
Abstract
Contamination can cause a rapid environmental change which may require populations to respond with evolutionary changes. To evaluate the effects of pulp mill effluents on population genetics, we sampled three-spined sticklebacks (Gasterosteus aculeatus) near four pulp mills and four adjacent reference sites and analyzed Amplified Fragment Length Polymorphism (AFLP) to compare genetic variability. A fine scale genetic structure was detected and samples from polluted sites separated from reference sites in multidimensional scaling plots (P < 0.005, 1000 permutations) and locus-by-locus Analysis of Molecular Variance (AMOVA) further confirmed that habitats are significantly separated (FST = 0.021, P < 0.01, 1023 permutations). The amount of genetic variation between populations did not differ between habitats, and populations from both habitats had similar levels of heterozygosity (polluted sites Nei’s Hs = 0.11, reference sites Nei’s Hs = 0.11). Still, pairwise FST: s between three, out of four, pairs of polluted-reference sites were significant. A FST-outlier analysis showed that 21 (8.4%) loci were statistically different from a neutral distribution at the P < 0.05 level and therefore indicated to be under divergent selection. When removing 13 FST-outlier loci, significant at the P < 0.01 level, differentiation between habitats disappeared in a multidimensional scaling plot. In conclusion, pulp mill effluence has acted as a selective agent on natural populations of G. aculeatus, causing a convergence in genotype composition change at multiple sites in an open environment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There is a widespread interest in the genetic structure and variability of natural populations, but not many studies have addressed the effects of contaminants in marine environments on population genetics (for review see e.g. Belfiore and Anderson 2001; Medina et al. 2007). The evolutionary response of populations can be surprisingly rapid, about a quarter of the rate of ecological change (Hairston et al. 2005) and environmental shifts caused by humans is often more rapid than naturally occurring changes and thus requires a faster response from populations (e.g. Hendry et al. 2008; Palumbi 2001; Stockwell et al. 2003). When a population is exposed to a new environment with a novel selective pressure caused by anthropogenic contaminants a population could respond in different ways. The population could decrease drastically causing a genetic bottleneck that result in lowered genetic diversity in the population (Bickham et al. 2000). Some alleles could be beneficial and increase in frequency in the new environment (Wirgin and Waldman 2004), or genetic variation could actually increase due to high mutation rate caused by genotoxic contaminants (Dubrova et al. 1996; Ellegren et al. 1997). In a recent meta-analysis of the impact of pollution on marine environments Johnston and Roberts (2009) noted that pollution was never associated with the complete exclusion of life from a location and that commonly 50–70% of species that were able to tolerate the contaminant load.
Rare alleles, which under normal circumstances are neutral or slightly deleterious, may increase individual tolerance to a novel environmental stressor (Bell and Collins 2008). Such alleles can accrue fitness benefits in the new environment and by natural selection later become common in the population. This appears to have happened when oceanic three-spined sticklebacks (Gasterosteus aculeatus, L) got trapped in freshwater habitats. The EDA allele responsible for lateral plate reduction is rare (1%) in oceanic populations of sticklebacks but is common in freshwater habitats. Due to the strong selection pressure favoring this allele in freshwater environment, its frequency increases dramatically in freshwater (to 100%) and is thus critical to the repeated occurrence of locally adapted freshwater stickleback populations (Barrett et al. 2008; Colosimo et al. 2005; Hohenlohe et al. 2010; Mäkinen et al. 2008).
In the Baltic Sea high concentrations of pollutants in the water and sediment (Elmgren 1989; Fonselius 1972) may drive evolution of resistance to contamination, and as in evolutionary change in general, genetic variation available in gene-loci involved in resistance is critical to rate of evolution and magnitude of resistance. When populations Attheyella crassa (Crustacea) were exposed to contaminated coastal Baltic Sea sediment mortality increased, levels of genetic variation decreased, and the experimental populations diverged genetically (Gardeström et al. 2008). These experiments highlight the complicated interaction between selection and random genetic drift caused by environmental change. To date a few studies have conclusively demonstrated the evolution of increased genetic tolerance to contaminants in natural populations (Williams and Oleksiak 2008; Wirgin and Waldman 2004).
