
Abstract
Nuclear magnetic resonance, mid and near infrared, and ultra violet (UV) spectra of wood contain information on its chemistry and composition. When solid wood samples are analysed, information on the molecular structure of the lignocellulose complex of wood e.g. crystallinity of polysaccharides and the orientation of the polymers in wood cell walls can also be gained. UV and infrared spectroscopy allow also for spatially resolved spectroscopy, and state-of-the-art mapping and imaging systems have been able to provide local information on wood chemistry and structure at the level of wood cells (with IR) or cell wall layers (with UV). During the last decades, these methods have also proven useful to follow alterations of the composition, chemistry and physics of the substrate wood after fungi had grown on it as well as changes of the interactions between the wood polymers within the lignocellulose complex caused by decay fungi. This review provides an overview on how molecular spectroscopic methods could contribute to understand these degradation processes and were able to characterise and localise fungal wood decay in its various stages starting from the incipient and early ones even if the major share of research focussed on advanced decay. Practical issues such as requirements in terms of sample preparation and sample form and present examples of optimised data analysis will also be addressed to be able to detect and characterise the generally highly variable microbial degradation processes within their highly variable substrate wood.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The main structural wood polymers—cellulose, hemicelluloses and lignin—are the most abundant biopolymers in the Earth's carbon cycle. These polymers form the lignocellulose complex in all woody tissues. Its highly ordered structure of cellulose microfibril aggregates embedded in a matrix of hemicelluloses and lignin provides the basis for its mechanical strength (Salmén and Burgert 2009) and for the resistance to microbial attack (Daniel 2003), to which also low molecular mass extractives contribute (Zabel and Morrell 1992).
Wood therefore is a remarkably durable material. In nature, only higher fungi have developed biochemical systems to degrade the lignocellulose complex and perform the conversion and mineralisation of wood to carbon dioxide and water. Most of these fungi belong to the Basidiomycetes. Although they are phylogenetically closely related (Binder and Hibbett 2002; Floudas et al. 2012) their strategies of degrading wood are diverse: While brown-rot fungi degrade primarily the wood polysaccharides and leave behind a polymeric but highly modified lignin, simultaneous white-rot fungi degrade all polymeric wood constituents at similar rates. Selective white-rot fungi, which lack the ability to degrade cellulose efficiently, cause extensive delignification of wood. Ascomycetes and Deuteromycetes may cause soft-rot decay that leads to softening of wet wood. Cavity formations in wood cell walls are most characteristic for this decay type. Extensive reviews on decay pattern, chemistry, and biochemistry of microbial wood degradation are available (Daniel 2003; Eriksson et al. 1990; Goodell 2003).
The natural processes occurring during fungal wood degradation may be utilised for industrial purposes and have a great potential for cellulose-producing and wood-processing industries as well as for high value-added conversion of lignocellulosic waste materials in Biorefineries. Particularly, the molecular mechanisms of selective white-rot fungi offer a series of applications in the field of biotechnology of renewable resources (Bajpai 2012).
However, Basidiomycetes are not only microorganisms of industrial interest. They are also considered as pest organisms on construction wood, and it is of particular importance to detect wood decay by sensitive methods as early as possible. Although the mechanical properties of wood suffer quite early in the decay (Winandy and Morrell 1993), it is difficult to trace the chemical alterations related to the mechanical ones because they are too small to be detected with conventional and standardised wet-laboratory methods. Moreover, the information related to molecular structure i.e. the interactions between and within the macromolecules of the lignocellulose complex is lost when samples are analysed with degradative chemical methods, and separation or isolation methods may lead to non-representative results.
Molecular spectroscopy—ultra violet (UV), near infrared (NIR) and mid infrared (MIR) spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy provide less invasive or even non-destructive methods that have been proven well suited to investigate the chemical composition as well as the molecular structure of wood and functional groups of its constituents. It is therefore the aim of this review to present an overview on how these methods could contribute to understand the action of wood degrading fungi on their actual substrate wood and what kind of chemical and microstructural alterations take place during decay. In addition, UV and IR microspectroscopy provide the opportunity to localise these alterations within the cellular and chemically and structurally highly variable substrate wood. In this review, mainly original research publications were considered which focussed on spectroscopic analyses of fungal wood degradation without applying means of chemical degradation and separation.
Potentials of spectroscopic methods to analyse fungal wood degradation processes
The (molecular) spectroscopic methods used in the context of fungal wood degradation were NMR, NIR and MIR spectroscopy and Fourier transform (FT)-IR and UV microspectroscopy. Although Raman spectroscopy would perfectly fit into the scope of this review, we were not able to find peer-reviewed literature on that topic.
The methods and their potential applications for wood and wood processing have been reviewed elsewhere (Gil and Pascoal Neto 1999; Maunu 2002; Moore and Owen 2001; Tsuchikawa 2007; Tsuchikawa and Schwanninger 2011). Thus, this review focus on practical issues such as sample form and preparation (Table 1) and intends to provide insights into the information that can be potentially gained from the spectral data also with the aid of advanced data analysis tools.
Nuclear magnetic resonance spectroscopy
Solid state NMR (13C cross polarisation magic angle spinning nuclear magnetic resonance spectroscopy, 13C CPMAS NMR) is a technique to analyse solid (wood) samples. The contents of the main wood components (cellulose, hemicelluloses, lignin and extractives) and their molecular structure can be assessed, because chemical shifts in 13C CPMAS NMR spectra are related to the molecular environment of carbon atoms within the sample. Chemical shifts for functional groups of lignin and carbohydrates have been reviewed and summarised by Gil and Pascoal Neto (1999).
It was NMR spectroscopy that allowed Atalla and Vanderhart (1984) to discover two crystal allomorphs in native cellulose I—cellulose Iα and cellulose Iβ. Furthermore, through spectral fitting of the C4 cellulose region, the quantification of the various cellulose structural features within wood cell walls and fibres is possible (Larsson et al. 1997; Wickholm et al. 1998). Thus, cellulose crystallinity and the contents of cellulose crystal allomorphs as well as hemicelluloses content can be evaluated from 13C CPMAS NMR spectra.
Low field NMR (synonyms: time resolved NMR, wide-line solid state NMR) can be applied to measure the mobility of molecules within wood cell walls (Gilardi et al. 1994).
Lu and Ralph (2003) developed a preparation method for analysing wood with solution state NMR. After extensive disintegration by ball-milling, wood can be dissolved in various solvents such as dimethyl sulfoxide and readily be acetylated in this solution. Although structural information related to polysaccharides is lost during this treatment, the method allows for representative analysis of lignin structures using 1 H–13C heteronuclear single quantum correlation (HSQC).
