Abstract
To clarify the relationship between Li+ transport rate in glyme-based electrolytes and Li deposition/dissolution behavior at Li metal negative electrode (NE) in Li-air batteries (LAB) systems, 1.0 M tetraglyme (G4) electrolytes were prepared containing a Li salt of LiSO3CF3, LiN(SO2CF3)2, or LiN(SO2F)2. Two aspects of Li+ transfer between the two phases, i.e., G4 electrolyte | Li metal NE, were evaluated, namely i) Li+ supplying rate and ii) Li+ charge transfer rate through solid electrolyte interphase (SEI) films. The former was investigated by self-diffusion coefficients D of Li+, anions, and G4 solvent together with ionic conductivity σ, viscosity, density, and apparent dissociation degree αapp of the Li salts estimated by the Nernst–Einstein equation. The latter was evaluated with Li | Li symmetric and LAB (Li | O2) cells containing the electrolytes. The Li deposition/dissolution reaction basically depended on the Li+ supplying rate in the Li | Li cell; however Li dendrites were formed. Conversely, the LAB cell performance was controlled by Li oxide layers formed on the NE, resulting in similar discharge/charge properties without Li dendrites. The effects of surface-oxidation was also confirmed with Li | Li cells containing O2 gas, where both SEI and charge transfer resistances were reduced.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
In recent years, rechargeable non-aqueous Li-air batteries (LABs) have received increasing attention as large-scale energy storage devices for long-range electric vehicles (EVs), because of their high energy density, more than five times greater than that of conventional Li-ion batteries (LIBs).1–4 However, some problems need to be addressed to enable the realization of this technology, including choking of the air electrode by Li2O2 generated during discharging and the high overpotential required by Li2O2 oxidation reaction during charging. These factors lead to oxidative deterioration of the electrolytes and the air electrode, which consists of porous carbon materials. At the Li metal negative electrode (NE), Li dendrite growth during discharge/charge cycles also poses serious safety problems.5–7 Conventional Li metal NE batteries (LMBs) contain carbonate-type electrolytes, including propylene carbonate- (PC) and ethylene carbonate (EC)-based electrolytes. The use of additives, such as ethylene sulfite (ES),8 vinylene carbonate (VC)9–11 and fluoroethylene carbonate (FEC),12–14 and replacing the Li metal NE with Li pre-doped Si,15 has been suggested as a way of avoiding the short circuiting of cells caused by Li dendrite growth. However, the additives commonly used to control the solid electrolyte interphase (SEI) film do not effectively work in LAB systems. In these systems the Li dissolution/deposition reaction is repeated many times and O2 gas is fed in from the air electrode, resulting in destabilization of the SEI films. Furthermore, carbonate-type electrolytes are decomposed by O2− radicals generated at the air electrode during the discharge process. Recently, ether-based electrolytes, based on 1,2 dimethoxyethane (DME or G1), diglyme (G2), triglyme (G3), or tetraglyme (G4) as a solvent, have been widely investigated for non-aqueous LAB systems.16 These ethers have high oxygen solubility and relatively low electric constants, resulting in lower reactivity toward O2− radicals than that of carbonate-based electrolytes.17 Hence, alternative ways of stabilizing the surface of Li metal NE in glyme electrolytes are currently in demand. Moreover, much more data is required on the Li dissolution/deposition behavior in glyme electrolytes for finding new stabilization methods to enable realization of next-generation LMBs, including LABs.
In this study, we investigated the Li dissolution/deposition reaction at the glyme electrolyte side, including the formation of SEI films on the Li metal NEs, to understand the factors affecting the Li dendrite growth.18 Figure 1 shows a schematic illustration of the interface between the Li metal NE and the glyme electrolyte used in LAB systems. The Li deposition/dissolution reaction is thought to be controlled by two aspects, namely: i) a Li+ supply process to the Li metal NE; and ii) a Li+ charge transfer process through the SEI films on the surface of the Li metal NE. Here, we evaluated the former aspect by measuring the self-diffusion coefficients D of Li+, the anions, and G4 solvent individually with a pulsed gradient spin echo nuclear magnetic resonance (PGSE-NMR) method.19,20 We also measured the viscosity η, density ρ, and ionic conductivity σ of the electrolytes. In addition, to consider the number of carrier ions per specific volume of electrolyte, the apparent dissociation degree αapp of the Li salts was also calculated from D and σ values by the Nernst–Einstein equation.19–22 For the latter aspect, the deposition/dissolution reaction was evaluated from the polarization curves of Li | Li symmetric cells with different glyme electrolytes. The Li+ charge transfer resistance at the Li metal NEs and the morphology of the deposited Li compounds were analyzed by electrochemical impedance spectroscopy (EIS) and scanning electron microscopy (SEM), respectively. In addition, the LAB (Li | O2) cell test was also conducted to investigate the effects of the anion species of Li salts and O2 gas from the air electrode on the Li metal NE. Changes in the morphology of the Li deposition compounds and the chemical composition of the SEI films were analyzed by SEM and X-ray photoelectron spectroscopy (XPS), respectively. To clarify the effects of O2 gas in more detail, Li | Li symmetric cells were also evaluated containing O2 gas in the cells. Both processes i) and ii), which affect the Li dissolution/deposition reaction at the Li metal NE, were considered and are discussed to provide new strategies for fast and stable Li+ transfer at the interface of Li metal NEs in LAB systems.
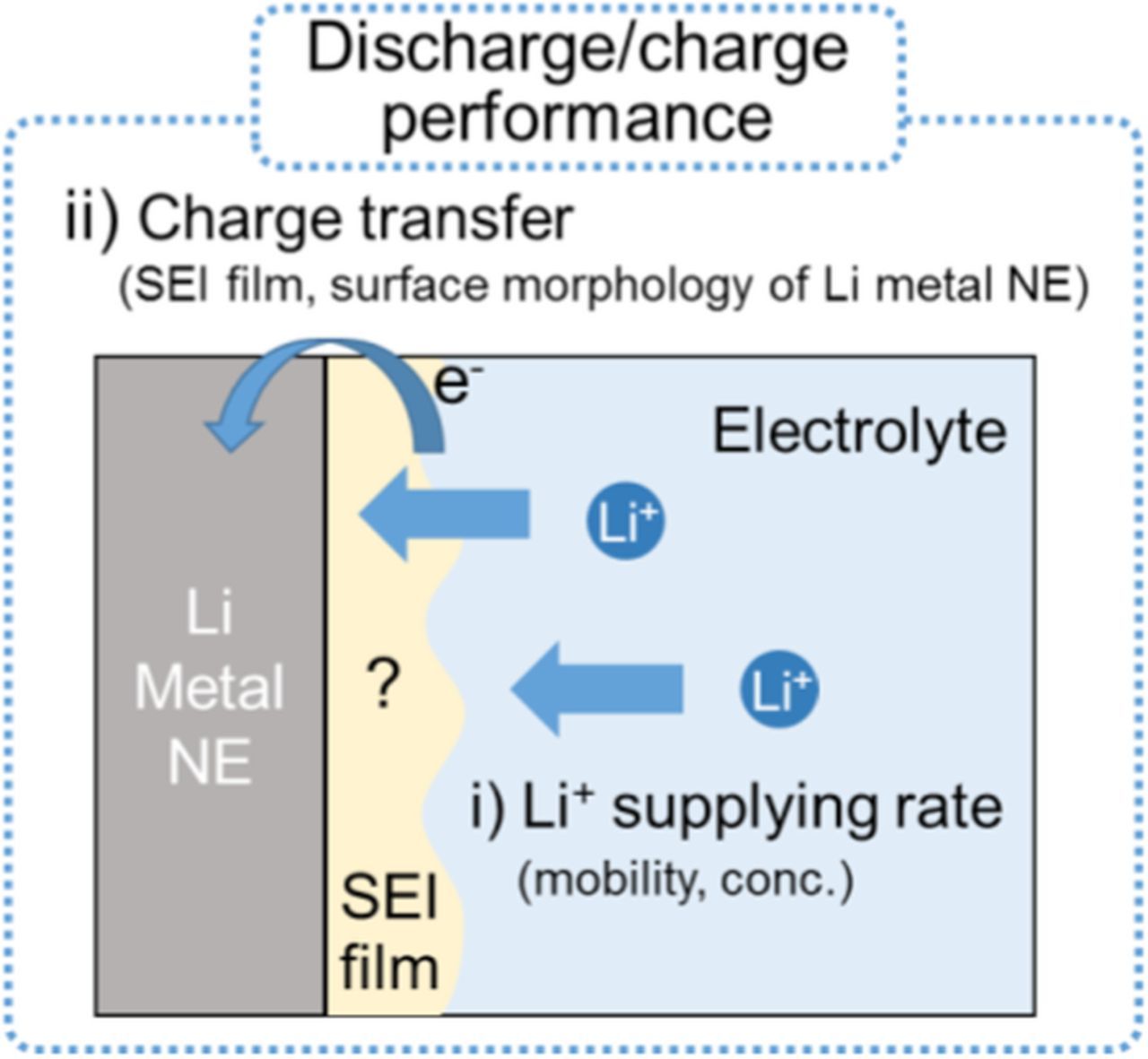
Figure 1. Schematic illustration for determination factors affecting Li dendrite growth at Li metal NE.
