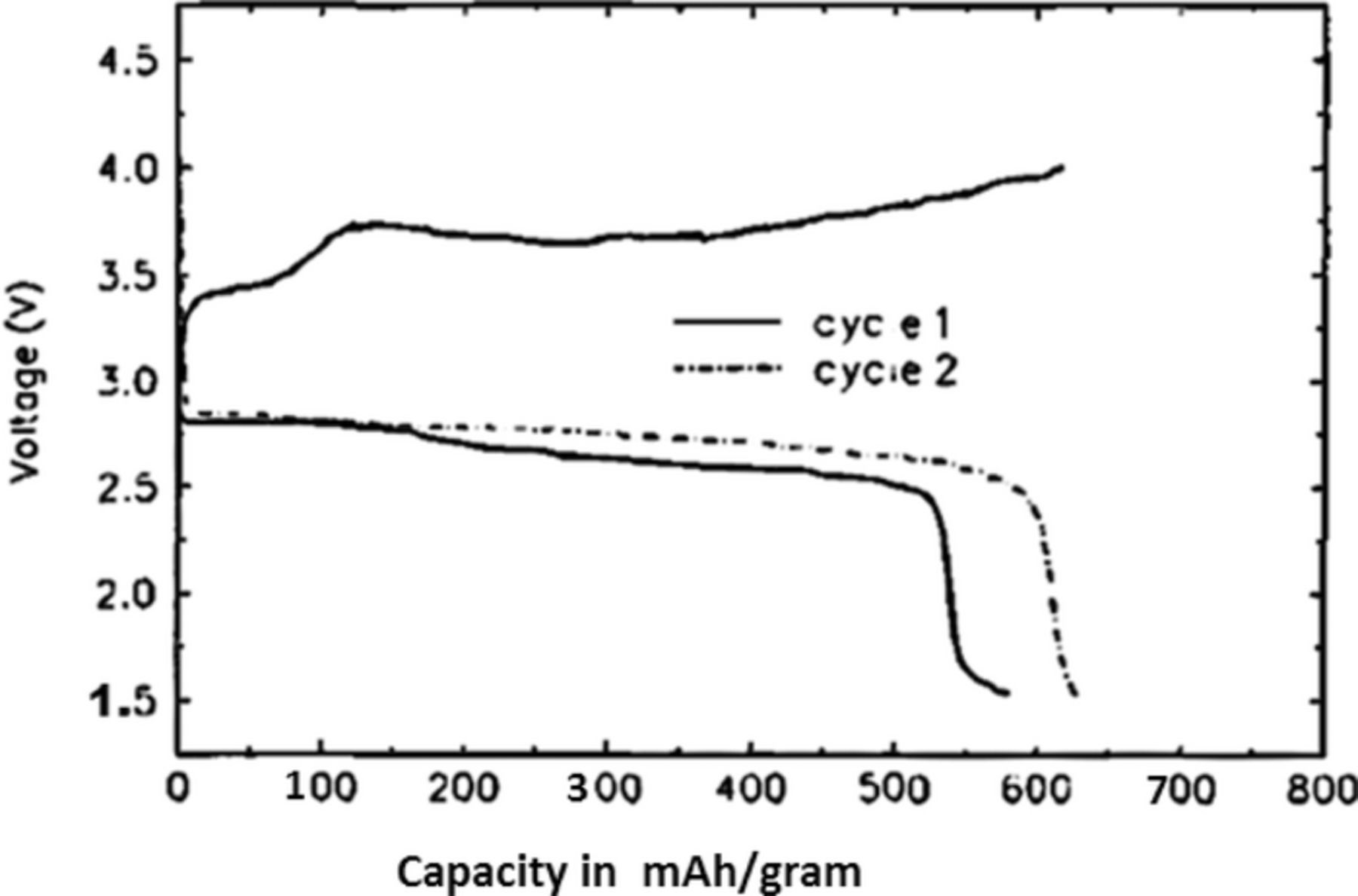
Figure 1. Galvanostatic discharge/charge cycling curves for Li/PAN-based polymer electrolyte/O2 cell at room temperature. The cell was discharged at 0.1 mA/cm2 and charged at 0.05 mA/cm2.1
Oxygen Reduction Reactions (ORR) and Oxygen Evolution Reactions (OER) in Non-aqueous Electrolytes
We have been conducting an in-depth study of the oxygen reduction reactions (ORR) in non-aqueous electrolytes in order to elucidate the ORR mechanism and to characterize the ORR products in the Li-air battery. The rechargeability of the Li-air battery embedded in oxygen evolution reactions (OER) has also been an objective of these studies.
Our investigations have revealed that the non-aqueous electrolyte, particularly the solvent, strongly influences the ORR mechanism whether the reaction is carried out on a catalyzed or an un-catalyzed porous carbon electrode. In this account I summarize these findings. This account is not intended as a comprehensive review of the Li-air battery but is meant to communicate the new perspective on reaction mechanisms and products that evolved from our studies of the ORR and OER reactions in the non-aqueous Li-air battery. While this overview provides a comprehensive account of the new perspective on the Li-air battery chemistry, much of the discussion presented here is based on various pieces of work we previously published. I acknowledge that there were prior investigations focusing on the effect of electrolyte on ORR in non-aqueous electrolytes5–8 although they did not provide the type of insight presented in this paper relating donor-acceptor properties of the electrolytes with ORR mechanism and the stability of the products formed.
ORR in Li+-containing non-aqueous electrolytes
The oxygen reduction reactions (ORR) and the subsequent oxidation reactions of the reduction products, (generally expressed as oxygen evolution reactions (OER)), we have determined from detailed electrochemical studies and product analysis in Li salt solutions in dimethyl sulfoxide (DMSO) are shown in equations 1–7.9,10
Cathodic (ORR) reactions
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn1.jpg)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn2.jpg)
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn3.jpg)
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn4.jpg)
Anodic (OER) reactions
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn5.jpg)
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn6.jpg)
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn7.jpg)
The reactions depicted in these equations do not indicate the fact the Li ions that charge compensate for the oxide ions, superoxide (O2−), peroxide (O22−) and monoxide (O2−), produced from the reduction of O2 on the porous electrode, are present in the electrolyte as solvated ions of the general formula, Li+(solvent)n. In reality, the reactions depicted in these equations initially proceed through these solvated Li ions. The solvated Li+ are eventually de-solvated when Li2O2 and Li2O crystalize on the carbon electrode. This modification of the ORR and OER reactions depicted in equations 1–7 will be discussed later in the section dealing with the role of solvation of Li+ on modulating the Lewis acidity of Li+ and thus controlling the stabilities of the ORR intermediates and final products.
The cyclic voltammetry (CV) data which allowed us to arrive at the sequence of ORR and OER reactions presented in equations 1–7 are displayed in Figure 2. A key step in the overall ORR process is the formation of the initial one-electron reduction product, lithium superoxide (LiO2). It was possible to demonstrate the existence of the short-lived lithium superoxide from the CV data through a voltage reversal study, depicted in Figure 2, in which the OER processes were examined as a function of step-wise voltage reversal. Even at the very beginning of the cathodic sweep starting at about 2.5 V, we observed two oxidation products following the formation of one reduction product. From a detailed analysis of the CV results using the Randles-Sevcik relationship and rotating disk electrode (RDE) experiments in the DMSO electrolyte,10–12 the initial ORR was established to be one-electron reduction of O2 to form LiO2. The LiO2 is either chemically decomposed (equation 2) or electrochemically reduced (equation 3) to form Li2O2. The CV data coupled with peak analysis also led to the identification of the ORR process depicted in equation 4 involving the formation of the four-electron O2 reduction product Li2O. The potential-dependent formation of both Li2O2 and Li2O from the reduction of O2 is now well-established.12 The Li2O2 was identified from its characteristic Raman spectral absorption at ∼790 cm−1 of the product formed by electrolysis of O2 on a carbon electrode in a DMSO/LiPF6 solution at 2.32 V. The formation of Li2O was confirmed from the Raman spectrum of the product formed on the carbon electrode at 2.12 V.12 The OER reactions shown in equation 5–7 are consistent with the sequential formation of the products in the anodic sweep following the incremental potential reversal steps associated with the cathodic CV scans. These ORR processes involving the initial formation of the one-electron reduction product LiO2 in the non-aqueous Li-air cell using different solvent-based electrolytes is now recognized.13,14
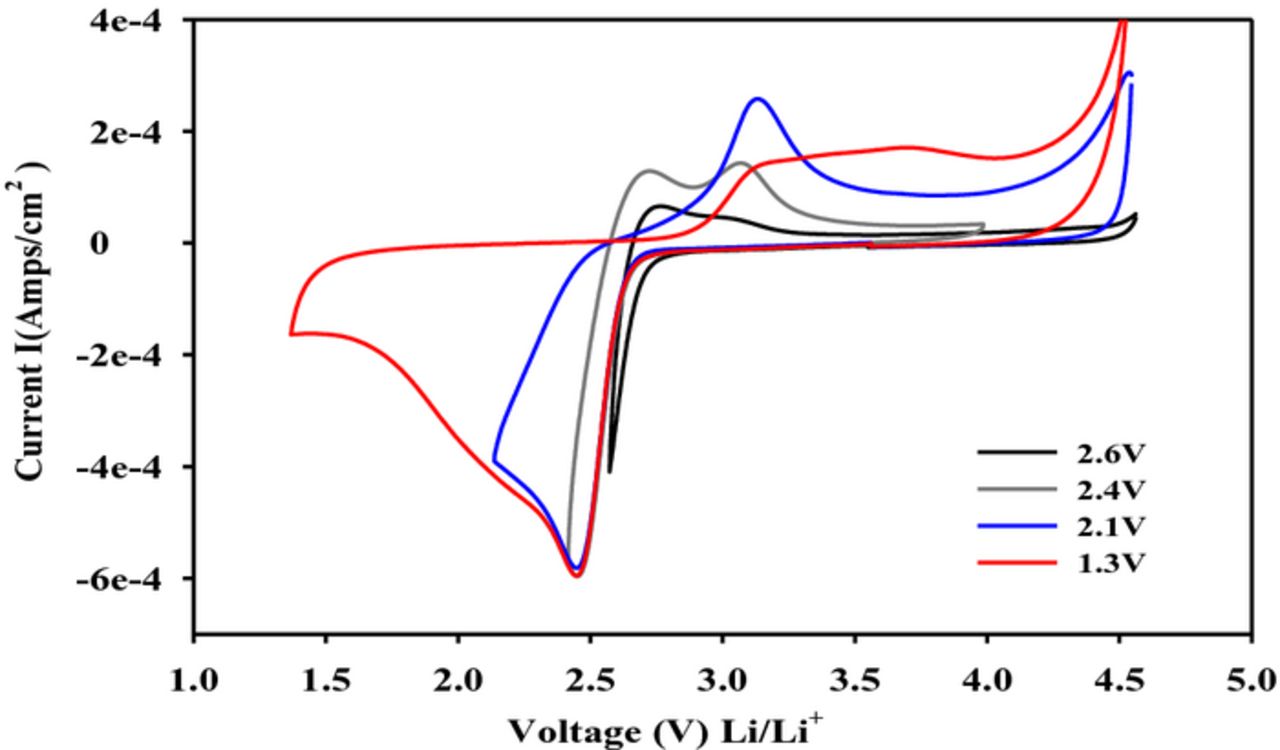
Figure 2. Cyclic voltammograms (CV) for the reduction of oxygen in 0.1 M LiPF6/DMSO at various potential windows. All scans were performed on a glassy carbon working electrode. Scan rate of 100 mV/sec.9
The ORR electrochemistry results obtained in an acetonitrile (AN)/LiPF6 solution showed one major difference from that in DMSO-LiPF6, namely the absence of a long-lived LiO2. This was established from the lack of an oxidation peak for LiO2 even when the cathodic sweep in the CV in the AN/LiPF6 solution was reversed soon after the commencement of O2 reduction at about 2.5 V (Figure 3). Although LiO2 is formed in AN/LiPF6, the life-time of the LiO2 in acetonitrile is apparently much shorter than in DMSO. It should be noted that in situ Raman studies of ORR in acetonitrile has revealed the existence of LiO2 in this solution and its decomposition has been studied by time-dependent analysis of the Raman spectra.15 The ORR and OER electrochemistry results in DMSO and AN-based electrolytes have clearly shown that the initial reduction product of O2 is LiO2 and that its life-time is solvent-dependent. The CV results in AN/LiPF6 solution, given in Figure 3, indicate that unlike in DMSO/LiPF6, there are no separate ORR steps for O2 reduction to LiO2 and its further reduction to Li2O2 (as described in equations 1 and 3). This is consistent with a very short life-time for LiO2 in AN, a conclusion supported by the lack of LiO2 oxidation peak following the incremental sweep reversal tests displayed in Figure 3. A similar electrochemistry with short life-times for LiO2 was also observed in the ether solvent-based electrolytes including dimethoxyethane (DME)/LiPF69,17 and tetraethylene glycol dimethyl ether (TEGDME)/LiPF6.9,16 The ORR in all of these electrolytes is electrochemically irreversible since the reduced and oxidized peaks do not follow the classical reversibility criteria. The ORR in the Li salt solutions is more aptly termed chemically reversible but electrochemically irreversible.