Mid infrared spectroscopy (MIR or IR or FT-IR)
IR is now a fast and convenient spectroscopic method applicable for the analysis of solid wood samples requiring a minimum of sample preparation. During recent years, also various technical solutions for spatially resolved spectroscopy became available—microspectroscopy and IR imaging microscopy. With these methods, chemical and microstructural information on the cellular and to some extent also on the subcellular level can be gained. Absorption bands in the MIR region of the electromagnetic spectrum derive from the excitation of fundamental molecular vibrations of chemical bonds. Vibration frequencies depend on the strength of the chemical bonds and on the masses of the involved atoms. Comprehensive assignments of infrared bands for wood are found in the literature (Faix 1991; Liang and Marchessault 1959a, b; Schwanninger et al. 2004b).
Information related to the molecular structure of the wood polymers are mainly found in the spectral region of O–H vibrations (Maréchal and Chanzy 2000; Niduszynski and Marchessault 1972) but also in the so-called fingerprint region where bands can be related to cellulose crystallinity (Barsberg 2010; Kataoka and Kondo 1998; O'Connor et al. 1958; Richter 1991; Richter et al. 1991) and to cellulose I crystal allomorphs (Åkerholm et al. 2004).
From the practical point of view, highest signal to noise ratios can be acquired in transmission mode using the potassium bromide (KBr) technique, where 1–2 mg of milled wood are diluted and homogenised in about 200–300 mg of KBr and pressed to a dense pellet. Alternatively, very thin sections of wood of 10–30 μm thickness can be measured in transmission mode. The latter sample preparation is also appropriate for transmission FT-IR microspectroscopy or FT-IR mapping or imaging microspectroscopy (Fackler et al. 2010).
More convenient in terms of sample preparation is a method, where IR radiation (diffusely) reflected from the sample surface is detected (diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy). However, dilution of a milled sample with KBr is also advantageous in this technique (Faix and Böttcher 1992; Ferraz et al. 2000). Nowadays, however, spectra from wood surfaces and spectra of milled wood are mostly acquired applying the (attenuated total reflection (ATR)-FT-IR technique, where the sample is pressed onto a crystal of high refractive index and the so-called evanescent wave contains the spectral information of the sample surface. ATR-FT-IR spectra can be acquired within seconds.
FT-IR microspectroscopic spectra can be recorded either in transmission, reflection, or with the ATR-technique typically using a Germanium crystal. With the most sensitive detectors of advanced equipment, spectra of high signal to noise ratio can be readily obtained even with a spatial resolution at the diffraction limit. Current state of the art are pixel sizes as small as several square micrometre (Griffiths 2009). Array detectors such as focal plane array detectors or linear array detectors allow for recording up to 16,384 (i.e. 128 × 128) spectra simultaneously. In this way, chemical images of wood surfaces and thin sections can be obtained within several minutes.
Near infrared spectroscopy
Near infrared spectroscopy is a rapid method that has been most commonly used for applications related to process control also in wood and cellulose fields (Tsuchikawa 2007; Tsuchikawa and Schwanninger 2012). In addition, NIR spectra may yield a lot of information on the chemical composition and on the molecular structure of wood (Fackler and Schwanninger 2010; Schwanninger et al. 2011; Tsuchikawa and Siesler 2003; Watanabe et al. 2006). In the NIR, absorption bands related to overtones of and combinations of fundamental vibrations in the MIR are found. The overtones of C–H and O–H vibrations have been mostly used for descriptive data analyses. The vibration frequency of O–H overtone vibrations is highly dependent on the presence of hydrogen bonds. Therefore, information related to crystal structure—cellulose crystal allomorphs Iα, Iβ have different NIR spectra—and the extent of crystallinity is also present in NIR spectra (Fackler and Schwanninger 2010; Inagaki et al. 2010). In the C–H first overtone region, lignin-derived aromatic C-H groups can be differentiated from aliphatic ones of different sources (Schwanninger et al. 2011).
NIR spectra are mostly acquired from wood surfaces or milled wood in diffuse reflectance mode using fibre optic probes or integrating spheres. However, also transmission spectra of up to several 100-μm-thick thin sections with intact cellular structure can be recorded. Transmission NIR spectra recorded in transversal and longitudinal direction to spruce wood tracheids allow for conclusions on the molecular structure within the orthotropic material wood, because polarised NIR radiation causes selective excitation of oriented molecular vibrations. Therefore, degradation or modification of wood polymers oriented in transversal and longitudinal direction can be assessed with this polarised transmission FT-NIR spectroscopy (Fackler and Schwanninger, 2010). Although the information contained in NIR spectra resembles to a large extent that also found in the MIR, NIR spectroscopy possesses several advantages over MIR: due to low absorption coefficients, bulk or thick samples of intact cellular structure can be measured, O-H groups of no, weak, and strong H-bonding are better separated than in the MIR with the weakly H-bonded ones being more pronounced than the strongly H-bonded ones (Watanabe et al. 2006).
UV microspectroscopy
UV microspectroscopy has been employed to detect and quantify aromatic structures derived from lignin and aromatic extractives that have an absorption peak near 280 nm or structures with conjugated double bonds. Lignin can be localised within wood cell walls with the diffraction limit of spatial resolution of several 100 nm (~250 nm).
Ways to extract complex information from complex spectra
As a compound material of the biopolymers cellulose, hemicelluloses and lignin and the presence of low molecular mass organic compounds (extractives), wood is a highly complex material. This complexity is also reflected in the MIR and NIR as well as NMR spectra which are characterised by overlapping bands and signals. Comprehensive analysis of the spectral data, therefore, is a challenge.
Degradative methods intend to separate components or selectively assess them qualitatively and quantitatively. In contrast, data gained from spectroscopic methods contain information of the entire sample in one spectrum. Therefore, it is necessary to literally analyseFootnote 1 the data instead of the sample.
In spectroscopy, the absorption of electromagnetic radiation by chemical groups follows Lambert–Beer’s law, which can be applied in UV microspectroscopy e.g. to quantify lignin in a very thin wood section. Because extinction coefficients depend on lignin chemistry (Fergus and Goring 1970), only semi-quantitative analyses can be attempted for wood. For 13C CPMAS NMR, quantitativeness of signals is assumed in some cases rendering information on the composition accessible. Thus, knowledge on the content of one characteristic compound or functional group acquired with reference methods may allow for quantification of another group in the sample. In UV and IR, extinction coefficients are highly dependent on the nature of the chemical group, and in addition, other interactions of electromagnetic radiation with matter such as scattering, diffraction, specular and diffuse reflectance have to be considered for real-life solid wood samples. Furthermore, absorption bands are often highly overlapped. For all these reasons, it is difficult to assess the quantitative information directly, and reference methods are required. IR spectroscopy being an indirect or secondary method requires external calibration that can be performed via univariate regression analysis using band height or area, ratios of bands (Pandey and Pitman 2004, Fackler et al. 2007b) or multivariate regression using several bands (multilinear regression) or all data points of a spectral region (e.g. partial least squares regression) (Ferraz et al. 2000, Schwanninger et al. 2004b).