Experimental
Preparation of electrolyte solutions
To prepare 1.0 M glyme-based electrolytes for LABs, LiOTf (LiSO3CF3, 99.0%, Kishida), LiTFSI (LiN(SO2CF3)2, 99.9%, Kishida), and LiFSI (LiN(SO2F)2, 99.0%, Kishida) were used as supporting salts, and dissolved in G4 (Japan Advanced Chemicals, H2O content: < 30 ppm) as a solvent in an Ar-filled dry box (GBJF100E805, Glovebox Japan Inc.).
Evaluation of the electrolyte properties
The σ values of the electrolytes were measured with a conductivity meter (S230 SevenCompact, Mettler Toledo).
The D values of Li+ (7Li), the anion (19F), and solvent G4 (1H) in the electrolytes were measured by PGSE–NMR with a JEOL tunable pulsed-field gradient (PFG) probe (1H resonance: 400 MHz).19–21 Each sample was prepared in a NMR microtube (BMS-005J, Shigemi, Tokyo) to a height of less than 5 mm to prevent convection effects. The PFG was calibrated with H2O (1H resonance). Measurements were performed by setting the same PFG strength for each nuclear species with a proper irradiation time δ. The accuracy of the D values was confirmed by obtaining the same values at different δ. Measurements of η and ρ were performed with a falling ball-type viscometer (Lovis2000ME, Anton Paar). All the electrolyte properties were measured at 30°C for the following electrochemical cell tests.
Electrochemical evaluation of Li | Li symmetric and LAB cells
The Li | Li symmetric cell was constructed with Li metal foil (thickness: 0.5 mm, Honjo Metal Co., Ltd.) used for both electrodes, a separator (Celgard 2400), and the G4-based electrolytes in an argon-filled glove box (Miwa, MDB-1BK-NT1). The polarization curves of the Li | Li symmetric cells were measured to evaluate the overpotentials for Li dissolution/deposition reactions at both Li metal electrodes and the changes occurring during cycling by using a VSP potentiostat (BioLogic Science Instruments). The applied current density and maximum discharge/charge capacity were 0.20 mA cm−2 and 0.50 mAh cm−2, respectively, in the cut off voltage range of −2.0 to 2.0 V. EIS analysis was also conducted during the Li dissolution/decomposition tests in Li | Li symmetric cells. The applied voltage and measured frequency range were 5 mV and from 100 kHz to 0.10 Hz, respectively.
For fabrication of the LAB (Li | O2) cell, Ketjen Black (KB, Lion Corp. Ltd., EC600JD)-loaded carbon paper (TGP-H-060, Chemix. Co. Ltd.) was used as an air electrode, i.e., the positive electrode (PE). The loading weight of KB was ca. 1 mg cm−2. The same separator and Li metal NE as that used in the Li | Li symmetric cell were used in this cell. The same applied current density (0.20 mA cm−2) and maximum discharge/charge capacity (0.50 mAh cm−2) were applied to the LAB cell in the cut off voltage range of 2.0 to 4.5 V.
To investigate the effects of O2 gas on the Li deposition/dissolution behavior, Li | Li symmetric cells containing O2 gas were fabricated based on the LAB-type cell with an inlet for O2 gas. The polarization curves of these cells were measured.
For all the electrochemical tests, the cycle number was set to 15 cycles and the operating temperature was 30°C.
Analysis of Li NE surface morphology and SEI film chemical composition
After 15 cycles of polarization and discharge/charge tests, the Li metal electrodes were removed from the Li | Li symmetric and LAB cells and rinsed with fresh G4 solvent without Li salts. After drying under vacuum, the Li metal electrodes were set in transfer vessels and the surface morphology and chemical composition of the SEI films were evaluated by SEM-energy dispersive X-ray spectroscopy (SEM-EDS, JSM-7800F, JEOL) and X-ray photoelectron microscopy (XPS, VersaProbe II, ULVAC-PHI), respectively.
Results and Discussion
Solution properties of the prepared electrolytes
As mentioned above, the Li dissolution/deposition behavior at the Li metal NE was influenced by the Li+ supply rate (Li+ diffusion rate and number per specific volume) and the surface conditions (chemical composition of the SEI film) of the Li metal NEs. Here, we first focus on the former factor of the Li+ supply rate to the surface of the Li metal NE.
Figure 2 shows the D values of the Li+, anion, and G4 solvent for 1.0 M of G4-based electrolytes containing LiOTf, LiTFSI, and LiFSI. For the LiTFSI and LiFSI electrolytes, the D values of Li+ and the G4 solvent were smaller than those of LiOTf. This result indicates that a larger amount of G4 strongly solvated Li+ leading to dissociation of the Li salts, and an increased Li+ diffusion radius. This finding was in good agreement with the difference between the D values of the anion and Li+. For the LiOTf electrolyte, the D values of TfO− and Li+ were almost the same as each other, implying the existence of a large number of ionic pairs consisting of Li+ and TfO−. On the contrary, the D value of TFSI− and FSI− was clearly higher than that of Li+ owing to the high dissociation of the Li salts.