It has been shown that G. aculeatus respond to anthropogenic disturbance (eutrophication) with plastic alterations in their behavior such as enhanced reproductive success, increase in courtship activity and increased cost of courtship for males (Candolin 2009). Hence, eutrophication relaxes opportunity for sexual selection on several traits and that could increase the opportunity for natural selection (Candolin 2009). Further, it has been known for over three decades that effluents from paper and pulp mills affect fish reproduction such as decreased gonad size, altered expression of secondary sex characteristics and reduction in fecundity (for review see Hewitt et al. 2008). For example, female mosquito fish, Gambusia sp., downstream of pulp and paper mill outlets in Florida had a masculinized development and decreased embryo production (Orlando et al. 2007). Female G. aculeatus exposed to pulp mill effluents has been shown to masculinization and induction of the male specific protein spiggin (Katsiadaki et al. 2002). Masculinization of eelpout, Zoarces viviparus, has been reported close to a pulp mill effluent in the Baltic Sea with a lowered percentage of females (Larsson et al. 2000). It has been suggested that androgenic steroids derived from microbial degradation of phytosterols originating from the wood, such as androstenedione, androstadienedione and progesterone, are responsible for the masculinisation of fish observed in the receiving waters of pulp and paper industry effluents (Denton et al. 1985). Pulp mill effluents have also been shown to exhibit estrogenic effects in fish. Elevated plasma vitellogenin levels have been observed in rainbow trout, Oncorhynchus mykiss, exposed in vivo to bleached mill effluents (Tremblay and van der Kraak 1999; van den Heuvel and Ellis 2002), and in juvenile whitefish, Coregonus lavaretus, exposed to effluents from a chlorine free bleaching process (Mellanen et al. 1999). Still, Pettersson et al. (2007) could not detect any estrogenic or androgenic disruption in juvenile three-spined stickleback at receiving waters of pulp mill in the Swedish Baltic Sea coast.
Patterns of genetic diversity between populations at multiple genetic loci are often used to detect loci under selection in genome scans (for a review see Luikart et al. 2003). Loci involved in local adaptations should show high FST values compared to the global average FST (e.g. Bonin et al. 2006; Excoffier et al. 2009; Wilding et al. 2001; Shimada et al. 2011). We have used AFLP (Vos et al. 1995) as it yields primarily neutral genetic markers from multiple locations in the genome, making it possible to find loci that are under selection and to detect adaptation to habitat differences and environmental change (e.g. Bensch and Åkesson 2005; Campbell and Bernatchez 2004; Wilding et al. 2001).
Here we have sampled G. aculeatus near four different paper pulp mills in the Baltic Sea (three in Baltic proper and one in the Gulf of Bothnia) and four adjacent reference sites. The aim of the study was to examine whether pollution from point sources could act as a selective agent and drive local adaptation. In this study we sought to test whether G. aculeatus sampled at polluted sites differ in their genotypic composition from fish sampled from nearby reference sites. And if polluted environments at different geographical sites constitute a selective regime more similar among polluted habitats. We also wanted to evaluate if levels of genetic variation within populations differ between contaminated and reference sites. And finally, investigate the possibly to identify genetic loci under directional selection using a genome scan approach.
Methods
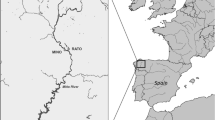
All samples (Table 1) were collected in 2003 in a nested way with one reference south of every polluted site. Our study thus contrast two habitat types; polluted sites located close to the main exhaust of the pulp mill (1–5 km) and reference sites less influenced by the point source located 7 to 50 km south of the reference site (Fig. 1 and Table 2). The sites were chosen based on information from local authorities’ environmental assessments schemes. At the sites in southern Sweden adult fish, both females and males, were sampled in late April during breeding season and in northern Sweden fry were sampled in August. Fishes were caught with drop nets and trap nets, depending on habitat conditions, in shallow water from 1 meter depth to 2 m dept. A small piece of the tail-fin was collected (2 × 2 mm) and stored in 99% ethanol and the fish were then released.