Nevertheless, IR spectroscopy is very sensitive to qualitative and quantitative differences within and between samples, and knowledge on the absolute amounts is not always important.
Other methods to visualise and accentuate the spectral information are deconvolution of overlapping bands, a method most frequently applied to 13C CPMAS NMR spectra (Popescu et al. 2010a), calculation of difference spectra and second derivatives, or two-dimensional correlation spectra, which was in the context of fungal wood degradation applied for NMR spectra (Yelle et al. 2008; Yelle et al. 2011) and for IR spectra (Popescu et al. 2010b).
Multivariate data analysis (MVA) offers great opportunities particularly in IR spectroscopy which as a very rapid method that allows the acquisition of high numbers of spectra within very short time. It is the aim of MVA to identify similarities and dissimilarities among spectra with e.g. principal component analysis (PCA) (Fackler and Schwanninger 2010; Fackler et al. 2007c; Jellison et al. 2002; Schwanninger et al. 2004a) or, as mentioned before, to relate the spectral data to reference data (calibration) for the prediction of the composition of new samples. MVA was also well applicable to analyse IR microscopy images (Fackler et al. 2010, 2011) and analysis of NIR spectra with PCA allowed for conclusions on brown-rot degradation that had not been a priori hypothesised (Fackler and Schwanninger 2010). MVA, however, has been rarely used for the analysis of NMR data in this context (Sivonen et al. 2003).
Observations of alterations due to fungal wood degradation processes
Changes of wood composition and its estimation
One aspect of fungal wood degradation is that the chemical composition of wood is altered while fungi are growing on it, because wood constituents are degraded at different rates. Observations concerning these changes due to brown-rot, white-rot and soft-rot degradation are the first ones reported for MIR of degraded wood (Takahashi and Nishimoto 1967). A summary of degradation of a large variety of different wood species by an even larger variety of fungal species are given in Table 2.
Provided that a higher number of degraded samples with a wider range of compositional differences were available, regression models to predict the chemical composition of degraded samples could be built. In particular, fast FT-IR methods can be used for this purpose. In mid infrared, univariate regression using ratios of bands has been often employed (Pandey and Pitman 2004). However, the relative intensity of reference bands may not be independent from alterations of the molecular structure (Barsberg 2010; Barsberg et al. 2011), and, thus, making use of the entire spectral information (multivariate calibration) is beneficial for regression models (Ferraz et al. 2000). Chemical properties such as lignin and polyose contents can be predicted from FT-MIR (Fackler et al. 2007b; Ferraz et al. 2000) and FT-NIR spectra (Fackler et al. 2007b; Schwanninger et al. 2004a) of fungal degraded samples as well as their physical properties: mass loss and density (Backa et al. 2001; Fackler et al. 2007b; Fackler et al. 2007c; Green et al. 2011; Kelley et al. 2002; Stirling et al. 2007; Sykacek et al. 2006) and the mechanical property compression strength (Green et al. 2011) could be analysed with NIR and MIR spectroscopy. Fackler et al. (2007a) connected mass loss and delignification, evaluated the selectivity of various strains of white-rot fungi on spruce and beech wood and found out that selectivity was more pronounced in the early phase when the mass loss was still low.
PCA of NIR spectra was useful to differentiate between the different decay types (Fackler et al. 2007c), different decay days (Schwanninger et al. 2004a), between brown-rot fungi of the order Boletales (Coniophora puteana, Serpula lacrymans) and Gloeophyllum trabeum (Gloeophyllales) and Postia placenta (Polyporales) (Jellison et al. 2002) and even between strains of the same species (Schwanninger et al. 2004a).
Changes of wood chemistry
A number of early MIR studies are available that indicate oxidative modification of lignin during white-rot of wood. Kirk and Chang (Kirk 1975; Kirk and Chang 1975) found that carboxyl groups were formed on spruce wood lignin by both the white-rot fungi Coriolus versicolor (Trametes versicolor) and Polyporus anceps and by the brown-rot fungus Lenzites trabea (G. trabeum) and that the bulk of these carboxyl groups was conjugated to aromatic rings. Faix et al. (1991) reported similar results from white-rot (T. versicolor, Pleurotus ostreatus and Lentinula edodes)-degraded beech wood. Increase of a C═O band was described as indicative for incipient brown-rot of pine wood (Körner et al. 1990).
Davis et al. (1994a, b, c) (Fig. 1) studied white-rot (P. chrysosporium, T. versicolor and Dichomitus squalens) and brown-rot (P. placenta) decay of a softwood (Colorado blue spruce, Picea pungens) and a hardwood (paper birch, Betula papyrifera) with 13C CPMAS NMR. Their results indicated a small preference for the degradation of amorphous polysaccharides compared to crystalline cellulose by white-rot fungi, which was even smaller on beech wood. They also reported a decrease of the syringyl (S) to guaiacyl (G) ratio of lignin as decay proceeded and suggested a direction of decay from the lumen towards the cell wall rather than the preference of the fungi for S structures over G structures, because in hardwoods, S structures are enriched in secondary cell wall layers, whereas the content of G structures is higher in middle lamellae and outer cell wall layers (Fengel and Wegener 1989). Major evidence for oxidative Cα-Cβ cleavage caused by white-rot fungi was found only in softwoods, but the authors did not rule out the possibility that oxidised structures were just metabolised more easily in hardwoods than in softwoods and therefore were not present in the degraded wood. The brown-rot fungus showed a clear preference for amorphous polysaccharides, with crystalline cellulose being accumulated as decay proceeded. This preference was smaller however on birch wood than on spruce wood. Softwood lignin lost methoxyl groups during brown-rot, and vanillic acid type structures were formed. Significant ring cleavage was not found. They also reported cleavage of β-O-4 linkages by the brown-rot fungus as the main reaction of the hardwood lignin structures. This mechanism of lignin cleavage of spruce wood by the brown-rot fungus G. trabeum was confirmed later with HSQC-solution state NMR spectroscopy (Yelle et al. 2008). Fackler and Schwanninger (2010) compared methyl ether and aryl ether cleavage by the polyporaceae G. trabeum and P. placenta by means of polarisation FT-NIR spectroscopy in samples of up to 16 % mass loss and concluded from the lack of relative orientation of the formed phenols that both routes to take place to similar extents. Yelle et al. (2008; 2011) related benzaldehydes, benzoic acids and arylglycerols to methoxyl in sound and brown-rot degraded spruce (Picea abies, 64 % mass loss) and aspen wood (Populus sp., 70 % mass loss). They also found that a strong preference for aryl ether cleavage in the softwood and only a slight preference on the hardwood at these advanced stages.

The 13C CPMAS NMR spectra show the preferential removal of amorphous carbohydrates (63 and 84 ppm) relative to crystalline cellulose (66 and 89 ppm) and accumulation of aromatic lignin structures (110–160 ppm) and a changed pattern of the respective region indicating lignin modification after brown-rot of Colorado blue spruce wood caused by P. placenta. Reproduced from Davis et al. (1994c) with permission from de Gruyter
In their imaging IR spectroscopy studies, Fackler et al. (2010, 2011) found a significant reduction of glycosidic linkages within polysaccharides in relation to carbohydrate ring structures in brown-rot (G. trabeum, P. placenta)- and in white-rot (T. versicolor)-degraded wood. These findings were highly indicative for incipient brown-rot and early white-rot and corroborated in a non-destructive way early findings by Cowling (1961) who found an increase of extractable polysaccharide degradation products in brown-rot wood.
Wood molecular structure
A preference for amorphous polysaccharides over crystalline cellulose is well known for many decay fungi. A closer look into the molecular structure of the degraded wood cell walls was provided by solid state NMR and by infrared methods.
Gilardi et al. (1994) studied white-rot (T. versicolor)- and brown-rot (C. puteana)-degraded pine and beech wood with wide-line solid state NMR. They found increased mobility of the molecular components of the cell wall in the degraded wood. Kim and Newman (1995) found an overall reduced surface to interior ratio of crystalline cellulose in brown-rot (G. trabeum)-degraded softwood wood (Pinus koraiensis) and concluded that crystallites of narrower dimensions—cellulose Iα crystallites located in middle lamellae and primary cell walls (Kataoka and Kondo 1998)—were degraded in preference to cellulose Iβ crystallites of secondary cell walls.
Popescu et al. (2010a) reported the apparent increase of crystallite size in lime (Tilia cordata) wood degraded by Trichoderma viride—a wood-inhabiting deuteromycete—and assigned this effect to higher mobility of less ordered polysaccharide chains after selective degradation of amorphous polysaccharides. The authors investigated the same degraded wood with 2D-correlation FT-MIR spectroscopy and found decreased carbohydrate content and decreased crystallinity. Selective degradation of the amorphous fraction (glucomannan and amorphous cellulose) was found for brown-rot (G. trabeum and P. placenta)-degraded spruce wood by means of polarised FT-NIR spectroscopy (Fackler and Schwanninger 2010). In a later study employing polarised FT-NIR in combination with deuterium exchange, the same authors (Fackler and Schwanninger 2011) found accessibility and hydrophilicity loss in dried brown-rot wood even before a substantial mass loss occurred (Fig. 2). This loss was most pronounced at amorphous polysaccharides in middle lamellae, primary cell walls and outer secondary cell walls and indicated structural changes within these cell wall regions during the incipient stage of degradation.

Second derivatives of FT-NIR transmission spectra of spruce wood in the OH first overtone region assigned to alcoholic and phenolic OH groups (6,928 cm-1) before (black line) and after (grey line) impregnation with deuterium oxide and differences (Δ NIR) between them (coloured line) demonstrate the accessibility of OH groups to deuterium oxide. The derivatives of the NIR spectra were vector normalised (VN) and show the lower number of OH groups due to polysaccharides degradation and a reduced accessibility of these groups in brown-rot wood with 16 % mass loss (a) compared to non-degraded spruce wood (b) Reproduced after Fackler and Schwanninger (2011) with permission from IM publications
Alterations of interactions of white-rot (Ceriporiopsis subvermispora) wood with water, namely increased hydrophilicity, was reported by Ferraz et al. (2004) and by Schwanninger et al. (2004a). Both groups employed NIR spectroscopy. The first group of authors reported that dried biodegraded wood absorbed water more rapidly than the non-decayed control; the second group reported more water adsorbed to cellulose even after thoroughly drying the milled samples and found significant differences between three strains.
Localisation of degradation
In the previous chapter, it was shown that some conclusions on the localisation of incipient and early wood degradation processes can be drawn without employing microscopy techniques, when the anisotropic nature of wood and the orientation and molecular structure of its macromolecular constituents in its cell wall layers are considered.
Microspectroscopic techniques, however, provide direct access to spatially resolved information, and IR imaging microscopy allowed for localisation of incipient and early decay processes in white-rot (T. versicolor and C. subvermispora)- and brown-rot (G. trabeum and P. placenta)-degraded spruce earlywood tracheids (Fackler et al. 2010, 2011) (Fig. 3). Incipient brown-rot processes could be localised in outer cell wall regions allocated to middle lamellae, primary cell walls and outer secondary cell walls, whereas white-rot processes were evenly distributed in the tracheid cells, even if the cells in one thin section were affected to very different extents. Indicative for incipient brown-rot processes was the reduction of glycosidic linkages when sugar moieties had not yet disappeared from wood. Cleavage of polysaccharides prior metabolisation of carbohydrates was to some minor extent also found during white-rot by T. versicolor and was not significant during selective white-rot by C. subvermispora.

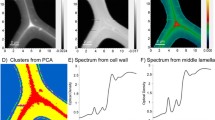
Localisation of early brown-rot degradation within a radial thin section of spruce wood with FT-IR imaging microscopy: a CCD camera image of 10 tracheids of a degraded spruce wood section from a sample degraded by the brown-rot fungus G. trabeum for 4 weeks. b FT-IR pseudo-colour spectral absorbance image. Zones of high total absorbance (red) show a high contribution of middle lamella (ML) and primary cell walls (P) and outer layers of secondary cell walls (S1). Those with low total absorbance (blue) can be assigned to regions with a high contribution of secondary cell walls (S2) and pits. c Example of a partial least squares discriminant analysis image gained through multivariate image analysis—degraded pixels are plotted yellow to light green and are found in ML/P/S1 regions of the section in 6.25 μm pixel resolution
Due to its much higher spatial resolution (~250 nm), UV microspectroscopy allows for investigating degradation processes on the subcellular level. Bauch et al. (1976) investigated changes of the lignin content of wood cell walls during white-rot, brown-rot and soft-rot (Table 2) and found a decrease of the lignin content in soft-rot-degraded secondary walls of hardwood fibres in vicinity to cavities.
Kleist and Schmitt (2001) studied brown-rot degradation of Sapelli wood and found that at high moisture content, C. puteana caused a soft-rot type of decay with rhomboidal cavities in secondary cell wall, but no differences in lignin content of adjacent cell wall regions could be evidenced by UV microscopy.