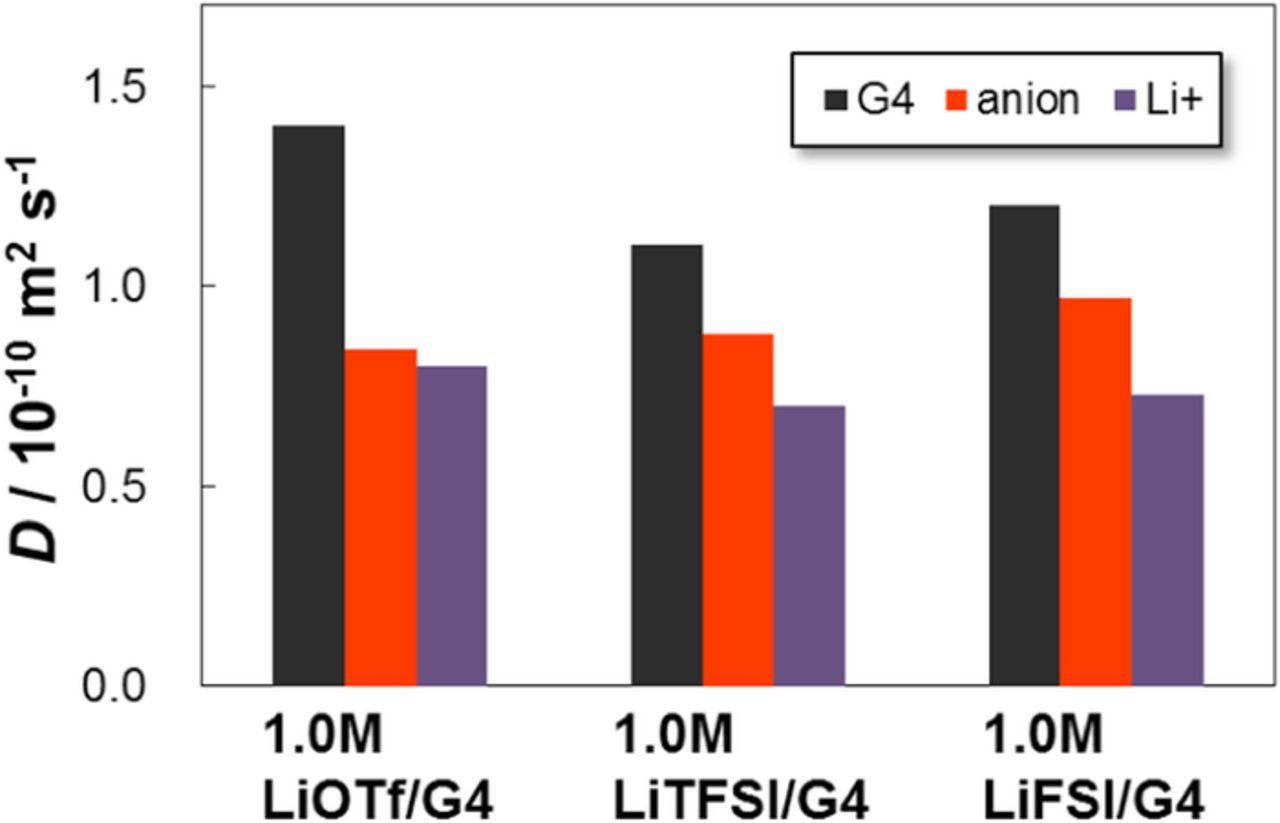
Figure 2. Self-diffusion coefficients D of ions and G4 solvent of G4-based electrolytes measured by PGSE-NMR at 30°C.
In fact, the LiTFSI and LiFSI electrolytes exhibited higher viscosity η and density ρ owing to the stronger interaction between the G4 solvent and Li+ (Table I). As a result, higher σ values of 3.02 and 3.33 mS cm−1 were obtained for the LiTFSI and LiFSI electrolytes, respectively. To confirm the degree of dissociation of the Li salts, the molar ionic conductivity ΛNMR and apparent dissociation degree αapp were calculated from the Nernst–Einstein equation as follows:19–22
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/164/12/A2872/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/164/12/A2872/revision1/d0002.gif)
where N is the number of isolated ions per specific volume, e is elementary charge, k is Boltzman's constant, T is temperature (K), Λimp is molar ionic conductivity calculated from σ and ρ values for the electrolytes by AC impedance method, respectively. The obtained ΛNMR and αapp values are summarized in Table I. The LiTFSI and LiFSI electrolytes exhibited higher αapp values (0.52 and 0.53) than that of LiOTf (0.14).
Table I. Solution Properties of G4-based Electrolytes at 30°C.
| η / | ρ / | σ/ | ΛNMR / | αapp / | |
|---|---|---|---|---|---|
| Electrolyte | mPa s | g cm−3 | mS cm−1 | mS cm2 mol−1 | − |
| 1.0 M LiOTf/G4 | 7.45 | 1.09 | 0.83 | 6.06 | 0.14 |
| 1.0 M LiTFSI/G4 | 9.99 | 1.16 | 3.02 | 5.84 | 0.52 |
| 1.0 M LiFSI/G4 | 9.61 | 1.12 | 3.33 | 6.28 | 0.53 |
Therefore, the LiTFSI and LiFSI electrolytes have advantages in terms of fast supply of Li+ to the Li Metal NE, owing in particular to the high dissociation degree of these Li salts.
Evaluation of Li deposition/dissolution in Li|Li symmetric cells
Figure 3 shows the polarization curves measured from Li | Li symmetric cells containing the different G4-based electrolytes. For the LiOTf electrolyte, the overpotential for the Li deposition/dissolution reaction gradually increased upon cycling. Conversely, the LiTFSI and LiFSI electrolytes exhibited relatively stable overpotentials; for the LiFSI electrolyte the value gradually decreased upon cycling. Thus, the LiFSI electrolyte, which had the higher supply rate σ owing to its higher value of αapp, exhibited a notable advantage in terms of the Li deposition/dissolution reaction.
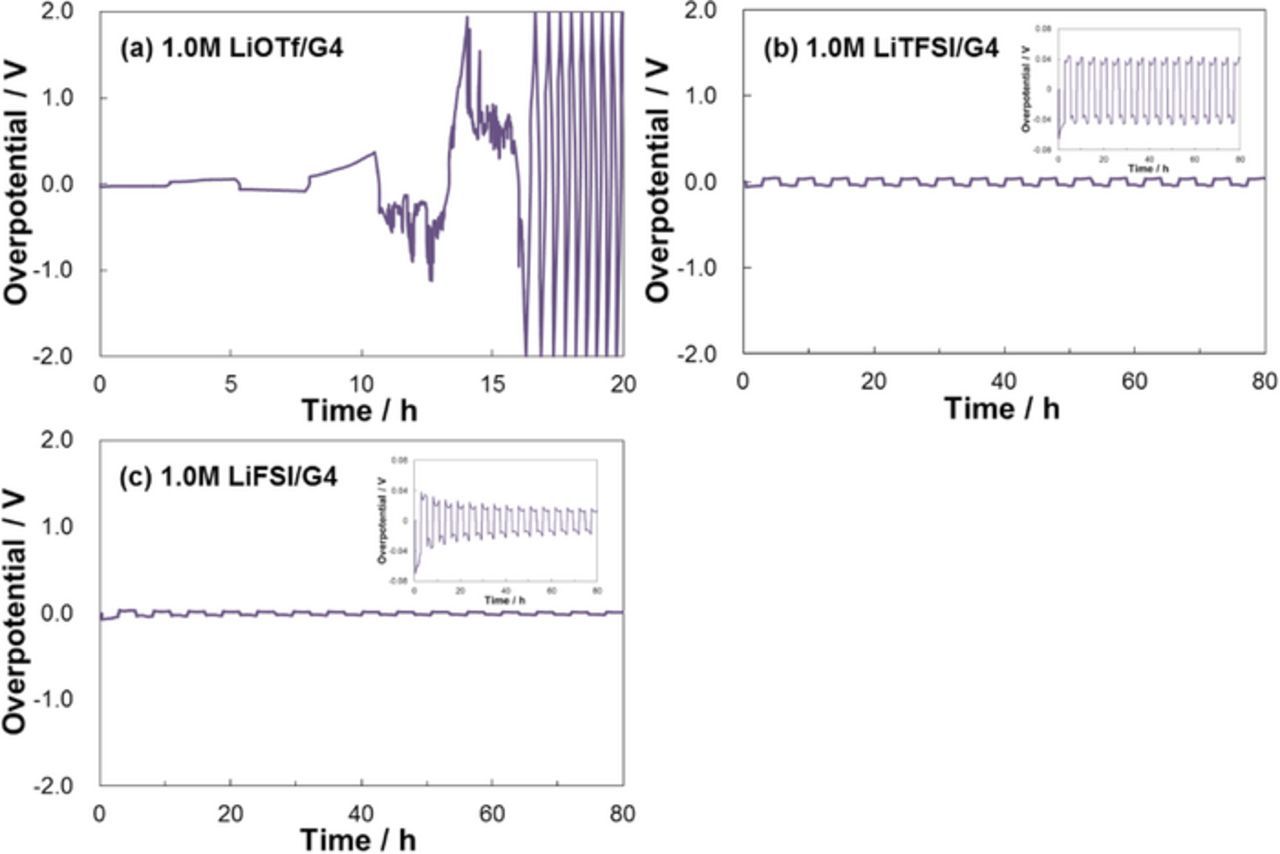
Figure 3. Polarization curves of the Li | Li symmetric cells with different G4-based electrolytes at 30°C.