Sampling sites along Sweden’s eastern coast in the Baltic Sea. Three different geographic areas (a, b and c) were sampled. In area a in the Gulf of Bothnia one pulp mill, Uta-P, and one reference sites, Uta, were sampled. In area b Mönsterås pulp mill we sampled, Mon-P, and one reference site, Mon. Further south, in area c, we sampled two pulp mills Nymölla, Nym-P and Mörrum mill, Mor-P, and two references, Nym and Mor
The Baltic Sea is a brackish inland sea with a salinity gradient stretching from south to north and lack visible tide and lack strong current patterns (Elmgren 2001). Salinity at sampling sites in Baltic proper (site Nym/Nym-P, Mor/Mor-P, Mons/Mons-P; see Fig. 1) were 6.7 psu at the time of sampling (SHARK database http://www.smhi.se/oceanografi/oce_info_data/SODC/download_sv.htm) in Gulf of Bothnia (site Uta/Uta-P) salinity is much lower, around 2 psu. All pulp mills in this study have been active for more than 40 years. The site at the Mönsterås pulp mill has been extensively studied. The effluent is subject to activated sludge treatment, and it enters the sea via a 5-km effluent tube with a 1.5 km diffuser at a water depth of 9–12 m, into fairly well ventilated waters. The final discharge is approximately 35 m3 per ton of pulp produced. The plume from the mill can reach at least 3.8 km south or 2.4 km north (1:500 v/v dilution) depending on different current conditions (Landner et al. 1994). The activated sludge plant and the ultra filtration plant had a notable effect at one of the pulp mills (Nymölla) on the emissions of organic matter into the Baltic Sea. Emissions of chemical oxygen demand (COD) have declined from 45000 metric tonnes/year in 1993 to 13000 in 2001 (HELCOM 2004). Due to changes in the productions process environmental impact of the discharge has been lowered continuously since mid 1980 and the impact is fairly local (0–3 km). Moreover, most of the effects on the local ecosystem seem to be related to eutrophication (Landner et al. 1994).
Genetic analysis
Total genomic DNA was isolated from a 2 × 2 mm tail fin clip as described by Laird et al. (1991). Approximately 25 mg of fin was incubated for 4 h in 56°C with 100 μl lysis buffer and 0.03 mg Pro K in a total volume of 103 μl. DNA from the supernatant was extracted with 99.5% ethanol, centrifuged and the pellet washed with 70% ethanol. DNA was dissolved in 1× TE buffer and kept at 4°C. The concentration of the DNA was determined using Nanodrop© ND-1000 and then diluted to the working concentration of 50 ng/μl.
AFLP markers were generated as described by Vos et al. (1995) with minor modifications as described by Bensch et al. (2002) and prior to AFLP analyses, samples were randomized in relation to sampling site to minimize between batch-variation in the PCR reactions (Bensch and Åkesson 2005). Genomic DNA (250 ng) was cut with EcoR1 (5′-G↓AATTC-3′ MBI Fermentas) and Tru1 (5′-T↓TAA-3′ MBI Fermentas) and after digestion E-adaptor (5′-CTCGTAGACTGCGTACC-3′, 3′-CATCTGACGCATGGTTAA-5′ MWG Biotech) and M-adaptor (5′-GACGATGAGTCCTGAG-3′, 3′-TACTCAGGACTCAT-5′ MWG Biotech) was ligated to fragments. In pre-amplifications we substituted 45% of the ddH2O with bovine serum albumin (1 μg μl−1) and used E-primer (5′-GACTGCGTACCAATTCT-3′) and M-primer (5′-GATGAGTCCTGAGTAAC-3′). PCR products were diluted 10 times in ddH2O and stored in −20°C. In selective amplification 12% ddH2O was substituted with bovine serum albumin (1 μg μl−1). We used three different primer combinations, each combination with tree additional bases at the 3′-ends, M-primer (5′-GATGAGTCCTGAGTAANNN-3′) and E-primer labeled in the 5′-end with HEX. (5′-GACTGCGTACCAATTCNNN-3′) and we used following primer combinations Etct/Mcta, Etct/Mcac and Etag/Mcac. Approximately 10% of the samples were duplicates. DNA fragments was separated on an ABI-3730XL capillary electrophoresis unit at Uppsala Genome Center with separation medium POP7TM Polymer (Applied Biosystems), size standard GeneScanTM 500 ROXTM (Applied Biosystems), injection time 15 s (1.6 kV), run time 1600 s and array length 50 cm. Data was scored in Genemapper 3.0 (Applied Biosystems); analysis range was set to 150–500 bp, bin width to 1 bp and locus selection threshold 200 RFU using default settings with normalization within project. We manually checked that duplicate samples yielded the same genotypes. Each primer combination was scored separately in Genemapper 3.0 and the three genotype matrixes were set together to one dataset.