Modern UV imaging systems allow for a clear visualisation of the wood composition that is derived from the aromatic absorbance band due to lignin (Fig. 4). Very recently, Lehringer et al. (2011) reported two types of degradation patterns, when Physisporinus vitreus, commonly described as a selective white-rot fungus, degraded spruce wood: selective delignification leading to reduced lignin content of cell walls and a soft-rot type of decay characterised by hyphal tunnelling with bore holes surrounded by cell wall regions with reduced lignin content.

UV microscopic images of Norway spruce latewood after zero (a), 7 (b), 9 (c), 12 (d) and 32(e) weeks of incubation with the white-rot fungus P. vitreus. The colour pixels represent different UV280 absorbances. Hyphal tunnelling (1), notches (2) and cavities (3) can be observed as proof for simultaneous degradation. The progress of selective delignification is visible by lowered absorbances in areas of the secondary cell wall layers. Reproduced from Lehringer et al. (2011) with permission from de Gruyter
Conclusions and outlook
Nuclear magnetic resonance, IR and UV spectroscopy provide valuable and less invasive or even non-destructive tools to study wood chemistry, physics and molecular structure in various stages of softwood and hardwood degradation by white-rot, brown-rot and soft-rot fungi. The methods reviewed here allow for the analysis and characterisation of fungal degradation processes on their actual substrate wood and do not rely on model systems of isolated wood constituents or synthesised model compounds. Some methods have been proven to be sensitive enough to detect and characterise early or even incipient stages of decay.
The detection of compositional changes in degraded wood, however, is due to high variability of the raw material and due to uneven degradation processes often restricted to degradation stages with significant mass loss and changes of the visual appearance of the substrate. Thus, many studies reviewed here were carried out with strongly degraded wood.
Microspectroscopy methods allow for studying extreme situations in unevenly degraded samples and therefore are applicable for detecting decay processes very early. To assess the significance of differences caused by fungal decay and resolve them from natural variations found in non-decayed wood, sound data and image analysis techniques are required; otherwise, early decay may remain undetected. Of the microspectroscopy methods, UV microspectroscopy provides the highest spatial resolution, whereas the advantage of infrared microspectroscopy is the higher information content.
Both NMR and MIR spectroscopy confirmed molecular mechanisms involving one-electron oxidants and consecutive formation of α-carbonyls for many species of white-rot fungi on a number of different wood species of both softwoods and hardwoods. One-electron oxidation, putatively by free hydroxyl radicals, also plays a significant role in lignin degradation by brown-rot fungi, during which methoxyl and aryl ether structures are cleaved, and oxidised structures are formed.
Among the wood polysaccharides, amorphous structures are degraded in preference to crystalline ones, albeit this preference depends on the species of the fungus and on the wood species. The accumulation of polysaccharide degradation products of lower molecular mass during early brown-rot preceding their uptake and respiration by the fungi has been suggested or demonstrated with non-destructive means independently with wide-line NMR spectroscopy and FT-IR imaging microscopy.
Infrared imaging microscopy allowed for the localisation of incipient brown-rot degradation processes in outer cell wall regions (middle lamellae, primary or outer secondary walls). These findings corroborated earlier NMR studies carried out under consideration of the molecular structure of wood cell walls: brown-rot fungi were found to degrade smaller cellulose Iα crystallites of primary cell walls and middle lamellae in preference to larger cellulose Iβ crystallites of secondary cell walls, and with polarised FT-NIR spectroscopy, it was found that less ordered and transversely aligned polysaccharides of outer cell wall regions were modified in preference to longitudinally aligned ones of secondary cell walls. In contrast to brown-rot, white-rot has been shown to be distributed quite evenly in softwood tracheids when investigated with IR microscopy; with UV microscopy, with its higher spatial resolution, it was possible to detect different morphologies and different extents of delignification within the cell walls.
The most sensitive markers for the detection of incipient decay assessed by the spectroscopic methods reviewed here were the depolymerisation of wood polysaccharides (Fackler et al. 2010) reflected as the loss of the IR band of the glycosidic bond of polysaccharides, and loss of accessibility of distinct OH groups in dried degraded wood (Fackler and Schwanninger 2011). It may be speculated that also wide-line NMR would be applicable to detect much smaller differences than shown by Gilardi et al. (1994), because differences of mobility of cell wall components and water were highly significant between the investigated specimens in advanced degradation stages.
Future potential of the methods
Nuclear magnetic resonance, IR, and UV spectroscopy proved to be successful and highly significant to study wood properties related to intermediate and advanced decay. Although it has been shown that the characteristics of fungal wood degradation may change during decay (Cowling 1961; Fackler et al. 2007a), particularly the NMR methods, unfortunately, have been scarcely employed to study incipient and early degradation processes. IR spectroscopy on the contrary had been challenged to its limitations with statistic means because the short data acquisition times characteristic for IR spectroscopy are well suited for multivariate data analysis of high numbers of spectra. IR spectroscopy has also a great potential for in situ studies of decay processes.
UV microscopy offers the highest spatial resolution and has got a high potential for further investigations of incipient and early decay processes in distinct wood cell wall layers of various cell types.
For future studies, it will be advantageous to combine knowledge on the alterations of wood chemistry and structure during decay gained through spectroscopic and microspectroscopic methods with that on biochemical processes taking place while wood is degraded by the fungi. This approach calls for interdisciplinarity to be able to connect spectroscopy to ultrastructural and immunochemical analyses with e.g. electron microscopy, X-ray diffraction or atomic force microscopy as well as to biochemistry and molecular biology.
Within the Basidiomycota, species of wood decay fungi are found in many orders, and comparative genomic analyses of 31 fungal genomes revealed that brown-rot fungi evolved at least four times from white-rot ancestors and therefore may differ from each other (Floudas et al. 2012). Spectroscopic studies of degraded wood, however, by now, rarely accounted for biological systematics within the wood-degrading Agirocomycetes but rather focussed on the decay type. Often, aggressive strains and wood species recommended for standardised decay tests have been investigated (e.g. European standard EN 113); other studies were related to industrial applications of white-rot fungi. More systematic comparative spectroscopic studies on the same substrate considering fungal phylogenies would be desirable to be able to find differences particularly among the brown-rot fungi.
Apparent slow and asynchronous growth of hyphae in lignocellulosic substrates leads to uneven decay caused by brown-rot and by white-rot fungi even in adjacent wood cells (Fackler et al. 2010, 2011) and different decay morphology—selective and simultaneous white-rot caused by Heterobasidium annosum—in the same piece of wood was reported very early by Hartig (1878) and demonstrated with UV microscopy by Lehringer et al. (2011). Experimental tools to identify the metabolic state of a hypha therefore would be required. The combination of spatially resolved biochemical data with spatially resolved microspectroscopy data could bring this research field further step forward.