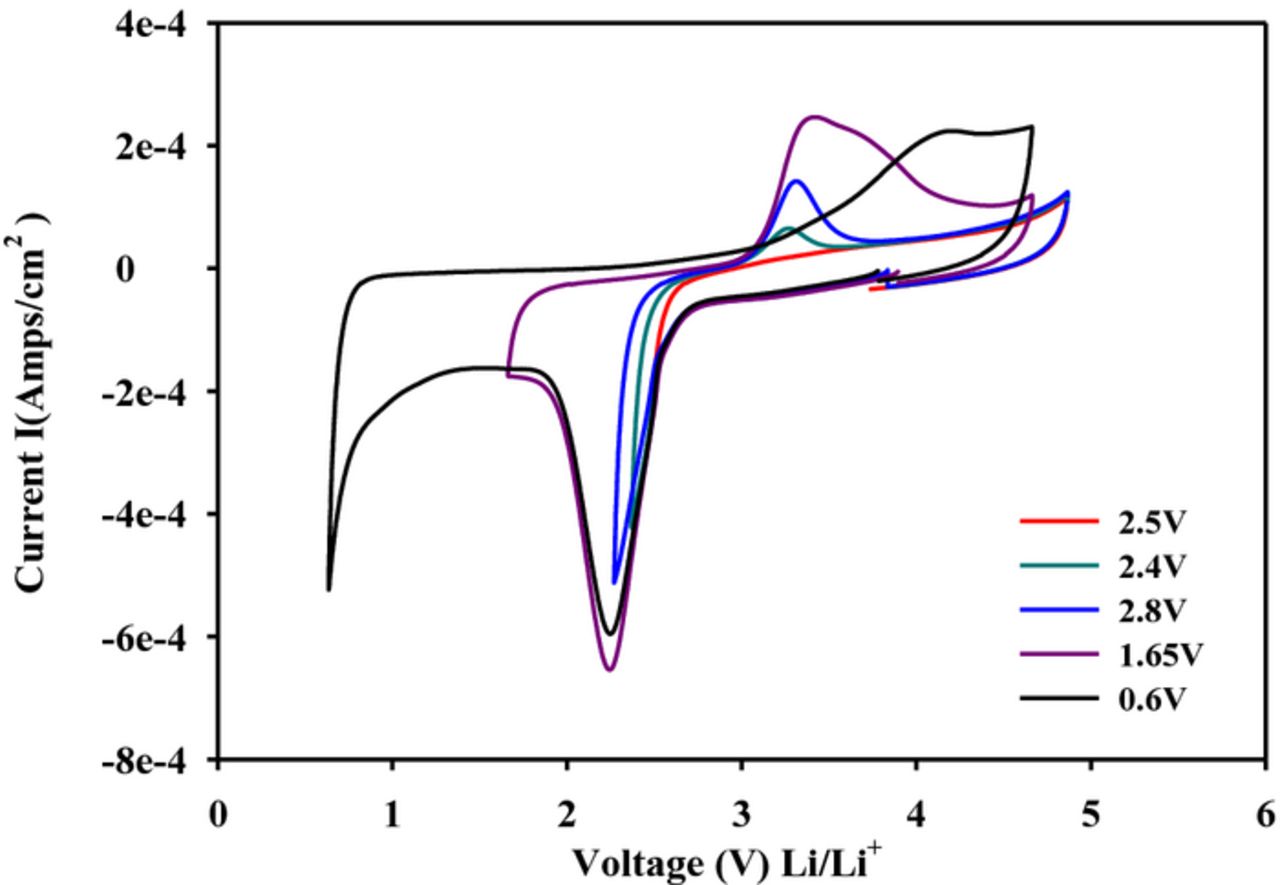
Figure 3. Cyclic voltammograms for the reduction of oxygen in 0.1 M LiPF6/MeCN at various potential windows. All scans used a glassy carbon working electrode. Scan rate was 100 mV/s.
The different life-times of LiO2 in the various organic electrolytes have led to one school of thought (primarily from quantitative determination of the O2 generated during OER following an ORR step in DME-based electrolytes13) that the initial ORR in the Li-O2 cell is a two-electron process to form Li2O2. Eventually, this line of thought has given way to the ORR processes described by the reactions in equations 1–4, supported by extensive electrochemistry results coupled with product analysis. We have demonstrated that the electrochemical and chemical pathways during ORR correlate well with the electron donor properties of the solvents and electron acceptor properties of the conducting cations present in the electrolytes. We have arrived at this conclusion by comparing the ORR results obtained from studies in the Li+-containing electrolytes detailed above with that discussed below in electrolytes containing other alkali metal and tetraalkylammonium salts in organic solvents, and investigations in room temperature ionic liquids.
ORR in electrolytes containing tetraalkyl ammonium salts, and salts of alkali metals other than lithium
If the lithium salts in the DMSO electrolytes is replaced with a tetrabutyl ammonium (TBA) salt such as (C4H9)4NPF6 (TBAPF6) or (C4H9)4NClO4 (TBAClO4), a remarkable change in the ORR happens as indicated by the CV data in Figure 4 (in red).5 This figure also shows the early part of the ORR in LiPF6 in DMSO (in blue) for comparison. The ORR in the TBA salt solution is reversible with the characteristic separation of about 60 mV between the anodic and catholic peaks, and a ratio of the charge under the anodic peak to cathodic peak (Qa/Qc) of close to unity. Randles-Sevcik analysis of the current-potential curves obtained from a series of sweep rates confirmed the reaction to be a one-electron process involving the formation of superoxide, O2−. The anion of the TBA salt had no effect.9,10 Clearly, the superoxide is stabilized as TBA–O2 in tertaalkylammonium salt solutions. Indeed, it is difficult to further reduce this one-electron O2 reduction product in TBA salt solutions in DMSO or acetonitrile. In acetonitrile/TBAPF6 evidence for a second reduction of the superoxide is observed only when a significant over-potential is applied bringing potential down to about 0.8 V (Figure 5).
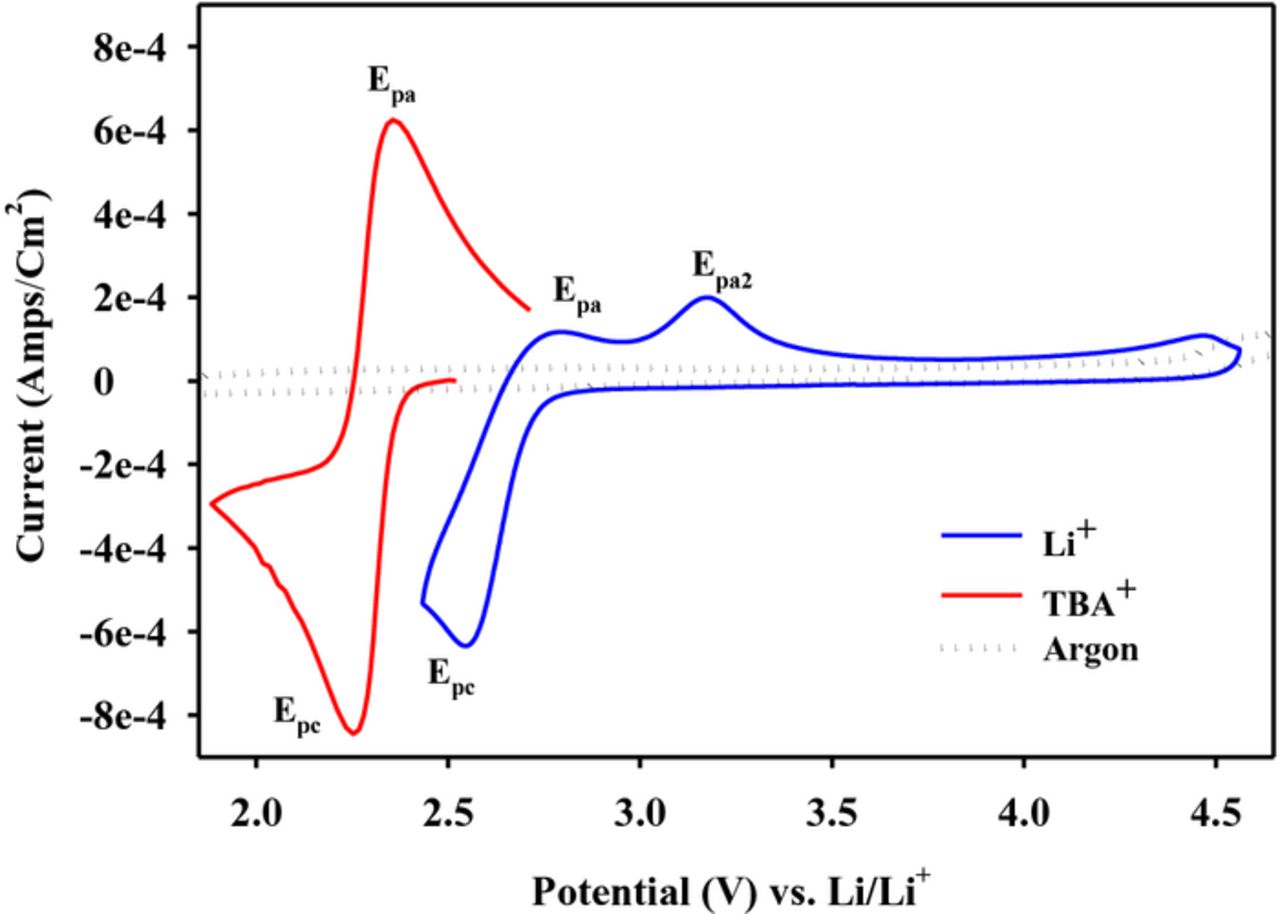
Figure 4. Cyclic voltammograms for O2 reduction in DMSO with 0.1 M TBAPF6 (Red); 0.1 M LiPF6 (Blue) and for the electrolyte purged with Argon (dotted). All scans used a glassy carbon working electrode at scan rate of 100 mV/s.
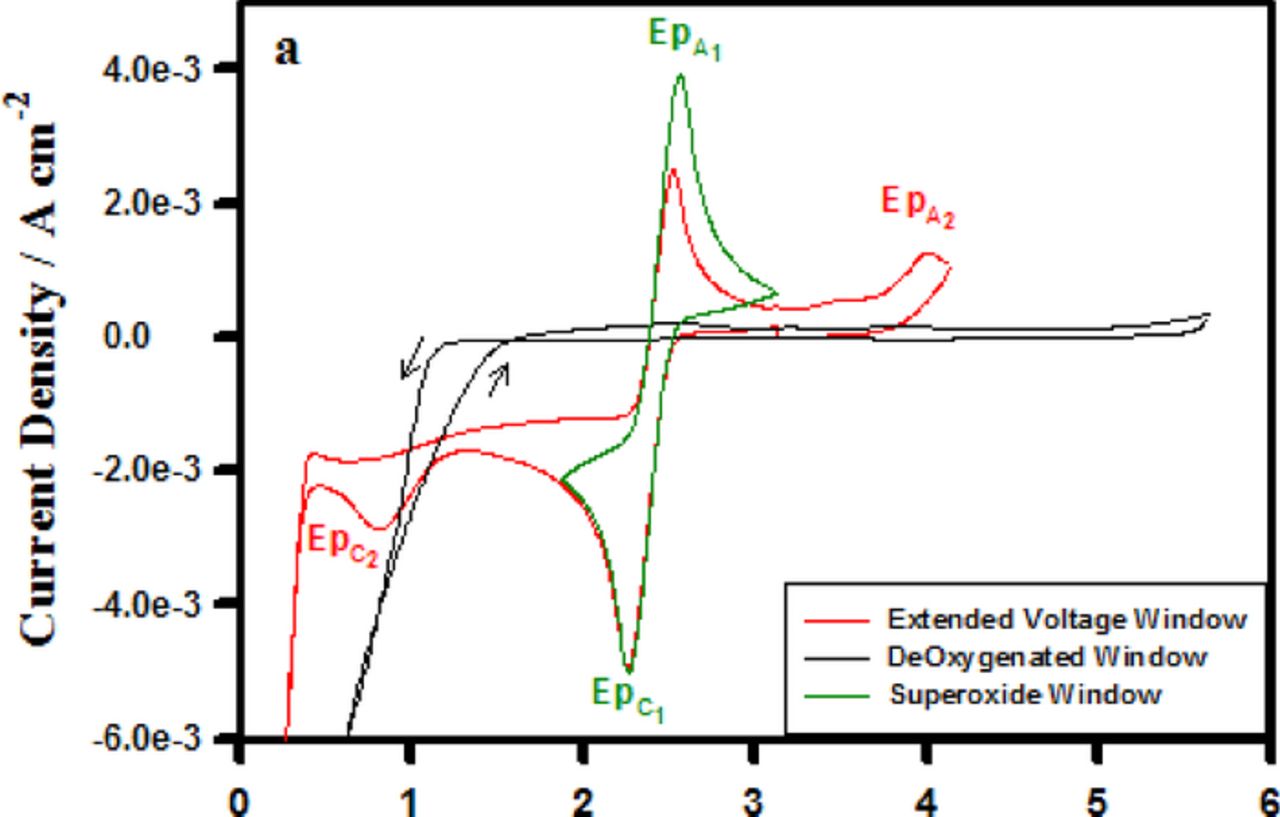
Figure 5. Cyclic voltammograms collected from a glassy carbon working electrode at a sweep rate of 100 mV/s in 0.1 M TBAPF6/CH3CN.
ORR studies in acetonitrile/ NaPF6 solution has revealed10 that the initial step in O2 reduction in this solution is also a one-electron process to form NaO2, which appears to decompose rapidly to Na2O2 and passivate the electrode surface. Because of the rapid passivation of the electrode, the ORR products are not oxidized well in this electrolyte. The sodium oxides cannot be oxidized even at high overvoltages, except in highly concentrated electrolyte solutions. In a KPF6/AN solution the voltammogram suggested that the ORR is quasi-reversible as verified by the increase in the anodic current in comparison to that observed in lithium and sodium salt solutions.10 Although the oxygen reduction is not as reversible as that in the tetraalkyl ammonium salt solutions, the initial reduction of O2 to KO2 and then the reduction of KO2 to K2O2 are observed as two distinct one-electron quasi-reversible CV peaks.10
ORR in ionic liquids
We studied ORR in two room temperature ionic liquids (RTILs), namely, 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide, (EMIImide), and 1-methyl-1-butyl-pyrrolidinium bis-(triflouromethanesulfonyl)imide (PYR14TFSI), depicted in Scheme 1.18,19 The electrochemistry results are summarized by the CV data presented in Figure 6.