Statistical analyses
Estimations of genetic variation within sites (He and Hs) were obtained from software AFLP-SURV 1.0 (Vekemans 2002) using the approach of Lynch and Milligan (1994) assuming Hardy–Weinberg equilibrium and using the Bayesian method with a non-uniform prior distribution of allele frequencies (Bensch and Åkesson 2005; Zhivotovsky 1999) and 1000 permutations to test the significance of the FST. Genetic distances, FST, were combined in a matrix with geographic distances to test for Isolation by distance with Isolation by Distance Web Service version 3.16 (Jensen et al. 2005) with a Mantel test. We did a principal coordinate analysis, PCoA, using the capscale procedure in the Vegan package (Oksanen et al. 2006) in R 2.5.1 (R Development Core Team 2007) and a Constrained PCoA with habitat (viz. polluted and reference sites) as a constraint. This is similar to a redundancy analysis but allows non-Euclidian dissimilarity indices (here we used Jaccard distances). Genetic structure was further investigated with the Bayesian approach in STRUCTURE 2.2. (Pritchard et al. 2000), burnin was set to 50000 with 70000 additional cycles. Each run was iterated 3 times, and number of clusters (K) set from 1 to 10, assuming admixture and uncorrelated allele frequencies. Structure Harvester on the Web, version 0.56.4 (Earl 2009) was used to evaluate K (Evanno et al. 2005). To test if data separates between the two different habitats we did a locus-by-locus Analysis of Molecular Variance (AMOVA) in the software Arlequin 3.0 (Excoffier et al. 2005) which assumes that loci are unlinked and correctly adjust sample sizes for each locus. We also tested FST values for each locus to find FST-outlier loci by simulations as implemented in the Arlequin package (version 5.5.1.2). Populations were grouped according to geography (Nym/Nym-P, Mor/Mor-P, Mons/Mons, Uta/Uta-P, Fig. 1) and we simulated 50 groups with 100 demes each for 50000 runs. This analysis simulates evolution in a hierarchical set of populations according to the Wright island model. Coalescent simulations are used to get a null distribution of locus-specific FST values and confidence intervals around the observed values to test if observed FST values can be considered as outliers in relation to the overall observed FST value for each loci (Excoffier et al. 2009). In order to further test the effect of different selection between polluted and reference sites identified FST-outlier loci were removed from the dataset and data was re-analyzed. To test the robustness of the FST-outlier analysis, data was divided into the two habitat types, and analyzed in random forest package in R 2.5.1 (R Development Core Team 2007), an algorithm for classification developed by Breiman (2001), to identify which loci that contributed most to the difference in genotypes between habitats.
Results
The three primer combinations resulted in a total of 248 polymorphic AFLP-loci which all were used for statistical analyses. Primer combination Etct/Mcta gave 80 loci, Etct/Mcac 79 loci and Etag/Mcac 89 loci. All duplicate samples gave a congruent genotype pattern.
The results showed a distinct genetic structure from AFLP-SURV analyses according to which 12% of the genetic variation was among populations (FST = 0.12, P < 0.001), between strictly polluted populations genetic variation were 13% (FST = 0.13, P < 0.001) and between reference sites 14% (FST = 0.14 P < 0.001) (Table 2). Genetic diversity within populations (He, Table 1) did not differ between the two different habitat types (Table 1). All pairwise FST: s except those between Uta-P–Uta and Mor-Nym were significant (Table 2). There were isolation by distance were genetic distances (FST) increased with geographic distance (r = 0.95, P = 0.02). This pattern was consistent also after removing 13 FST-outlier loci (see below) (r = 0.92, P = 0.01). Polluted and reference sites showed isolation by distance to a similar degree, but the isolation by distance patterns were not significant when tested for each habitat group separately.
Data showed a separation on habitat in the PCoA (P < 0.005, tested with a permutation test, Fig. 2 a) where the first axis separates population on a geographic scale and explains 60% of the genetic variation. The second axis separates populations on habitat, explaining 12% of the variation, polluted sites in the Baltic proper forms one group and the references in Baltic proper forms another group. When 13 FST-outlier loci (see below) were removed, the separation on habitat disappeared and data mainly separated on geography (Fig. 2b). The constrained PCoA, using habitat as constraint, showed a significant difference in genotype composition between polluted and reference sites (permutation test P < 0.005, Fig. 2c). Structure analyses showed that K = 6 had the highest probability (Mean LnP K6 = −16659 SD 18.9, Fig. 3a). In one cluster, cluster 3 (Fig. 3b), the two sites from Gulf of Bothnia (Uta and Uta-P) are dominant. Cluster 1, even though not exclusively, consisted mostly of fish collected at polluted sites and cluster 2 was more frequent in reference sites.

