
Abstract
Bacterial cellulose (BC) exhibits unique properties such as high purity compared to plant-based cellulose; however, commercial production of BC has remained a challenge, primarily due to the strain properties of cellulose-producing bacteria. Herein, we developed a functional and stable BC production system in genetically modified (GM) Escherichia coli by recombinant expression of both the BC synthase operon (bcsABCD) and the upstream operon (cmcax, ccp Ax). BC production was achieved in GM HMS174 (DE3) and in GM C41 (DE3) by optimization of the culture temperature (22 °C, 30 °C, and 37 °C) and IPTG concentration. BC biosynthesis was detected much earlier in GM C41 (DE3) cultures (3 h after IPTG induction) than those of Gluconacetobacter hansenii. GM HMS174 (DE3) produced dense fibres having a length of approximately 1000–3000 μm and a diameter of 10–20 μm, which were remarkably larger than the fibres of BC typically produced by G. hansenii.
Similar content being viewed by others

Introduction
Bacterial cellulose (BC) is a biomaterial with unique properties and receiving a growing attention for industrial applications. BC has many advantages over plant-based cellulose such as high purity, which allows a purification process that is straightforward [1, 2]. In addition, it exhibits excellent mechanical and chemical properties including a high crystallinity, high tensile strength, high water holding capacity, high modulus of elasticity, and biocompatibility [3,4,5]. These properties enable BC to be used in a wide range of industrial applications such as medical products, electronics, paper, food, and cosmetics. BC provides all requirements for wound dressing material such as high porosity and providing a barrier against infection [6, 7].
Currently, plant-derived cellulose has a significant economic value with $200 billion per year worldwide. In 2016, the price of wood pulp, the main source of plant-based cellulose, was as low as $0.96 per kg. However, the extraction process of cellulose from wood causes both environmental hazards and requires high energy [8]. Recent developments have demonstrated the use of cellulose-producing bacteria to alleviate the demand on plant cellulose; nonetheless, the average cost of bacterial cellulose remains at $35 per pound ($77 per kg), which is still 80 times higher than plant-derived cellulose [9].
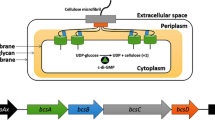
Gluconacetobacter hansenii (formerly known as Gluconacetobacter xylinus) is the most efficient BC producer among cellulose producer species [10]. Biosynthesis of cellulose is encoded by the bacterial cellulose synthesis (bcs) operon in G. hansenii. This operon is essential for a complete BC biosynthesis and consists of four subunits (bcsA, bcsB, bcsC, and bcsD). The first gene of the bcsABCD operon, bcsA, encodes the catalytic subunit of cellulose synthase. The second gene, bcsB, encodes the regulatory subunit of cellulose synthase that binds to c-di-GMP, which plays an important role as the second messenger to activate the cellulose synthesis process [11]. Although the functions of bcsC and bcsD have not been completely clarified yet, it has been suggested that bcsC is responsible for the formation of pores in the membrane to secrete cellulose, as it encodes a protein similar to the proteins involved in the formation of membrane channels or pores [12]. Inactivation of bcsA, bcsB, or bcsC completely inhibits BC synthesis, indicating that these genes are crucial for the biosynthesis. In contrast, the inactivation of bcsD reduces BC production by 40%, which suggests that bcsD is involved in the crystallization of cellulose into nanofibrils [12,13,14].
Immediately upstream of the bcs operon, two more genes are encoded: cmcax and ccpAx. The endo-β-1,4-glucanase enzyme, which has cellulose hydrolyzing activity and enhances cellulose synthesis, is encoded by cmcax [15,16,17]. Based on an electron microscopic analysis, it has been suggested that endo-β-1,4-glucanase could have an influence on the ribbon assembly. CcpAx is also essential for BC production and plays a critical role in locating the bcs complex on the cell membrane [15]. It has been suggested that the functions of CcpAx could be related to extracellular transport of cellulose chains from the sites of polymerization within the cell membrane and the crystallization of cellulose fibrils [18, 19].
The progress made in BC production has been extensively discussed and the challenges of commercial scale BC production have been reviewed in our previous study [20]. Several strategies including the designing of advanced reactors and utilization of various carbon sources have been developed for enhanced production of BC [21]. So far, the most extensively studied bacteria are G. hansenii. However, the challenges in BC production are mainly related to the strain properties of G. hansenii: (1) slow growth compared to many other bacteria (such as E. coli), (2) low productivity, (3) susceptibility to culture conditions, and (4) the spontaneous mutation of cellulose-producing bacteria into cellulose non-producing mutants. Although BC biosynthesis is a highly complex process, novel cellulose-producing strains needs to be developed for enhanced BC production [21, 22].
Escherichia coli, as a well-characterized cell factory, represents a strong candidate for accomplishing BC production in new platforms due to its rapid growth kinetics. E. coli B and E. coli K12 lines are used almost equally as hosts for recombinant protein production. E. coli BL21, a B line derivative, is one of the most commonly used strains in research. This is due to the fact that BL21 strains are deficient in the production of proteases Lon and OmpT, leading to reduced recombinant protein degradation and higher yields [23,24,25]. HMS174 (DE3), an engineered successor of the E. coli K-12 wild-type, provides a recA1 mutation in a K-12 background. This mutation has a positive effect on plasmid stability [26]. After lysogenization with the DE3 prophage, it became a popular strain for the overexpression of heterologous proteins [27]. However, overexpression of membrane-associated proteins is often toxic to the cells and this can inhibit biomass formation and severely reduce product yields. C41 (DE3), derived from BL21 (DE3), allows high expression of a wide variety of toxic proteins that have previously been difficult or even impossible to express in bacteria [28,29,30]. The mutations of this strain that were reported to be related to lacUV5 operon or expression of T7 RNA polymerase prevented cell death associated with expression of many toxic recombinant proteins [31].
In this study, simultaneous expression of the bacterial cellulose synthase operon (bcsABCD) and its upstream operon (cmcax and ccp Ax) was performed by co-expression plasmids, pBCS and pCMP, respectively. The design of the vector constructs compatible with different E. coli strains has allowed us the BC production to be examined in these strains previously mentioned: BL21 (DE3), HMS174 (DE3), and C41 (DE3). Plasmid DNA maintenance and replication commonly create a metabolic burden in E. coli and the response of cells under this energy limiting condition is extremely complex, leading to the alterations of physiology and metabolism of host cells [32]. In return, this results in lower yields of plasmid DNA, particularly a decrease in process outcome. The maintenance of recombinant plasmids is greatly affected by culture conditions [33]. Herein, we developed genetically engineered strains to investigate BC biosynthesis in E. coli platforms and examined the effect of temperature and inducer concentration on the plasmid stability, the cell growth, and the BC biosynthesis of these strains.
Materials and methods
Bacterial strains and plasmids
Bacterial strains and plasmids used in this study are described in Table 1.
Construction of expression vectors
The co-expression system was developed using backbones of two compatible Duet Vectors (Novagen): petDuet-1 and pCDFDuet-1. pETDuet-1 has the ColE1 replicon and bla gene for ampicillin/carbenicillin resistance, and pCDFDuet-1 has the CloDF13 replicon and aadA gene for streptomycin resistance. Each of these vectors was designed to express multiple genes and each gene was controlled by a separate T7lac promoter that provides a tight control of the expression by IPTG (isopropyl β-D-1-thiogalactopyranoside) induction. Each expression unit has own optimal ribosomal binding side located downstream of the promoter. The sequences for cmcax, ccpAx, bcsA, bcsB, bcsC, and bcsD of G. hansenii ATCC 53582 were obtained from literature. Access numbers for the upstream region (cmcax and ccpAx) and bcsABCD operon are AB091058 and X54676, respectively [12, 34]. In an effort to enhance the efficiency and accuracy of translation, these target sequences were optimized according to codon usage of E. coli by Genewiz, Inc.
Cloning was performed by the restriction digest procedure; ccpAx was cloned into pCDFDuet-1 via 5′ using BglII and 3′ XhoI as ligation sites, generating the intermediate construct ccpAx_pCDFDuet-1 for the next step. Following that, cmcax was inserted into ccpAx_pCDFDuet-1 via 5′ BamHI and 3′ Hindlll as ligation sites to create the final construct pCMP (Fig. 1a). The sequences of bcsA and bcsB were co-inserted into pETDuet-1 via 5′ BamHI and 3′ AflII to generate the intermediate construct. After that, bcsC and bcsD were co-inserted into this intermediate construct via 5′ FseI and 3′ XhoI to generate pBCS (Fig. 1b). E. coli strains were then transformed with the constructed plasmids by chemical co-transformation. The selection of transformations was accomplished by plating the cells onto LB agar plates supplemented by 25 mg/ml carbenicillin and 25 mg/ml streptomycin (Calbiochem).

Strategy used for the construction of pCMP (a) and pBCS (b). pCMP expression unit for the upstream operon (cmcax and cppAx) with T7 promoter and lac operator, streptomycin resistance region, CloDF13 replicon (CDF ori), lac I region. pBCS expression unit for bcs operon with T7 promoter and lac operator (bcsA, bcsB, bcsC, and bcsC), f1 replicon, ampicillin resistance region, pBR322-derived ColE1 replicon, lac I region
Verification of the inserted fragments in pCMP and pBCS
To verify whether the target fragments were cloned in the correct orientations, the restriction enzyme digestion tests were performed for both plasmids. BamH I and Hind III cleaved pCMP at two edges of the sequences encoding cmcax, and resulted in two linear fragments with the length of 1042 and 4819 bp. Mlu I cleaved the pBCS from two sites that were located on the sequences of bcsC and lacI genes, while Xho I cleaved it at the end of the sequence encoding bcsD on this plasmid. These generated three linear fragments with length of 3071, 4304, and 7219 bp. The fragments are visualized on agarose gel to detect the DNA bands corresponded to their size. Unrestricted plasmids were also visualized to verify the total size, which also showed bands corresponding to the total size of pCMP (5.9 kbp) and pBCS (14.6 kbp) (Online Resource, Fig. ESM1).
Fermentation of genetically modified (GM) E. coli cells
GM E. coli pre-cultures were cultured in flasks at 37 °C and 180 rpm, in Lysogeny Broth (LB) medium [10 g/L tryptone (Sigma), 5 g/L yeast extract (Sigma), 10 g/L NaCl (Sigma)]. The culture media were supplemented by both streptomycin (50 mg/ml) and carbenicillin (50 mg/mL) for the maintenance of pCMP and pBCS, respectively. Although pBCS carries a β-lactamase (bla) gene, which confers resistance to ampicillin, also confers resistance to carbenicillin. Carbenicillin was used in GM cultures, because it tends to be more stable than ampicillin. 4 g/L glucose (Sigma) was used as a culture supplement not only to prevent basal expression of recombinant proteins prior to the IPTG induction but also as a carbon source for BC biosynthesis. The simultaneous expression of recombinant BC proteins in genetically modified (GM) E. coli strains was initiated by inducing the strong T7lac promoter with IPTG. Following the IPTG induction, the culture temperature was shifted to 30 °C or 22 °C, if necessary. The effect of IPTG induction on plasmid stability and BC production in each GM cell line was examined at different culture temperatures (22 °C, 30 °C, and 37 °C). The experimental conditions used in this study are summarized in Online Resource, Table ESM1. Optical density (OD) of the cells was measured at 600 nm using spectrophotometer to monitor cell growth.
Determination of Dry Cell Weight (DCW)
GM cells were cultured overnight in LB medium at 180 rpm and 37 °C. The cells were then centrifuged at 5000 x g for 10 min and washed with distilled water for three times, and 50 ml of cells were re-suspended in 100 ml of distilled water and dried at 80 °C. Cell dry weight (mg/ml) for each sample was determined. The remaining 50 mL of cells were used to prepare diluted cell culture samples; OD600 nm values of the samples were obtained a spectrophotometer. A calibration curve was prepared by plotting optical density vs. DCW (g/L).
Plasmid stability test
Plasmid-harboring GM cells were distinguished from the total viable cells using the plasmid-borne antibiotic resistance. The diluted samples were plated on selective (supplemented with 50 mg/mL streptomycin and 50 mg/mL carbenicillin) and non-selective (without antibiotics) LB plates. The colony forming units (cfu) were counted after cultivation at 37 °C overnight [30]. The percentage of plasmid-harboring colonies among total viable colonies on the plates represents the plasmid stability.
q-PCR assays
The culture samples were collected at specific time intervals by centrifugation at 10,000×g for 10 min at 4 °C. The samples were treated with RNAprotect Bacteria Reagent (Qiagen) to stabilize RNA before extraction of RNA. RNeasy Mini Kit (Qiagen) was used to purify the total RNA. The QuantiTect Reverse Transcription (Qiagen) protocol was followed for the synthesis of cDNA. q-PCR analysis was conducted by following QuantiTect SYBR Green Protocol [35]. Total reaction volume was 20 µL, which contained 12.5 mL SYBR Green PCR Master Mix, 0.5 mL of forward primer (0.3 µM) and 0.5 mL of reverse primer (0.3 µM) (Invitrogen, UK), 1 µl of cDNA (100 ng), and 5.5 mL of sterile water. q-PCR analysis was conducted in duplicate measurements for each time point. Gradient PCR was performed to select the annealing temperature for the primer pairs. A melting curve was generated for each reaction to ensure the specificity of each PCR product. serC was used as housekeeping gene [36]. Negative controls (no template controls) were used to serve as a general control for nucleic acid contamination and primer dimer formation. The un-induced cells right before the IPTG induction (T = 0) as positive controls were used as positive control to monitor the response of the GM cells to IPTG. Then, the data were normalised to the expression levels in positive controls.
The denaturation temperature was set at 95 °C for 3 min, followed by 95 °C for 20 s. In the next step, different annealing temperatures were employed in each column of the thermocycler. The annealing temperatures ranged from 58 to 60 °C for 30 s, and 72 °C for 30 s. The amplification last for 50 cycles. The oligonucleotide sequences used in this study can be found in Online Resources, Table ESM2.
Extraction of proteins and SDS PAGE analysis
Periplasmic proteins were extracted by the osmotic shock procedure [37]. In cases where induction with IPTG was needed, GM E. coli cells were grown until the optical density at 600 nm of the cell culture reached 1.0 and 0.2 mM IPTG was added to culture medium. Grown cells were harvested by centrifugation for 10 min at 5000 x g, 4 °C and re-suspended in 20% sucrose solution (w/v) in 30 mM Tris–HCl (pH 8.0) with 1 mM ethylenediaminetetraacetic acid (EDTA). Following a second centrifugation at 10,000×g for 20 min, the pellet was rapidly re-suspended in ice-cold 5 mM MgCl2 and gently incubated for 20 min. Cells were removed from the periplasmic protein extract by centrifugation at 10,000×g, 4 °C for 20 min. Supernatant contained the periplasmic proteins.
The insoluble fractions were extracted by treatments with BugBuster Reagent (Sigma). rLysozym solution was added to a final concentration of 1 KU/ml. The suspension was centrifuged at 5000g for 15 min at 4 °C to collect the insoluble fraction. After re-suspension of the insoluble fractions in half of the original culture volume of 1:10 diluted BugBuster, the sample was again mixed by vortex and centrifuged as in previous step. This step was repeated a few times. The sample was centrifuged at 16,000g for 15 min at 4 °C and the supernatant removed. Protein concentrations of the extracted proteins were determined using the method described by Bradford (1976) prior to the SDS PAGE analysis [38].
Protein samples were mixed with 2× Sample Buffer containing SDS at a 1:1 ratio and boiled for 5 min to denature the proteins. The marker and the samples were loaded into the wells of 12% SDS polyacrylamide gels. Separation of proteins based on size was achieved by allowing them to run at 120 V for approximately 90 min.
Recovery of bacterial cellulose
Following 24 h culturing, the cultures first filtered to recover the BC particles. The samples were treated with 1% NaOH solution (w/v) at 70 °C for 20 min to remove bacteria and other impurities. The samples were then rinsed with deionized water several times until neutral pH and then dried in microcentrifuge tubes at 50◦C to constant weight to obtain dry weight.
Scanning electron microscopy (SEM) analysis
The morphology and the microstructural features of samples produced by GM E. coli strains were investigated using the field-emission scanning electron microscope (FESEM) LEO 1525 (Zeiss, Germany) operating at 5 kV. Prior to analysis, samples were dried until constant weight and placed onto double-sided carbon tape mounted onto an aluminum stub. They were gold coated for 2 min at 20 mA using an Emitech K575X Peltier (Ashford, UK) cooled sputter coater.
Fourier transform infrared (FTIR) analysis
FTIR spectroscopy was used to characterize cellulose samples produced by bacteria. ATR-IR spectra were recorded using a Spectrum One FTIR-spectrometer (Perkin Elmer, Massachusetts, USA). The spectra were collected at a resolution of 2 cm−1 in the range of 600 and 4000 cm−1.
Statistical analysis
Each fermentation was repeated three independent times and each analysis was replicated at three times. Statistical analysis was performed using one-way analysis of variance (ANOVA) and statistical significance was assigned to P < 0.05.
X-ray diffraction (XRD) analysis
Crystallographic analysis of the samples was conducted using a Philips X’Pert Pro diffractometer (PANalytical, The Netherlands) equipped with a Nickel-filtered Cu-Kα radiation (λ = 1.5406 Å). The operating voltage and current were 40 kV and 40 mA, respectively. The diffraction spectra were collected over a range of 2θ values from 5° to 40° in increments of 0.04°. The relative crystallinity index was calculated by Segal’s method [39]:
where I 002 is the peak intensity corresponding to the (110) plane and Iam is the peak intensity of amorphous fraction at 2θ = 18°.
Results
Optimization of IPTG concentration and temperature for BC biosynthesis
In plasmid-based expression systems, plasmid replication and maintenance can be easily affected by the inducer concentration and the fermentation temperature. Consequently, these factors can severely affect the process outcome. Appearance of plasmid-free cells is not totally prevented even when antibiotic selection is used, as the concentration of antibiotics often decreases during long-term cultivation as a result of dilution and/or enzymatic degradation by the growing cells [30, 40,41,42,43].
All IPTG-induced groups of GM BL21 (DE3) exhibited a dramatic decrease in plasmid stability compared to un-induced GM cells within 1 h after the induction at both 37 °C and at 30 °C (P < 0.05) (Online Resource, Fig. ESM2, a&b). The use of elevated growth temperatures (> 30 °C) has been previously described as one of the reasons for plasmid instability [44]. However, the final plasmid stability of the induced cells was calculated to be only 5% at 22 °C and production of extracellular BC was not detected in GM BL21 (DE3) under any conditions (Online Resource, Fig. ESM2, c).
When induced GM HMS174 (DE3) cells were cultured at 30 °C, the plasmid stability was significantly improved compared that of 37 °C (Fig. 2a, b). The final percentage of plasmid-bearing cells induced by 0.025 mM IPTG and 0.2 mM IPTG was determined to be 97 and 82%, respectively. Nevertheless, BC production was not detected at 30 °C. The plasmid stability in this GM strain remained above 85% at 22 °C (0.025 mM IPTG) and BC formation was detected with a concentration of 23.0 ± 0.02 mg/L (Fig. 2c). This suggested that the effect of the metabolic stress caused by recombinant protein expression was eliminated by lowering both the culture temperature and the IPTG concentration. Therefore, BC biosynthesis in GM C41 (DE3) was investigated at 22 °C initially (Fig. 2f). The GM C41 (DE3) cells induced by 0.2 mM IPTG produced 8.5 ± 0.03 mg/L cellulose (83% plasmid-bearing), while those induced by 0.025 mM IPTG produced 13.5 ± 0.02 mg/L (96% plasmid-bearing). As C41 (DE3) has been proved previously that it was a superior strain in overexpression of membrane proteins and plasmid stability compared to other E. coli strains, higher IPTG concentrations were also examined [28,29,30]. At 30 °C, BC production in the cultures induced by 0.05 mM IPTG and by 0.2 mM IPTG (both > 90% plasmid-bearing) was 21.0 ± 0.02 mg/L and 31.1 ± 0.03 mg/L, respectively. Increasing the concentration of IPTG to 1.0 mM did not stimulate BC production, because the plasmid stability dropped dramatically (Fig. 2e). At 37 °C, the final plasmid stability was calculated to be higher than 93% in both of the induced cultures (Fig. 2d). However, BC production was not observed in any of the GM C41 (DE3) cells cultured above 30 °C or induced by above 0.2 mM IPTG, proving that high IPTG concentrations/temperatures contribute to the stress level of the host.

Effect of IPTG induction on plasmid stability in HMS174 (DE3) at 37 °C (a), 30 °C (b), and 22 °C (c), and in GM C41 (DE3) at 37 °C (c), 30 °C (d), and 22 °C (f). The dashed arrows indicate IPTG addition
The growth kinetics of the induced GM lines was slightly slower compared to the un-induced GM lines at 37 °C. However, lowering the culture temperature (to 30 or 22 °C) closed the gap between the growth of the un-induced and the induced groups (data not shown). Dry cell weights (DCW) and product concentrations are presented in Table 2 under the conditions that BC production was detected. GM C41 (DE3) was identified as the best performer, as it exhibited the highest productivity with 34.3 ± 0.22 mg cellulose/g cells.
Effect of IPTG concentration on the expression of cmcax, ccpAx, and bcsABCD
Confirmation of the expression of the recombinant genes (cmcax, ccpAx, bcsA, bcsB, bcsC, and bcsD) in GM cells was performed by q-PCR analysis. For GM BL21 (DE3), all target genes highly expressed even 15 min after IPTG induction (Online Resource, Fig. ESM3, a&b). The gene expression levels of GM BL21 (DE3) induced by 0.2 mM IPTG were much higher than those induced by 0.025 mM IPTG (Online Resource, Fig. ESM3, c). For an effective expression of membrane proteins, transcription of expression plasmids should be induced slowly with low concentrations of IPTG to prevent accumulating of inactive target proteins [45,46,47,48]. Elevated levels of gene expression in GM BL21 (DE3) suggest that BC may not be detected as a result of aggregation of the encoded proteins as inclusion bodies. The expression of recombinant genes in GM BL21 (DE3) was detected even after the dramatic decrease in plasmid stability (18 h). This could be because the GM cells was retaining only one of the plasmids, which would not allow the expression of the complete target sequence in a single cell for the successful biosynthesis of BC.
Expression of the recombinant genes in GM HMS174 (DE3) was investigated under culture conditions in which BC was detected (0.025 mM IPTG, 22 °C). In contrast to the q-PCR data obtained for BL21 (DE3), the gene expressions in GM HMS174 (DE3) were observed 3 h after induction at lower but significant levels (between 1.41 and 2.71 fold), which is more desirable to allow the expression of target proteins in an active form (Online Resource, Fig. ESM4, a).
Under the conditions that the highest BC production detected (30 °C, 0.05 mM IPTG) in GM C41 (DE3), the gene expressions 3 h after IPTG induction were as low as (0.25–2.12 fold) those detected in GM HMS174 (DE3) (Online Resource, Fig. ESM4, b). After 18 h of induction, bcs genes expressed between 1.61- and 2.19-fold, while upstream genes cmcax and ccpAx expressed 5.02- and 3.32-fold, respectively. All target genes expressed at higher levels in the presence of 1.0 mM IPTG compared to those expressed in the presence of 0.05 mM IPTG as expected (Online Resource, Fig. ESM4, c).
SDS–PAGE analysis of bcs proteins
BC is synthetized on the cytoplasmic side of the inner membrane and is transported through the periplasm before being secreted into the extracellular space [49, 50]. Therefore, bcs proteins are located in periplasm or in cell membrane [19, 49,50,51,52,53]. Membrane proteins are difficult to solubilize and often found in insoluble protein fraction. Therefore, both periplasmic and insoluble fractions of GM cells were analyzed by SDS PAGE. Two protein bands corresponding to the sizes of BcsC and BcsD were detected in the insoluble fraction of GM BL21 (DE3), while the other proteins were detected in the periplasmic fraction (Fig. 3a, b). Membrane proteins are often difficult to solubilize and most insoluble protein fractions contain membrane proteins along with inclusion bodies [54, 55] Therefore, it is difficult to predict whether BcsC (an outer membrane protein) detected in insoluble fraction was in a form of aggregation or properly folded. Detection of BcsD in the insoluble fraction could be the reason for GM BL21 (DE3) not being able to produce BC, as BcsD is a periplasmic protein that is responsible for the crystallization of BC.
CcpAx and Cmcax were detected slightly above their size region (4 kDa) due to the additional sequences (105 bp including His-tag for cmcax and 96 bp for cppAx) on the cloning sites of pCMP in upstream of these proteins after translation starts. A protein band corresponding to the size of BcsC, an outer membrane protein, was detected in the insoluble fraction of GM HMS174 (DE3) and GM C41(DE3), whereas the other proteins were detected in periplasmic fraction of both (Fig. 3c–f). Clear bands corresponding to the target proteins were detected when GM cells were induced with 0.2 mM IPTG. However, SDS–PAGE analysis did not reveal a significant difference in between the induced fractions and un-induced fractions in the conditions where lower concentrations of IPTG (0.025, 0.05 mM) resulted into BC synthesis (data not shown). The concentration of target proteins could be below the detection limit of SDS–PAGE analysis due to the slow induction, but still well enough to cause the activity.

Periplasmic (a) and insoluble (b) fractions of native BL21 and GM BL21 (DE3) induced by 0.2 mM IPTG. For a&b, Line 1 shows the molecular weight marker, Lines 2, 3, 4 show the extracts of native cells, un-induced GM cells, and induced GM cells (18 h), respectively. Periplasmic (c) and insoluble (d) fractions of native HMS174 (DE3) and GM HMS174 (DE3) induced by 0.2 mM IPTG. For c&d, Line 1 shows the molecular weight marker, Line 2 shows the extracts of native cells. Lane 3 and Lane 4 show the extracts of induced GM cells collected 3 h and 18 h after the induction. Periplasmic (e) and insoluble (f) fractions of native C41 (DE3) and GM C41 (DE3) induced by 0.2 mM IPTG. For e&f, Line 1 shows the molecular weight marker, Line 2 shows the extracts of native cells. Lane 3 and Lane 4 show the extracts of induced GM cells collected 3 h and 18 h after the induction
Direct detection of BC in GM E. coli HMS174 (DE3) and GM C41 (DE3) cell cultures
Since BC is an extracellular material secreted into the culture medium, it was directly detected 18 h after the induction in cultures of HMS174 (DE3). In GM C41 (DE3) cultures, BC production was observed even 3 h after the induction (Fig. 4b, d). BC production was not found in the cultures of native HMS174 (DE3) and native C41 (DE3) used as negative controls (Fig. 4a, c). The shaking speed is one of the most important parameters that can significantly influence the morphology of BC in cell cultures. Typically, BC produced by G. hansenii is in the form of a dense and flat gel-like layer that covers the surface of the culture medium under static condition, which is more preferred to avoid the shear stress generating the mutation of G. hansenii into non-cellulose-producing state in high agitation [56,57,58]. Under agitated culture conditions, many particles with various sizes (10 µm to 1 mm) and various shapes (spherical, ellipsoidal, fibrous, or stellate) were generated in BC cultures to form a well-dispersed suspension previously [57, 59, 60]. Here, BC was detected in the form of dispersed fibre-like structures inside the culture media of both GM HMS174 (DE3) and C41 (DE3). A less dense BC was produced by GM strains unlike the structure of original BC produced under agitated conditions. This was possibly due to the fact that BC was exposed to the vigorous agitation in GM cultures not only to provide enough oxygen to the cells but also to prevent the cells from precipitating, as E. coli tends to settle to the bottom of cell cultures in the absence of agitation unlike G. hansenii.

Liquid culture images of native HMS174 (DE3) (a), GM HMS174 (DE3) (b), native C41 (DE3) (c), and GM C41 (DE3) (d). GM strains were cultured at 22°C and induced by 0.025 mM IPTG
Morphology characterization of BC
The morphology of BC produced by GM cells was characterized by SEM and light microscopy. SEM images showed that BC produced by GM HMS174 (DE3) has smooth fibres that formed a dense and random network, which is similar to the general morphology of BC produced by G. hansenii (Fig. 5a–c). However, the fibres exhibited an extraordinary structure in terms of size. The lengths of the fibres were approximately between 1000 and 3000 μm with diameters ranging from 10 to 20 μm. The diameter of BC produced by G. hansenii is in nanoscale, typically ranging from 10 to 100 nm, while its length can be between 100—several 1000 nm [3, 61, 62]. The fibres produced by GM HMS174 (DE3) were extremely longer than those in BC typically produced by G. hansenii.

Scanning electron micrographs showing the microstructure of BC produced by GM HMS174 (DE3) (a–c) and GM C41 (DE3) (d–f) 18 h after the induction at different resolutions. Light microscope images of BC fibres and cell aggradations 18 h after the induction produced by GM C41 (DE3) induced with 0.05 mM IPTG at different resolutions (g, h). Length of BC particles that were produced by GM C41 (DE3) under the same conditions (i)
BC produced by GM C41 (DE3) shows a more random network and an irregular pattern compared to that of GM HMS174 (DE3) (Fig. 6d–f). It exhibits a biofilm-like structure, which is rigid, but contains fragile connections between cells [63]. Fibres with a length of approximately 500 μm were detected on SEM images. Microscope images of BC produced by GM C41 (DE3) revealed more clearly that these fibres are embedded into a biofilm-like structure/cell–cell interactions and exhibit a very smooth structure with a length of up to 2 cm (Fig. 5g–i).
FTIR analysis of BC
The spectra of BC produced by GM strains are characterized by very strong absorption bands located between 900 and 1243 cm−1 attributed to the C–O and C–O–C stretching vibration of glucose (Fig. 6a) [64,65,66,67]. The band assigned to the stretching of –OH groups present in cellulose is identified between 3500 and 3100 cm−1 [68]. The spectral band located between 2800 and 2900 cm−1 is assigned to C-H stretching vibrations of BC (including -CH2 and -CH3), whereas the spectral bands located between 1420 and 1278 cm−1 are corresponding to the in-plane bending of C-H groups [66, 69]. Cellulose synthesis is correlated with the biofilm formation in E. coli [70,71,72], which is an extracellular polymeric substance composed of extracellular DNA, proteins, and polysaccharides. Another noticeable region of the spectra appeared around 1517 and 1537 cm−1, which corresponds to amide I groups in this substance [73]. Furthermore, the band at 1640 cm−1 is assigned to –OH bending.

ATR-FTIR spectra (a) and XRD (b) of BC biosynthesized by G. hansenii ATCC 53582, GM HMS174 (DE3), and GM C41 (DE3)
XRD analysis of BC
The XRD spectra of BC produced by GM strains are presented in Fig. 6b. BC biosynthesized by G. hansenii ATCC 53582 exhibits the typical spectrum of cellulose I planes, which correspond to [110], [110], and [200] positioned at 14.5°, 16.6°, and 22.8°, respectively [74,75,76]. The spectrum of BC produced by GM strains showed one broad band at around 21° along with two weak peaks at 14.4° and 16.6°, which exhibited clearly more amorphous structure. The degree of crystallinity of BC biosynthesized by G. hansenii ATCC 53582 is estimated to be as high as 85%, while it was estimated to be 47 and 46% for GM HMS174 (DE3) and GM C41 (DE3), respectively. The ratio of the crystalline and amorphous regions depends on various conditions such as cellulose source, microorganism, medium composition, and fermentation conditions [76]. Accordingly, the absence of sharp peaks in XRD spectrum of BC produced by GM may be due to the differences in export mechanisms of bacterial species or to the fermentation conditions.
Discussion
New cellulose-producing strains have recently been explored by different approaches. It has been shown that the facultative anaerobe Enterobacter sp. FY-07 is able to produce cellulose under aerobic and anaerobic conditions. The gene constitution and arrangement of Enterobacter sp. FY-07 BC synthesis gene cluster bcsIII, comprising bcsA, bcsB, bcsC and bcsD, are the same as G. hansenii ATCC 53582. This suggests that the same BC synthesis mechanism of G. hansenii ATCC 53582 exists in Enterobacter sp. FY-07 [77]. Authors obtained cellulose production also in E. coli DH5α harboring bcsIII and its upstream; however, the study was focused on BC production in Enterobacter sp. FY-07 and level of BC production in E. coli DH5α was not examined. When the gene (AKI40_0895) upstream of bcsIII in Enterobacter sp. FY-07 was knocked out, the BC biosynthesis ability of mutant Enterobacter sp. FY-07 (Δ hyp) decreased sharply. Similarly, in G. hansenii ATCC 53582, the gene in the location corresponding to AKI40_0895 encodes a cellulose complementing factor (CcpAx), which was also essential for BC production. In another study, bcs genes were constructed using Gibson assembly and transformed into Komagataeibacter xylinus for overexpression [78]. Although there were no significant differences between the growth of wild-type and engineered strains, the engineered K. xylinus strains demonstrated faster BC production, generating 2–4-fold higher production compared to wild-type strain. Komagataeibacter rhaeticus, which is also a cellulose-producing strain, has been used to develop a modular genetic toolkit that enables rational reprogramming of the cell for controlled BC production recently [79]. In another study, the reconstitution of cellulose synthase in E. coli was investigated [80]. However, this study did not achieve complete BC biosynthesis, since the important proteins for crystallization and localization were not included in the developed system: BcsC, BcsD, and CcpAx. In addition, no contracts were codon-optimized to eliminate the variations in codon usage between different bacteria in any of these studies, which can have a significant influence on translation efficiency. Here, we included not only bcsABCD operon but also its upstream operon in separate vector constructs to test the effect of the operons separately. BC production was not detected when only bcsABCD operon was expressed in the initial trials (data not shown).
The recombinant co-expression of complex genetic circuits comes with the cost of low evolutionary stability, especially when most of these proteins are membrane-associated [28, 30, 81, 82]. A number of biological restraints, primarily replication of plasmids and culture conditions, are responsible for limiting the final biomass and the product concentration in such systems. To overcome plasmid instability, genome-based expression systems that integrate the target DNA fragments into the chromosome have been established for E. coli; however, plasmid-based systems are still preferred, because the cloning procedures are simpler and faster for such large DNA sequences [26, 83, 84]. In plasmid-based systems, it is necessary to carefully optimize process conditions for continuous and stable production of a recombinant product.
The metabolic stress caused by the maintenance of recombinant plasmids ultimately resulted in plasmid instability in BL21 (DE3) expression systems previously [40,41,42]. This phenomenon was observed in also GM BL21 (DE3). Decreasing culture temperature and slow induction of the target genes (with lower IPTG concentrations) have been defined as favorable for recombinant expression systems to prevent inclusion body formation [45,46,47, 85,86,87]. However, these were not able to stop the dramatic plasmid loss in GM BL21 (DE3), since the overexpression of multiple membrane proteins in BL21 (DE3) is usually toxic and often results in high overexpression yields [26]. The high gene expression proved by q-PCR in GM BL21 (DE3) supports this theory.
A comparative study demonstrated that HMS174 (DE3) exhibited a high plasmid stability, while a dramatic plasmid loss clearly occurred in BL21 (DE3) [26]. In addition, overexpression of many membrane proteins in BL21 (DE3) was not successful, whereas this was achieved in C41 (DE3) with high plasmid stability previously [28, 29]. Herein, stable expression systems (plasmid stability above > 85%) were established in both GM C41 (DE3) and GM HMS174 (DE3) when the bioprocess conditions (temperature and IPTG concentration) were carefully optimized. Both of these GM strains were able to biosynthesize BC at lower temperature (≤ 30 °C) and lower IPTG concentrations (≤ 0.2 mM). Gene expression levels revealed that the target genes were slowly expressed 3 h after IPTG induction in these strains. This suggests that the accumulation of inactive proteins may be eliminated by slow induction to allow the production of active proteins [85, 86]. Due to the absence of significant expression levels of the target protein by SDS–PAGE analysis at low IPTG (0.025, 0.05 mM) also supported the possibility of slow expression (data not shown). The BC produced by GM HMS174 (DE3) revealed a notable structure constituted of dense fibres with a length reaching 3000 µm with a width ranging between 10 and 20 µm. The mechanical properties of BC are affected by the length of the fibres and more importantly the aspect ratio (length/width ratio) of the fibres [88, 89]. Herein, we produced fibres with high aspect ratio (> 150), which are, therefore, potentially suitable for high efficiency composite materials for various applications as high aspect ratio is desirable for reinforcement [88, 90]. BC formation was detected even 3 h after IPTG induction in GM C41 (DE3) which allows shortening the fermentation time for BC production. The BC obtained previously in agitated cultures of G. hansenii exhibited a lower degree of crystallinity compared to that of static cultures [60]. Here, the BC produced by GM cells showed also a lower degree of crystallization due to the vigorous agitation (180 rpm).
The volumetric productivity of G. hansenii ATCC 53582, the donor organism for bcs genes herein, was reported approximately to be between 6.0–18.45 mg/L h previously [34, 91,92,93]. This is calculated to be 1.73 mg/L h for GM E. coli C41 (DE3) and 1.36 mg/L h for GM E. coli HMS174 (DE3) in our study (Table 3). Although the volumetric productivity appears lower in GM E. coli strains, BC productivities should be reported after normalising to carbon source concentrations in culture media as it highly differs in between E. coli and G. hansenii cultures (4 g/L glucose supplement in LB medium for GM E. coli cultures and 20 g/L of glucose in HS medium for G. hansenii cultures). The yield of the cellulose from glucose is calculated to be 0.44 mg/g.h in GM E. coli C41 (DE3) and 0.34 mg/g.h in GM E. coli HMS174 (DE3). Both were obtained in the range of normalised productivity of G. hansenii ATCC 53582 cultures (0.30–0.92 mg/g h). A potent cellulose producer, Gluconacetobacter xylinum BRC5, produced 13.88 mg/L h (0.69 mg/g h) cellulose in agitated flasks initially [94]. When G. xylinum BRC5 was cultured in fed-batch conditions in a jar bioreactor under both pH and dissolved O2 control, the volumetric productivity increased significantly and reached to the highest BC productivity obtained so far with 0.3 g/L h (15 mg/g h) [95]. Therefore, volumetric yields in GM strains should be further investigated when using a bioreactor system with an efficient O2 supply and pH control, as E. coli cells grows highly fast when compared to acetic acid bacteria which could potentially result in higher yields. In addition, the fermentation period for GM E. coli strains was only 18 h, whereas it was reported to be around 168 h for G. hansenii cultures, which could be an advantage for the reducing the energy consumption for BC production processes.
Conclusion
We achieved recombinant biosynthesis of bacterial cellulose in E. coli platforms by the co-expression of bcsABCD and its upstream operon. The optimization of bioprocess conditions resulted in the functional and stable biosynthesis of BC with remarkable fibre structure, as early as 3 h after the induction. The system developed in this study can be potentially used to contribute to future bioprocess design for bacterial cellulose production by further investigations.
Abbreviations
- BC:
-
Bacterial cellulose
- Bcs :
-
Bacterial cellulose synthesis
- Bp:
-
Base pair
- DCW:
-
Dry cell weight
- FTIR:
-
Fourier transform infrared
- GM:
-
Genetically modified
- IPTG:
-
Isopropyl β-D-1-thiogalactopyranoside
- KU:
-
Kilo unit
- Kbp:
-
Kilo base pair
- LB Medium:
-
Lysogeny broth medium
- q-PCR:
-
Quantitative polymerase chain reaction
- RQ:
-
Relative quantification
- SEM:
-
Scanning electron microscopy
References
Jonas R, Farah LF (1998) Production and application of microbial cellulose. Polym Degrad Stab 59:101–106. https://doi.org/10.1016/s0141-3910(97)00197-3
Vandamme EJ, De Baets S, Vanbaelen A, Joris K, De Wulf P (1998) Improved production of bacterial cellulose and its application potential. Polym Degrad Stab 59:93–99. https://doi.org/10.1016/s0141-3910(97)00185-7
Klemm D, Heublein B, Fink HP, Bohn A (2005) Cellulose: Fascinating biopolymer and sustainable raw material. Angew Chemie-International Ed 44:3358–3393. https://doi.org/10.1002/anie.200460587
Helenius J, Brouhard G, Kalaidzidis Y, Diez S, Howard J (2006) The depolymerizing kinesin MCAK uses lattice diffusion to rapidly target microtubule ends. Nature 441:115–119. https://doi.org/10.1038/nature04736
Hutchens SA, Benson RS, Evans BR, O’Neill HM, Rawn CJ (2006) Biomimetic synthesis of calcium-deficient hydroxyapatite in a natural hydrogel. Biomaterials 27:4661–4670. https://doi.org/10.1016/j.biomaterials.2006.04.032
Fontana JD, De Souza AM, Fontana CK, Torriani IL, Moreschi JC, Gallotti BJ, De Souza SJ, Narcisco GP, Bichara JA, Farah LFX (1990) Acetobacter cellulose pellicle as a temporary skin substitute. Appl Biochem Biotechnol 24–25:253–264. https://doi.org/10.1007/BF02920250
Czaja W, Krystynowicz A, Bielecki S, Brown RM (2006) Microbial cellulose - the natural power to heal wounds. Biomaterials 27:145–151. https://doi.org/10.1016/j.biomaterials.2005.07.035
Brown RM (1999) Cellulose structure and biosynthesis. Pure Appl Chem 71:767–775. https://doi.org/10.1351/pac199971050767
Thomas S (2013) Handbook of biopolymer-based materials : from blends and composites to gels and complex networks
Bielecki S, Krystynowicz A, Turkiewicz M, Kalinowska H (2005) Bacterial cellulose, WILEY-VCH Verlag GMBH & Co. KGaA,
Wong HC, Fear AL, Calhoon RD, Eichinger GH, Mayer R, Amikam D, Benziman M, Gelfand DH, Meade JH, Emerick AW, Bruner R, Benbassat A, Tal R (1990) Genetic organization of the cellulose synthase operon in Acetobacter xylinum. Proc Natl Acad Sci U S A 87:8130–8134. https://doi.org/10.1073/pnas.87.20.8130
Saxena IM, Kudlicka K, Okuda K, Brown RM (1994) Characterization of genes in the cellulose-synthesizing operon (acs operon) of Acetobacter xylinum - implications for cellulose crystallization. J Bacteriol 176:5735–5752
Brown RM, Saxena IM (2000) Cellulose biosynthesis: A model for understanding the assembly of biopolymers. Plant Physiol Biochem 38:57–67. https://doi.org/10.1016/s0981-9428(00)00168-6
Hu S-Q, Gao Y-G, Tajima K, Sunagawa N, Zhou Y, Kawano S, Fujiwara T, Yoda T, Shimura D, Satoh Y, Munekata M, Tanaka I, Yao M (2010) Structure of bacterial cellulose synthase subunit D octamer with four inner passageways. Proc Natl Acad Sci 107:17957–17961. https://doi.org/10.1073/pnas.1000601107
Standal R, Iversen TG, Coucheron DH, Fjaervik E, Blatny JM, Valla S (1994) A new gene required for cellulose production and a gene encoding cellulolytic activity in Acetobacter xylinum are colocalized with the bcs operon. J Bacteriol 176:665–672
Tonouchi N, Thara N, Tsuchida T, Yoshinaga F, Beppu T, Horinouchi S (1995) Addition of a small amount of an endoglucanase enhances cellulose production by Acetobacter xylinum. Biosci Biotechnol Biochem 59:805–808
Koo HM, Song SH, Pyun YR, Kim YS (1998) Evidence that a beta-1,4-endoglucanase secreted by Acetobacter xylinum plays an essential role for the formation of cellulose fiber. Biosci Biotechnol Biochem 62:2257–2259. https://doi.org/10.1271/bbb.62.2257
Sunagawa N, Tajima K, Hosoda M, Kawano S, Kose R, Satoh Y, Yao M, Dairi T (2012) Cellulose production by Enterobacter sp CJF-002 and identification of genes for cellulose biosynthesis. Cellulose 19:1989–2001. https://doi.org/10.1007/s10570-012-9777-2
Sunagawa N, Fujiwara T, Yoda T, Kawano S, Satoh Y, Yao M, Tajima K, Dairi T (2013) Cellulose complementing factor (Ccp) is a new member of the cellulose synthase complex (terminal complex) in Acetobacter xylinum. J Biosci Bioeng 115:607–612. https://doi.org/10.1016/j.jbiosc.2012.12.021
Lee KY, Buldum G, Mantalaris A, Bismarck A (2014) More than meets the eye in bacterial cellulose: biosynthesis, bioprocessing, and applications in advanced fiber composites. Macromol Biosci 14:10–32
Islam MU, Ullah MW, Khan S, Shah N, Park JK (2017) Strategies for cost-effective and enhanced production of bacterial cellulose. Int J Biol Macromol 102:1166–1173. https://doi.org/10.1016/j.ijbiomac.2017.04.110
Chawla PR, Bajaj IB, Survase SA, Singhal RS (2009) Microbial cellulose: fermentative production and applications. Food Technol Biotechnol 47:107–124
Gottesman S (1989) Genetics of proteolysis in Escherichia coli*. Annu Rev Genet 23:163–198. https://doi.org/10.1146/annurev.ge.23.120189.001115
Gottesman S (1996) Proteases and their targets in Escherichia coli. Annu Rev Genet 30:465–506. https://doi.org/10.1146/annurev.genet.30.1.465
Shiloach J, Kaufman J, Guillard AS, Fass R (1996) Effect of glucose supply strategy on acetate accumulation, growth, and recombinant protein production by Escherichia coli BL21 (lambdaDE3) and Escherichia coli JM109. Biotechnol Bioeng 49:421–428. https://doi.org/10.1002/(sici)1097-0290(19960220)49:4<421::aid-bit9>3.0.co;2-r
Marisch K, Bayer K, Cserjan-Puschmann M, Luchner M, Striedner G (2013) Evaluation of three industrial Escherichia coli strains in fed-batch cultivations during high-level SOD protein production. Microb Cell Fact 12:58. https://doi.org/10.1186/1475-2859-12-58
Mairhofer J, Krempl PM, Thallinger GG, Striedner G Finished Genome Sequence of Escherichia coli K-12 Strain HMS174 (ATCC 47011). Genome Announc, 2
Miroux B, Walker JE (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260:289–298. https://doi.org/10.1006/jmbi.1996.0399
Wagner S, Baars L, Ytterberg AJ, Klussmeier A, Wagner CS, Nord O, Nygren PA, van Wijk KJ, de Gier JW (2007) Consequences of membrane protein overexpression in Escherichia coli. Mol Cell Proteomics 6:1527–1550. https://doi.org/10.1074/mcp.M600431-MCP200
Dumon-Seignovert L, Cariot G, Vuillard L (2004) The toxicity of recombinant proteins in Escherichia coli: a comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr Purif 37:203–206. https://doi.org/10.1016/j.pep.2004.04.025
Wagner S, Klepsch MM, Schlegel S, Appel A, Draheim R, Tarry M, Högbom M, van Wijk KJ, Slotboom DJ, Persson JO, de Gier J-W (2008) Tuning Escherichia coli for membrane protein overexpression. Proc Natl Acad Sci 105:14371–14376. https://doi.org/10.1073/pnas.0804090105
Sorensen HP, Mortensen KK (2005) Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol 115:113–128. https://doi.org/10.1016/j.jbiotec.2004.08.004
Goyal A, Prasad KN, Prasad A, Gupta S, Ghoshal U, Ayyagari A (2009) Extended spectrum beta-lactamases in Escherichia coli & Klebsiella pneumoniae & associated risk factors. Indian J Med Res 129:695–700
Kawano S, Tajima K, Uemori Y, Yamashita H, Erata T, Munekata M, Takai M (2002) Cloning of cellulose synthesis related genes from Acetobacter xylinum ATCC23769 and ATCC53582: Comparison of cellulose synthetic ability between strains. DNA Res 9:149–156. https://doi.org/10.1093/dnares/9.5.149
Qiagen (2011) QuantiTect® SYBR® Green PCR Handbook. In: Sample Assay Technol. https://www.qiagen.com/us/resources/resourcedetail?id=38083588-73db-4d2e-9ed2-9f66b154ab3e&lang=en.Accessed 21 Oct 2017
Herold S, Siebert J, Huber A, Schmidt H (2005) Global expression of prophage genes in Escherichia coli O157:H7 strain EDL933 in response to norfloxacin. Antimicrob Agents Chemother 49:931–944. https://doi.org/10.1128/aac.49.3.931-944.2005
Nossal NG, Heppel LA (1966) The release of enzymes by osmotic shock from Escherichia coli in Exponential Phase. J Biol Chem 241:3055–3062
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29:786–794. https://doi.org/10.1177/004051755902901003
Collins T, Azevedo-Silva J, da Costa A, Branca F, Machado R, Casal M (2013) Batch production of a silk-elastin-like protein in E. coli BL21(DE3): key parameters for optimisation. Microb Cell Fact 12:21. https://doi.org/10.1186/1475-2859-12-21
Yu H, Shi Y, Sun X, Luo H, Shen Z (2003) Effect of poly(beta-hydroxybutyrate) accumulation on the stability of a recombinant plasmid in Escherichia coli. J Biosci Bioeng 96:179–183
Zhao J-B, Wei D-Z (2007) Use of a derivative of Escherichia coli BL21(DE3) for Efficient production of three different recombinant proteins. Biotechnol Prog 23:1043–1048. https://doi.org/10.1021/bp070155b
Balbás P (2001) Understanding the art of producing protein and nonprotein molecules in Escherichia coli. Mol Biotechnol 19:251–267 https://doi.org/10.1385/MB:19:3:251
Corchero JL, Villaverde A (1998) Plasmid maintenance in Escherichia coli recombinant cultures is dramatically, steadily, and specifically influenced by features of the encoded proteins. Biotechnol Bioeng 58:625–632
Sevastsyanovich Y, Alfasi S, Overton T, Hall R, Jones J, Hewitt C, