
Abstract
We report here a considered novel study on the structural, FTIR spectra and optical properties of pure and co-doped Zn0.90-xFe0.1MxO with ((M = Cu, Ni and (x = 0.00, 0.10) and (0.00 < y < 0.20)) at different sintering temperatures Ts (Ts = 850 °C for series I and 1000 °C series II). Although the ZnO wurtzite structure is conformed for all samples, some secondary lines with little intensity are formed. But the number of these lines is higher for series I than for series II. The (c/a) value and U-parameter are almost constant for all samples, while Zn–O bond length L is slightly increased. The porosity and crystallite size are decreased by Fe, and also for (Fe + Cu) samples, and their values for series I are lower than for series II. The residual stress is tensile for most samples. Interestingly, the Young’s, rigid and bulk modulus, Poisson’s ratio and Debye temperature, obtained from FTIR analysis, are increased by Fe addition with a further increase for Fe + Ni) samples for both series. A ductile nature is obtained for pure, Fe and (Fe + Cu) samples; whereas a brittle nature is approved for (Fe + Ni) samples. On the other hand, the energy gap (Eg), residual lattice dielectric constant (εL) and carrier density N are increased by Fe addition, followed by a further increase for (Fe + Cu) samples, while the vice is versa for the inter-atomic distance R. For example, Eg was increased from 3.153 eV for pure ZnO to 3.974 eV for (Fe + Cu) samples (i.e., 0.821 eV more), while it was decreased to 2.851 eV for (Fe + Ni) samples (i.e., 0.302 eV less). A direct behavior is obtained between Eg and both elastic modulus (Y, β), lattice and micro strains (εL, εm), dislocation density (δ), residual stress (σ) and carrier density N, whereas a reverse behavior is obtained between Eg and both crystallite size (D), porosity (PS) and inter-atomic distance (R). These results are explained in terms of the generated blocked states of the conduction band as indicated by the Burstein Moss effect. These novel findings reveal that the co-doping has intense ZnO and moderate metal oxide modes in the ZnO matrix structure, which makes ZnO co-doped with (Fe + Cu) more suitable for gas sensors and optoelectronic devices. In contrast, ZnO co-doped with (Fe + Ni) samples is strongly recommended for altering plastic deformation. To our knowledge, the present investigation can be considered the first study and probably has never been discussed elsewhere, which highlights the present investigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
N-type ZnO has been early used as an additive in many artificial products such as gas sensors, optoelectronic and spintronics. The ZnO wurtzite structure is formed when the O−2 atoms arranged in a close packed type with Zn2+ occupying half the tetrahedral sites. The unit cell of ZnO (a = b ~ 0.325 nm and c ~ 0.522 nm) is generally formed with wide band gap of 3.2 eV and 60 meV exciton energy [1, 2]. The synthesis of ZnO at high temperature exhibits intrinsic defects due to oxygen vacancies which usually introduce donor states and consequently highlights its properties [3, 4]. Additionally, the properties of ZnO could be also controlled through replacement of divalent Zn2+ ions by 3-D transition metals through individual doping by one element or through co-doping simultaneously with two different elements [5,6,7,8,9].
As compared to individual doping, it is approved that the co-doping improve as possible the solubility of ZnO, and therefore the stability of the expected defects improve the activation rate and mobility of charge carriers as a result of lowering the ionization energy of acceptors and donors [10,11,12]. Furthermore, the transition from diluted magnetic insulator (DMI) to diluted magnetic semiconductor (DMS) is switched [13]. For example, the co-doping of Cu and Mn in place of Zn support the transformed ZnO from nanorods-like shape to nanoparticles-like shape [14]. It also gave change to the ZnO structural analysis according to the co-doping specifications relative to Zn such as doping content, type (magnetic or non-magnetic), ionic radius and valance state [15,16,17,18]. But, unfortunately, an appropriate formation of secondary phases such as ferrite at higher content of co-doping may be expected [13,14,15,16,17,18]. In addition, it has been reported that the density was decreased from 5.25 g/cm3 for ZnO with the increase of secondary phases, which disagrees with that obtained from XRD analysis [19].
The broadening of the diffraction peaks of ZnO obtained from XRD analysis have shown that ZnO is not completely perfect crystal due to its crystalline finite size. This deviation can be evaluated in terms of the values of c/a, U-parameter for tetrahedral distances, Zn–O bond length (L), crystallite size (Dhkl), micro-strain (є), lattice strain (ε) and residual stress (σ). However, crystallite size is a measure of the size of a coherently diffracting domain. Lattice strain is arising from crystal imperfections through lattice dislocation, while residual stress is a measure of the stress due to plastic deformation [20, 21]. So, the evaluation of the above structural parameters may be necessary for most of practical applications [22].
The vibration of lattice in solids has been considered early as the subject of many applications such as optical Kerr shutter (OKS), switching devices and detectors. The Fourier-transform infrared (FTIR) spectroscopy is generally based on interference between two coherent optical beams for recording the transmittance over a wide range of wave numbers likes middle infrared (400–4000 cm−1) [23,24,25,26]. The analysis of FTIR helps us for approximate relation between the spin vacancy and the carrier concentration. For example, the absorption bands of ZnO are due to metal oxides arising from inter-atomic vibrations, stretching and deformation of O–H assigned to the water adsorption on the metal surface and some of other bands due to Zn–O stretching and deformation vibrations [27, 28]. In addition, elastic and stiffness constants, elastic modulus and Debye temperature can be evaluated with the help of FTIR analysis.
Consequently, evaluation of ZnO optical constants is required because ZnO has significant importance in most of optoelectronic applications such as filters, modulators, gas sensor, solar cells, photodiode, switches, etc. [29,30,31,32]. Exciton energy Eexc for larger Urbach energy such as ZnO (~ 100 meV) is usually provided by studying the photon energy (hc/λmax) against maximal absorption. Exciton is a quasi-particle formed upon the electron optical excitation from VB into CB, and thereby created charge vacancy in the VB; the positively charged hole induces an attractive Coulomb interaction toward the electron in CB. Hence, they can form a bound pair state (e–h) of finite binding energy. However, exciton energy, band gap energy, charge carrier concentration, dielectric lattice constant and inter-atomic distance can be determined from UV absorption analysis [33]. But unfortunately, further research still needed in order to take more advantage especially in case of ZnO co-doped by two different elements.