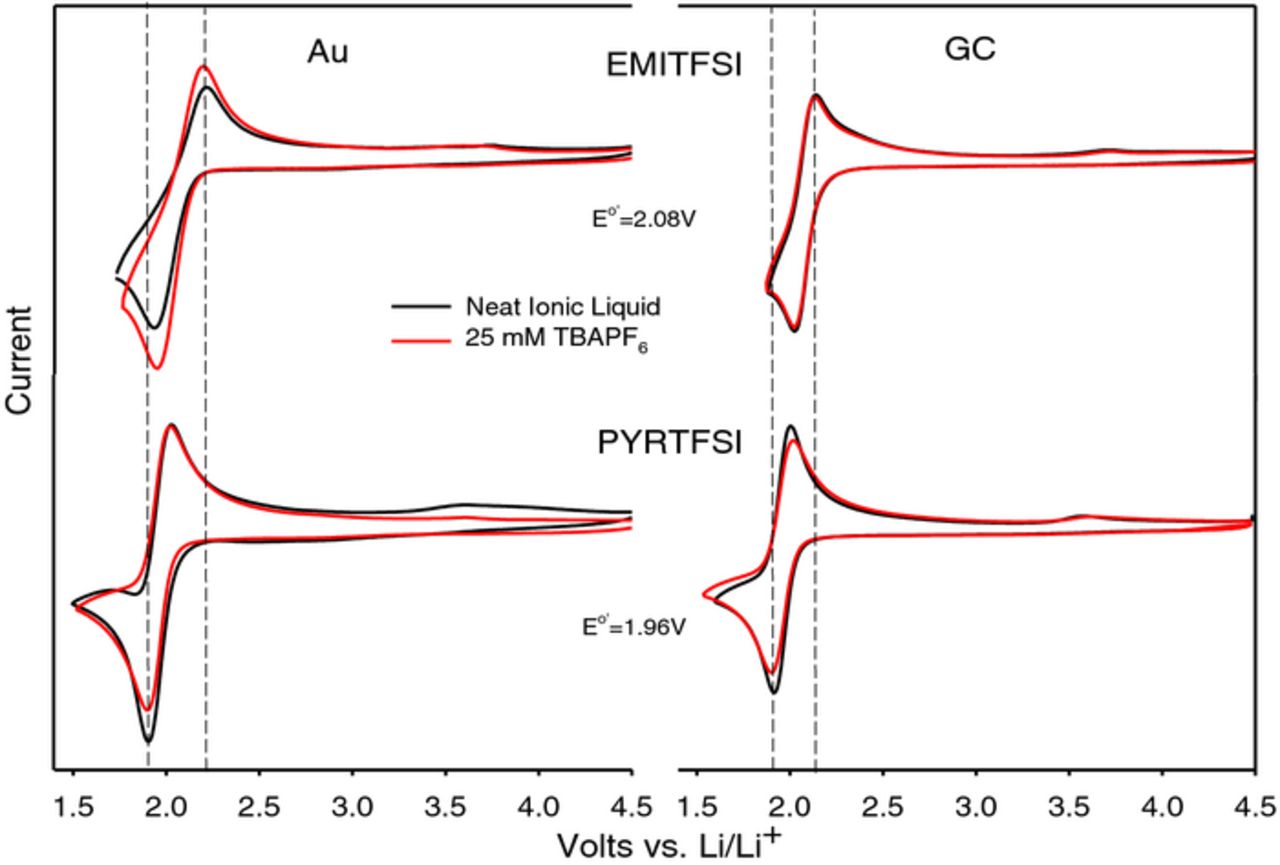
Figure 6. Cyclic Voltammograms on Au and Glassy Carbon electrodes in O2-saturated EMITFSI and PYRTFSI both with (red) and without (black) added 0.025 M TBAPF6. Scan rate = 100 mV/s19
Scheme 1. The two ionic liquids used with their cations and the TFSI anion.
The CVs of O2 reduction and subsequent oxidation (i.e; ORR and OER) in the two ionic liquids showed a behavior identical to that in TBA salt solutions in organic solvents; i.e; reversible one-electron reduction leading to the formation of superoxide, O2−. In fact, the addition of TBAPF6 to the ionic liquids showed little change in the overall CV on either gold or a glassy carbon electrode as depicted in Figure 6. Clearly, the ionic liquid cations stabilize the superoxide in the same manner as TBA+. The ORR CV in RTIL undergoes a significant change when a small amount of Li salt is added as shown in Figure 7 in which the ORR data in EMITFSI doped with .025 M LiTFSI is compared on both GC and Au electrodes. The electrochemical irreversibility peak profile is substantially similar to that found in Li+-conducting organic electrolytes.9,10 Apart from the anodic peak A1 (which is very minor here) seen on Au due to the oxidation of superoxide ion-paired with EMI+ (EMI+–O2−), oxidation peaks at A2 and A3 in Figure 7 are at the same positions on both GC and Au during the first cycle. The appearance of two oxidation processes following what appears to be a single reduction reaction is similar to that found in DMSO/Li salt solutions and it supports the view that the initial one-electron ORR is accompanied by chemical reactions. A most significant observation is that the ORR performance on these surfaces deviates during subsequent scans. The first five repetitive cycles on GC showed diminishing peak currents indicative of electrode surface passivation, as well as loss of the shoulder on the anodic profile at 3.7 V. On Au on the other hand, little loss in current occurs during subsequent cycles revealing a chemically reversible (but electrochemically irreversible) reaction on this electrode. The unique ability of the Au electrode to support chemically reversible ORR observed here for the first time. It was subsequently extended to Li-air cells in organic electrolytes with the demonstration of long cycle life for a Li-O2 cell, albeit at low utilization, with a porous gold electrode in DMSO electrolytes.20
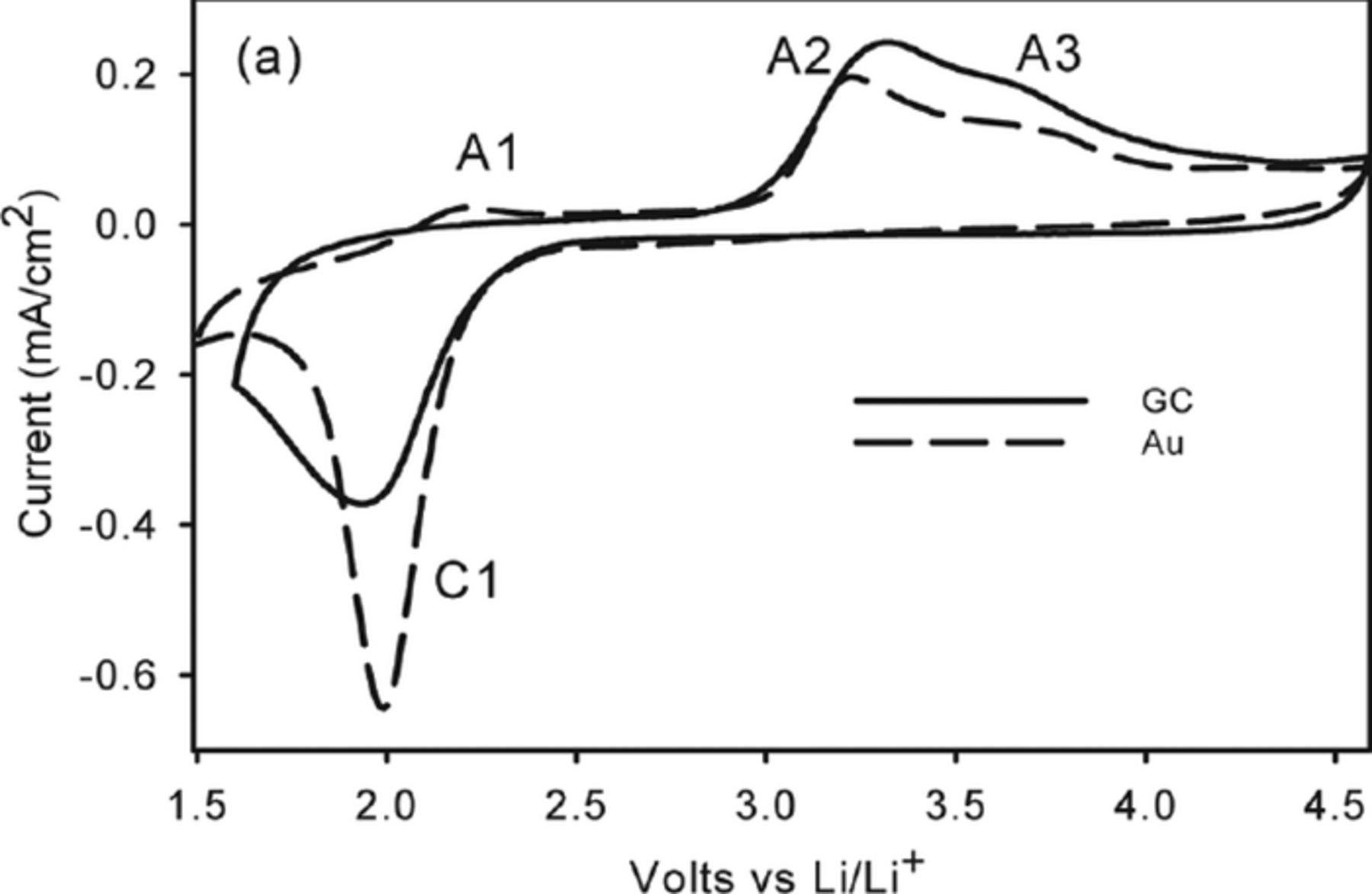
Figure 7. Cyclic Voltammograms in 0.025 LiTFSI doped O2 saturated EMITFSI on Au and GC at 100 mV/s.18
We have also studied the influence of Na and K ions on ORR in ionic liquids.19 The reversibility of ORR in presence of these ions was intermediate between those in Li and TBA salt solutions in the RTIL with a good correlation between the relative size of the cation (or its charge density) and the ORR mechanism as well as the stability of the ORR intermediates and the final products formed. These studies have led to our formulation of the general concept of the Hard Soft Acid Base (HSAB) theory of ORR in non-aqueous electrolytes. This concept satisfactorily explains the dependency of relative stability of the initial ORR product, superoxide, on the Lewis acidity of the conducting cation and the electron donor property of the organic solvent.
Solvation and Lewis Acidity of Cations of Conducting Salts in Non-aqueous Electrolytes
The ORR and OER results presented above clearly indicate a strong correlation between the nature of the conducting cations present in the non-aqueous electrolytes and the nature of ORR products formed, and the potentials at which they can be subsequently oxidized. An obvious difference among the various cations studied is their relative ionic radii (Table II). Since the ORR products, whether it is O2− or O22−, do not differ substantially in their ionic size but only in their relative Lewis basicity, a property of the cation that influences the stability of the ORR products is its Lewis acidity.
Table II. Crystallographic ionic radii of cations used in both ORR and NMR studies.
| Cation | Ionic radius (pm) | Ref |
|---|---|---|
| Li+ | 60 | 21 |
| Na+ | 96 | 21 |
| K+ | 133 | 21 |
| EMI+ | 239 | 22 |
| PYR+ | 330 | 23 |
| TBA+ | 494 | 21 |
We measured the relative Lewis acidity of the various cations including those of the two ionic liquids taking advantage of the fact that the conducting salt cations (M+) in non-aqueous electrolytes are generally solvated by the solvent by forming acid-base complexes, M+(solvates)n X−, where X is the anion of the conducting salt.24,25 The value of n for the solvates Li+(solvates)n X− in Li salt solutions is believed to be around four. Li-ion solvation by ethers24 and organic carbonates25 had been previously established by nuclear magnetic resonance (NMR) studies. We has been suggested that the 13C NMR chemical shift and spin lattice relaxation times (T1) of the C=O functional group in propylene carbonate (PC) due to its interaction (or formation of the acid-base complex) with each of the cations studied would provide information of the relative acidity of the cations. Although the Li+ to PC molar ratio in a 1M Li salt solution was much less than four, only one 13C signal was seen in the Li salt solution because of the rapid exchange with Li+ of the solvated and unsolvated PC molecules at room temperature in the NMR time scale.24 Our results are presented in Figure 8. The Li+ with relatively high positive charge density or strong Lewis acidity formed a stronger complex with C=O as depicted in Structure I, and hence exhibited a larger 13C=O chemical shift relative to PC. The very large IL and TBA cations with their low charge densities exhibited the least Lewis acidity and, therefore, the least chemical shift change.
