
Abstract
Stop codon mutations in the gene encoding the prion protein (PRNP) are very rare and have thus far only been described in two patients with prion protein cerebral amyloid angiopathy (PrP-CAA). In this report, we describe the clinical, histopathological and pathological prion protein (PrPSc) characteristics of two Dutch patients carrying novel adjacent stop codon mutations in the C-terminal part of PRNP, resulting in either case in hereditary prion protein amyloidoses, but with strikingly different clinicopathological phenotypes. The patient with the shortest disease duration (27 months) carried a Y226X mutation and showed PrP-CAA without any neurofibrillary lesions, whereas the patient with the longest disease duration (72 months) had a Q227X mutation and showed an unusual Gerstmann-Sträussler-Scheinker disease phenotype with numerous cerebral multicentric amyloid plaques and severe neurofibrillary lesions without PrP-CAA. Western blot analysis in the patient with the Q227X mutation demonstrated the presence of a 7 kDa unglycosylated PrPSc fragment truncated at both the N- and C-terminal ends. Our observations expand the spectrum of clinicopathological phenotypes associated with PRNP mutations and show that a single tyrosine residue difference in the PrP C-terminus may significantly affect the site of amyloid deposition and the overall phenotypic expression of the prion disease. Furthermore, it confirms that the absence of the glycosylphosphatidylinositol anchor in PrP predisposes to amyloid plaque formation.
Similar content being viewed by others

Introduction
Transmissible spongiform encephalopathies, or prion diseases, are fatal neurodegenerative disorders that can occur as sporadic, inherited or acquired disorders. These diseases are pathogenetically characterized by the accumulation of protease-resistant PrPSc, an abnormal structural conformer of the normal cellular prion protein, PrPC. In humans, prion diseases are classified into three major groups, showing a wide phenotypic heterogeneity: Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS) and fatal insomnia (FI). While in most cases of CJD and in FI PrPSc accumulates in brain parenchyma without significant amyloid deposition, PrP-amyloid constitutes the dominant type of abnormal PrPSc accumulation in GSS. This striking phenotypic difference has a significant correlate at the molecular level. Indeed, while in CJD and FI abnormal prion protein deposits include both full-length PrPSc and truncated fragments in the 12–21 kDa range, in GSS the dominant PrPSc forms comprise 7–10 kDa unglycosylated fragments, truncated at both the C-terminal and N-terminal end, lacking the glycosylphosphatidylinositol (GPI) anchor [11, 24, 28].
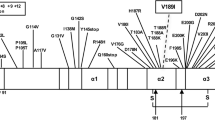
To date, GSS has only been described in association with prion protein gene (PRNP) mutations and is inherited in an autosomal dominant manner. In its classic form, GSS is characterized by a progressive cerebellar syndrome accompanied by extrapyramidal and pyramidal signs and cognitive decline, which may evolve into severe dementia [11]. There is considerable heterogeneity, however, in age at onset, disease duration and clinical symptoms, both within and between families with the same mutation. Thus, in some individuals, marked extrapyramidal signs with Parkinsonism may occur early in the course of the disease, whereas in others pyramidal signs, pseudobulbar signs or dementia predominate. Neuropathological features associated with GSS vary substantially, but always include multicentric amyloid plaques, which can be either the only significant lesion or be associated with neurofibrillary tangles or spongiform changes [11]. About 10-point mutations in PRNP affecting various codons are known to be associated with GSS. In addition, a GSS-like phenotype has been described in patients with octapeptide repeat insertions of 8 or 9 additional 24 base pair repeats [7].
Pure prion protein cerebral amyloid angiopathy (PrP-CAA) is another form of hereditary prion protein amyloidosis, albeit with a disease phenotype different from GSS. In this rare variant, which thus far has only been reported in two patients with nonsense mutations at codon 145 (Y145X) or 163 (Y163X) of PRNP, PrPSc depositions are found in blood vessels, without co-localization of Aβ [9, 27].
We describe here two cases of inherited prion disease carrying two novel consecutive stop codon mutations in the PRNP C-terminus, resulting in a PrP-CAA in one case, and in an unusual GSS phenotype with many neurofibrillary tangles and relative sparing of the cerebellum in the other.
Patients and methods
Patient consent
In the Netherlands, all CJD autopsies are performed after informed consent, including explicit permission to use tissues for research.