As a comparative novel investigation, it is necessary to look more on the effects of co-doping, such as Ni and Cu as magnetic and non-magnetic elements, on the structural, FTIR spectra and optical properties of Zn1-x-yFexMyO (M = Cu, Ni) and (x = 0.00, 0.10) and (0.00 ≤ y ≤ 0.20). However, the present study approved that co-doping by the two different ions has dramatic changes in the properties of ZnO as compared to individual doping even for Cu as non-magnetic element. Furthermore, these changes are strongly controlled by the type and content of co-doping, and also on Ts. Interestingly, the solubility of co-doping through the ZnO lattice could be extended to 0.30, which is better than the reported elsewhere (0.15). Further, the residual stress is found to be tensile for most of the considered samples. Furthermore, Young’s, rigid and bulk modulus, Poisson’s ratio and Debye temperature can be increased by addition of co-doping up to 0.30 for (Fe + Ni) samples for both series. On the other hand, the exciton energy (Eex), energy gap (Eg), residual lattice dielectric constant (εL), and carrier density N are increased by Fe, followed by an increase/decrease for (Fe + Cu)/(Fe + Ni) samples, while the vice is versa for the inter-atomic distance R. For example, Eg was increased from 3.153 eV for ZnO to 3.974 eV for (Fe + Cu) samples (i.e., 0.821 eV more), while it was decreased to 2.851 eV for (Fe + Ni) samples (i.e. 0.302 eV less). These novel finding may be related to the oxygen vacancies which make ZnO co-doped with (Fe + Cu) samples highly suitable for gas sensors during the reactions between ZnO defects and atmospheric oxygen. In addition, increasing the N and εL for the same samples is required for optoelectronic applications. In contrast, improving the elastic constants for (Fe + Ni) samples up to 0.30 is highly recommended for altering plastic deformation, which highlights the present investigation. On the other hand, the room temperature ferromagnetic (RTFM) required for spintronic investigation is expected, but it will be done after COVID-19 is over. To our knowledge, the present work can be considered the first study and probably has never been discussed elsewhere.
2 Experimental details
Zn1-x-yFexMyO co-doped samples with (M = Cu, Ni) and (x = 0.00, 0.10 and 0.00 ≤ y ≤ 0.20) were synthesized by using conventional solid-state reaction method. The powders of ZnO, Fe2O3, CuO and NiO were thoroughly mixed into 8-samples and calcined in air for 16 h at 950 °C. The resulting powder of each sample was divided into two parts and separately pressed into 16 pellets, 8 pellets for each series as (x = 0.00, y = 0.00 for S1), (x = 0.10, y = 0.00for S2), (x = 0.10, y = 0.05, M = Cu for S3), (x = 0.10, y = 0.10, M = Cu for S4), (x = 0.10, y = 0.20, M = Cu for S5), (x = 0.10, y = 0.05, M = Ni for S6), (x = 0.10, y = 0.10, M = Ni for S7) and (x = 0.10, y = 0.20, M = Ni for S8). The two series of pellets (3 mm thick and about 1 cm in diameter) were separately sintered at 850 °C and 1000 °C for 16 h, and then slowly cooled to room temperature. Finally, the sintered pellets are individually grinding for XRD, FTIR and optical measurements. For simplicity, the samples called as (S1(850), (S2(850), (S3(850), (S4(850), (S5(850 °C) (S6(850), (S7(850), (S8(850)series I and (S1(1000) (S2(1000) (S3(1000) (S4(1000), (S5(1000)(S6(1000)(S7(1000), (S8(1000) for series II. The density of the samples ρexp was measured by using Archimedes principle in terms of their mass in air and acetone; \( \rho _{{\exp }} = \frac{{m_{a} }}{{m_{a} - m_{\ell } }} \) (\(\rho_{\ell }\) = 0.784 g/cm3). The phase purity of the samples was tested by XRD using Cu-Kα radiation of wave length 1.5418 Ẳ at 40 kV and 30 mA settings, and (20°–70°) diffraction angles with a step of 0.06°. FTIR absorption spectra of the samples were carried out using spectrum 400-FT-1R/FT-NIR spectrometer in the range of (400–4000 cm−1) with 4 cm−1 resolution and 2 (cm/s) scanning speed. The samples in powder form were homogenized in spectroscopic grade of KBr carrier in an agate mortar and pressed in to ~ 2 mm pellets. The optical properties were measured against wavelength (200–1000 nm) using a JascoV-570 (Japan) computer programmable double beam with UV–visible-NIR spectrophotometer at standard incidence with a scan speed of 1000 mm min−1. A supplementary attachment model ISN-470 is given in the case of reflectivity measurement.
3 Results and discussion
3.1 XRD and density analysis
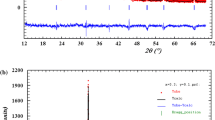
The XRD patterns of the samples for both series are shown in Fig. 1a, b. The diffraction peaks are well identified and it is seen that most of them belonging to the well-known hexagonal (wurtzite) structure of ZnO. The broadening and the sharp of the peaks can be considered as the evidence for good crystallizations of the samples. Interestingly, there are some of unknown lines with relatively lower intensities formed at angles 2θ = 25.06°, 39.74°, 42.16° and 45.34° for series I, and at 2θ = 42.28° and 45.04° for series II, as listed in Table 1. These lines are slightly shifted to higher 2θ angles, widths ∆θ and intensities as the content of co-doping increases. However, the number of these unknown peaks is higher for (Fe + Cu) samples as compared to (Fe + Ni) samples, while the vice is versa for the intensity. Furthermore, the number and intensity of the peaks are higher for series I than for series II, which indicates better phase purity as the Ts increases to 1000 °C. The average intensities Iavg of the most intense peaks (D100), (D002) and (D111) related to ZnO are listed in Table 1. It is noted that no exact trend for the behavior of Iavg of Fe doped samples because it increased for series I and decreased for series II. In contrast, Iavg for co-doped (Fe + Ni) samples are higher than (Fe + Cu) samples and they are decreased as Ts increases. To avoid solubility, the (0.05–0.10) of doping content has been taken as the limit for the 3D-transition metals in place of Zn within ZnO lattice [34, 35]. By looking on the present data, one can say that solubility limit could be extended for the co-doped ZnO up to 0.30.