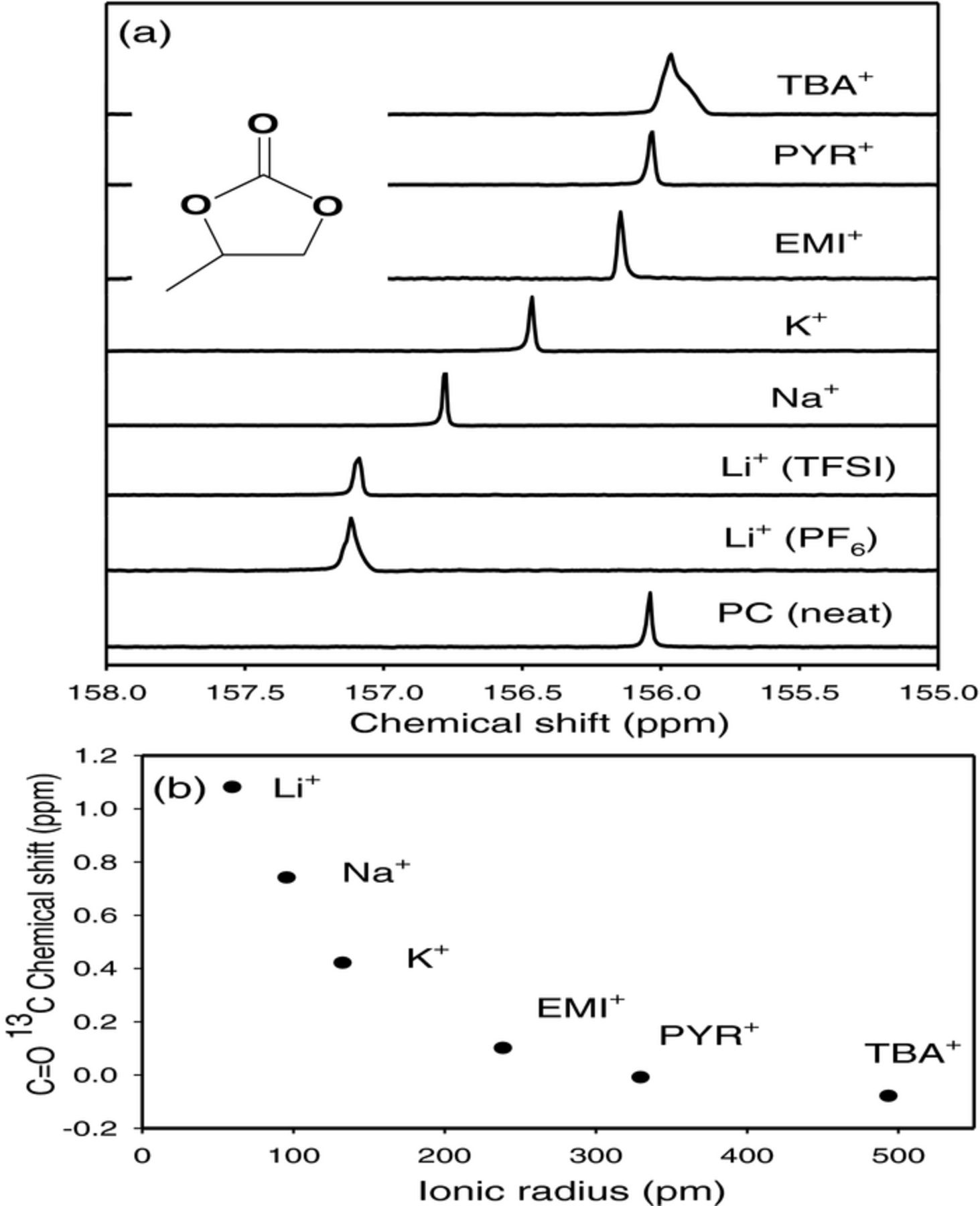
Figure 8. (a) 13C NMR chemical shifts of the propylene carbonate (PC) carbonyl carbon, concentrated with various 1 M salt solutions. 1 M LiPF6 (Li+), 1 M LiTFSI (Li+), NaPF6 (Na+), KPF6 (K+), EMITFSI (EMI+), PYRTFSI (PYR+), and TBAPF6 (TBA+). The structure of PC is shown in the inset. (b) C(=O) 13C chemical shifts relative to neat PC as a function cation ionic radius.19
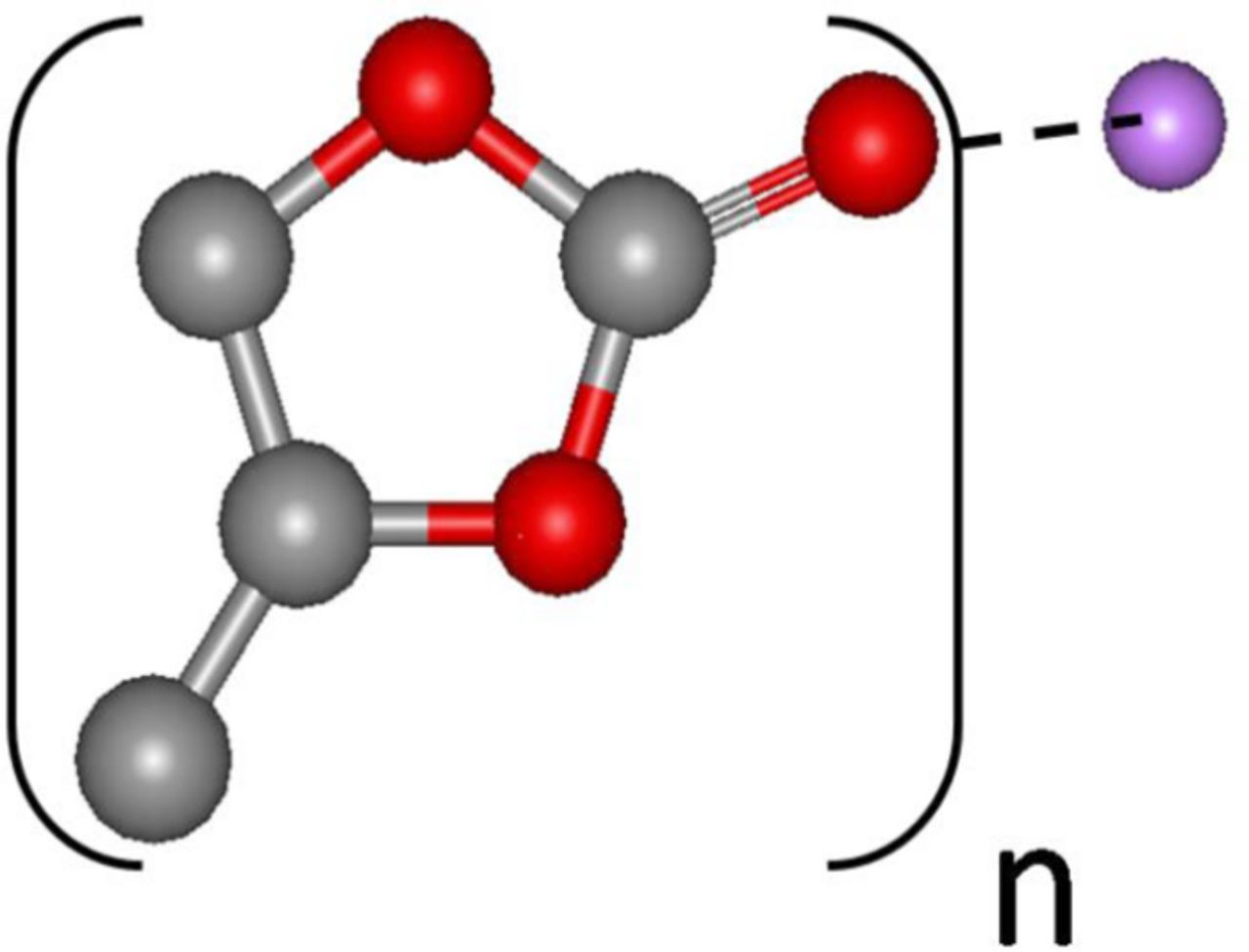
Structure I. Propylene carbonate (in brackets) complexed with Li+; C, gray; O, red; and Li+, purple. The hydrogen atoms are omitted.
The data in Figure 8 show that in LiTFSI and NaPF6 salt solutions, Li+ and Na+ ions are strongly solvated to the carbonyl oxygen atoms of PC while K+ and the three large cations, TBA+, EMI+, and PYR+ are weakly coordinated. Comparing LiPF6 and LiTFSI, the counter anion was found to have little effect on the 13C shift. Since all of the ions studied have the same single positive charge, the radius (and, hence, ionic volume) is roughly inversely proportional to the charge density and polarizability, which are the measures of Lewis acidity.
The 13C NMR chemical shifts of the C=O provide a measure of the complexing ability of the Lewis base C=O (by donating the electron pair on the oxygen atom) with the Lewis acid cations, and hence the relative acidity of the cations. This effect is illustrated in Figure 8b with a cascading chemical shift as a function of ionic radius for these cations. The cations of the RTILs have similar weak acidity as TBA+ with similar 13C chemical shifts closer to neat PC, while the 13C peaks are farther downfield for Li+ and Na+ solutions indicating their strong Lewis acidity. The K+ has an intermediate Lewis acidity among the cations studied and its electrochemistry clearly supports it. The chemical shift data was complemented by the 13C=O spin relaxation times (T1) with the shortest T1 in solutions of Li salts indicating again the strongest solvation of Li+, reflecting the same trend in Lewis acidity of the cations exhibited by the chemical shift data.19
The HSAB Theory of Oxygen Reduction Reactions in Non-aqueous Electrolytes
Pearson's Hard Soft Acid Base (HSAB) theory26 states that hard acids prefer to be associated with hard bases and soft bases prefer soft acids. The ions present in the solutions in our studies were TBA+, EMI+, and PYR+, Li+, Na+, K+, PF6−, and the electrochemically generated ions superoxide (O2−), peroxide (O22− and monoxide (O2−). The 13C nmr data revealed that TBA+, EMI+, and PYR+ are soft acids while K+ and Na+ are increasingly harder acids with Li+ being the strongest Lewis acid. The organic solvents in which the non-aqueous electrolytes are formed are Lewis bases whose basicity is characterized by the Gutmann Donor Numbers (DN); the higher the value of DN, the greater is the electron donating property or basicity of the solvent.23 The DN for the solvents we have used in our various studies27 follows the order, acetonitrile (MeCN) (14.1) < tetraethyleneglycoldimethyl ether (TEGDME) (16.6) < (DME) (20.0) < (DMSO) (29.8). Among the oxides formed from the reduction of oxygen, the superoxide O2− has a relatively large radius and low charge density; which makes it a moderately soft base. In keeping with the HASB theory, the naked soft acid TBA+ or the ionic liquid cations EMI+ and PYR+ stabilize the soft base O2− in these electrolytes with the formation of an ion pair complex of the type in Scheme 2.
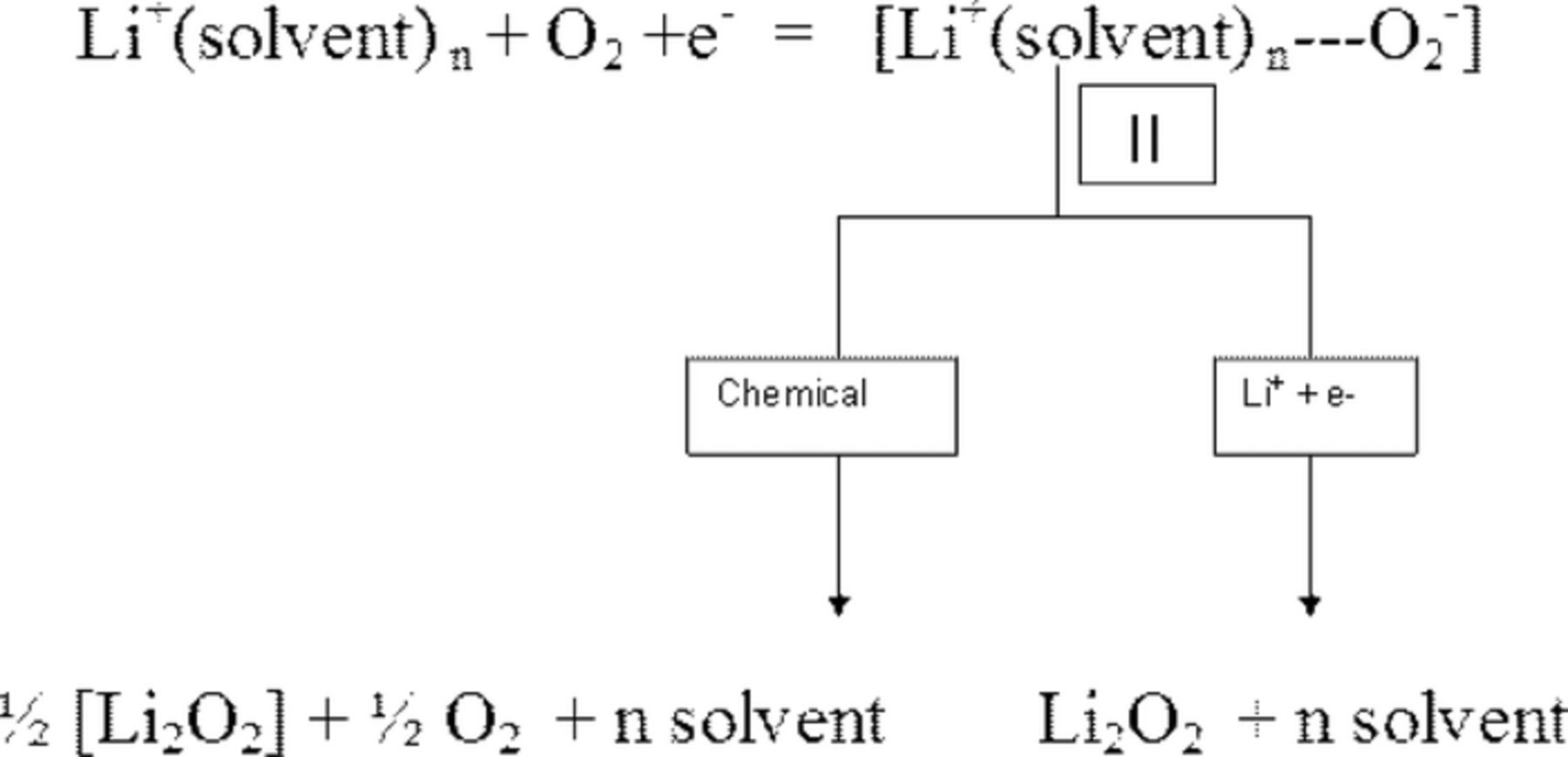
Scheme 2. ORR in organic electrolytes.
Reversibility of the O2/O2− redox couple in TBA+ and ionic liquid solutions is a result of this stable solution species II. As O2− radical is strongly coordinated to TBA+ or the ionic liquid cation as depicted in II, further reduction of superoxide to peroxide (O22−) is hindered.
In electrolytes formed in organic solvents, the Lewis acidity of the alkali cations would be modulated (lowered) due to their solvation by the solvents in proportion to their basicity as discussed above. Therefore, it is the net Lewis acidity of the solvated Li+ ions, Li+(solvent)n, that determines the overall ORR mechanism and products formed.
We have made an attempt to correlate the Donor Number of several family of solvents used in preparing Li+-conducting electrolytes with the solvation energy of the solvates (complexes) (i.e.; Li+(solvent)n) formed between Li+ and the solvents using Drago's equation,28
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn8.jpg)
In this equation, ΔH is the enthalpy (in kcal/mole) of formation of a Lewis acid-base adduct, EA, CA and RA are parameters characteristic of the acid, and EB, CB and TB are parameters characteristic of the base. The EAEB term defines electrostatic energy based on inherent electrostatic bonding capabilities (dipole-dipole interactions) and CACB term defines inherent covalent bonding capabilities. The RATB term represents the energy accompanying the transfer of electron density from an electron rich base to an electron poor acid. For additional explanation the reader should refer to the original paper of Drago.28 These parameters for Li+ are EA = 11.72, CA = 1.45 and RA = 24.21 and for (CH3)2SO these are EB = 2.4, CB = 1.47 and TB = 0.65, all values in kcal/mole. The enthalpy of formation calculated for a 1:4 solvate between Li+ and DMSO is 184 kcal/mole whereas that for a complex formed from Li+ and CH3CN is 161.4 kcal/mole, about 12% lower. Clearly, the higher solvation energy involved in the formation of the complex Li+(DMSO)n translates to lower Lewis acidity for the Li+ in this solvent than for the Li+ in AN forming Li+(AN)n. The higher solvation energy calculated for the solvate formed between DMSO and Li+ (Table III) is consistent with that expected from its high donor number of 29.8 compared with a donor number of 14.1 for CH3CN.