Notes
Analysis: greek “ανάλυση – analyse”, breaking up
References
Åkerholm M, Hinterstoisser B, Salmén L (2004) Characterization of the crystalline structure of cellulose using static and dynamic FT-IR spectroscopy. Carbohydr Res 339:569–578
Atalla RH, Vanderhart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223:283–285
Backa S, Brolin A, Nilsson T (2001) Characterization of fungal degraded birch wood by FT-IR and Py-GC. Holzforschung 55:225–232
Bajpai P (2012) Biotechnology for pulp and paper processing. Springer, New York
Barsberg S (2010) Prediction of vibrational spectra of polysaccharides—simulated IR spectrum of cellulose based on density functional theory (DFT). J Phys Chem B 114:11703–11708
Barsberg S, Sanadi AR, Jorgensen H (2011) A new density functional theory (DFT) based method for supporting the assignment of vibrational signatures of mannan and cellulose—analysis of palm kernel cake hydrolysis by ATR-FT-IR spectroscopy as a case study. Carbohydr Polym 85:457–464
Bauch J, Seehann G, Fitzner H (1976) Microspectrophotometrical investigations on lignin of decayed wood. Mat Organ Beih 3:141–152
Binder M, Hibbett DS (2002) Higher-level phylogenetic relationships of homobasidiomycetes (mushroom-forming fungi) inferred from four rDNA regions. Mol Phylogenet Evol 22:76–90
Boonstra MJ, Pizzi A, Rigolet S (2006) Correlation of 13C-NMR analysis with fungal decay tests of polymeric structural wood constituents. I. Basidiomycetes. J Appl Polym Sci 101(4):2639–2649
Cowling EB (1961) Comparative biochemistry of the decay of sweetgum sapwood by white-rot and brown-rot fungi. USDA Technical Bulletin No. 1258
Daniel G (2003) Microview of wood under degradation by bacteria and fungi. In: Goodell B, Nicholas DD, Schultz TP (eds) Wood deterioration and degradation. Advances in Our Changing World, vol 845. ACS Symposium Series, pp 34–72
Davis MF, Schroeder HR, Maciel GE (1994a) Solid-state 13C nuclear magnetic resonance studies of wood decay. I. White rot decay of Colorado Blue Spruce. Holzforschung 48:99–105
Davis MF, Schroeder HR, Maciel GE (1994b) Solid-state 13C nuclear magnetic resonance studies of wood decay. II. White rot decay of Paper Birch. Holzforschung 48:186–192
Davis MF, Schroeder HR, Maciel GE (1994c) Solid-state 13C nuclear magnetic resonance studies of wood decay. III. Decay of Colorado Blue Spruce and Paper Birch by Postia placenta. Holzforschung 48:301–307
Eriksson K-EL, Blanchette RA, Ander P (1990) Microbial and enzymatic degradation of wood and wood components. Springer Series in Wood Science, Springer, Berlin
Fackler K, Schwanninger M (2010) Polysaccharide degradation and lignin modification during brown-rot of spruce wood: a polarised Fourier transform near infrared study. J Near Infrared Spectrosc 18:403–416
Fackler K, Schwanninger M (2011) Accessibility of hydroxyl groups of brown-rot degraded spruce wood to heavy water. J Near Infrared Spectrosc 19:359–368
Fackler K, Gradinger C, Hinterstoisser B, Messner K, Schwanninger M (2006) Lignin degradation by white rot fungi on spruce wood shavings during short-time solid-state fermentations monitored by near infrared spectroscopy. Enzyme Microb Technol 39:1476–1483
Fackler K, Schmutzer M, Manoch L, Schwanninger M, Hinterstoisser B, Ters T, Messner K, Gradinger C (2007a) Evaluation of the selectivity of white rot isolates using near infrared spectroscopic techniques. Enzyme Microb Technol 41:881–887
Fackler K, Schwanninger M, Gradinger C, Hinterstoisser B, Messner K (2007b) Qualitative and quantitative changes of beech wood degraded by wood rotting Basidiomycetes monitored by Fourier transform infrared spectroscopic methods and multivariate data analysis. FEMS Microbiol Lett 271:162–169
Fackler K, Schwanninger M, Gradinger C, Srebotnik E, Hinterstoisser B, Messner K (2007c) Fungal decay of spruce and beech wood assessed by near infrared spectroscopy in combination with uni- and multivariate data analysis. Holzforschung 62:223–230
Fackler K, Stevanic JS, Ters T, Hinterstoisser B, Schwanninger M, Salmén L (2010) Localisation and characterisation of incipient brown-rot decay within spruce wood cell walls using FT-IR imaging microscopy. Enzyme Microb Technol 47:257–267
Fackler K, Stevanic JS, Ters T, Hinterstoisser B, Schwanninger M, Salmén L (2011) FT-IR imaging microscopy to localise and characterise simultaneous and selective white-rot decay within spruce wood cells. Holzforschung 65:411–420
Faix O (1991) Classification of lignins from different botanical origins by FTIR spectroscopy. Holzforschung 45(Supplement):21–27
Faix O, Böttcher JH (1992) The influence of particle size and concentration in transmission and diffuse reflectance spectroscopy of wood. Holz als Roh- und Werkst 50:221–226
Faix O, Bremer O, Schmidt O, Stevanovic T (1991) Monitoring of chemical changes in white-rot degraded beech wood by pyrolysis-gas chromatography and Fourier transform infrared spectroscopy. J Anal Appl Pyrolysis 21:147–162
Fengel D, Wegener G (1989) Wood—chemistry, ultrastructure, reactions. De Gruyter, Berlin
Fergus BJ, Goring DAI (1970) Location of guaiacyl and syringyl lignins in birch xylem tissue. Holzforschung 24(4):113–117. doi:10.1515/hfsg.1970.24.4.113
Ferraz A, Baeza J, Rodriguéz J, Freer J (2000) Estimating the chemical composition of biodegraded pine and eucalyptus wood by DRIFT spectroscopy and multivariate analysis. Biores Technol 74:201–212
Ferraz A, Parra C, Freer J, Baeza J, Rodriguéz J (2001) Characterization of white zones produced on Pinus radiata wood chips by Ganoderma australe and Ceriporiopsis subvermispora. World J Microbiol Biotechnol 16:641–645
Ferraz A, Mendonca R, Guerra A, Ruiz J, Rodriguéz J, Baeza J, Freer J (2004) Near-infrared spectra and chemical characteristics of Pinus taeda (Loblolly pine) wood chips biotreated by the white-rot fungus Ceriporiopsis subvermispora. J Wood Chem Technol 24(2):99–113