a XRD patterns of the samples sintered at Ts = 850 °C (series I) b XRD patterns of the samples sintered at Ts = 1000 °C (series II)
To further confirm that co-doping is localized in the unit cell of ZnO, the behavior of lattice parameters a and c against (Fe + M) content for both series is shown in Fig. 2a–b. It is evident that both of them is increased by Fe addition for both series, followed by a decrease as M increases. The values of a, c and (c/a) are in the range of (3.215–3.246 Ǻ), (5.149–5.208 Ǻ) and (1.599–1.605 Ǻ), in consistent with those ideal values of ZnO (3.250, 5.206 and 1.602 Ǻ) [36, 37]. Anyhow, the average ionic radii of the co-doped ions for (Fe2+ + Cu2+) = (0.70 Ǻ + 0.73 Ǻ) = 0.0715 Ǻ and (Fe2+ + Ni2+) = (0.70 Ǻ + 0.70 Ǻ) = 0.70 Ǻ are relatively lower than that of the host Zn2+ (0.74 Ẳ) at the same tetrahedral fold-coordination. Therefore, the decrease of the c-parameter is logic for (Fe + Cu) and (Fe + Ni) samples, but it is not accurate for the Fe samples. However, it has been reported that this behavior may be due to slight shift of 2\(\theta \) angles by the doping that signifies either expansion or shrinkage of lattice constants. Therefore, the lower expansion induced by Fe followed by higher shrinkage by (Fe + M) may be expected as reported [38].

a a-parameter versus (Fe + M) content at different Ts for the samples. b c-parameter versus (Fe + M) content at different Ts for the samples
The values of U-parameter calculated by; U = listed in Table 2 is 0.380 for all samples as well as the reported for ZnO [39]. In fact, (c/a) < 1.633 and U > 0.375, and therefore the deviation than the ideal values are 2.1% for (c/a) and 1.33% for U-parameter. This behavior nearly specifies that the four tetrahedral distances stay almost constant through the distortion of the tetrahedral angles and thereby support the samples lattice stability [40].The Zn–O bond lengths (L) given by [41];\(L = \left[ {\left( {\frac{{a^{2} }}{3}} \right) + (0.5 - U)^{2} c^{2} } \right]^{\frac{1}{2}}\) are between (1.961–1.974 Å) for series I, (1.961–1.982 Å) for series II. In addition, their values are generally increased by Fe and it is higher for (Fe + Ni) samples than (Fe + Cu) samples which indicating an increase for the number of electrons contributed in the bond formation for (Fe + Cu) samples [42].
The theoretical density ρth calculated from XRD and given by; \(\rho_{{{\text{th}}}} = \frac{NW}{{N_{A} V}}\), where N = 2 for ZnO, NA is Avogadro's number, W is the atomic weight of the sample and V is the volume of unit cell. However, the values of ρth and ρexp listed in Table 2 are gradually increased by Fe, followed by an increase/decrease with more addition of (Cu/Ni) up to 0.20, and their values for series I are higher than series II. It is also noted that ρth of ZnO samples are (5.34/5.25) g/cm3 for the two series, which is smaller than the ρexp (5.76/5.70) g/cm3 and consistent with the reported elsewhere [43]. This has been explained in terms of imperfection and stoichiometric ratio of the real ZnO [44]. The porosity calculated by; PS = [1—(ρexp/ρth)], versus (Fe + M) and at different Ts is shown in Fig. 3a. It is clear that PS decreased by Fe and their values for series I are lower than series II. Interestingly, it is gradually increased for (Fe + Ni) samples, but it is decreased for (Fe + Cu) samples. Furthermore, it changes from positive to negative for M = Cu = 0.10 and 0.20, (S4 and S5) for both series. This behavior indicated that the co-doping by (Fe + Cu) and also sintering at lower Ts (850 °C) are responsible for decreasing the number of pores of ZnO, and the vice is versa for (Fe + Ni) sintered at 1000 °C [45].

a Dhkl versus (Fe + M) content at different Ts for the samples. b PS versus (Fe + M) content at different Ts for the samples
The crystallite size Dhkl is given by [46];
K = 0.91 for ZnO, λ = 1.5418 Å and β is the half-maximum line width. The Dhkl for the most intense peaks (D100), (D002) and (D111) versus (Fe + M) and at different Ts is shown in Fig. 3b. It is found that Dhkl values are decreased by Fe for both series and their values are generally higher for series II than series I. A further decrease/increase is obtained for (Fe + Cu)/(Fe + Ni) samples, which is different than expected because the atomic radius of Cu is greater than that of Ni. Maybe, some other reasons are also related to such behavior, such as decreasing the porosity and c-parameter of Cu samples as compared to Ni samples. However, the vice is versa for the behavior of dislocation density δ calculated by (1/Dhkl2) and shown in Fig. 4a. The micro-lattice strain єm calculated by; \(\in_{m} = \frac{\beta \cos \theta }{4}\) and listed in Table 2 is increased by Fe followed by an increase/decrease for (Fe + Cu)/(Fe + Ni) samples, and it is generally higher for series I than series II. This behavior indicated that Ni helps Fe samples for both series to be good qualities of crystalline and free from the lattice defects as compared to Cu [47,48,49].

a δ versus (Fe + M) content at different Ts for the samples. b n versus (Fe + M) content at different Ts for the samples
The number of unit cells existent in each particle of ZnO is given by [50, 51]:
where Vun and Vuc are the volumes of ZnO crystallite-like shape and hexagonal unit cell-like shape, respectively. The behavior of n against (Fe + M) at different Ts shown in Fig. 4b is typically similar to the behavior of Dhkl.
3.2 FTIR analysis
Figures 5 and 6 show FTIR spectra against wave number ύ for all samples. The values of ύ against absorption peaks are listed in Table 3 for all samples. Firstly, there is a clear peak recorded at 3642.42, 3642.72 and 3642.45 cm−1 for S1, S6 and S8 of series I, shifted to 3647.52 cm−1 for S1 of series II and completely disappear for the other samples. Two successive peaks at 3421.36 and 3451.52 cm−1 are obtained for the samples of series I, and at 3443.59 and 34,422.76 cm−1 for (Fe + Ni) samples of series II. In contrast, they disappeared for pure, Fe and (Fe + Cu) samples of series II. However, these peaks are due to O–H stretching vibrations as reported [52, 53]. The functional peak (1630 cm−1) is also recorded, but at about 1637.44 cm−1 and 1640.61 cm−1 for S3 and S4 of series I, and disappeared for the other samples. This peak is usually attributed to H–O–H bonding vibrations and C=C stretching [54, 55]. This behavior indicates that addition of Cu beside Fe helps for the evaluated bending and stretching vibrations especially at lower Ts (850 °C). Some of other peaks are obtained at 1426.86 cm−1, 1426.51 cm−1 and 1425.62 cm−1 for S1, S2 and S3 of series I. It is slightly shifted to 1429.57 cm−1and 1436 cm−1 for S4 and S5 (Fe + Cu) samples, and strongly shifted to higher values for (Fe + Ni) samples (1472.55 cm−1, 1469 cm−1 and 1479.24 cm−1 for S6, S7 and S8). In contrast, it is completely absent for all samples of series II, which may due to increasing Ts up to 1000 °C. There are also some peaks between 678.96 and 1277.91 cm−1 for all samples, but no exact trend for the behavior of ν against the variable parameters. However, the presence of weak bands in the range of (650–1500 cm−1) is an evidence for incorporation of Fe3+ ions into ZnO matrix upon Fe-doping. The shift of ν is due to addition of either Cu or Ni beside Fe. These peaks are correspond to the C=C, C=O, C–N and O–H stretching and C–H bending vibration [56, 57].