Table III. Donor Numbers of several solvents and Li+ solvation energy.
| Donor Number of the solvent | Li+-solvation Energy, kcal/ mole | |
|---|---|---|
| DMSO | 29.8 | 183.3 |
| Diethyl ether | 19.2 | 167.4 |
| MeCN | 14.1 | 161.8 |
Since the formation of the Li+-(solvent)n complexes would lower the acidity of Li+ roughly in proportion to the Donor Number of the solvent, the Li+-solvent bond strength in the Li-solvates would follow the solvent DN scale as DMSO > MeCN > DME > TEGDME. Since superoxide is a moderately soft base it has low affinity for the hard acid Li+ present in Li+- conducting electrolytes. Consequently, the superoxide formed as the first reduction product of O2 will want either to decompose or undergo a fast second reduction to form the hard base, peroxide (O22−), as shown in equations 2 and 3. The peroxide, O22−, is a strong Lewis base which wants to be associated with the strong base Li+. Similarly, the ultimate reduction product of O2, the monoxide O2− is a hard base with a strong affinity for Li+. Consequently, based on the HSAB theory, the stable O2 reduction reaction products expected in Li+-containing electrolyte solutions are Li2O2 and Li2O. In DMSO solutions of LiPF6 or other Li salts, because of its high DN, the Li+ Lewis acidity is decreased more than in other solvents having lower DN. As a result, the superoxide, O2−, formed in DMSO/Li salt solutions has an increased affinity for the solvated Li+, Li+(DMSO)n, and with that the O2− is stabilized for a longer period in this solution, in a structure of the type III, reminiscent of the TBA+–O2 complex in structure II. This explains the distinct O2/O2− couple seen in the CV of O2 in DMSO/LiPF6 solutions in the same way in TBA salt solutions and ionic liquids. Our results suggest that depending on the basicity of the solvent as measured by its DN, the superoxide formed as the first reduction product of oxygen will be stabilized to varying lengths of time before transforming to O22− via a chemical or an electrochemical reaction.
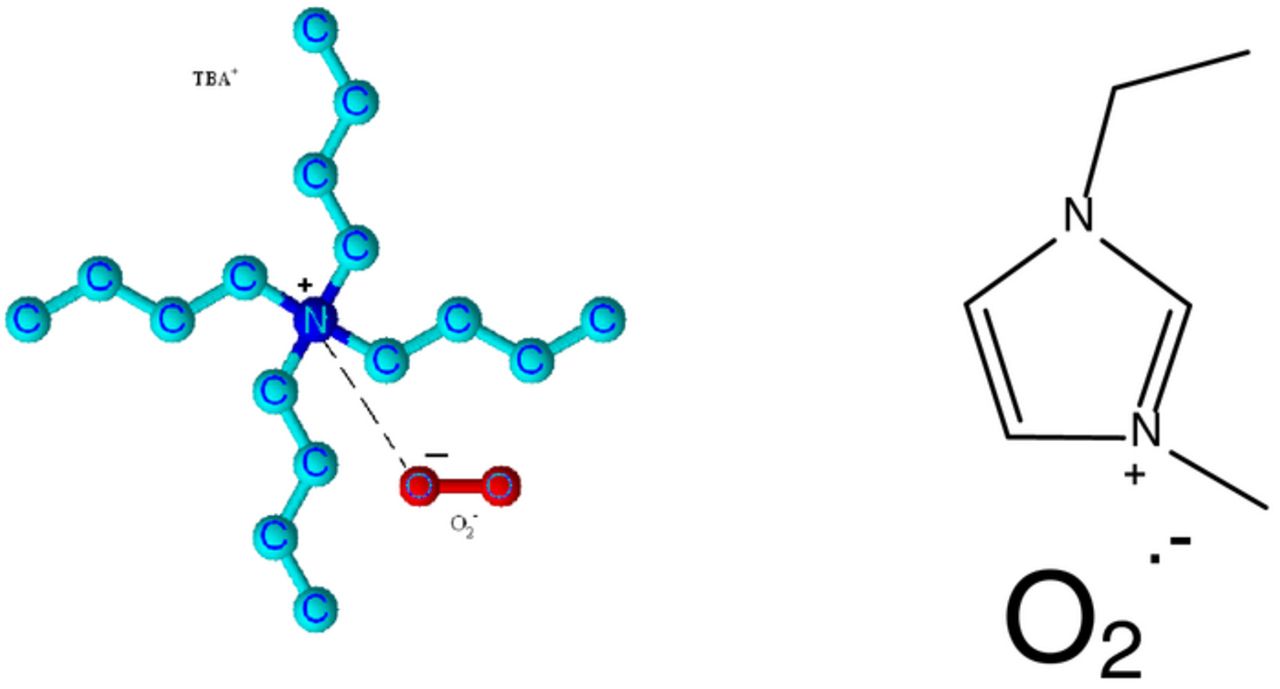
Structure II. Schematic representation of ion pair between TBA+ and O2− (TBA+- - -O2−) (Alkyl hydrogen atoms are omitted in structure); and between ionic liquid cation and O2−, EMI+- - -O2−.
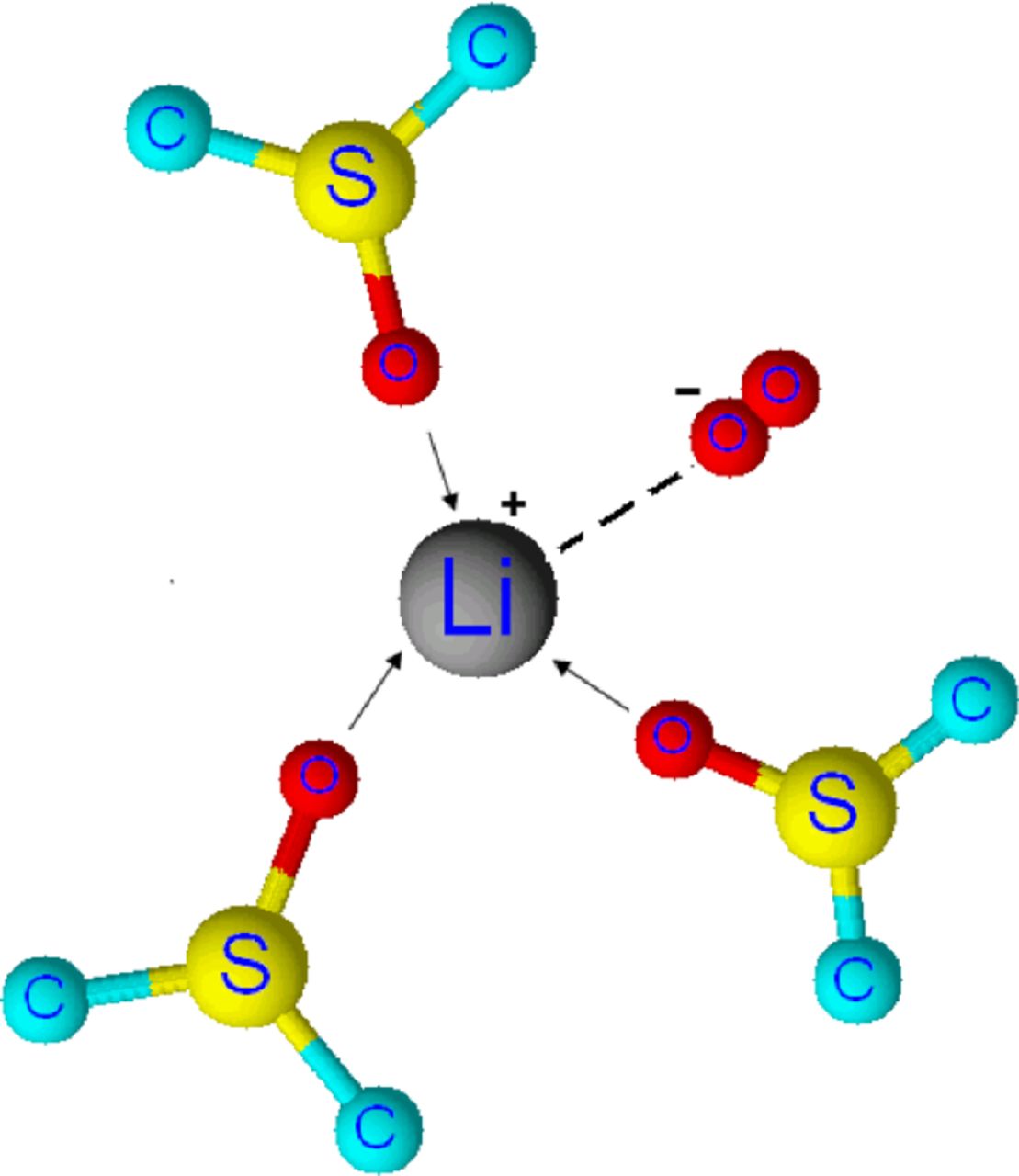
Structure III. Schematic representation of ion pair between solvated Li+ and O2− in DMSO9 (The hydrogens on the methyl groups attached to the S atoms are omitted in the structure).
The solvation of ions gleaned from NMR data and the O2 electrochemistry characterized in organic electrolytes and room temperature ionic liquids lend support to the view that the ORR reactions in depicted earlier in equations 1–4 are more appropriately represented as shown equations 9–12 involving solvated Li+, Li+(solvent)n.
The multi-step electrochemical reduction of O2 in Li+-containing organic electrolyte solutions can be schematically represented as shown in Scheme 2.
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn9.jpg)
![Equation ([10])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn10.jpg)
![Equation ([11])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn11.jpg)
![Equation ([12])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn12.jpg)
High DN solvents (probably DN > 25) provide increased stability for complex II seen in Scheme 2 because of the decreased Lewis acidity of the hard acid Li+ via solvation. In such electrolytes a distinct O2/O2− reversible couple may be seen in presence of Li+. In solvents with low DN, the general tendency is for the O2− to quickly decompose or to undergo fast electrochemical reduction to O22−.
The influence of cation acidity on the oxygen electrode voltammetry is depicted below in Figure 10. The soft Lewis acid TBA+-based electrolyte enables nearly an order of magnitude larger oxygen reduction current than does the Li+-based electrolyte. This result is explained based on the formation of the insoluble Li2O2 via equation 10 at the disk interface in the Li+-based electrolyte which occurs as soon as the dissolved oxygen begins to be reduced to superoxide. The passivation film is imaged in the micrograph shown in Figure 10 which was collected from the disk electrode after holding it at the O2 → O−2 reduction peak potential. The buildup of the insoluble product at the electrode surface prevents the reduction wave magnitude from reaching its diffusion controlled limit as it does in case (a) in TBA-based electrolyte in which the TBAO2 product is soluble.
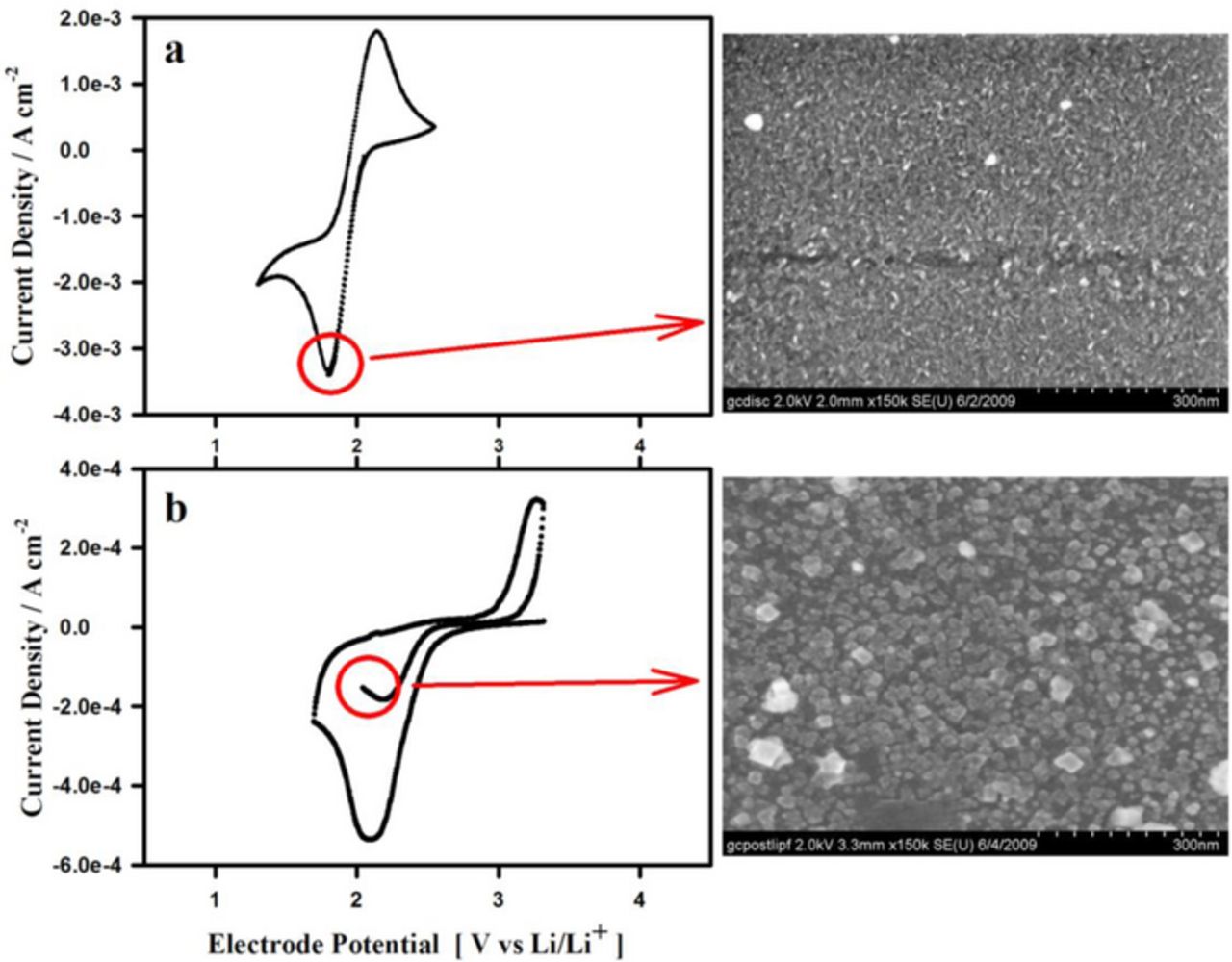
Figure 10. Scanning electron micrographs of a glassy carbon working electrode magnified 150 × 103 times after holding at the cathodic peak in two different electrolytes separately, (a) 0.1 M TBAPF6 in CH3CN and (b) 0.1 M LiPF6 in CH3CN.39
Li2O2 or Li2O as Stable ORR Products in Non-aqueous Li-Air Cells
Whether or not ORR ultimately leads to the formation of Li2O2 involving an overall two-electron reduction of O2 or to the formation of Li2O in a four-electron O2 reduction depends on the conditions under which the electrochemistry is carried out. In cells in which the potential is scanned as in a cyclic voltammetry experiment, the three products LiO2, Li2O2 and Li2O can be selectively formed by controlling the potential at which the reduction is performed.12 Evidence for all the three products have been obtained from Raman spectra when the O2 reduction is conducted potentiostatically at 2.6, 2.3 and 2.1 V versus Li/Li+, respectively. The formation and chemical decomposition of LiO2 as in shown equations 1 and 2 are well-established. Our microelectrode studies have established that the one-electron reduction of LiO2 to Li2O2 also occurs under potential sweep conditions. Indeed, Raman spectra confirmed the formation of Li2O2 and L2O at 2.3 V and 2.1 V versus Li/Li+ under constant potential reduction on a carbon cloth electrode.12
In Li-air cells discharged at constant current, the product almost always is Li2O2. In these cells the discharge potential is kept more or less constant with a continuous supply of O2 to the cell. Under those conditions, the discharge appears to proceed exclusively according to reactions 1 and 2 or more precisely 9 and 10. The Li2O2 produced is stored in the pores of the carbon electrode and the end of cell discharge is reached when these pores are choked by the insoluble Li2O2. Recent in situ Raman spectral evidence gathered from Li-air cell cathode showed the presence of LiO2 in the electrode along with Li2O2 confirming that that the more likely reaction pathway is via reactions 9 and 10.
Influence of Li Salt Anion on ORR Mechanism and Products
As we have seen Li+ is usually solvated by four solvent molecules in non-aqueous electrolytes to form solvent-separated ion pairs; e.g., Li+(DMSO)4PF6−, in LiPF6/DMSO electrolyte. Some anions such as trifluoromethanesulfonate (CF3SO3−) (triflate) have electron donating atoms or groups which can complex with the Li+ as well. Such a co-ordination would further modulate the Lewis acidity of Li+ to the extent that the ORR mechanism and the properties of the ORR products could be modified as a result. Ir spectral studies we have carried out suggest that triflate and (triflimide), (formulated as bis(trifluoromethanesulfonyl) imide), (N(CF3SO2)2‑, anions can replace some of the solvent molecules in the co-ordination sphere of Li+ in its solvates and form direct ion pairs with it as depicted in Structure IV. In fact the donor numbers for the anions presented in Table IV lend further support for the ability of the triflate ion (with a high donor number of 16.90) to form strong internal coordination with Li+. The high donor capability of the triflate anion has two possible advantages in the Li-air cathode reaction; i) it stabilizes the superoxide ions by Li(triflate)x(n-1)+(DMSO)y cations in a complex of the structure Li(triflate)x(n-1)+(DMSO)y—O2− in which Li+ is softer than in the solvates without the anion co-ordination.
Table IV. Donor number of some anions.29
| Anion | Donor Number |
|---|---|
| PF6− | 2.5 |
| ClO4- | 8.44 |
| CF3SO3− | 16.9 |
| N(CF3SO2)2− | 5.4 |
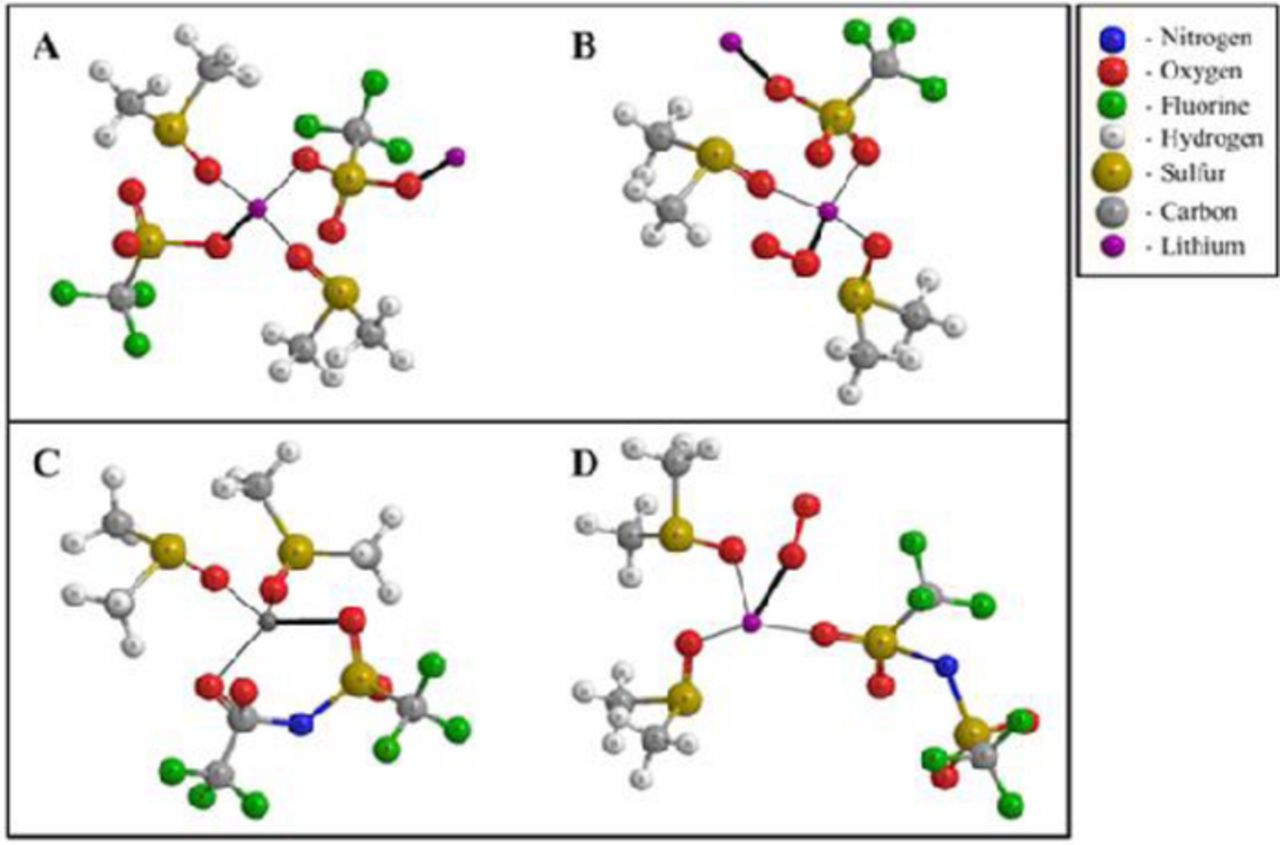
Structure IV. Schematic representation of solvation and contact ion pair formation in A) LiCF3SO3/ DMSO and C) LiN(CF3SO2)2/DMSO electrolytes. Ion pair between solvated Li+ and superoxide ion in B) LiCF3SO3/DMSO, D) LiN(CF3SO2)2/DMSO electrolytes.12
The stabilized superoxide in turn mitigates its rapid chemical decomposition/electrochemical conversion to peroxide; ii) the anion and the solvent help solvate the Li+ in Li2O2 to form Li2(triflate)x(n-1)+(DMSO)y–O2 leading to some solubility for the peroxide in the LiCF3SO3/DMSO electrolyte and dissolving it from the electrode surface to mitigate electrode passivation.
FT-IR spectra recorded in 1 M LiPF6/TEGDME, and 1 M LiCF3SO3/TEGDME also strongly support TEGDME complexed with Li+ in a curled structure of the type shown in Structure V. The PF6− ion has a very low donor capability (DN = 2.5) compared to the donor capability of the triflate anion (DN = 16.9). As a result, the absence of contact ion pairs with PF6− ion evidenced by IR spectral data is understood. Thus in LiPF6/TEGDME solutions the Li ions are primarily coordinated to the solvent molecules. IR spectral data revealed that in Li triflate/ TEGDME solutions, some of the five coordination bonds due to solvent are replaced by the sulfonyl oxygen atoms of the triflate ions through the lone pair electrons on the oxygen atoms in the anion. Thus, the overall Lewis acidity of Li+ in Li triflate solutions is slightly lower than in LiPF6 solutions leading to differences in the ORR electrochemistry in the two solutions.
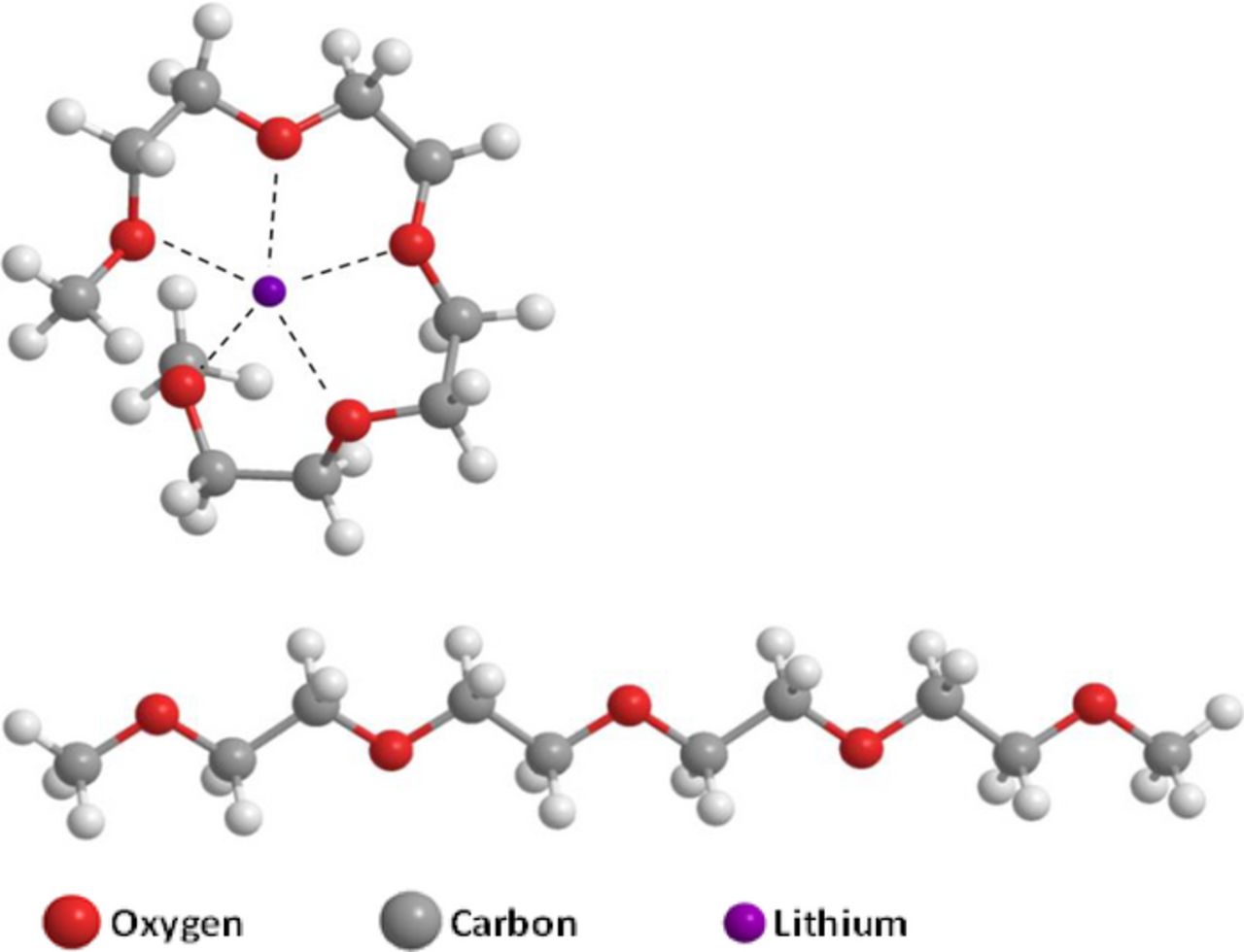
Structure V. The linear and curled forms of TEGDME molecule,. The curled form is shown as a five co-ordinate complex with Li+.
Predicating OER from HSAB Theory
Although hard acids such as Li+ and Na+ have a strong electrostatic attraction toward soft bases such as O2−, they cannot remain as stable LiO2 or NaO2 because of the stability founded in the HSAB theory. In terms of Lewis acid hardness, the cations investigated in this work listed in Table I rank in the manner, TBA+ < PYR+ < EMI+ < K+ < Na+ < Li+.
As depicted in Figure 9, the oxygen evolution reaction (OER) overpotentials of the cation–O2− ion pairs increase as a function of cation hardness.19 According to HSAB theory, stronger covalent bond character is more favorable for soft/soft interactions resulting in improved equilibrium of the redox species at the electrode surface and better reversibility. As the cation hardness increases, it imparts instability in the cation–O2− ion pair which coupled with chemical decomposition forms the peroxide and forces redox potentials farther from Nernstian-like conditions.
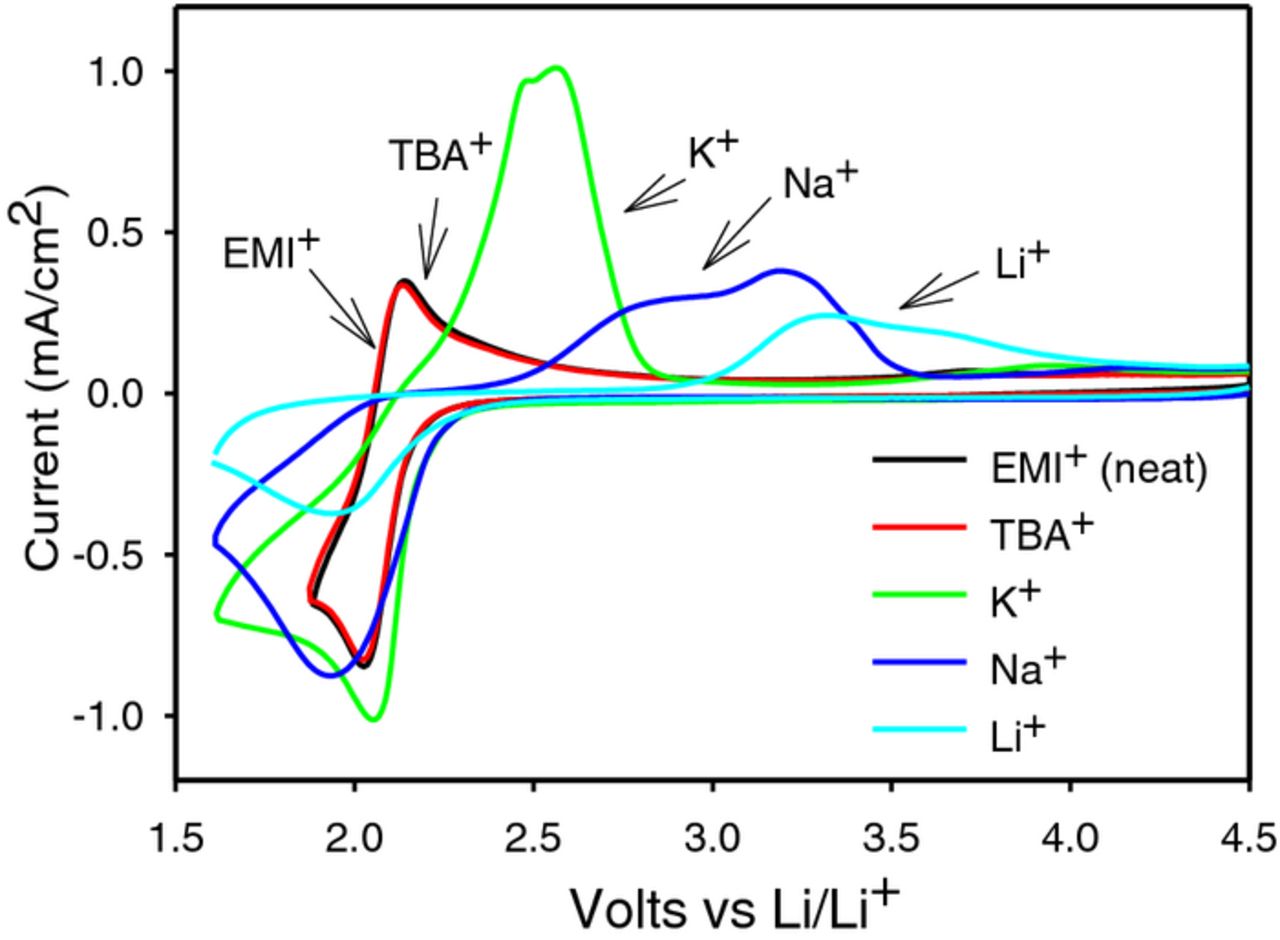
Figure 9. Cyclic Voltammogram of neat EMITFSI along with various salts at 0.025 M concentration on a GC electrode at 100 mV/s.19
The strongly ion-paired O2−–M+ and M+–O22−–M+ (where M = Li and Na) ORR products are oxidized at the highest anodic potentials.