a Principal coordinate analysis based on 248 Amplified Length Polymorphism (AFLP) loci. A total of eight sites, four polluted sampled outside pulp mills (Mons-P, Mor-P, Nym-P and Uta-P) and four close-by reference sites, all sites are significantly separated (P < 0.005) tested with a permutation test. The first principal coordinate axis explains 59.6% of the variation and the second principal coordinate axis 12% of the variation. Site names in the plot indicate the centriole of every population. b Principal coordinate analysis were loci identified as FST-outliers and thereby under positive selection (comparing pairs of polluted with reference sites) has been removed. Site names in the plot indicate the centriole (mid-point) of populations. c Constraint principal coordinate analysis with constrain on habitat. Habitat type (polluted and reference) differed significantly with a permutation test (P < 0.005). In the plot * indicates centrioles of grouped reference sites respective grouped polluted sites

Results from structure analyses a shows plotted output data from structure, with L(K) mean (±SD) are plotted for K 1–10, were K is genetic cluster with (no prior information and under the assumption of admixture and correlated allele frequencies). The figure shows that K 6, six clusters, has the highest probability. b Assignment of individuals from the eight sampling sites to each of the six clusters. Each shade of grey, black and white illustrates one cluster. Y-axis show the proportion of every population assigned to the six genetic clusters
The locus-by-locus AMOVA showed that the two habitats were significantly separated (FST = 0.021, P < 0.01, 1023 permutations). The FST-outlier analysis detected 13 AFLP loci that were indicated to be under positive selection with FST values above the 1% quantile (Fig. 4), most of the loci (11) were from one primer combination (Etct/Mcac) the other two loci originated from the Etag/Mcac primer combination. Further, 21 loci were identified as FST-outliers above the 5% quantile. 28 loci were under stabilizing selection, having lower FST than expected (P < 0.01, Fig. 4). There was some congruence between the FST-outlier analysis and the classification analysis performed with random forest, and 5 of the 20 loci ranked as the most important for separation between the two habitats was also identified by the FST-outlier analysis to be above the 0.95 quantile (Fig. 5). Four of the five most informative loci in the classification analysis were significant (P < 0.05) in the FST-outlier analysis. However, an unexpected high number of the most significant FST-outlier loci were not identified by the classification analysis.

FST-outlier analysis for the 248 polymorphic AFLP loci after grouping the matched polluted reference pairs in four groups according to geography (see text). The contour lines indicates the P < 0.05 and P < 0.01 quantiles corresponding to the 95 and 99% confidence intervals. Observations outside these ranges are considered as having significantly higher or lower FST than expected under a neutral model indicating directional or stabilizing natural selection respectively

Output from the classification analysis by Random Forest analysis indicating the highest ranked loci affecting the classification of individuals. Data was divided into the two habitat categories (polluted and references) and the 20 loci of highest importance for separating the categories are ranked. Indicated loci was significant in the FST-outlier analysis above the 0.95 (*) and 0.99 (**) level
Discussion
We found that the genetic composition of multiple populations of G. aculeatus have responded to the directional selection pressure from the effluents of paper pulp mills in the Baltic Sea. Pairs of polluted and clean sites in proper Baltic all had significant FST values between them (Table 2). Populations sampled at polluted sites were separated from populations from nearby reference sites (Fig. 2a). The genetic difference between polluted and reference sites was further confirmed when habitat was set as a constraint, removing the genetic variation caused by geography (Fig. 2c). Yet, most of the genetic structure in our sample could be explained by geographic distance, and nearly all sites in proper Baltic even though separated by as short distance as 7 km were significantly differentiated (Table 2). This clear geographic structure is in contrast with the findings for microsatellite data presented by Mäkinen et al. (2006) and Cano et al. (2008) who found a very weak population structure in the Baltic Sea even when sites in proper Baltic and Gulf of Bothnia were included.
Genetic variation was not different in the populations from polluted sites as populations from both habitats had similar levels of heterozygosity (Table 1). Neither were there any differences in genetic separation between populations when comparing polluted and reference sites as the global FST in both groups are similar to each other. Loss of genetic variation has been shown in several studies (e.g. Murdoch and Hebert 1994; Street and Montagna 1996). In this study, we sampled wild populations in an environment without physical barriers to migration and it is most likely that there is gene-flow between polluted and clean sites which could compensate for the loss of genetic variation in neutral loci in populations at polluted sites. In wild populations of Fundulus heteroclitus living in heavily PCB contaminated harbors as compared to both moderately contaminated sites and clean sites the same pattern were observed and genetic diversity did not differ between sites even thought the fish in contaminated habitats was shown to differ to great extent in PCB-tolerance (McMillan et al. 2006).
We identified FST-outlier loci that were statistically different from a neutral distribution and therefore were indicated to be under different selection pressures at polluted and reference sites (Fig. 5). When these loci were removed from our dataset the differentiation in the PCoA according to habitat disappeared (Fig. 2a and b). This suggests that the selective pressure is in fact caused by habitat difference at these, or closely linked loci. Williams and Oleksiak (2008) identified several outlier loci when comparing populations of F. heteroclitus from three polluted sites with references and saw that when excluding positively selected loci the pattern of isolation by distance in samples disappeared. We did not observe the same pattern in our study––isolation by distance was present both in the whole dataset, and when removing the FST-outlier loci. The incidence of loci indicated to be under directional selection in our study was 8.4% (21 out of 248) at the P < 0.05 level (Fig. 5). This number of loci under directional selection is similar to the number earlier reported for nine-spined sticklebacks (Pungitius pungitius) in the Baltic Sea region in relation to salinity difference (Shikano et al. 2010). These numbers are relatively high compared to earlier studies of directional selection by genome scan methods (Shikano et al. 2010 and references therein). To identify genes subject to directional selection and responsible for local adaptation in the three-spined stickleback Shimada et al. (2011) genotyped microsatellite markers located within, or closely linked to (<6 kb) target genes. They investigated directional selection for 157 genes with known physiological functions and found a high incidence (17%) of significant footprints of directional selection.
Not all loci identified by the FST-outlier analysis are likely to confer adaptation to a pulp mill influenced environment, as only a minority of the outlier loci was also highlighted by the classification analysis. The precise factors responsible for the putative selection will require further research, as will the role of the potential marker loci identified. It would be possible to look further into the AFLP-loci under selection. It is feasible to convert the AFLP fragment to a codominant genetic marker (Bensch et al. 2002) and further to use it to find candidate genes that could be involved in processes important for the organism’s adaptation to the habitat. Moreover, although most AFLP generated markers are expected to be unlinked (Vos et al. 1995) we cannot exclude the possibility of linkage between some of the identified loci. Linkage analysis of AFLP markers cannot be done reliably in the absence of pedigree data.
Our study showed that marine anthropogenic contamination can act as a selective agent and that is possible to identify genetic loci involved in this differentiation. Moreover, this differentiation in genotypic composition has taken place in an open environment, a sea, and over a time period no longer than 45 years. However, we cannot rule out the possibility that non-toxic effects linked to pulp mill effluents such as eutrophication, changes in vegetation, or changed predation pressure also are of importance and may be responsible for the observed changes in genotype frequencies between the polluted and reference sites.
References
Barrett RDH, Rogers SM, Schluter D (2008) Natural selection on a major armor gene in three spine stickleback. Science 322:255–257
Belfiore NM, Anderson SL (2001) Effects of contaminants on genetic patterns in aquatic organisms: a review. Mutat Res 489:97–122
Bell G, Collins S (2008) Adaptation, extinction and global change. Evol Appl 1:3–16
Bensch S, Åkesson M (2005) Ten years of AFLP in ecology and evolution: why so few animals? Mol Ecol 14:2899–2914
Bensch S, Helbig AJ, Salomon M, Seibold I (2002) Amplified fragment length polymorphism analysis identifies hybrids between two subspecies of warblers. Mol Ecol 11:473–481
Bickham JW, Dandhu S, Hebert PND, Chikhi L, Athwal R (2000) Effects of chemical contaminants on genetic diversity in natural populations: implications for biomonitoring and ecotoxicology. Mutat Res 463:33–51
Bonin A, Taberlet P, Miaud C, Pompanon F (2006) Explorative genome scan to detect candidate loci for adaptation along a gradient of altitude in the common frog (Rana temporaria). Mol Biol Evol 23:773–783
Breiman L (2001) Random forests. Mach Learn 45:5–32
Campbell D, Bernatchez L (2004) Generic scan using AFLP markers as a means to assess the role of directional selection in the divergence of sympatric whitefish ecotypes. Mol Biol Evol 21:945–956
Candolin U (2009) Population responses to anthropogenic disturbance: lessons from three-spined sticklebacks Gasterosteus aculeatus in eutrophic habitats. J Fish Biol 75:2108–2121
Cano JM, Mäkinen H, Merilä J (2008) Genetic evidence for male-biased dispersal in the three-spined stickleback (Gasterosteus aculeatus). Mol Ecol 17:3234–3242
Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G Jr, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM (2005) Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 37:1928–1933
Denton TE, Howell WM, Allison JJ, McCollum J, Marks B (1985) Masculinization of female mosquito fish by exposure to plant sterols and Mycobacterium smegmatis. Bull Environ Contam Toxicol 35:627–632
Dubrova YE, Nesterov VN, Krouchinsky NG, Ostapenko VA, Neumann R, Neil DL, Jeffreys AJ (1996) Human minisatellite mutation rate after the Chernobyl accident. Nature 380:683–686
Earl DA (2009) Structure Harvester v 0.3. Available from website: http://users.soe.ucsc.edu/~dearl/software/struct_harvest/. Accessed September 2010
Ellegren H, Lindgren G, Primmer CR, Møller AP (1997) Fitness loss and germline mutations in barn swallows breeding in Chernobyl. Nature 389:593–596
Elmgren R (1989) Man’s impact on the ecosystem of the baltic sea: energy flow today and the turn of the century. Ambio 18:326–332
Elmgren R (2001) Understanding human impact on the Baltic ecosystem: changing views in recent decades. Ambio 30:222–231
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Excoffier L, Hofer T, Foll M (2009) Detecting loci under selection in a hierarchically structured population. Heredity 103:285–298
Fonselius SH (1972) On eutrophication and pollution in the Baltic Sea. Marine pollution and Sea Life. Fishing News (Books) Ltd., London, pp 23–28
Gardeström J, Dahl U, Kotsalainen O, Maxson A, Elfwing T, Grahn M, Bengtsson B-E, Breitholtz M (2008) Evidence of population genetic effects of long-term exposure to contaminated sediments–A multi-endpoint study with copepods. Aquat Toxicol 86:426–436
Hairston NG, Ellner SP, Geber MA, Yoshida T, Fox JA (2005) Rapid evolution and the convergence of ecological and evolutionary time. Ecol Lett 8:1114–1127
HELCOM (2004) Thematic report; status of the hot spots in Denmark, Finland, Germany and Sweden; based on the seventh HELCOM PITF regional workshop, Germany, January 2002, and the eighth HELCOM PITF regional workshop, Sweden, May 2002. Baltic Sea Environ. Proc. No. 99
Hendry AP, Farraugia TJ, Kinnison MT (2008) Human influences on rates of phenotypic change in wild animal populations. Mol Ecol 17:20–29
Hewitt LM, Kovacs TG, Dubé MG, MacLatchy DL, Martel PH, McMaster ME, Paice MG, Parrott JL, van den Heuvel MR, van der Kraak GJ (2008) Altered reproduction in fish exposed to pulp and paper mill effluents: roles of individual compounds and mill operating conditions. Environ Toxicol Chem 27:682–697
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA (2010) Population genomics of parallel adaptation in three spine stickleback using sequenced RAD tags. PLoS Genet 6(2): e1000862
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13. v. 3.16 http://ibdws.sdsu.edu/. Accessed September 2010
Johnston EL, Roberts DA (2009) Contaminants reduce the richness and evenness of marine communities: a review and meta-analysis. Environ Pollut 157:1745–1752
Katsiadaki I, Scott AP, Hurst MR, Matthiessen P, Mayer I (2002) Detection of environmental androgens: a novel method based on enzyme-linked immunosorbent assay of spiggin, the stickleback (Gasterosteus aculeatus) glue protein. Environ Toxicol Chem 21:1946–1954
Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A (1991) Simplified mammalian DNA isolation procedure. Nucleic Acids Res 19:4293
Landner L, Grahn O, Härdig J, Lehtinen K-J, Monfelt C, Tana J (1994) A field study of environmental impacts at a bleached kraft pulp mill site on the Baltic sea coast. Exotoxicol Environ Saf 27:128–157
Larsson DGJ, Hällman L, Förlin L (2000) More male fish embryos near a pulp mill. Environ Toxicol Chem 19:2911–2917
Luikart G, England PR, Tallmon D, Jordan S, Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–994
Lynch M, Milligan BG (1994) Analysis of population genetic structure with RAPD markers. Mol Ecol 3:91–99
Mäkinen HS, Cano JM, Merilä J (2006) Genetic relationships among marine and freshwater populations of the European three-spined stickleback (Gasterosteus aculeatus) revealed by microsatellites. Mol Ecol 15:1519–1534
Mäkinen HS, Cano JM, Merilä J (2008) Identifying footprints of directional and balancing selection in marine and freshwater three-spined stickleback (Gasterosteus aculeatus) populations. Mol Ecol 17:3565–3582
McMillan AM, Bagley MJ, Jackson SA, Nacci DE (2006) Genetic diversity and structure of an estuarine fish (Fundulus heteroclitus) indigenous to sites associated with a highly contaminated urban harbor. Ecotoxicology 15:539–548
Medina MH, Correa JA, Barata C (2007) Micro-evolution due to pollution: possible consequences for ecosystem responses to toxic stress. Chemosphere 67:2105–2114
Mellanen P, Soimasuo M, Holmbom B, Oikari A, Santti R (1999) Expression of the vitellogenin gene in the liver of juvenile whitefish (Coregonus lavaretus L-s.l.) exposed to effluents from pulp and paper mills. Ecotoxicol Environ Saf 43:133–137
Murdoch MH, Hebert PDN (1994) Mitochondrial DNA diversity of brown bullhead from contaminated and relatively pristine sites in the great lakes. Environ Toxicol Chem 13:1281–1289
Oksanen J, Kindt R, Legendre P, O’Hara B (2006) Vegan: community ecology package version 18-3. R package URL http://www.R-project.org. Accessed November 2010
Orlando EF, Bass DE, Caltabiano LM, Davis WP, Gray LE, Guillette LJ (2007) Altered development and reproduction in mosquito fish exposed to pulp and paper effluent in the Fenholloway River, Florida. Aquat Toxicol 84:399–405
Palumbi SR (2001) Evolution explosion: how humans cause rapid evolutionary change. W.W. Norton & Company, New York
Pettersson M, Hahlbeck E, Katsiadaki I, Asplund L, Bengtsson B-E (2007) Survey of estrogenic and androgenic disruption in Swedish coastal waters by the analysis of bile fluid from perch and biomarkers in the three-spined stickleback. Mar Pollut Bull 54:1868–1880
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Development Core Team (2007) R: a language and environment for statistical computing, R Foundation for statistical computing. Vienna. ISBN 3-900051-07-0. URL http://www.R-project.org
Shikano T, Ramadevi J, Shimada Y, Merilä J (2010) Utility of sequenced genomes for microsatellite marker development in non-model organisms: a case study of functionally important genes in nine-spined sticklebacks (Pungitus pungitus). BMC Genome 11:334
Shimada Y, Shikano T, Merilä J (2011) A high incidence of selection on physiologically important genes in the three-spined stickleback, Gasterosteus aculeatus. Mol Biol Evol 28:181–193
Stockwell CA, Hendry AP, Kinnison MT (2003) Contemporary evolution meets conservation biology. Trends Ecol Evol 18:94–101
Street GT, Montagna PA (1996) Loss of genetic diversity in Harpacticoida near offshore platforms. Mar Biol 126:271–282
Tremblay L, van der Kraak G (1999) Comparison between the effects of the phytosterol β-sitosterol and pulp and paper mill effluents on sexually immature rainbow trout. Environ Toxicol Chem 18:329–336
van den Heuvel MR, Ellis RJ (2002) Timing of exposure to a pulp and paper effluent influences the manifestation of reproductive effects in rainbow trout. Environ Toxicol Chem 21:2338–2347
Vekemans X (2002) AFLP-SURV version 1.0. Distributed by the author. Laboratorie de Génétique et Ecologie Végétale, Université Libre de Bruxelles, Belgium
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Friters A, Pot J, Paleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Wilding CS, Butlin RK, Grahame J (2001) Differential gene exchange between parapatric morphs of Littorina saxatilis detected using AFLP markers. J Evol Biol 14:611–619
Williams LM, Oleksiak MF (2008) Signatures of selection in natural populations adapted to chronic pollution. BMC Evol Biol 8:282
Wirgin I, Waldman JR (2004) Resistance to contaminants in North American fish populations. Mutat Res Fund Mol Mech Mut 552:73–100
Zhivotovsky LA (1999) Estimating population structure in diploids with multilocus dominant DNA markers. Mol Ecol 8:907–913
Acknowledgments
We gratefully thank Prof. Juha Merilä, Prof. Richard Handy and an anonymous referee for valuable comments that greatly improved this manuscript. We also thank Prof. B-E Bengtsson for field sampling in northern Sweden and J Lind for artwork (map) and valuable comments on earlier version of this manuscript. The study was in part founded by grants from the Baltic Sea Foundation (to MG) and the County council of Stockholm.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lind, E.E., Grahn, M. Directional genetic selection by pulp mill effluent on multiple natural populations of three-spined stickleback (Gasterosteus aculeatus). Ecotoxicology 20, 503–512 (2011). https://doi.org/10.1007/s10646-011-0639-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10646-011-0639-8