a, b FTIR spectra of the samples sintered at Ts = 850 °C (series I)

a, b FTIR spectra of the samples sintered at Ts = 1000 °C (series II)
Among of absorption peaks observed between (404–518 cm−1) which corresponds to the active modes of ZnO related to hexagonal structure [58, 59]. The number of peaks is higher for (Fe + Cu) samples as compared to (Fe + Ni) samples for both series, see S8 for each series. This means that ZnO structure is almost the same for (Fe + Cu) samples, but it is slightly deviates than its ideal case for (Fe + Ni) samples, which is consistent with XRD analysis. Thus, the present data revealed that the co-doping has intense ZnO and moderate metal oxide modes in the ZnO matrix structure [60, 61].
Elastic properties can be correlated with thermodynamic properties of solids such as ZnO ceramic samples through Debye temperature θD calculated using the relation [33];
where c is the velocity of light, \(\overline{\Delta v}\) is the average value of the waven umbers for the most identified absorption bands related to ZnO which are between (421.54–485.54 cm−1). Figure 7 shows the behavior of θD against (Fe + M) content, and in which θD are between (611.90–698.21 K) for all samples. However, θD is generally increased by Fe for both series, and sudden increase up to maximum values, followed by a decrease is observed for (Fe + Cu) samples. In contrast, a sudden decrease followed by an increase is observed for (Fe + Ni) samples. On the other hand, no trend or exact sequence could be recorded for θD against Ts. However, this behavior is attributed to the shift in the wave number of FTIR bands to higher/lower values against (Fe + M) content as listed in Table 3.

θD versus (Fe + M) content at different Ts for the samples
To describe the strength of interaction between tetrahedral cations and the surrounding four oxygen (O−2) ions of ZnO, the force constant Kt can be calculated using; Kt = 0.076 W\(\left( {\overline{\Delta v} } \right)^{2}\) [62, 63]. Also, the stiffness constants S11 and S12 are calculated in terms of c- parameter and Poisson's ratio γ using; S11 = (Kt/c), S12 = (S11γ/1 − γ) and γ = 0.324 (1–1.043PS) [64]. The value of γ gives the degree directionality of the bond presents in the samples. If (γ > 0.26), it exhibits brittle nature, whereas it exhibits ductile nature if (γ < 0.26) [65]. It is evident from Table 3 that (γ > 0.26) for pure, Fe and (Fe + Cu) samples (ductile nature). In contrast (γ < 0.26) for (Fe + Ni) samples (brittle nature). Furthermore, the values of γ for series I are higher than series II. The general increase of S11 listed in Table 3 for all doped samples may be attributed to the formation of co-doped clusters, which usually makes S11 ≥ 2S12. However, S12values are increased by Fe, followed by increase/decrease for (Fe + Cu)/(Fe + Ni) samples. Although no exact trend for S11 and S12 against Ts, their behaviors is related to the change of porosity.
For more illustration on the ZnO structure defects, the residual stress \(\sigma \) is calculated by [66, 67];
where \({S}_{13}\)= 104.2 GPa, \({S}_{33}\)= 213.8 GPa, \({S}_{11}\)= 208.8 GPa, \({S}_{12}\)= 119.7 GPa are the elastic stiffness constants of hexagonal ZnO and εL is the c-axis strain given by; \(\varepsilon_{L} = \frac{{c - c_{ \circ } }}{{c_{ \circ } }}\) and listed in Table 2 (co = 5.2066 Ẳ for the unstrained ZnO) [68]. However, similar values of \(\sigma \) listed in Table 2 indicated that σ is positive (tensile-like behavior) for series I, and for the samples of (Fe + M) = 0.20 and 0.30 of series II. In contrast, it is negative (compressive-like behavior) for pure, Fe and (Fe + Ni) = 0.15 samples of series II. This result indicated that the increase of Ts from 850 to 1000 °C is able to eliminate the tensile stress and turn it to compressive stress. This is may be related to the number of pores and the lattice defects discussed above [69]. The observed deviation in lattice spacing induces lattice stress and strain in ZnO. In case of co-doping, a mutual interaction between the different atoms and their segregation in the grain boundaries may be contribute to longitudinal stress and strain as reported [70].
Young Y and bulk modulus β are determined by [71];
Figure 8 shows the behavior of Y against (Fe + M), whereas the values of β are listed in Table 4. Anyhow, both of them are increased by Fe, followed by a decrease/increase for (Fe + Cu)/(Fe + Ni) samples. Further, their values for pure, Fe and (Fe + 0.05 M) samples are higher for series II than series I, but above that no exact trend against Ts is obtained. The increase/decrease may be related to the shrinkage/expand of inter-atomic bond length L for the samples listed in Table 2. This is subsequently controls the bond strength as well as the lattice energy [72, 73].

Young’s Modulus versus (Fe + M) content at different Ts for the samples
3.3 Optical measurements
Due to covid-19, Fig. 9a shows optical reflectance (R) against wave lengths λ for only the samples of series II (Ts = 1000 °C). R is gradually decreased as λ increases and there is a hump close to (~ 400 nm) for all samples, which is attributed to a significant decrease of R as λ decreases. The vice is versa for the behavior of A shown in Fig. 9b. This behavior is due to increasing free carrier density as λ shifted toward near-infrared ~ 400 nm (exciton peaks) [74]. The fluctuation of A and R close to shorter wave lengths may be due to either the motion of electron–hole pairs or the possible interference between reflected lines occurs at high photon energy. Interestingly the (R/A) are increased/decreased by Fe addition to ZnO, and a significant further increase/decrease is also obtained for (Fe + Cu) samples, while the vice is versa for (Fe + Ni) samples. This behavior may be related to the oxygen vacancies which making ZnO highly suitable for the use of oxygen sensor during the reactions between ZnO defects and atmospheric oxygen [75, 76]. However, the relative changes of R and A for (Fe + Cu) samples as compared to (Fe + Ni) samples may be related to either change of ZnO particle size or band gap or both of them. As discussed above, the crystallite size is decreased for (Fe + Cu) samples, whereas it is increased for (Fe + Ni) samples, whereas the energy gap will be presented in the next paragraph.