Influence of Water on ORR in Non-Aqueous Electrolytes
Water is a contaminant in Li battery electrolytes and efforts are made to scrupulously dry them prior to use in Li cells. Ultimately, the Li-air battery is expected to operate with O2 accessed from the atmosphere and in those cells some ingression of water into the cell is a possibility. Accordingly, it is important to understand how water affects the ORR and OER mechanisms and the nature of the products formed. Figure 11 provides some insight into the behavior of the O2/O2− couple in the TBA+-based electrolyte upon H2O addition. The water concentrations were 10 mM, 300 mM, and 3 M which correspond to water mole fractions (χ H2O) of 0.06%, 1.6% and 16% respectively. A variety of prior spectroscopic studies have shown30–32 that in acetonitrile-water mixtures of χH2O < 20%, all of the water is associated with acetonitrile in a 1:1 CH3CN:H2O complex via hydrogen bonding with the acetonitrile nitrogen atom, CH3CN—H-O-H. The hydrogen-bonded complex found in solutions with a low mole fraction of water molecules is believed to function as a charge transfer mediator promoting an outer Helmholtz plane electro-reduction of the superoxide complex. The blue curve in Figure 11 is a measurement of the superoxide couple after the addition of 10 mM H2O. At this concentration a mild change in the response is seen as a suppression of the magnitude of the superoxide oxidation signal labeled EpA1. In the case of the dry electrolyte (Figure 5), the superoxide oxidation peak current magnitude is approximately 4 mA cm−2,while after the 10 mM addition the magnitude is lowered to 2.5 mA cm−2. This decrease in the EpA1 signal is correlated with the onset of the reduction wave shown at 1.75 V which is the electrochemical reduction of the superoxide to peroxide. This process, in the presence of 10 mM CH3CN-H2O, has been shifted to lesser overpotentials compared to the dry electrolyte voltammogram in Figure 5 where such process is not initiated until 1.1 V. The hydrogen- bonded complex with its positive dipole oriented toward the electrode surface probably facilitates the reduction of the superoxide as a charge transfer mediator, allowing the electrochemical process to occur at lesser over-potentials. An illustration of this ORR process is provided in Figure 12.
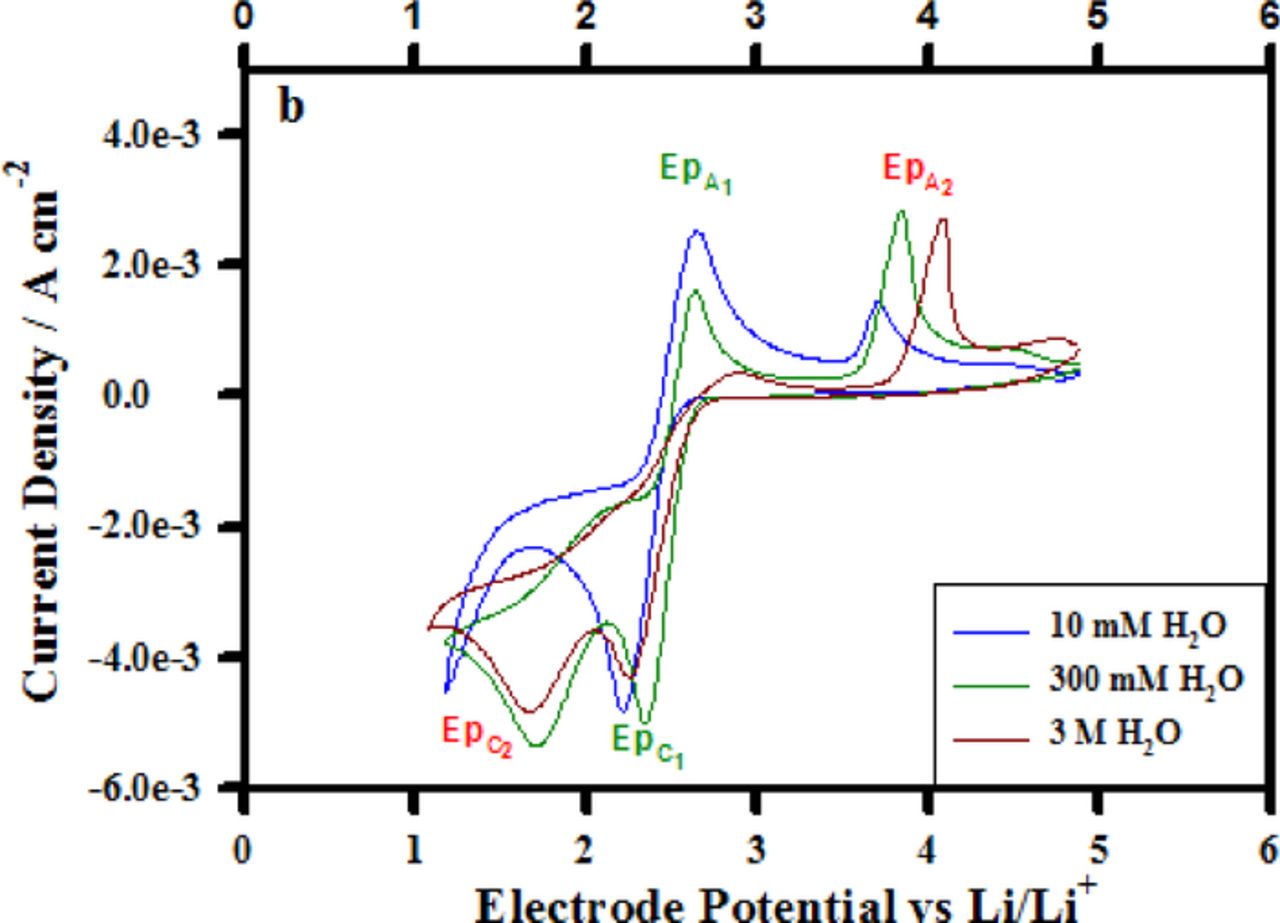
Figure 11. Cyclic voltammograms collected from a glassy carbon working electrode at 100 mV s−1 in (a) dry 0.1 M TBAPF6 in CH3CN and (b) water spiked 0.1 M TBAPF6 in CH3CN.
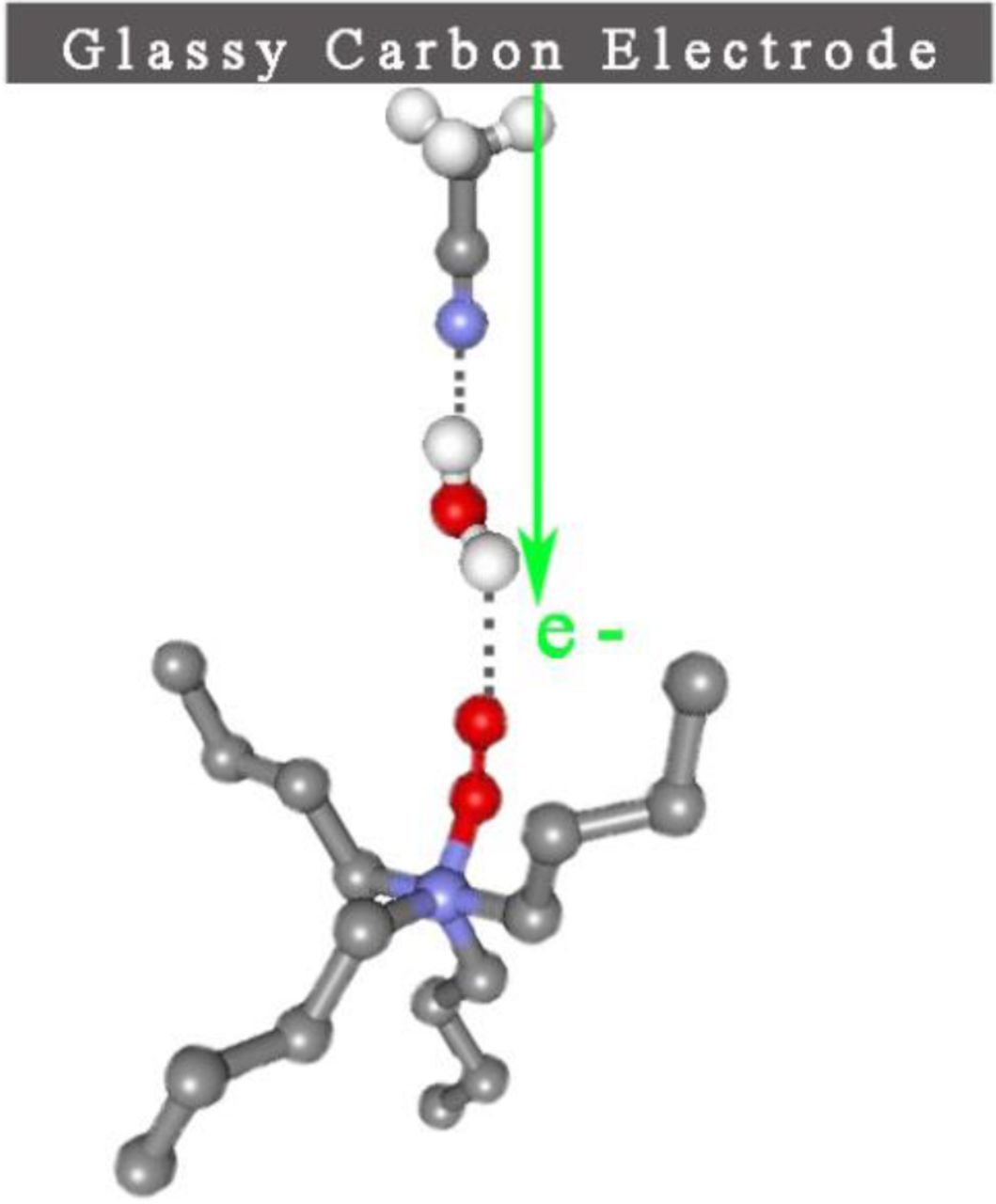
Figure 12. Schematic diagram showing the hydrogen bonded water-acetonitrile complex functioning as a mediator of the outer sphere charge transfer reduction of O−2—TBA+.
Increasing the CH3CN-H2O charge transfer mediator concentration from 10 mM to 300 mM (χH20 = 1.6%) shifts the onset of the Epc2 process further to 2.0 V vs. Li/Li+ and also decreases the EpA1 signal magnitude further as a more substantial quantity of the superoxide has been electrochemically reduced.
At 3 M H2O (χH20 = 16%) two additional changes in the oxygen voltammogram are observable. First the EpA1 signal is almost completely absent, only a relatively small magnitude signal which is shifted to greater anodic over-potential remains. This change is consistent with a near complete depletion of the superoxide concentration. However, comparing the magnitude of EpC2 signals for the 300 mM H2O and 3 M H2O there is no increase in the amount of electrochemically formed peroxide as a result of increasing the charge transfer mediator concentration. Therefore, the almost complete absence of the EpA1 signal cannot be explained solely as an effect of the electrochemical reduction of the superoxide to peroxide as it was in the 300 mM circumstance. Considering that water clusters containing hydronium cations are expected to be present in acetonitrile-water mixtures at approximately χH2O = 20% in conjunction with the aforementioned observation leads to the conclusion that the electrochemically formed superoxide must also be consumed in a chemical reaction in addition to the measured electrochemical reduction (EpC2). This chemical reaction is presumably a reaction involving the hydronium ion present in solution as an effect of the water cluster formations which occur in the χ H2O = 16% solution. A possible reaction sequence is shown in equations 13 and 14 below
![Equation ([13])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn13.jpg)
![Equation ([14])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn14.jpg)
The second major change observed at the 3 M concentration is a shift to greater anodic over-potential required to induce the peroxide oxidation process labeled as EpA2. The peroxide reduction product is a harder base than is the superoxide product and since the hydronium ion is a harder acid (due to its smaller ionic radius) than the TBA cation, the peroxide reduction product will preferentially pair in solution with the hydronium ions present in the 3 M H2O solution. Therefore, in this solution the peroxide is more stable in accordance with HSAB theory as it is paired with the harder acid hydronium cation and requires more electrochemical force to drive the oxidation process depicted in equation 15.