Floudas D, Binder M (2012) The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336:1715–1719
Gil AM, Pascoal Neto C (1999) Solid-state NMR studies of wood and other lignocellulosic material. Annu Rep NMR Spectrosc 37:75–117
Gilardi G, Abis L, Cass AEG (1994) Wide-line solid-state NMR of wood: proton relaxation time measurements on cell walls biodegraded by white-rot and brown-rot fungi. Enzyme Microb Technol 16(8):676–682
Gilardi G, Abis L, Cass AEG (1995) Carbon-13 CP/MAS solid state NMR and FT-IR spectroscopy of wood cell wall biodegradation. Enzyme Microb Technol 17:268–275
Goodell B (2003) Brown-rot fungal degradation of wood: our evolving view. In: Goodell B, Nicholas DD, Schultz TP (eds) Wood deterioration and degradation. Advances in Our Changing World, vol 845. ACS Symposium Series, pp 97–118
Green B, Jones PD, Nicholas DD, Schimleck LR, Shmulsky R (2011) Non-destructive assessment of Pinus spp wafers subjected to Gloeophyllum trabeum in soil block decay tests by diffuse reflectance near infrared spectroscopy. Wood Sci Technol 45:583–595
Griffiths PR (2009) Infrared and Raman instrumentation for mapping and imaging. In: Salzer R, Siesler HW (eds) Infrared and Raman Spectroscopic Imaging. Wiley-VCH, Weinheim, pp 3–64
Hartig R (1878) Die Zersetzungserscheinungen des Holzes der Nadelholzbäume und der Eiche in forstlicher botanischer und chemischer Richtung. Springer Verlag, Berlin
Hortling B, Forsskåhl I, Janson J, Sundquist J, Viikari L (1992) Investigations of fresh and biologically decayed birch. Holzforschung 46(1):9–19
Inagaki T, Siesler HW, Mitsui K, Tsuchikawa S (2010) Difference of the crystal structure of cellulose in wood after hydrothermal aging degradation: a NIR spectroscopy and XRD study. Biomacromolecules 11:2300–2305
Irbe I, Andersons B, Chirkova J, Kallavus U, Andersone I, Faix O (2006) On the changes of pinewood (Pinus sylvestris L.) Chemical composition and ultrastructure during the attack by brown-rot fungi Postia placenta and Coniophora puteana. Int Biodeterior Biodegrad 57(2):99–106
Irbe I, Andersone I, Andersons B, Noldt G, Dizhbite T, Kournosova N, Nuopponen M, Stewart D (2011) Characterisation of the initial degradation stage of scots pine (Pinus sylvestris L.) sapwood after attack by brown-rot fungus Coniophora puteana. Biodegradation 22:719–728
Jellison J, Kelley SS, Goodell B, Hui D, Ostrofsky A (2002) Differences in pH, electrical resistance, cation composition and NIR spectra of red spruce wood during early stages of brown rot degradation. The International Research Group on Wood Protection:IRG/WP 02-10449
Karklins VZ, Kreicberga SN, Ekabsone M (1975) Infrared spectroscopy of wood and its main components. VII. Use of IR-spectra to determine lignin in samples of birch wood destroyed by the fungus Fomitopsis pinicola. Khim Drevesny 2:53–57
Kataoka Y, Kondo T (1998) FT-IR microscopic analysis of changing cellulose crystalline structure during wood cell wall formation. Macromolecules 31:760–764
Kelley SS, Jellison J, Goodell B (2002) Use of NIR and pyrolysis-MBMS coupled with multivariate analysis for detecting the chemical changes associated with brown-rot biodegradation of spruce wood. FEMS Microbiol Lett 209:107–111
Kim YS, Newman RH (1995) Solid state 13C NMR study of wood degraded by the brown rot fungus Gloeophyllum trabeum. Holzforschung 49:109–114
Kirk TK (1975) Effects of a brown-rot fungus, Lenzites trabea, on lignin in spruce wood. Holzforschung 29:99–107
Kirk TK, Chang H (1975) Decomposition of lignin by white rot fungi. II Characterization of heavily degraded lignins from decayed spruce. Holzforschung 29(2):56–64
Kleist G, Schmitt U (2001) Characterisation of a soft rot-like decay pattern caused by Coniophora puteana (Schum.) Karst. in Sapelli wood (Entandrophragma cylindricum Sprague). Holzforschung 55(6):573–578
Koenig A, Sleighter RL, Salmon E, Hatcher PG (2010) NMR structural characterisation of Quercus alba (white oak) degraded by the brown rot fungus, Laetiporus sulphureus. J Wood Chem Technol 30:61–85
Körner S, Pecina H, Wienhaus O (1990) Investigations on the identification of the beginning brown-rot fungus infestation of wood by means of IR spectroscopy. Holz als Roh- und Werkst 48:413–416
Körner I, Faix O, Wienhaus O (1992) Attempt to determine brown-rot breakdown of Scots pine wood with the aid of FTIR spectroscopy. Holz als Roh- und Werkst 50:363–367
Larsson PT, Wickholm K, Iversen T (1997) (1997) A CP/MAS 13C NMR investigation of molecular ordering in celluloses. Carbohydr Res 302:19–25
Lehringer C, Koch G, Adusumalli R-B, Mook WM, Richter K, Militz H (2011) Effect of Physisporinus vitreus on wood properties of Norway spruce. Part 1: Aspects of delignification on surface hardness. Holzforschung 65:711–719
Levin L, Villalba L, Da Re V, Forchiassin F, Papinutti L (2007) Comparative studies of loblolly pine biodegradation and enzyme production by Argentinean white rot fungi focused on biopulping processes. Process Biochem 42:995–1002
Liang CY, Marchessault RH (1959a) Infrared spectra of crystalline polysaccharides. I. Hydrogen bonds in native celluloses. J Polym Sci 37:385–395
Liang CY, Marchessault RH (1959b) Infrared spectra of crystalline polysaccharides. II. Native celluloses in the region from 640 to 1,700 cm-1. J Polym Sci 39:269–278
Lu F, Ralph J (2003) Non-degradative dissolution and acetylation of ball-milled plant cell walls: high-resolution solution-state NMR. Plant J 35:535–544
Maréchal Y, Chanzy H (2000) The hydrogen bond network in Iβ cellulose as observed by infrared spectrometry. J Mol Struct 523:183–196
Martinez AT, González AE, Valmaseda M, Dale BE, Lambregts MJ, Haw JF (1991) Solid-state NMR studies of lignin and plant polysaccharide degradation by fungi. Holzforschung 45(Supplement):49–54
Maunu SL (2002) NMR studies of wood and wood products. Prog Nucl Magn Reson Spectrosc 40:151–174
Moore KM, Owen NL (2001) Infrared spectroscopic studies of solid wood. Appl Spectrosc Rev 36(1):65–86