a Reflectance versus wave length for the samples. b Absorbance versus wave length for the samples. c Exciton energy versus (Fe + M) content for the samples
However, the wave lengths against Amax are 322, 316, 264, 258, 280, 324, 334 and 340 nm for the samples (S1–S8), respectively. The values of Amax are used for obtaining the exciton energy Eexc against (Fe + M) content as shown as in Fig. 4c. It is evident that Eexc is slightly increased by Fe followed by significant increase for (Fe + Cu) samples, whereas it is gradually deceased for (Fe + Ni) samples. They are increased from 3.855 eV for S1 to 4.812 eV for S5 (0.957 eV more), but they are decreased to 3.651 eV for S8 (0.204 eV less). This means that the required energy for creating electron–hole pair is increased/decreased by Cu/Ni co-doped with Fe to ZnO and consequently less/more pairs could be formed.
The absorption coefficient α is determined using the formula, α = 2.303 ln (A/t), where t is taken by 0.85 cm, and it is defined by the distance traveled by the photons through the samples solutions. After that the optical band gap Eg is calculated using the following Taucs equation [31, 75];
A is constant, ν is the frequency of photons, h is the constant of Planck and m is a parameter that characterizes electronic transition (m = 1/2) for direct transition permitted by ZnO. However, Eg is estimated from the linear portion of the plot between (αhυ)2 and hυ to α = 0, shown in Fig. 10a, b [76, 77]. The behavior of Eg against (Fe + M) content is shown in Fig. 10c, and the related values are also listed in Table 5. The values of Eg are 3.153, 3.403, 3.751, 3.902, 3.974, 3.133, 2.952 and 2.851 eV for all samples of series II, respectively. It is clear Eg was increased from 3.153 eV for S1 to 3.974 eV for S5 of (Fe + Cu) samples (0.821 eV more), while it decreased to 2.851 eV for S8 (Fe + Ni) samples (0.302 eV less).

a (αhν)2 versus photon energy for pure (Fe + Cu) samples. b (αhν)2 versus photon energy for pure (Fe + Ni) samples. c Eg versus (Fe + M) content for the samples
The high values of Eg for the (Fe + Cu) samples are probably related to the electronic transition from the bottom of VB to the top of CB. This is consistent with the higher absorption values (homo–lumo), which could take place at higher photon energy. However, the increase in the Eg for the doped samples could be explained in terms of the Burstein Moss effect [78, 79]. Based on this effect, the lowest energy necessary for an electron to travel from the VB to the CB is determined by the following equation [80, 81];
where Eg is the optical absorption edge energy, Eb is the energy difference between the valence and conduction band edges, and ∆EBM is the Eg widening caused by the CB lowest blocked states. In our case, the doping of ZnO with Fe or (Fe + Cu) samples may produce high carrier concentration N as obtained in the next paragraph. As a result, more blocked states of CB will causes an increase in the ∆EBM which in turn causes an increase in the Eg values as obtained. In contrast, we believed that the decrease in Eg for (Fe + Ni) samples may be due to the formation of some defects in the host materials, thereby creating some of energy states in the forbidden gap inside the band structure, which is consistent with the reported elsewhere for ZnO [80, 81]. On the other hand, it has been also reported that the crystalline size and oxygen vacancies defects have a significant effects on the Eg of ZnO. Anyhow, ZnO is usually characterized by the structure disorder and usually contains some of defects which produce some of localized blocked states [82, 83]. So, we believe that these defects are higher for (Fe + Cu) samples, and therefore Eg was increased.
The real part of dielectric constant (εʹ) is determined by using the equation [84, 85];
where N is concentration of the free carriers, m* is effective mass and Єo is permittivity of free space. €L is the residual lattice dielectric constant which represents the high-frequency component of the relative permittivity at (λ = 0) and n is refractive index calculated from reflectance R as follows [86];
The behavior of (n2 − k2) against λ2 shown in Fig. 11 for the samples is almost linear with positive slope for each sample. However, the values of (N/m*) and ЄL are then calculated from the slope of linear parts and extrapolation of the plot to λ2 = 0, respectively. The values of (N/m*) and ЄL listed in Table 5 are generally increased by Fe, followed by an increase/decrease for (Fe + Cu)/(Fe + Ni) samples up to 0.30. They are increased from 15.72 and 3.69 \(\times\) 1055 (g cm−3) for S2 to 23.96 and 221.4 \(\times\) 1055 (g cm−3) for S5, whereas they are decreased to 1.96 and 2.46 \(\times\) 1055 (g cm−3) for S8. By looking on the present data, one can see that both of Eg, (N/m*), and ЄL are gradually changed in the same manner according to co-doping contents. However, the increase/decrease of ЄL and (N/m*) can be achieved by the change of oxygen deficient in ZnO. This is an indication of the significant changes in the band structure for the co-doped samples as obtained for Eg behavior (Fig. 12). In other words, the electronic polarization of ZnO may be changed by such substitution. For more clarification, free carrier concentration N is calculated using m* = 3.82 × 10 −25 g for ZnO [87], and therefore the inter-atomic distance (R) can be also calculated; R = (0.86/N(1/3)) [86, 88]. From the values of N and R listed in Table 5, it is clear that N has a similar behavior of (N/m*), while the vice is versa for inter-atomic distance R. Based on the above data, we could obtain a direct behavior between the Eg and both of elastic modulus (Y, β), lattice and micro strains (εL, εm), dislocation density (δ), residual stress (σ) and carrier density N, whereas a reverse behavior is obtained between Eg and both of crystallite size (D), porosity (PS), number of unit cell exist in ZnO particle (n) and inter-atomic distance (R).