Patient 1
A 55-year-old woman was referred to the neurologist because of increasing cognitive impairment, forgetfulness and decreased concentration over the previous 12 months. The last week before admission, the patient had been markedly affected by headaches and both acoustic and visual hallucinations. On admission, she presented with aphasia, in particular with difficulty in finding words. She was disoriented and showed impaired memory and visuospatial functioning. There were no pyramidal or extrapyramidal signs, myoclonic jerks or cerebellar symptoms. Brain magnetic resonance imaging showed hyperintensity of the white matter, but no atrophy or abnormalities in the basal ganglia. The 14-3-3 test in cerebrospinal fluid was positive. An electroencephalogram (EEG) showed generalized slowing with a typical pattern of periodic synchronous wave complexes. During admission, she developed hyperactive tendon reflexes and apraxia. After 2 months, she was transferred to a nursing home, where she remained for 13 consecutive months. During this period, she became increasingly agitated and subsequently developed signs of Parkinsonism as a result of neuroleptic treatment. She became akinetic and mute with myoclonic jerks towards the end of the disease course. Death occurred at the age of 57 years, 27 months after onset of symptoms.
Of note was the patient’s mother had been diagnosed with “probable CJD” on the basis of comparable symptoms and signs. Death occurred at the age of 75, 18 months after onset of symptoms. Postmortem examination was not performed.
Patient 2
A 42-year-old woman was referred to the neurologist for the evaluation of a slowly progressive hypokinetic rigid syndrome with cognitive decline. For the last two-and-a-half years, she had experienced difficulties with finding words and memory disturbance, and others had also noticed personality changes. Her medical history was otherwise unremarkable. She suffered from a cramping, stiff feeling in her legs and arms, right more than left. On neurological examination, there was a masked face, a clear dysarthria and variably raised muscle tone in arms and legs with slight cogwheeling. All reflexes were normal and eye movements were intact. There were neither hallucinations, nor pyramidal or cerebellar signs. A CT scan of the brain showed moderate atrophy of the left frontal lobe and less severe atrophy of the left temporal lobe. EEG and 14-3-3 test were not performed. SPECT scan of the brain showed hypoperfusion in the left frontal and temporal cortex extending into the parietal cortex. She was clinically diagnosed with frontotemporal dementia. Over the following 3 years her condition gradually deteriorated. She suffered from a tremor in both her right hand and right foot, developed epileptic seizures and became completely dependent on care. During the last 2 months before death, she became mute and the frequency of epileptic seizures and spasms increased. She died at the age of 45 years, 72 months after clinical onset. In her family, one of her father’s sisters had died at the age of 42 years with comparable symptoms.
Neuropathology
The brains from both patients were removed 24 h after death. Samples of tissue from several brain regions of patient 2 were frozen at −80°C, whereas the whole brain from patient 1 was fixed in formalin. A complete neuropathological examination was performed, including gross brain examination and microscopic examination of paraffin-embedded sections from the grey and white matter of all lobes of the brain, central nuclei (caudate, putamen, pallidum and thalamus), cerebellum and brainstem. For histological examination, formalin-fixed sections were decontaminated for 1 h in 98% formic acid and embedded in paraffin. Paraffin-embedded sections were cut at 5 μm and stained routinely with haematoxylin–eosin and combined Luxol-periodic acid-Schiff (PAS) stains. In addition, sections were stained with methenamine silver for the presence of senile plaques. The monoclonal antibody 3F4 (1:400, overnight at 4°C, Signet Laboratories, Dedham, MA, USA) was used for PrP immunohistochemistry. Pretreatment protocols for PrP staining involved antigen retrieval by autoclaving in citric acid buffer pH 6.0 at 121°C for 10 min followed by incubation with Proteinase K (10 μg/mL, for 5 min at room temperature). Immunolabelling was visualized using PowerVision labelled with horseradish peroxidase (Immunologic, Duiven, The Netherlands) and diaminobenzidine as chromogen (DAB, Dako, Carpentaria, CA, USA). Other immunohistochemical stains included antibodies against Aβ (Aβ1–17, 1:400, Dako, Glostrup, Denmark and Aβ17–24 4G8, 1:5,000, Senetek, Napa, CA, USA), phosphorylated tau (AT8 1:250, Innogenetics, Gent, Belgium), 3R tau (RD3, 1:6,400, Upstate, Temecula, CA, USA) and 4R tau (RD4, 1:200, Upstate, Temecula, CA, USA).
Genetic analysis
Genomic DNA was extracted from peripheral blood (patient 1) or from frozen brain tissue (patient 2). Full sequence analysis of the open reading frame (ORF) of PRNP was carried out. The PRNP ORF was initially amplified as previously described [4]. The codon 129 genotype was then examined by digestion with the restriction endonuclease Xce I (Fermentas). Finally, the fragment encoding the PRNP ORF was purified by denaturing high pressure liquid chromatography peak captured and run on a ABI 310 Sequencer as previously described [3].
Biochemical studies
PrPSc was extracted from the temporal, occipital and cerebellar cortices of patient 2 and characterized as previously described [24, 25]. In particular, PrPSc typing was performed both on crude homogenates [25], and after purification of the protein in sarkosyl [24]. The following mouse monoclonal antibodies recognizing different human PrP epitopes were used as described [22]: 3F4 (residues 108–111), 12B2 (residues 89–93), Sha31 (residues 145–152), and SAF60 (residues 157–161). For PrP deglycosylation, denatured proteins were incubated with peptide N-glycosidase F (PNGase F) following the instruction from the manufacturer (New England Biolabs Inc., Beverly, MA, USA).
Results
Genetic analysis
DNA analysis (Fig. 1) revealed two novel mutations (678 C>A in patient 1 and 679 C>T in patient 2), resulting in premature stop codons (Y226X and Q227X). Both patients were heterozygous for methionine and valine at codon 129 and carried the mutation on the valine allele.

Sequence analysis of PRNP of patients 1 and 2. Short segments of PRNP of patient 1 with the Y226X mutation (upper part of figure) and of patient 2 with the Q227X mutation (lower part of figure) are shown. The insertion of either adenine (Y226X) or thymine (Q227X) causes a premature stop codon
Neuropathology
Patient 1 (Y226X mutation)
Gross examination of the brain (weight 1,300 g) was unremarkable. The results of microscopic examination are summarized in Table 1. The main finding was a diffuse and severe amyloid angiopathy involving small to medium-sized arteries and arterioles of the cerebral and cerebellar cortex and leptomeninges (Fig. 2a, c). Amyloid deposition also involved the capillaries, as in CAA type 1 [30], occasionally extending into the adjacent parenchyma, corresponding to dyshoric angiopathy. Anti-PrP antibodies intensely stained the CAA lesions (Fig. 2b, d, g). In addition, there was a faint, scattered, synaptic staining in the grey matter of the cerebral cortex and parenchymal plaques were observed in all brain regions examined. Immunohistochemical stainings with both monoclonal anti-Aβ antibodies (Fig. 2h), Dako 1-17 and 4G8, were negative. Focal, dot-like tau-accumulations were noted around blood vessels with amyloid depositions throughout the cerebral cortex (Fig. 2f) and the hippocampus. Neurofibrillary tangles were absent.

Histopathologic features of PRNP-Y226X. Sections are stained with haematoxylin–eosin (a, c), Luxol-periodic acid-Schiff reaction (e), 3F4 anti-PrP antibody (b, d, g), anti-phosphorylated tau (AT8) (f) and anti-Aβ (h). Original magnification ×100 (d), ×200 (a, b, c, h) and ×400 (e, f, g). Amyloid angiopathy involving small to medium-sized arteries, arterioles (arrow) and capillaries of the cerebral (a, b, g) and cerebellar cortex and leptomeninges (c, d). Spongiform changes in the parietal cortex with amyloid depositions surrounding blood vessels (e). Focal tau accumulations around blood vessels with amyloid depositions in the temporal cortex (f). Absence of staining with anti-Aβ antibodies (h)
Patient 2 (Q227X mutation)
Gross examination of the brain (weight 990 g) showed atrophy of the frontal lobe and caudate nucleus, with marked ventricular dilation. The substantia nigra was slightly pale. Microscopic examination revealed scattered status spongiosus in the second layer of the cerebral cortex. Numerous multicentric and unicentric plaques were present in all layers of the cerebral cortex, with frontotemporal accentuation (Fig. 3a). Plaques were also present in the subcortical nuclei, hippocampus, entorhinal cortex and focally in the subcortical white matter (Table 1). The cerebellum was relatively spared and only showed sparse amyloid depositions (Fig. 3c). Several plaques were located alongside capillaries, but no amyloid angiopathy was seen. The substantia nigra showed marked loss of pigmented neurons. Immunohistochemical staining with anti-tau, anti-RD3 and RD4 antibodies revealed numerous tangles, neuropil threads and also dystrophic neuritis around the plaques (Fig. 3e). The neurofibrillary tangles and neuropil threads were seen throughout the cerebral cortex (Fig. 3f), including the occipital lobe and striate area, which in Alzheimer’s disease would correspond to Braak stage VI [1]. The plaques themselves showed positive staining with mAbs against PrP (Fig. 3b, d), whereas stainings with anti-Aβ antibodies and methenamine silver were negative (data not shown).

Histopathologic features of PRNP-Q227X. Sections are stained with haematoxylin–eosin (a), Luxol-periodic acid-Schiff reaction (c), 3F4 anti-PrP antibody (b, d) and anti-tau antibody RD4 (e, f). Original magnification ×200 (a, b, c, d) and ×400 (e, f). Presence of numerous multicentric and unicentric plaques in the frontal cortex (arrows) (a, b). Relative sparing of the cerebellum with sparse amyloid depositions (arrow) (c, d). Dystrophic neurites surrounding PrPSc plaques and neuropil threads (e) and neurofibrillary tangles (f)
Biochemical studies
Western blot analysis of PK-treated brain homogenates from patient 2 demonstrated the presence of a PrPSc fragment with a molecular weight of approximately 7 kDa. The analyses of partially purified (P3) PrP preparations in sarkosyl showed that the fragment was already visible in the PK-untreated preparations (Fig. 4). Furthermore, there were neither PrPSc fragments in the 19–21 kDa range (i.e. type 1 or type 2) nor protease-sensitive abnormally insoluble full-length PrPSc (i.e. full-length PrP, which remains visible in the P3 fraction but disappears completely after PK digestion), as previously described in GSS P102L [24] (Fig. 4). The 7 kDa fragment was detected by all antibodies directed to epitopes located between PrP residues 89 and 112, while weak staining was observed with the antibody Sha31 (residues 145–152) and absent with the antibody SAF60 (residues 157 and 161) (data not shown). Furthermore, the electrophoretic mobility of the fragment did not change after N-linked deglycosylation (Fig. 4). Thus, epitope mapping combined with relative molecular mass analyses indicated that the fragment is unglycosylated and truncated at both the N- and C-terminal ends (around residues 145–150), just as the previously described fragment characterized in GSS cases carrying different PRNP mutations [26].

PrPSc typing and characterization of PRNP-Q227X. Immunoblot analysis of partially purified, detergent insoluble, abnormal PrP obtained from the frontal cortex of a sCJDVV2 case (lanes 1 and 3) and patient 2 (lanes 2, 4, 5). Samples are without (lanes 1, 2) or with a Proteinase K (PK) digestion step (lanes 3–5). The sample run in lane 5 was also deglycosylated with PNGaseF. Immunoblot was probed with 3F4. The smears in lanes 2, 4 and 5 likely represent incompletely solubilized (i.e. not monomeric) PrP-amyloid. Approximate molecular masses are in kilodaltons
Discussion
We describe two female Dutch patients with novel and almost identical stop codon mutations in the C-terminal part of PRNP, resulting in either case in hereditary prion protein amyloidoses, but with strikingly different clinicopathological phenotypes. The Y226X mutation was associated with pure PrP-CAA, also involving small arterioles and capillaries, without any neurofibrillary tangles, whereas the Q227X mutation resulted in a GSS disease phenotype with numerous multicentric amyloid plaques and neurofibrillary lesions in the cerebral grey matter and, remarkably, absence of PrP-CAA. Of note, the latter case showed relative sparing of the cerebellum, a rather unique feature in GSS, which has been only described before in patients carrying the A117V-129V haplotype [19]. This feature correlated with an atypical clinical phenotype for GSS characterized by progressive dementia with aphasia and personality changes without any evidence of ataxia or pyramidal signs. A clinical phenotype resembling frontotemporal dementia has been only reported before in GSS patients carrying the Q217R-129V genotype [33], and in familial CJD subjects carrying the T183A-129M haplotype [21].
Neurofibrillary degeneration is not uncommon in GSS with long disease duration, having been described in patients harbouring the A117V-129V, F198S-129V, D202N-129V, P105L-129V and Q217R-129V haplotypes [8, 10, 20, 26, 34]. Furthermore, it has been found in association with the Y145X, Y163X and Q227X mutations (Table 2), as well as in variant CJD, a bovine-derived acquired form of prion disease also characterized by extensive amyloid deposition [12, 17]. Interestingly, our patient with the Y226X mutation only showed focal tau accumulations in the absence of tangles, possibly because of the relatively short disease duration in this case. The mechanism of neurofibrillary degeneration in prion diseases still remains a largely unexplained phenomenon, although the pathological features in our patient with the Q227X mutation support previous findings indicating that tau accumulation and tangle formation almost invariably develop in the presence of parenchymal PrP amyloid formation, paralleling what occurs in Alzheimer’s disease [15]. These findings suggest a relationship between amyloid fibrils and the formation of abnormal tau deposits, irrespective of the chemical type of amyloid, possibly contradicting the hypothesis that monomeric molecules, such as Aβ, are the direct cause of tau accumulations [14].
The term CAA is used to describe the pathological process during which an amyloid protein is progressively deposited in blood vessel walls with subsequent degenerative vascular changes [31]. Deposition of Aβ in blood vessels, by far the most common cause of CAA in humans, has been also documented in CJD and GSS [13, 16, 18, 29]. In contrast, vascular deposits of PrP-amyloid are very rare and usually co-localize with Aβ [23, 32]. PrP-CAA, without co-localization of Aβ, has thus far only been described in a single Japanese patient carrying a Y145X stop codon PRNP mutation [9] and more recently in a patient carrying a Y163X mutation [27] (Table 2). Although this observation strongly suggests a link between the formation of truncated PrPSc on one hand and PrP-CAA on the other, its interpretation is difficult being limited to single cases only. A third patient with a similar stop-codon (Q160X) also presented with early onset dementia but, unfortunately, information on the pathological phenotype in this case is still lacking [6].
The Y226X and Q227X mutations differ from the previously reported truncating mutations since they result in two C-terminally truncated proteins lacking almost only the GPI anchor. Strikingly, the two proteins only differ in a single tyrosine residue at the C-terminus. Data obtained from cell culture models indicate that glycosylated anchorless full-length PrPC has a longer half-life than normal PrPC, is predominantly secreted and does not localize to the plasma membrane, although it may remain associated to some intracellular membranes without a transmembrane topology [2]. Chesebro and colleagues [5] have recently generated transgenic mice expressing full-length prion protein lacking the GPI anchor. Interestingly, when infected with scrapie these mice show extensive PrPSc depositions in the form of amyloid plaques, rather than the usual non-amyloid form of PrPSc, but lack clinical signs [5]. In contrast, scrapie-infected transgenic mice heterozygous for both wild-type and anchorless PrP, developed clinical signs and died faster than the wild-type mice, reflecting a situation similar to that in our patients.
Our observations confirm that the absence of a GPI-anchor in PrPC predisposes to amyloid plaque formation also in humans and therefore validate this animal model. Interestingly, the “GPI-anchorless” transgenic mice also showed perivascular depositions of PrP, similar to the Y145X, Y163X and Y226X mutations in familial prion disease. Why, in contrast, the Q227X PrP does not result in a prion CAA in the patient described here remains obscure, although it could be hypothesized that the two C-terminal tyrosines somehow prevent or influence the passage of the protein through the vessel wall. Further studies are required to elucidate these pathogenetic mechanisms in patients with pure PrP-CAA and stop codon mutations in PRNP.
References
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404
Campana V, Caputo A, Sarnataro D, Paladino S, Tivodar S, Zurzolo C (2007) Characterization of the properties and trafficking of an anchorless form of the prion protein. J Biol Chem 282:22747–22756
Capellari S, Cardone F, Notari S et al (2005) Creutzfeldt-Jakob disease associated with the R208H mutation in the prion protein gene. Neurology 64:905–907
Capellari S, Vital C, Parchi P et al (1997) Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology 49:133–141
Chesebro B, Trifilo M, Race R et al (2005) Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308:1435–1439
Finckh U, Muller-Thomsen T, Mann U et al (2000) High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 66:110–117
Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66:213–239
Ghetti B, Dlouhy SR, Giaccone G et al (1995) Gerstmann-Straussler-Scheinker disease and the Indiana kindred. Brain Pathol 5:61–75
Ghetti B, Piccardo P, Spillantini MG et al (1996) Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci USA 93:744–748
Ghetti B, Tagliavini F, Masters CL et al (1989) Gerstmann-Straussler-Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology 39:1453–1461
Ghetti B, Tagliavini F, Takao M, Bugiani O, Piccardo P (2003) Hereditary prion protein amyloidoses. Clin Lab Med 23:65–85, viii
Giaccone G, Mangieri M, Capobianco R et al (2008) Tauopathy in human and experimental variant Creutzfeldt-Jakob disease. Neurobiol Aging 29:1864–1873
Gray F, Chretien F, Cesaro P et al (1994) Creutzfeldt-Jakob disease and cerebral amyloid angiopathy. Acta Neuropathol 88:106–111
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8:101–112
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Ikeda SI, Yanagisawa N, Allsop D, Glenner GG (1994) Gerstmann-Straussler-Scheinker disease showing beta-protein type cerebellar and cerebral amyloid angiopathy. Acta Neuropathol 88:262–266
Ironside JW (2000) Pathology of variant Creutzfeldt-Jakob disease. Arch Virol Suppl 16:143–151
Keohane C, Peatfield R, Duchen LW (1985) Subacute spongiform encephalopathy (Creutzfeldt-Jakob disease) with amyloid angiopathy. J Neurol Neurosurg Psychiatry 48:1175–1178
Mallucci GR, Campbell TA, Dickinson A et al (1999) Inherited prion disease with an alanine to valine mutation at codon 117 in the prion protein gene. Brain 122(Pt 10):1823–1837
Mohr M, Tranchant C, Steinmetz G, Floquet J, Grignon Y, Warter JM (1999) Gerstmann-Straussler-Scheinker disease and the French-Alsatian A117V variant. Clin Exp Pathol 47:161–175
Nitrini R, Teixeira da Silva LS, Rosemberg S et al (2001) Prion disease resembling frontotemporal dementia and parkinsonism linked to chromosome 17. Arq Neuropsiquiatr 59:161–164
Notari S, Strammiello R, Capellari S et al (2008) Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem 283:30557–30565
Paquet C, Privat N, Kaci R et al (2008) Cerebral amyloid angiopathy with co-localization of prion protein and beta-amyloid in an 85-year-old patient with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol 116:567–573
Parchi P, Chen SG, Brown P et al (1998) Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc Natl Acad Sci USA 95:8322–8327
Parchi P, Notari S, Weber P et al (2009) Inter-laboratory assessment of PrPSc typing in Creutzfeldt-Jakob disease: a Western blot study within the NeuroPrion Consortium. Brain Pathol 19:384–391
Piccardo P, Dlouhy SR, Lievens PM et al (1998) Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 57:979–988
Revesz T, Holton JL, Lashley T et al (2009) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118:115–130
Tagliavini F, Prelli F, Ghiso J et al (1991) Amyloid protein of Gerstmann-Straussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. EMBO J 10:513–519
Tateishi J, Kitamoto T, Doh-ura K, Boellaard JW, Peiffer J (1992) Creutzfeldt-Jakob disease with amyloid angiopathy: diagnosis by immunological analyses and transmission experiments. Acta Neuropathol 83:559–563
Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del TK, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293
Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke 18:311–324
Vogelgesang S, Glatzel M, Walker LC, Kroemer HK, Aguzzi A, Warzok RW (2006) Cerebrovascular P-glycoprotein expression is decreased in Creutzfeldt-Jakob disease. Acta Neuropathol 111:436–443
Woulfe J, Kertesz A, Frohn I, Bauer S, George-Hyslop PS, Bergeron C (2005) Gerstmann-Straussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia. Acta Neuropathol 110:317–319
Yamada M, Itoh Y, Inaba A et al (1999) An inherited prion disease with a PrP P105L mutation: clinicopathologic and PrP heterogeneity. Neurology 53:181–188
Acknowledgments
The tissue was obtained with help from The Netherlands Brain Bank (NBB), the Netherlands. We are indebted to Dr. W. Kamphorst of the NBB for his expertise and excellent support. The monoclonal antibodies Sha31 and SAF60 were a generous gift from Dr. Jacques Grassi (Service de Pharmacologie et d′Immunologie, CEA/Saclay, France). This work was supported in part by the Gino Galletti Foundation, the Rijksinstituut voor Volksgezondheid en Milieu and the Koninklijke Nederlandse Akademie van Wetenschappen.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Jansen and P. Parchi contributed equally to this article.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jansen, C., Parchi, P., Capellari, S. et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP . Acta Neuropathol 119, 189–197 (2010). https://doi.org/10.1007/s00401-009-0609-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0609-x