The Li charge transfer resistance Rct for the Li dissolution/deposition reaction was evaluated by EIS analysis of the Li | Li symmetric cells together with the SEI film resistance RSEI. The obtained data were analyzed with the equivalent circuit, shown in Fig. 4. The Nyquist plots are shown in Figs. 5a–5c and the estimated cell resistance values are summarized in Fig. 5d. The Rct for the cell with the LiFSI electrolyte was smaller than that for the cell with LiTFSI, and decreased with increasing cycle number. This result was in good agreement with the overpotential results. In addition, the LiFSI electrolyte exhibited a lower SEI film resistance RSEI (< 5 Ω cm2) compared with that of LiTFSI (∼10 Ω cm2) during cycling. Therefore, LiFSI also stabilizes the Li metal NE at the SEI film. Conversely, the LiOTf electrolyte cell did not work regardless of the similar RSEI values before the Li | Li symmetric cell test. This result is likely caused by the formation of an unstable SEI film derived from the LiOTf salt and/or a slow Li+ supply rate. However, the main cause is considered to be the low dissociation degree of LiOTf. In general, stronger interaction between Li+ and anion increases the reduction potential of anion.23,24 Li+ adjacent to anion shares some negative charge and makes either the reduction of anion easier, resulting in reductive decomposition.
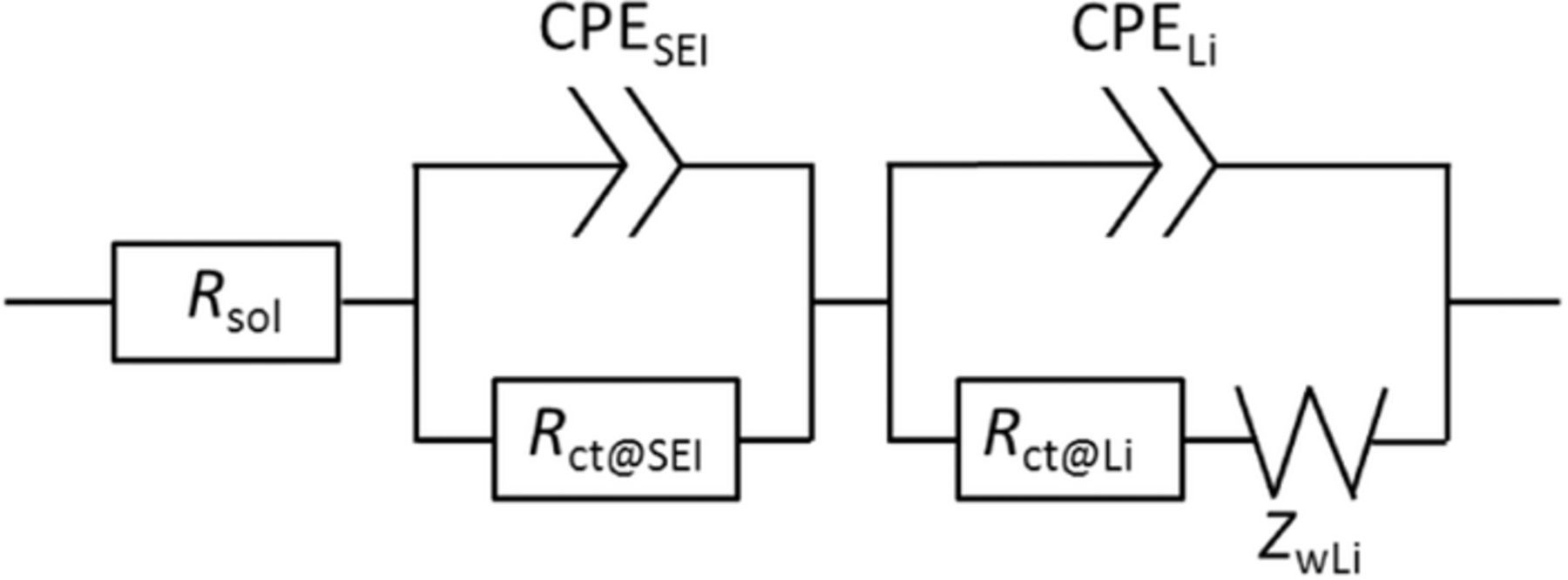
Figure 4. Equivalent circuit diagram for the Li | Li symmetric cells with different G4-based electrolytes at 30°C.
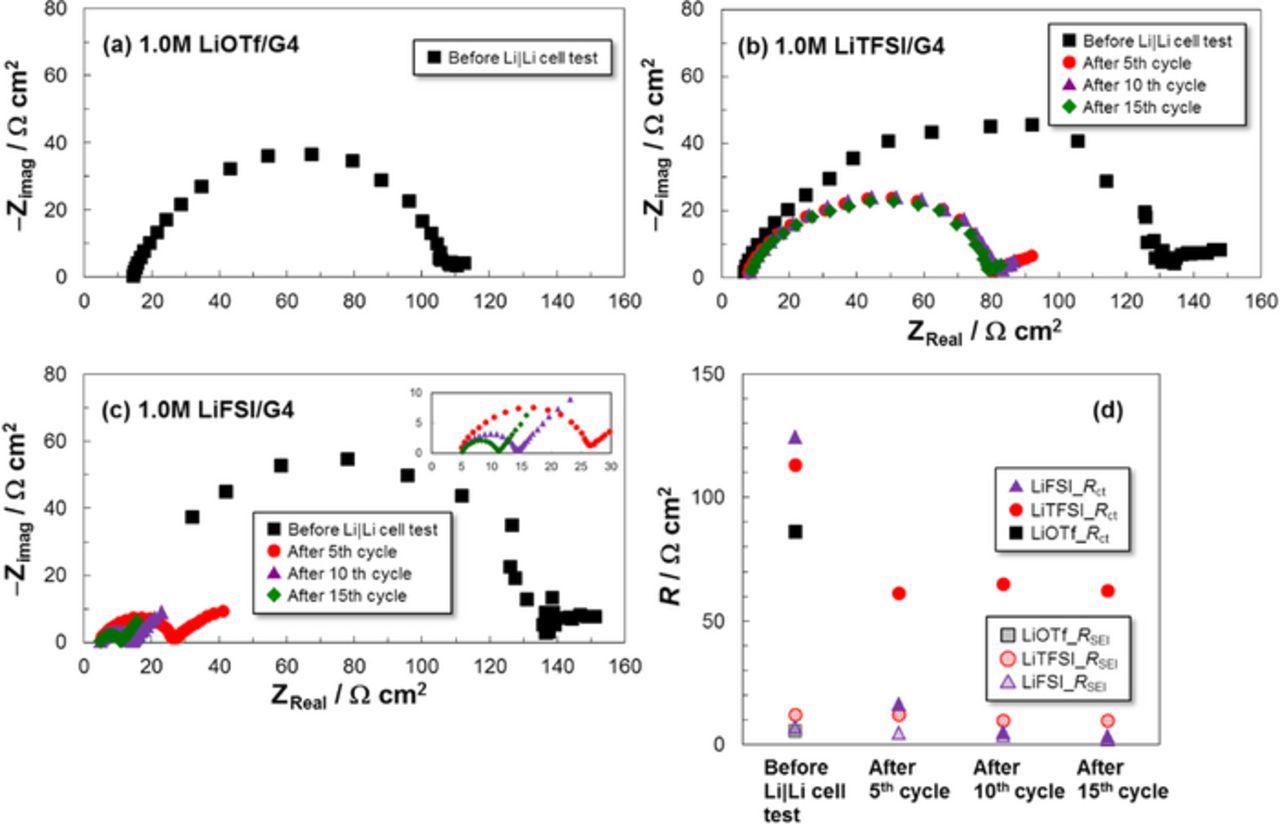
Figure 5. Nyquist plots and obtained cell resistances (RSEI and Rct) of the Li | Li symmetric cells with different G4-based electrolytes at 30°C.