The relation between (n2 − k2) and λ2 for the samples

a Dielectric loss versus (Fe + M) for the samples. b Carrier concentration (N) versus (Fe + M) content for the samples. c Inter-atomic distance (R) versus (Fe + M) content for the samples
Based on the above results, it is approved that co-doping of (Fe + M) up to 0.30 is well dissolved into the crystalline lattice of ZnO. During sintering at high temperature, the clusters of (Fe + Cu) samples may behave deep donors, and therefore the density of the intrinsic donors is reduced [89, 90]. Furthermore, the higher valence state of the co-doping (Fe3+/Cu2+/3) compared to divalent Zn2+ leads to more deep acceptor levels which trap the electrons from the conduction band and then widening the energy gap as obtained. In addition, Cu2+/3+ to Fe3+ may be helps for more oxygen vacancies, and therefore, the charge of carrier density and also the ability of electron migration are enhanced [91,92,93,94]. Further, the strong participation between Cu and Fe clusters may be makes this process more effective and also increase the width of localized states formed below the conduction band, and then more carriers are generated. Consequently, the ЄL and N are increased, while R is decreased. Furthermore, Cu is nearly non-magnetic, and addition of Cu beside Fe will decreases the localization of the carriers as compared to individual doping by only Fe or Cu. This of course strongly recommended (Fe + Cu) samples for gas sensor optoelectronic applications. In contrast, the (Fe + Ni) samples should have a similar behavior of Fe samples due to divalent of Ni2+ ion as well as Zn2+, but unfortunately this could not be obtained. Therefore, we believed that the participation of clusters for (Fe + Ni) samples probably work as extrinsic donors, but with less deep acceptor levels which trap less of electrons from the CB and consequently narrowing the energy band gap as obtained. Furthermore, Fe (3.36 µB) and Ni (3.2 µB) are magnetic ions, and therefore, addition of Ni beside Fe localized the charge carriers below the CB, and then both of Eg, ЄL and N are decreased as reported elsewhere [95,96,97,98].
4 Conclusion
The structural, FTIR spectra and optical constants of pure and co-doped Zn0.90-xFe0.1MxO are well investigated. The ZnO wurtzite structure is confirmed for all samples and the solubility limit of the co-doped samples could be extended to 0.30, but at higher Ts. Further, a decrease in both porosity and crystallite size was obtained for (Fe + Cu) samples. Residual stress is a tensile-like behavior for most considered samples. Among the absorption peaks are attributed to the ZnO hexagonal structure and the addition of Cu beside Fe helps with the evaluated bending and stretching vibrations, especially at lower Ts. A ductile nature is approved for pure, Fe and (Fe + Cu) samples, whereas a bright nature is obtained for (Fe + Ni) samples. Interestingly, the Young’s rigid and bulk modulus, Poisson’s ratio and Debye temperature can be increased by up to 0.30, for (Fe + Ni) samples. On the other hand, energy gap (Eg), residual lattice constant (ЄL) and carrier density N could be increased for (Fe + Cu) samples, while the vice is versa for inter-atomic distance R. These results are explained in terms of the generated blocked states of the conduction band as indicated by the Burstein Moss effect. This novel study perhaps recommended (Fe + Ni) samples for altering plastic deformation, whereas (Fe + Cu) samples are recommended for gas sensors and optoelectronic devices. Regardless of the type of sample, a direct behavior could be reported between Eg and both elastic modulus (Y, β), lattice and micro strains (εL, εm), dislocation density (δ), residual stress (σ) and carrier density N, whereas a reverse behavior is obtained between Eg and both crystallite size (D), porosity (PS) and inter-atomic distance (R). Moreover, the room temperature ferromagnetic (RTFM) required for spintronic investigation is expected for (Fe + Ni) samples, but it will be done later on after COVID-19 is over. To our knowledge, the present work can be considered the first study and has never been discussed elsewhere, which highlights the present investigation.
References
M. Matsouka, Jpn. J. Appl. Phys. 10(6), 736 (1971)
K. Mukae, K. Tsuda, I. Nagasawa, Jpn. J. Appl. Phys. 16(8), 1361 (1977)
D.R. Clarke, J. Am. Ceram. Soc. 82(3), 485 (1999)
A. Sedky, M. Abu-Abdeen, A.A. Almulhem, Phys. B 388, 266 (2007)
H. Morkoç, Ü. Özgür, Zinc Oxide: Fundamentals, Materials and Device Technology, 1st edn. (Wiley-Vch, Weinheim, Germany, 2009)
M. Mustaqima, C. Liu, ZnO-based nanostructures for diluted magnetic semiconductor. Turk. J. Phys. 38, 429–441 (2014)
I. Djerdj, Z. Jaglicic, D. Arcon, M. Niederberger, Co-doped ZnO nanoparticles: minireview. Nanoscale 2, 1096–1104 (2006)
Z. Yang, A perspective of recent progress in ZnO diluted magnetic semiconductors. Appl. Phys. A 112, 241–254 (2013)
C. Liu, F. Yun, H. Morkoç, Ferromagnetism of ZnO and GaN: a review. J. Mater. Sci. Mater. Electron. 16, 555–597 (2005)
J. Zhang, K. Tse, M. Wong, Y. Zhang, J. Zhu, A brief review of co-doping. Front. Phys. 11, 117405 (2016)
