
Abstract
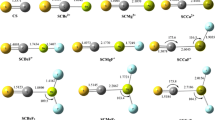
The structure and properties of the monocarbides ScC, TiC, VC, CrC, MnC, FeC, CoC, NiC, CuC, ZnC and their negatively and positively charged ions together with 3d-metal monoxide cations are calculated by density functional theory (DFT) and hybrid DFT methods. In addition to the spectroscopic constants, the computed properties include the electron affinities, ionization energies, and dissociation energies. These results along with our previous results for the neutral and negatively charged 3d-metal monoxides allow a detailed comparison of similarity and differences in the bonding of the metal oxides and carbides. These results are compared with results obtained using other theoretical approaches and with experiment. Chemical bonding, analyzed using the natural bond orbital scheme, was found to be rather different in the 3d-metal monocarbides and monoxides.
Similar content being viewed by others

Author information
Authors and Affiliations
Corresponding author
Additional information
Acknowledgement.G.L.G. was supported by grant no. NCC2-5415 to the University of Virginia.
Rights and permissions
About this article
Cite this article
Gutsev, G., Andrews, L. & Bauschlicher, Jr., C. Similarities and differences in the structure of 3d-metal monocarbides and monoxides. Theor Chem Acc 109, 298–308 (2003). https://doi.org/10.1007/s00214-003-0428-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00214-003-0428-4