Figure 6 shows SEM images of the Li metal electrodes after the Li | Li symmetric cell tests, i.e. after 15 cycling of polarization, with different G4 electrolytes. For the LiOTf electrolyte, almost no Li deposition and no Li dendrite formation were observed on the Li metal electrode. Conversely, many Li dendrites were found for the LiTFSI and LiFSI electrolytes. This result indicates that the core of Li dendrites was easily generated and grew in the G4 electrolytes having higher σ and αapp. Furthermore, as these values increased the average diameter of dendrites decreased. A decrease of Rct was thought to enhance the formation of the Li dendrite core and increase the surface area of the Li metal electrode through the growth of fine Li dendrites. This finding concurs with the reduction of Rct for the LiFSI electrolyte upon cycling.
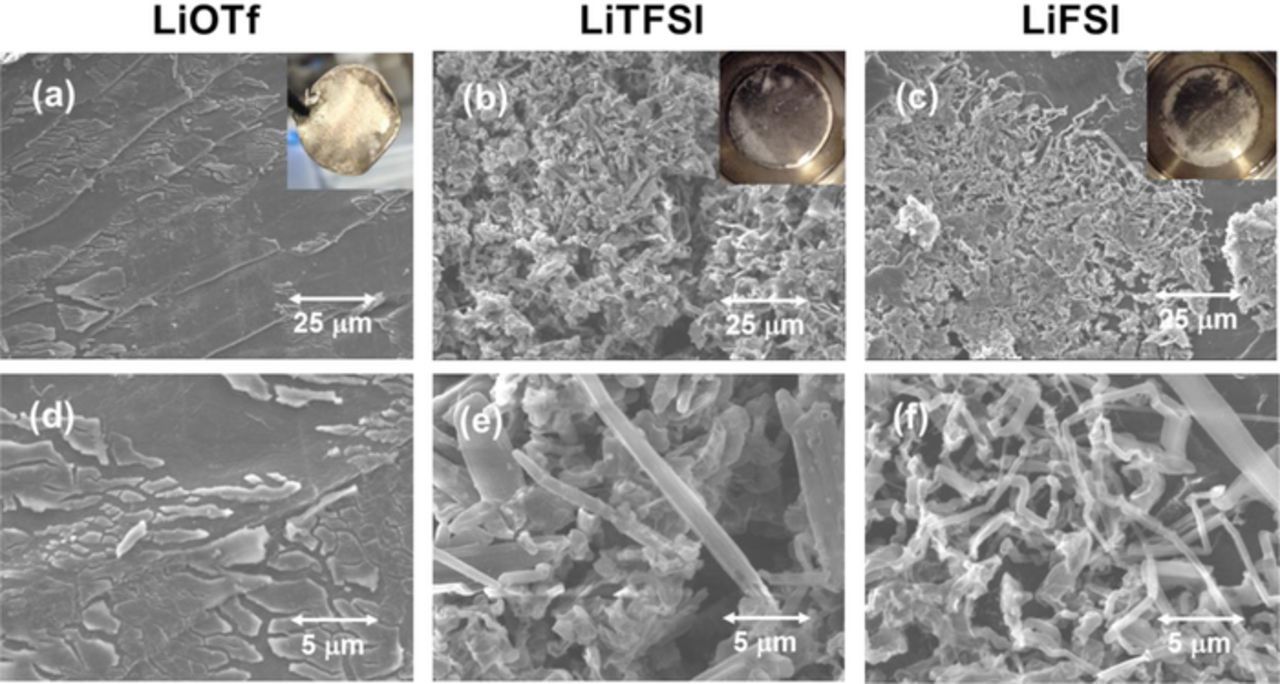
Figure 6. SEM images of the Li metal NEs after the Li | Li symmetric cell tests with different G4-based electrolytes at 30°C. (a, d) 1.0 M LiOTf/G4, (b, e) 1.0 M LiTFSI/G4 and (c, f) 1.0 M LiFSI. Images (d, e, f) show magnifications of (a, b, c), respectively.
LAB cell performance with different Li salt electrolytes
Figures 7a–7c and 7d–7f show the discharge/charge curves and cycling performance for LAB cells with different G4 electrolytes. Although the 1st discharge curves showed quite similar behavior regardless of the type of electrolyte, the overpotential at the 1st charge cycle were relatively low compared with those at the following cycles, especially for the electrolyte containing LiTFSI. After the 1st cycle, the discharge curves for the LiOTf and LiFSI electrolyte cells deteriorated with further cycling. The discharge/charge curves are influenced by the PE reaction at the air electrode; however, we confirmed that the selection of the Li salt also affects the cell performance. Namely, the LiOTf electrolyte exhibited relatively good LAB operation regardless of the low σ value. For the LiFSI electrolyte, oxidative decomposition was confirmed at the end of 1st charging over 4.05 V, which rose the overpotential and decreased the charge capacities at the following cycles. This degradation is clearly shown in the cycle performance results of the LAB cells (Fig. 7f). Therefore, O2 gas introduced from the air electrode markedly influenced the LAB electrode reactions including the Li dissolution/deposition reaction at the Li metal NEs. The LiOTf electrolyte was particularly strongly affected.
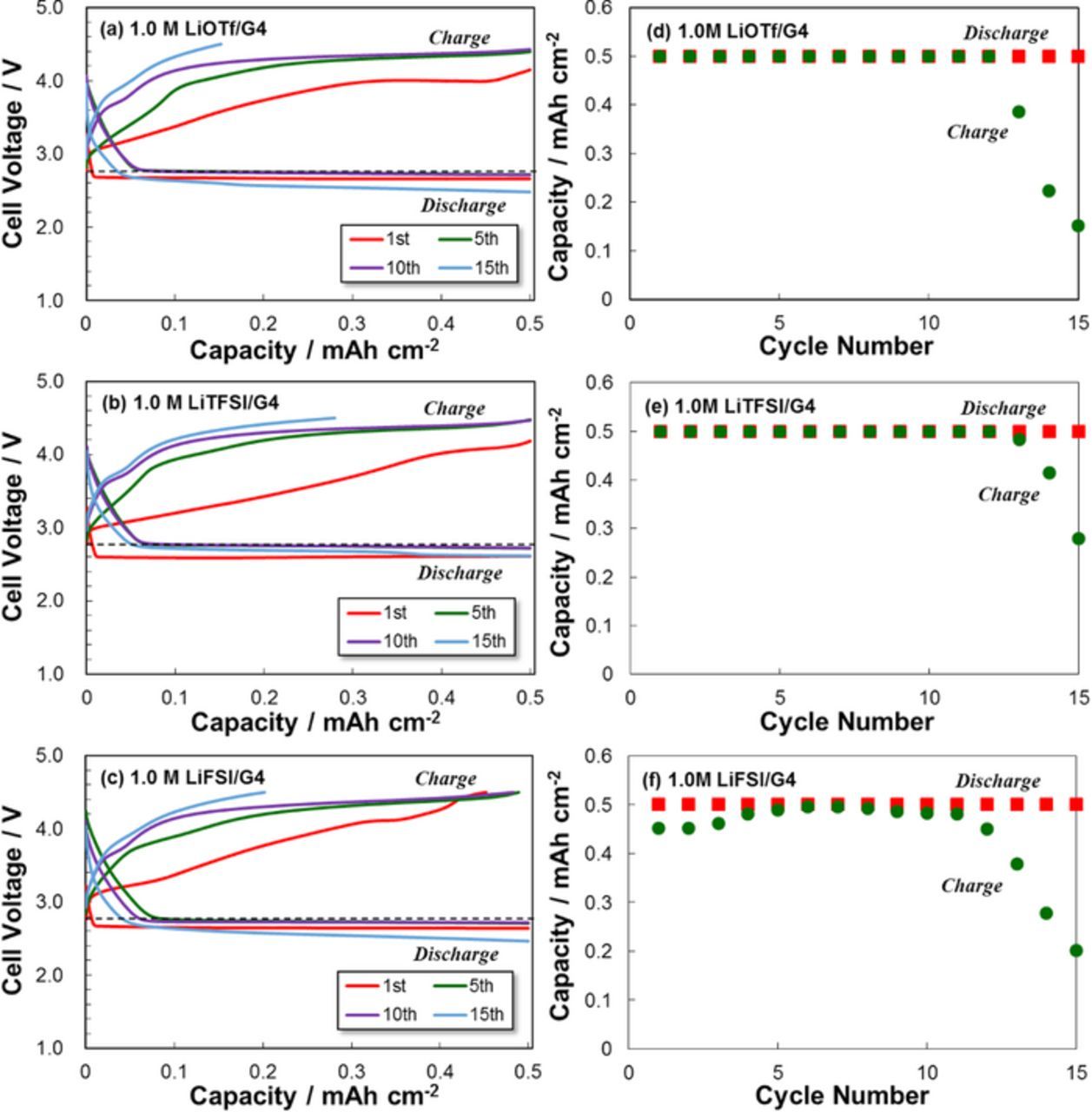
Figure 7. Discharge/charge curves and cycling performance of LAB (Li | O2) cells with different G4-based electrolytes at 30°C.
The LiTFSI and LiOTf electrolyte cells maintained lower cell voltages, particularly at the end of charging, and were relatively stable during cycling (Fig. 8). Conversely, for the LiFSI electrolyte the higher cell voltage due to the oxidative decomposition of FSI− anion at charging led to deteriorate the cell performance, particularly the cycling performance, despite this electrolyte having the highest σ value.
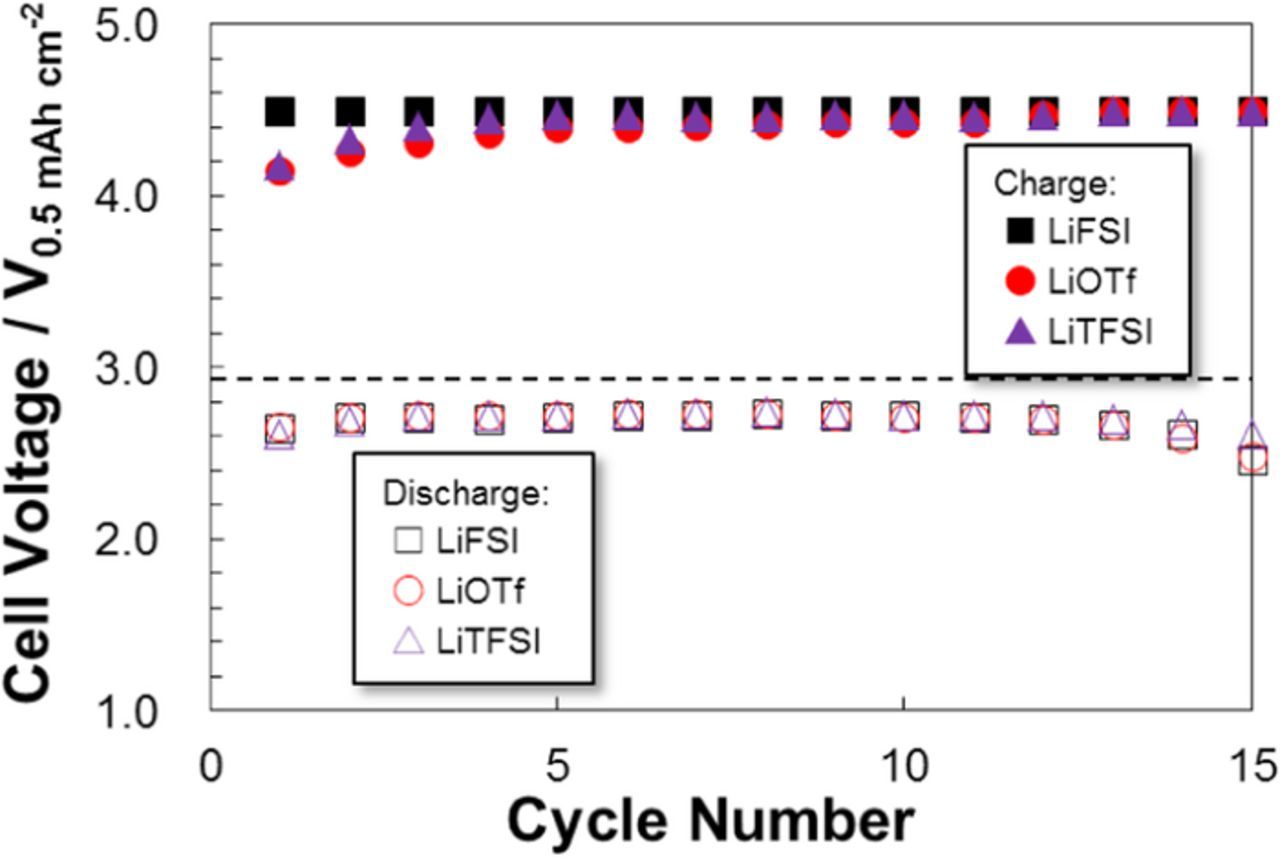
Figure 8. Change in cell voltages at the end of discharge and charge processes for LAB (Li | O2) cells with different G4-based electrolytes at 30°C.
To confirm and investigate the effects of O2 gas introduction into the LAB cells, SEM observations and the EDS analysis were conducted for the Li metal NEs after 15th charging (Fig. 9). For all electrolytes, the surface of the Li metal NEs was clearly oxidized by O2 and chemical compounds including carbon were also deposited. The Li dendrites almost disappeared, even for the LiFSI electrolyte. Only a few ribbon-like Li dendrites were observed for the LiTFSI electrolyte.

Figure 9. SEM images and EDS analyses of the Li metal NEs for the Li | Li symmetric cells with different G4-based electrolytes at 30°C. (a, d, g) 1.0 M LiOTf/G4, (b, e, h) 1.0 M LiTFSI/G4, and (c, f, i) 1.0 M LiFSI/G4.
Figure 10 shows XPS data for Li metal NEs after 15 discharge/charge cycles. For all electrolytes, a relatively large amount of carbon species, including Li2CO3, HCO2Li, CH3CO2Li and plolyethers/ethers were detected in the SEI films on the Li metal NE. These carbon species were derived from electrolyte decomposition.25,26
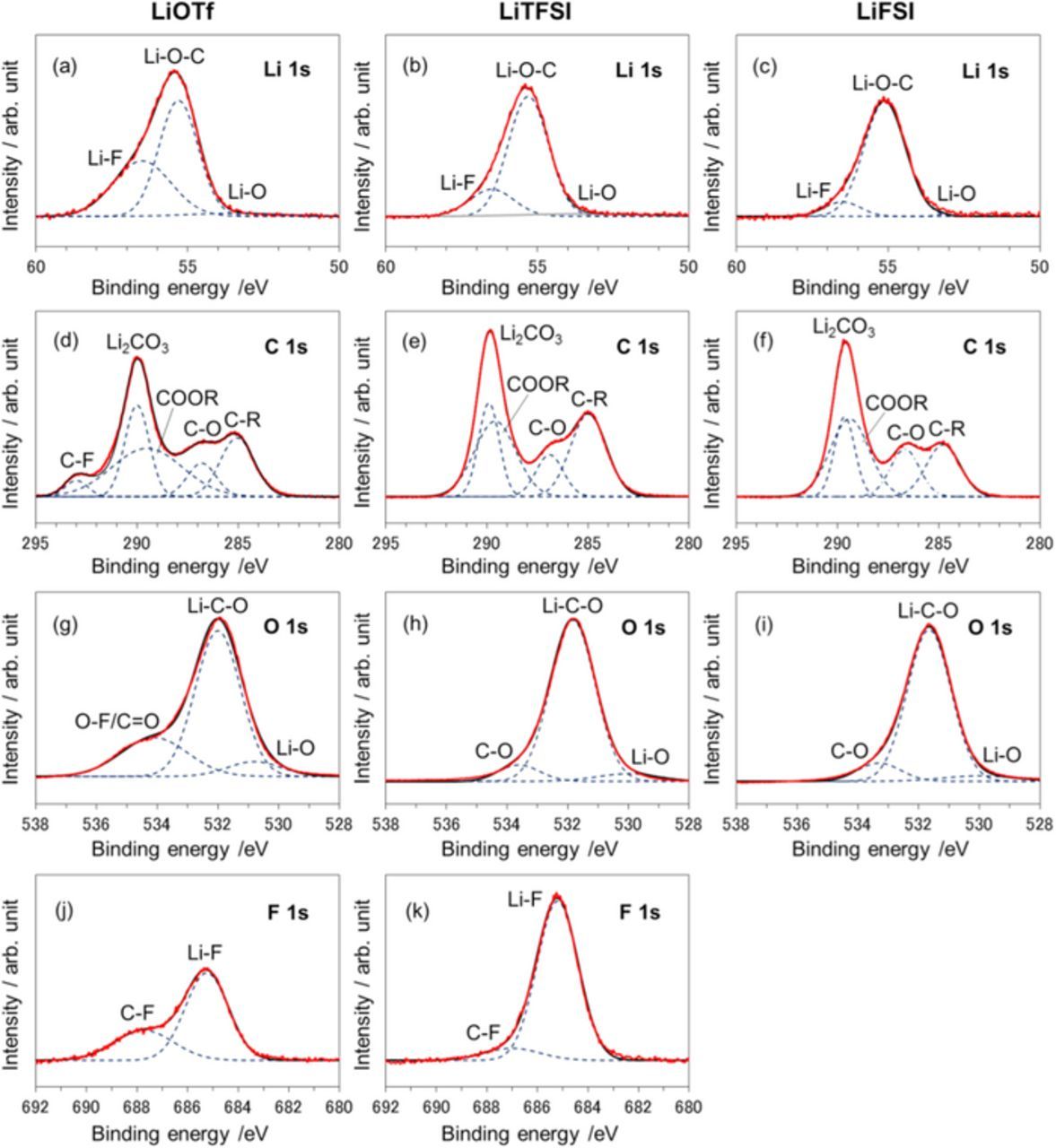
Figure 10. XPS analysis of the surface of the Li metal NEs in LAB (Li | O2) cells with different G4-based electrolytes after the 15th discharge/charge cycle. (a, d, g, j) 1.0 M LiOTf/G4, (b, e, h, k) 1.0 M LiTFSI/G4 and (c, f, i) 1.0 M LiFSI/G4.
The LiTFSI electrolyte produced a relative large amount of LiF. A LiF-rich SEI film covered the surface of the Li metal NE, which suppressed surface-oxidation and reduced the overpotential during discharge/charge cycles. This observation is supported by the fact that ribbon-like Li dendrites were only observed for the LiTFSI electrolyte. Although a LiF peak at 685.3 eV27,28 for F 1s was also confirmed for the LiOTf electrolyte, the strength of the XPS peak was low, and a C-F peak derived from CF3 (687.7 eV) became larger, indicating a different type of reductive decomposition of the anions. In the case of the LiFSI salt, an LiF peak was not detected because of the fewer F atoms present in the FSI− anion than in the anions of other electrolytes. A relative small LiF peak confirmed for the Li | Li symmetric cell disappeared, indicating the covering by a large amount of Li2O and Li2CO3 components as shown in SEM images. All XPS data implied that the surface of the Li metal NE was oxidized, and Li2CO3 and polymer-like SEI components formed a coating as a by-product of electrolyte decomposition in the operation of the LAB (Li | O2) cell. These facts support the similar LAB cell performance compared with data of Li | Li symmetric cells.
The cell performance deteriorates owing to contributions from the Li metal NE and the air PE, although it is not clear which of these factors affect the deterioration the most. We used Li | Li symmetric cells, containing O2 to investigate the effects of O2 gas on the Li metal NE performance and the differences among the anions.
Effects of O2 gas on the deposition/dissolution reaction in Li | Li symmetric cells
As mentioned above, the Li dissolution/deposition reaction is strongly influenced by the atmosphere in the cell (i.e., Ar or O2). These effects were clearly confirmed for the LiOTf and LiFSI electrolytes. Although the Li dissolution/deposition reaction did not occur in the Li | Li symmetric cell with the LiOTf electrolyte, in the LAB cell this electrolyte exhibited relatively good performance, which was notably different from the behavior of the LiTFSI electrolyte. However, the LiFSI electrolyte deteriorated in the LAB cell. From SEM, EDS and XPS analyses, the surface of the Li metal NEs in the LAB cells was oxidized by O2 introduced from the air electrode, and this influenced the cell performance. To confirm and elucidate the effects of O2 gas on the Li metal NEs, the Li dissolution/deposition reaction was also investigated in Li | Li symmetric cells containing O2 gas.
Figure 11 shows the polarization curves for O2 gas containing Li | Li symmetric cells. For all electrolytes similar Li dissolution/deposition behaviors were confirmed and the overpotentials were reduced by the introduction of O2 gas. The LiOTf electrolyte cell also operated stably and the overpotential decreased with cycling. This result indicates that proper surface-oxidation effectively stabilized the Li metal NEs and promoted the smooth Li dissolution/deposition reaction. Recently, similar effects of surface-oxidation of Li metal NE have been reported for LAB and LMB systems using N,N-dimethylacetamide (DMA)- and glyme-based electrolytes containing a LiNO3 salt, respectively, where NO3− anions oxidize and stabilize the surface of the Li metal NE by formation of Li2O passivating layer, and enable to improve the cycle performances.29–31 Namely, the Li oxide layer containing Li2CO3 also stabilizes the surface of the Li metal NE to accelerate the Li dissolution/deposition and lower the overpotential of the air-electrode. As a result, the LAB cell containing LiOTf electrolyte also exhibited good cell operation. The Li | Li symmetric cell based on the LiFSI electrolyte exhibited a similar stable Li dissolution/deposition reaction. This finding indicates that the surface of the Li metal NE was oxidized by O2 and the cell performance depends on the Li oxide layer. Furthermore, the FSI− anion was stable at the Li metal NE in the LAB cell and was likely oxidized at the air electrode.
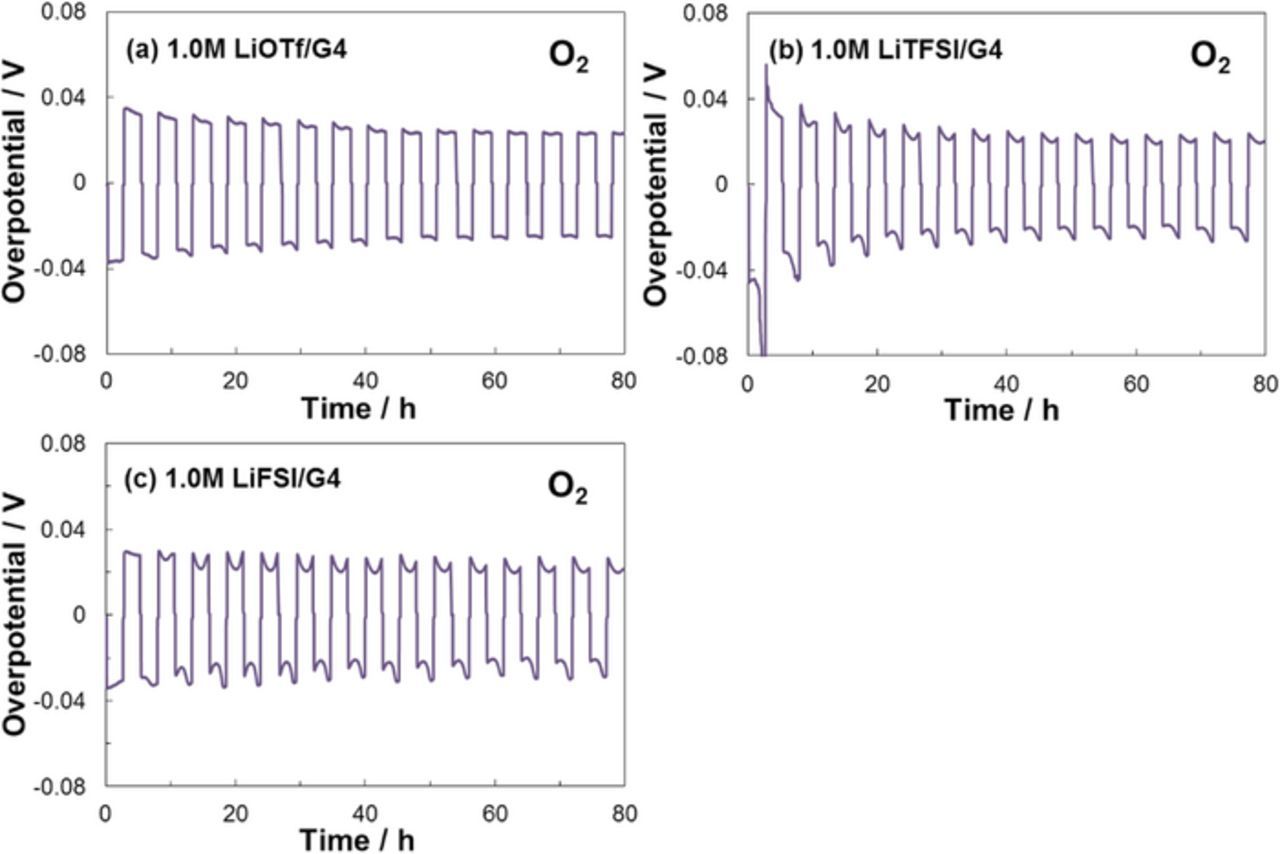
Figure 11. Polarization curves of the Li | Li symmetric cells with different G4-based electrolytes containing O2 gas at 30°C.
Figure 12 shows the results of EIS analysis for the Li | Li symmetric cells containing O2 gas. The Nyquist plots were analyzed with the equivalent circuit shown in Fig. 4, and compared with the results shown in Fig. 5. For all the electrolytes, both Rct and RSEI values were successfully reduced by the introduction of O2 gas and the Rct values clearly decreased with increased cycling. The lowest cell resistances were confirmed for the LiTFSI electrolyte. These results explain the LAB cells exhibiting similar cell performance, including those based on the LiOTf electrolyte. The best cell performance came from the cell containing the LiTFSI electrolyte.
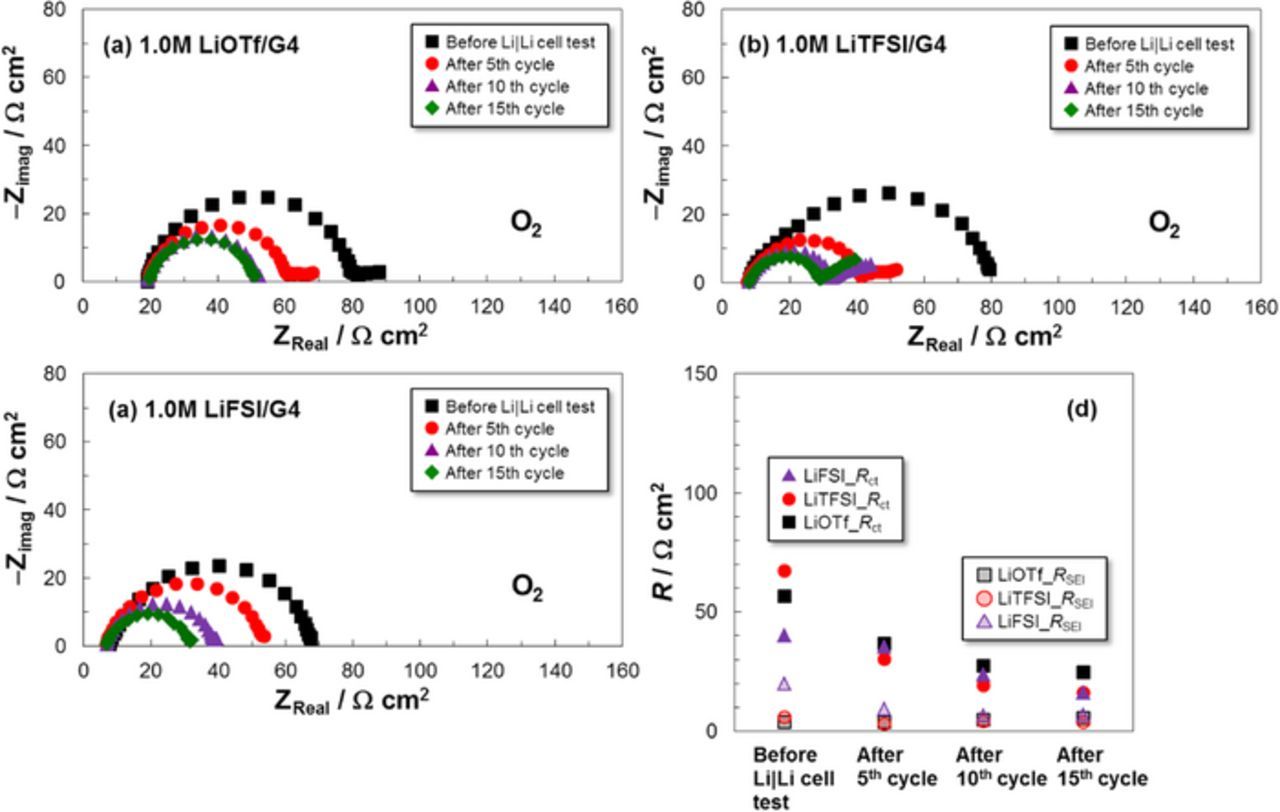
Figure 12. Nyquist plots and obtained cell resistances (RSEI and Rct) of the Li | Li symmetric cells with different G4-based electrolytes containing O2 gas at 30°C.
Conclusions
We examined factors contributing to smooth Li deposition/dissolution reactions at the Li metal NE of LAB systems. We used 1.0 M G4-based electrolytes containing Li salts including LiOTf, LiTFSI, and LiFSI as model glyme electrolytes. The Li+ supply rate to the Li metal NE and the Li dissolution/deposition reaction at the Li metal NE were investigated separately. The LiTFSI and LiFSI electrolytes exhibited advantages in both aspects. The Li+ conductivity σ of the electrolytes was high and the overpotential for the Li deposition/dissolution reaction in a Li | Li symmetric cell was lower than that of a cell with the LiOTf electrolyte. For the LiFSI electrolyte, the overpotential gradually decreased with repeated cycling. However, this trend was slightly altered for the LAB (Li | O2) cell. Our results suggested that O2 gas from the air electrode affected the performance. From SEM observations of the Li metal NEs after the LAB cell tests, none of the Li metal NEs exhibited Li dendrites on their surface. The XPS analysis of the SEI films on the Li metal NE after 15 discharge/charge cycles confirmed the presence of LiF from the LiTFSI electrolyte. Thus, this electrolyte formed an LiF-rich SEI film, which likely suppressed the surface-oxidation of Li metal NE. In fact, ribbon-like Li dendrites were confirmed only for the LiTFSI electrolyte. The Li deposition/dissolution reaction was enhanced by the introduction O2 gas for all electrolytes. This finding indicates that O2 gas from the air electrode crosses over into the glyme electrolytes and reacts with the Li metal NE to stabilize the surface and promote a smooth Li dissolution/deposition reaction. This result is in good agreement with those for LiNO3-based electrolytes, in which the NO3− anion acts as a mild oxidant for the Li metal NE to form a stable oxide film.29–31 Thus, the Li dissolution/deposition reaction rate depends on the formation of an Li oxide layer, containing Li2CO3, on the Li metal NEs. This suggests that control of the oxidation state of the Li metal NE surface is key to enhance and maintain good LAB performance. Proper surface-oxidation also effectively suppresses Li dendrite growth. As the further investigation, EIS analysis using a three-electrode LAB (Li | O2) cell to separate the effects of deterioration of the Li metal NE and the air electrode (PE) and examinations using the other electrolyte systems is now underway to promote smooth and fast Li deposition/dissolution reactions for advanced LAB systems.
Acknowledgments
This work was partly supported by each of JST Project for the Next Generation Batteries Area in Advanced Low Carbon Technology Research and Development (ALCA-SPRING) and MEXT Programs for the Development of Environmental Technology using Nanotechnology and the Scientific Technology Human Resource Development grant, Japan.