H. Katayama-Yoshida, T. Nishimatsu, T. Yamamoto, N. Orita, Codoping method for the fabrication of low-resistivity wide band-gap semiconductors in p-type GaN, p-type AlN and n-type diamond: Prediction versus experiment. J. Phys. Condens. Matter 13, 8901–8914 (2001)
J. Wojnarowicz, M. Omelchenko, J. Szczytko, T. Chudoba, S. Gierlotka, A. Majhofer, A. Twardowski, W. Lojkowski, Curr. Comput.-Aided Drug Des. 8(11), 410 (2018). https://doi.org/10.3390/cryst8110410
D. Karmakar, C. Rao, J.V. Yakhmi, A. Yaresko, V.N. Antonov, R.M. Kadam, S.K. Mandal, R. Adhikari, A.K. Das, T.K. Nath, N. Ganguli, I. Dasgupta, G.P. Das, Phys. Rev. B 81, 184421 (2010)
H.B. Wang, H. Wang, C. Zhang, F.J. Yang, J.X. Duan, C.P. Yang, H.S. Gu, M.J. Zhou, Q. Li, Y. Jiang, J. Nanosci. Nanotechnol. 9(5), 3308–3312 (2009)
J. Wojnarowicz, S. Kusnieruk, T. Chudoba, S. Gierlotka, W. Lojkowski, W. Knoff, M.I. Lukasiewicz, B.S. Witkowski, A. Wolska, M.T. Klepka et al., Paramagnetism of cobalt-doped ZnO nanoparticles obtained by microwave solvothermal synthesis. Beilstein J. Nanotechnol. 6, 1957–1969 (2015)
J. Wojnarowicz, R. Mukhovskyi, E. Pietrzykowska, S. Kusnieruk, J. Mizeracki, W. Lojkowski, Microwave solvothermal synthesis and characterization of manganese-doped ZnO nanoparticles. Beilstein J. Nanotechnol. 7, 721–732 (2016)
D. Neena, A.H. Shah, K. Deshmukh, H. Ahmad, D.J. Fu, K.K. Kondamareddy, P. Kumar, R.K. Dwivedi, V. Sing, Influence of (Co-Mn) co-doping on the microstructures, optical properties of sol-gel derived ZnO nanoparticles. Eur. Phys. J. D 70, 53 (2016)
S.S. Abdullahi, Y. Köseog, S. Güner, S. Kazan, B. Kocaman, C.E. Ndikilar, Synthesis and characterization of Mn and Co codopedZnO nanoparticles. SuperlatticesMicrostruct. 83, 342–352 (2015)
M.W. Barsoum, Fundamentals of Ceramics (Taylor & Francis Group, New York, NY, USA, 2002)
P. Bindu, S. Thomas, J. Theor. Appl. Phys. 8, 123 (2014). https://doi.org/10.1007/s40094-014-0141-9
M.S. Niasari, F. Davar, M. Mazaheri, Preparation of ZnO nanoparticles from [bis (acetylacetonato) zinc (II)]–oleylamine complex by thermal decomposition. Maters. Lett. 62, 1890–1892 (2008)
T. Ungar, Characterization of nanocrystalline materials by x-ray line profile analysis. J. Mater. Sci. 42, 1584–1593 (2007)
F.R. Sale, F. Mahloojchi, Ceram. Int. 14, 229 (1988)
R. Yanru, L. Hanpeng, L. Mingzhu, T. Qingyun, S. Lihua, L. Zhenjin, M. Xianren, Physica 156(5), 799 (1988)
J. Hwang, T. Timusk, G.D. Gu, Nature 427, 714 (2004)
M. Norman, Nature 427, 692 (2004)
H. Kumar, R. Rani, Int. Lett. Chem. Phys. Astron. 14, 26 (2013)
W.R.W. Abdullah, A. Zakaria, M.S.M. Ghazali, Int. J. Mol. Sci. 13, 5278 (2012)
Q. Shen, T. Toyoda, Jpn. J. Appl. Phys. 39, 511 (2000)
Q. Shen, T. Toyoda, Jpn. J. Appl. Phys. 39, 3146 (2000)
S. Abdalla, K. Easawi, T.A. El-Brolossy, G.M. Yossef, S. Negm, H. Talaat, Rev. Sci. Instrum. 74(1), 848 (2003)
T.A. El-Brolossy, S. Abdalla, O.E. Hassanein, S. Negm, H. Talaat, J. Phys. IV France 125, 685 (2005)
A. Sedky, S.A. Amin, M. Mohamed, Appl. Phys. A 125, 308 (2019)
A. Sedky, Braz. J. Phys. 44(4), 305 (2014)
L. Chow, O. Lupan, G. Chai, H. Khallaf, L.K. Ono, B. Roldan Cueny, I.M. Tiginyanu, V.V. Ursaki, V. Sontea, A. Schulte, Sens. Actuat A Phys. 189, 399 (2013)
K.S. Shankar, S. Kar, G.N. Subbanna, A.K. Raychaudhuri, Sol. State Commun. 129, 479 (2004)
Ü. Özgür, A. Ya, I. Alivov, C. Liu, A. Teke, A. ReshchikovM, S. Doğan, V. AvrutinC, S.J. Cho, H. Morkoçd, J. Appl. Phys. 98, 041301 (2005)
A. Sedky, S.B. Mohamed, Mater. Sci.-Pol. 32(1), 16 (2014)
S. Aksoy, Y. Caglar, S. Ilican, M. Caglar, Chem. Eng. Civ. Eng. Mech. Eng. 227, (2010)
U. Seetawan, S. Jugsujinda, T. Seetawan, A. Ratchasin, C. Euvananont, C. Junin, C. Thanachayanont, P. Chainaronk, Mater. Sci. Appl. 2, 1302 (2011)
J. Wojnarowicz, T. Chudoba, S. Gierlotka, K. Sobczak, W. Lojkowski, Curr. Comput. Aided Drug Des. 8, 179 (2018)
S. Kusnieruk, S. Wojnarowicz, A. Chodara, T. Chudoba, S. Gierlotka, W. Lojkowski, J. Beilstein, Nanotechnol. 7, 1586–1601 (2016)
M.A. Aksan, M.E. Yakinci, J. Alloy Compd. 433, 22 (2007)
A. Sedky, Adv. Mater. Sci. Eng. 2, 1 (2018)
F.K. Shan, Z.F. Liu, G.X. Liu, W.J. Lee, G.H. Lee, I.S. Kim, J. Electroceram. 13, 195 (2004)
X. Li, X. Cao, L. Xu, L. Liu, Y. Wang, C. Meng, Z. Wang, J. Alloys Compd. 675, 90 (2016)
A. Sedky, A.M. Ali, M. Mohamed, Opt. Quantum Electron. 52(1), 1–21 (2020)
W. Muhammad, N. Ullah, M. Haroona, B.H. Abbas, RSC Adv. 9, 29541 (2019)
T. Srinivasulu, K. Saritha, K.T. Ramakrishna Reddy, Modern Electron. Mater. 3(2), 76–851 (2017)
K. Raja, P.S. Ramesh, D. Geetha, Structural, FTIR and photoluminescence studies of Fe doped ZnOnanopowder by co-precipitation method. Spectrochem. Acta A Mol. Biomol. Spectrosc. 131, 183–188 (2014)
M. Huleihel, A. Salman, V. Erukhimovitch, J. Ramesh, Z. Hammody, S. Mordechai, J. Biochem. Biophys. Methods 50, 111–121 (2002)
H. Fabian, M. Jackson, L. Murphy, P.H. Watson, I. Fichtner, H.H. Mantsch, Biospectroscopy 1, 37–45 (1995)
B.R. Wood, M.A. Quinn, B. Tait, M. Ashdown, T. Hislop, M. Romeo, D. McNaughton, Biospectroscopy 4, 75–91 (1998)
M.M. Ba-Abbad, A.A.H. Kadhum, A.B. Mohamad, M.S. Takriffand, K. Sopian, The effect of process parameters on the size of ZnO nanoparticles synthesized via the sol–gel technique. J. Alloy. Compd. 550, 63–70 (2013)