Nicholas DD, Schultz TP (1987) Detection of incipient brown rot decay in wood by Fourier transform infrared spectrometry. The International Research Group on Wood Protection:IRG/WP/2275
Niduszynski I, Marchessault RH (1972) The crystalline structure of poly-b, D(1–4')mannose: Mannan I. Can J Chem 50:2130
O'Connor RT, DuPré EF, Mitcham D (1958) Applications of infrared absorption spectroscopy to investigations of cotton and modified cottons. Part I: physical and crystalline modifications and oxidation. Textile Res J 28:382–392
Okino EYA, Santana MAE, Resck IS, Alves MVS, Falcomer VAS, da Cunha JBM, dos Santos PHO (2008) Liquid chromatography and solid state CP/MAS 13C NMR techniques for chemical compound characterizations of cypress wood Cupressus glauca Lam. exposed to brown- and white-rot fungi. Carbohydr Polym 73(1):164–172
Pandey KK, Nagveni HC (2007) Rapid characterisation of brown rot and white rot degraded pine and rubberwood by FTIR spectroscopy. Holz als Roh- und Werkst 65:477–481
Pandey KK, Pitman AJ (2003) FTIR studies of the changes in wood chemistry following decay by brown-rot and white-rot fungi. Int Biodeterior Biodegrad 52:151–160
Pandey KK, Pitman AJ (2004) Examination of the lignin content in a softwood and a hardwood decayed by a brown-rot fungus with the acetyl bromide method and Fourier transform infrared spectroscopy. J Polym Sci A 42:2340–2346
Popescu C-M, Larsson PT, Tibirna C-M, Vasile C (2010a) Characterisation of fungal degraded lime wood by X-ray diffraction and cross-polarization magic-angle-spinning 13C-nuclear magnetic resonance spectroscopy. Appl Spectrosc 64(9):1054–1060
Popescu C-M, Popescu M-C, Vasile C (2010b) Structural changes in biodegraded lime wood. Carbohydr Polym 79:362–372
Richter U (1991) Strukturänderungen von Cellulose durch Alkalibehandlung: physikalische und chemische Beurteilungsmethoden. PhD, T. H. Darmstadt, Darmstadt
Richter U, Krause T, Schempp W (1991) Untersuchungen zur Alkalibehandlung von Cellulosefasern. Teil 1. Infrarotspektroskopische und röntgenographische Beurteilung der Änderung des Ordnungszustandes. Angew Makromol Chem 185(186):155–167
Salmén L, Burgert I (2009) Cell wall features with regard to mechanical performance. A review. Holzforschung 63:121–129
Schmutzer M, Schwanninger M, Fackler K, Messner K, Gradinger C (2008) Comparison of methods to evaluate the potential of fungal growth on decay of spruce after short-time treatment. Int Biodeterior Biodegrad 61:319–324
Schwanninger M, Hinterstoisser B, Gradinger C, Messner K, Fackler K (2004a) Examination of spruce wood biodegraded by Ceriporiopsis subvermispora using near and mid infrared spectroscopy. J Near Infrared Spectrosc 12:397–409
Schwanninger M, Rodrigues J, Pereira H, Hinterstoisser B (2004b) Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose. Vib Spectrosc 36:23–40
Schwanninger M, Rodrigues J, Fackler K (2011) A review of band assignments in near infrared spectra of wood and wood components. J Near Infrared Spectrosc 19:287–308
Sivonen H, Nuopponen M, Maunu SL, Sundholm F, Vuorinen T (2003) Carbon-thirteen cross-polarization magic angle spinning nuclear magnetic resonance and Fourier transform infrared studies of thermally modified wood exposed to brown and soft rot fungi. Appl Spectrosc 57:266–273
Stirling R, Trung T, Breuil C, Bicho P (2007) Predicting wood decay and density using NIR spectroscopy. Wood Fiber Sci 39:414–423
Sykacek E, Gierlinger N, Wimmer R, Schwanninger M (2006) Prediction of natural durability of commercial available European and Siberian larch by near-infrared spectroscopy. Holzforschung 60:643–647
Takahashi M, Nishimoto K (1967) Studies on the mechanism of wood decay (2). Changes in infrared spectra of Buna and Sugi wood as decay proceeds. Wood Res 42:1–12
Tsuchikawa S (2007) A review of recent near infrared research for wood and paper. Appl Spectrosc Rev 42:43–71
Tsuchikawa S, Schwanninger M (2012) A review of recent near infrared research for wood and paper (Part 2). Appl Spectrosc Rev (in print): doi:10.1080/05704928.05702011.05621079
Tsuchikawa S, Siesler HW (2003) Near-infrared spectroscopic monitoring of the diffusion process of deuterium-labeled molecules in wood. Part I: Softwood. Appl Spectrosc 57:667–674
Watanabe A, Morita S, Ozaki Y (2006) Temperature-dependent structural changes in hydrogen bonds in microcrystalline cellulose studied by infrared and near-infrared spectroscopy with perturbation-correlation moving-window two-dimensional correlation analysis. Appl Spectrosc 60:611–618
Wickholm K, Larsson PT, Iversen T (1998) Assignment of noncrystalline forms in cellulose I by CP/MAS 13C NMR spectroscopy. Carbohydr Res 312:123–129
Wienhaus O, Niemz P, Wagenfuehr A (1989) Characterization of microbiologically damaged wood. Part 2. Investigations to measure the decay of firwood by brown rot fungi using infrared spectroscopic techniques. Holztechnologie 30:151–153
Winandy JE, Morrell JJ (1993) Relationship between incipient decay, strength, and chemical composition of douglas-fir heartwood. Wood Fiber Sci 25:278–288
Yelle DJ, Ralph J, Li F, Hammel KE (2008) Evidence for cleavage of lignin by a brown rot basidiomycete. Environ Microbiol 10:1844–1849
Yelle DJ, Wei D, Ralph J, Hammel KE (2011) Multidimensional NMR analysis reveals truncated lignin structures in wood decayed by the brown rot basidiomycete Postia placenta. Environ Microbiol 13(4):1091–1100
Zabel RA, Morrell JJ (1992) Wood microbiology—decay and its prevention. Academic Press, San Diego
Acknowledgments
The Austrian Science Fund (Project Number V117-N17 granted to K.F.) is gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fackler, K., Schwanninger, M. How spectroscopy and microspectroscopy of degraded wood contribute to understand fungal wood decay. Appl Microbiol Biotechnol 96, 587–599 (2012). https://doi.org/10.1007/s00253-012-4369-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4369-5