![Equation ([15])](https://content.cld.iop.org/journals/1945-7111/162/2/A3021/revision1/jes_162_2_A3021eqn15.jpg)
Although the presence of the hydrogen bonded water-acetonitrile complex in the electrolyte certainly is shown to lower the stability of the O−2—TBA+ complex by facilitating a mediated outer Helmholtz plane reduction, the effect is not as drastic as that observed in the Li+-based electrolytes. The hydrogen bonded complex with its positive dipole oriented toward the electrode surface probably facilitates the reduction of the superoxide as a charge transfer mediator allowing the electrochemical process to occur at lesser over-potentials as illustrated in Figure 12. A more detailed discussion of the effect of water on ORR and OER in organic electrolytes is presented in the Ph. D thesis of my student Matt Trahan submitted to Northeastern University from which this account of the effect of water is taken.33
Stability of Solvents Toward O2−
The superoxide being an anion radical is very reactive toward polar solvents and will react with the non-aqueous solvents used in the Li-air battery. The reversibility revealed by the cyclic voltammograms of O2 in organic electrolytes containing tetrabutyl annonium salts can be used as a first order measure of this reactivity. This is demonstrated by the CV data in Figure 13 for ORR in TBAPF6/DMSO and TBAPF6/Propylene carbonate (PC) solutions. The former exhibits reversible behavior at sweep rates ranging from 20–500 mV/sec whereas the CV for O2 reduction in PC exhibits highly irreversible behavior with the ratio of the anodic to cathodic charges being less than 0.2. Data similar to this have been used to identify reactive and stable solvents for the Li-air battery.34,35 From similar studies and based on Li-air cell data we have identified the following solvents as the most stable among a series of organic solvents and ionic liquid that have been investigated to date. These are organic sulfones and sulfoxides, especially dimethyl sulfoxide (10, 11, organic ethers16,36 such as polyethylene glycol dimethyl ethers16,33 with TEGDME as a prominent example, and room temperature ionic liquids with the two important candidates of EMITFSI and PYRTFSI.18,19
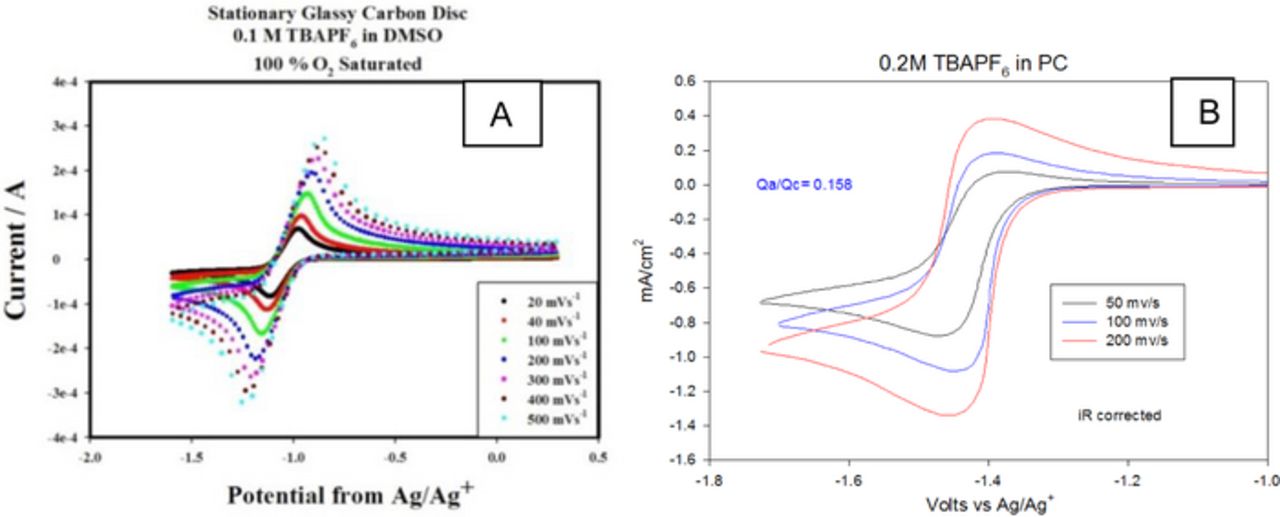
Figure 13. Cyclic voltammograms of O2 in (a) TBAPF6/DMSO and (b) TBAPF6/Propylene carbonate (PC) solutions.
Solvent-Directed ORR Catalysis
As we have seen from the above discussion, in electrolytes based on high donor solvents such as DMSO, the O2− is stabilized by the formation of stable ion pairs of the type Li+(DMSO)n—O2−. As a result, the activation energy for electron transfer is lowered to provide a higher voltage for the Li-air cells utilizing DMSO-based electrolytes.38,39 Thus, high donor number solvents homogeneously catalyze O2 reduction reaction through an outer Helmholtz plane (OHP) process as shown in Figure 14.38 The homogeneous ORR catalysis is so dominant that cathode catalysts such as CoPC showed little ORR catalytic activity in DMSO-based electrolytes. In the absence of a catalyst in the carbon cathode, there appears to be no homogeneous catalysis in low DN solvent-based electrolytes such as CH3CN and TEGDME, despite an outer Helmholtz layer (OHP) ORR process.38 In these low DN solvent-based electrolytes, catalysts such as CoPC, Pt and Au promote an inner Helmholtz plane (IHP) ORR process due to the strong adsorption of O2 as well as the ORR intermediates O−2 and O2 −2 onto the catalyst surface (Figure 15). The result is the catalyzed formation of Li2O2 as well as Li2O, the four-electron reduction product of O2 to O2−.37 The adsorption of the intermediate reduction products LiO2 and Li2O2 onto the catalyst surface may lower the propensity for these materials to react with the solvents leading to improved stability for the Li-air battery in some catalyzed cathode cells.
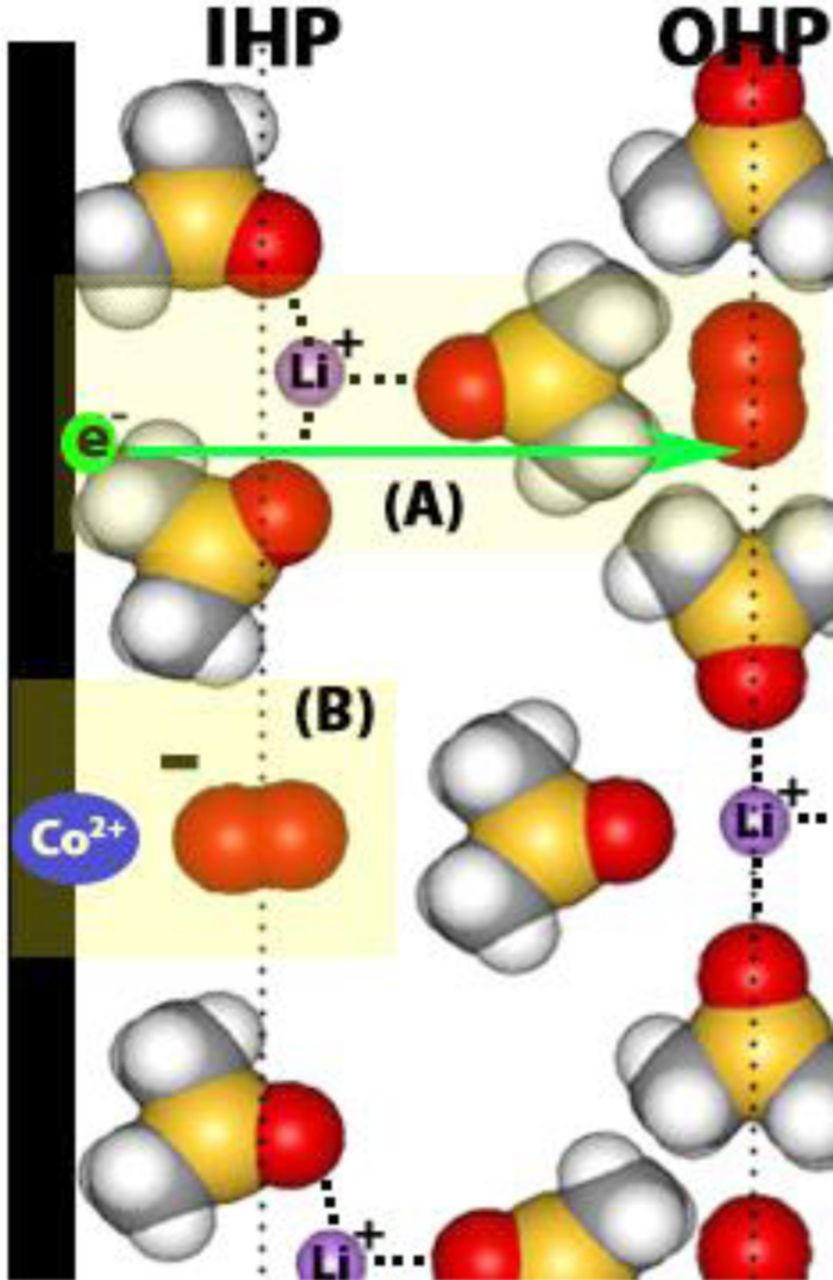
Figure 14. A schematic representation of the electrochemical double layer at the interface of a Co600 catalyzed carbon electrode and a Li+-DMSO electrolyte. Highlighted section A shows the outer Helmholtz plane (OHP) one electron charge transfer. Highlighted section B shows the one electron product superoxide chemisorbed to the Cobalt catalyst in the inner Helmholtz plane (IHP) promoting further electrochemical reduction of the oxygen radical.38
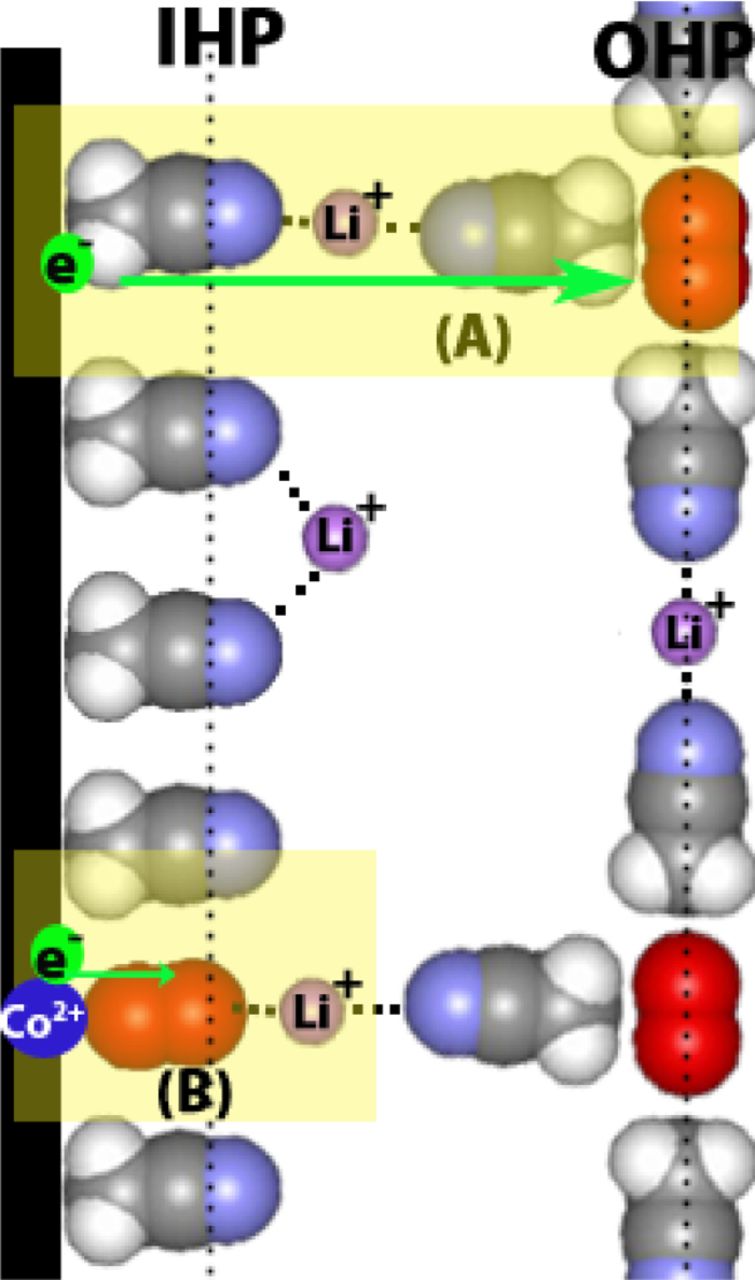
Figure 15. A schematic representation of the electrochemical double layer at the interface of a Co600 catalyzed carbon electrode in a Li+-(CH3CN)n electrolyte. Highlighted section A shows the outer Helmholtz plane (OHP) one = electron charge transfer as it occurs on an uncatalyzed surface. Highlighted section B shows inner Helmholtz plane (IHP) charge transfer process facilitated by the presence of a catalyzed surface.38
Conclusions
Organic electrolyte plays a critical role on the ORR and OER processes in the non-aqueous Li-air battery. In all organic electrolytes, O2 reduction initially proceeds through the one-electron reduction product, superoxide, O2−. The degree of stability of the superoxide formed is determined by the stabilization afforded to it by the solvated Li ions thorough ion pairs of the type Li+(solvent)n–O2−. High donor number (DN) solvents with DN greater than about 25 appear to provide a higher degree of stability for the superoxide with the result that it's redox reactions can be observed in the cyclic voltammogram. On the other hand, in low DN solvents, the superoxide has shorter lifetimes which result in its rapid decomposition to Li2O2, in accordance with the Hard Soft Acid Base (HSAB) theory. The HSAB concept is also useful as a qualitative metric to measure the rechargeability of the ORR products in the Li-air battery. The stronger is the ionic bond strength between the O2 reduction product and the conducting salt cation, the poorer it appears to be its rechargeability. The reactivity of the superoxide with solvents can be used to screen solvents and identify stable solvents for use in the Li-air battery. The three groups of solvents we have identified with reasonably good stability for use in non-aqueous Li-air cells are organic sulfones and sulfoxides, polyethylene glycol dimethyl ethers and room temperature ionic liquids. Our data also suggest that organic solvents affect ORR catalysis with high donor number solvents such as DMSO enabling homogeneous ORR catalysis whether or not a catalyst is present in the porous carbon cathode, while low donor number solvents exemplified by TEGDME enable heterogeneous catalysis in presence of catalysts such as metal phthalocyanines and noble metals in the porous carbon electrode. In the latter case the four-electron reduction of O2 to Li2O is possible.
Acknowledgment
Financial support of our work by US Army CERDEC is gratefully acknowledged. The author acknowledges the significant contributions of his graduate students, Cormac O'Laoire, Chris Allen, Matt Trahan, Iromie Gunasekara and Jaehee Hwang to the work reported here.