J. Santhoshkumar, S.V. Kumar, S. Rajeshkumar, Resour. Effic. Technol. 3, 459–465 (2017)
A. Sanmugam, D. Vikraman, S. Venkatesan, H.J. Park (2017) J. Nanomater. Article ID 7536364, (1–8).
P. Mitra, S. Mondal, Structural and morphological characterization of ZnO thin films synthesized by successive ion layer adsorption and reaction. Prog. Theor. Appl. Phys. 1, 17–31 (2013)
S.A. Mazen, H.M. Zaki, S.F. Mansour, Int. J. Pure Appl. Phys. 3, 40 (2007)
D. El-Said Bakeer, Appl. Phys. A 126, 443 (2020). https://doi.org/10.1007/s00339-020-03625-z
A.V. Anupama, V. Rathod, V.M. Jali, B. Sahoo, J. Alloys Compd. 728, 1091 (2017)
K.B. Modi, P.Y. Raval, S.J. Shah, Inorg. Chem. 54, 1543 (2015)
M. Chaari, A. Matoussi, Z. Fakhfakh, Mater. Sci. Appl. 2, 76 (2011)
T.P. Rao, M.C.S. Kumar, A. Safarullaa, V. Ganesan, S.R. Barman, C. Sanjeeviraja, Phys. B 405(9), 2226 (2010)
H.C. Ong, A.X.E. Zhu, G.T. Du, Appl. Phys. Lett. 80, 941 (2002)
C. Wang, P. Zhang, J. Yue, Y. Zhang, L. Zheng, Phys. B 403, 2235 (2008)
D.I. Rusu, G.G. Rusu, D. Luca, Acta Phys. Pol. A 119(6), 850 (2011)
M. Mukhtar, L. Munisa, R. Saleh, Mater. Sci. Appl. 3, 543 (2012)
N. Srinivasan, J.C. Kannan, Mater. Sci. Pol. 33, 205 (2015)
K.B. Modi, J.D. Gajera, M.P. Pandya, H.G. Vora, H.H. Joshi, Pramana 62, 1173 (2004)
S.M. Patange, S.E. Shirsath, S.P. Jadhav, J. Mol. Struct. 1038, 40 (2013)
B. Rajesh Babu, T. Tatarchuk, Mater. Chem. Phys. 207, 534 (2018)
S.M. Patange, S.E. Shirsath, K.S. Lohar, S.G. Algude, S.R. Kamble, N. Kulkarni, D.R. Mane, K.M. Jadhav, J. Magn. Magn. Mater. 325, 107 (2013)
N. Varalaxmi, K.V. Sivakumar, Ind. J. Appl. Res. 4, 537 (2014)
A. Sedky, A. Ali, H.H. Somaily, H. Algarni, Opt. Quantum Electron. 55(243), 1–21 (2021)
A.A. Othman, M.A. Othman, E.M.M. Ibrahim, M.A. Ali, Ceram. Int. 43, 527 (2017)
H.M. Ali, H.A. Mohamed, S.H. Mohamed, Eur. Phys. J. Appl. Phys. 31, 87 (2005)
R.A. Ismail, A.K. Ali, M.M. Ismail, K.I. Hassoon, ApplNanosci 1, 45–49 (2011)
S.J. Darzi, A.R. Mahjoub, A. Nilehi, Phys. E Low-Dimens. Syst. Nanostruct. 42(1), 76 (2009)
A. El-Denglawey, J. Lumin. 194, 381 (2018)
J. Yu, X. Zhao, Q. Zhao, Mater. Chem. Phys. 69, 25 (2001)
S. Sagadevan, K. Pal, Z.Z. Chowdhury, M.E. Hoque, J. Sol-Gel Sci. Technol. 83, 394–404 (2017)
H. Abdel-Khalek, E. Shalaan, M. Abd- El Salam, A.M. El-Sagheer, A.M. El-Mahalawy, J. Mol. Struct. 1178, 408 (2019)
H.M. Zeyada, M.M. EL-Nahass, I.S. El-Ashmawi, A.A. Habashi, Curr. Appl. Phys. 13(9), 19601 (2013)
T.K. Kundu, N. K., P. Barik and S. Saha, Int. J. Soft Comput. Eng. (IJSCE). ISSN: 2231-2307, volume 1, issue NCRAMT2011 (2011).
C.P. Lin, H. Chen, A. Nakaruk, P. Koshy, C.C. Sorrell, Energy Pro. 34, 627 (2013)
H.M. Ali, A.M.A. Hakeem, Eur. Phys. J. Appl. Phys. 72, 10301 (2015)
A. El-Denglawey, Non-Cryst. Solids 357, 1757 (2011)
E.R. Shaaban, M.M. Soraya, M. Shapaan, H. Shokry Hassan, M.M. Samar, J. Alloy Compd. 693, 1052 (2017)
A.A. Zaki, A.A. El-Amin, Opt. Laser Technol. 97, 71 (2017)
G. Patwari, P.K. Kalita, R. Singha, Mater. Sci. Pol. 34(1), 69–78 (2016). https://doi.org/10.1515/msp-2016-0030
S.S. Abdullahi, Y. Köseoğlu, S. Güner, S. Kazan, B. Kocaman, C.E. Ndikilar, Superlattices Microstruct. 83, 342–352 (2015)
Y. Bu, Z. Chen, J. Power Sources 272, 647 (2014)
Y. Hong, J. Li, H. Bai et al., Effect of octahedron tilt on the structure and magnetic properties of bismuth ferrite. J. Adv. Ceram. 9, 641–646 (2020). https://doi.org/10.1007/s40145-020-0398-1
K. Beppu, Y. Hirai, T. Kato, S. Ishizuka, T. Ina, T. Wada, Appl. Phys. Lett. 117, 043901 (2020). https://doi.org/10.1063/5.0013375
H. Cao, P. Lu, N. Cai, X. Zhang, Z. Yu, T. Gao, S. Wang, J. Magn. Magn. Mater. 352, 66–71 (2014)
D. Sharma, R. Jha, Transition metal (Co, Mn) co-doped ZnO nanoparticles: effect on structural and optical properties. J. Alloys Compd. 698, 532–538 (2017)
J. Li, Xu. Tongtong, L. Liu, Y. Hong, Z. Song, H. Bai, Z. Zhou, Microstructure, magnetic and low-frequency microwave absorption properties of doped Co–Ti hexagonal barium ferrite nanoparticles. Ceram. Int. 47(13), 19247–19253 (2021). https://doi.org/10.1016/j.ceramint.2021.03.191
H. Bai, J. Li, Y. Hong, Z. Zhou, J. Adv. Ceram. 9(4), 511–516 (2020). https://doi.org/10.1007/s40145-020-0384-7
Acknowledgements
Author's acknowledgment the Deanship of Scientific Research, King Faisal University for their support under Grant Number [17122004].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Al-Naim, A.F., Sedky, A., Afify, N. et al. Structural, FTIR spectra and optical properties of pure and co-doped Zn1-x-yFexMyO ceramics with (M = Cu, Ni) for plastic deformation and optoelectronic applications. Appl. Phys. A 127, 840 (2021). https://doi.org/10.1007/s00339-021-04915-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-021-04915-w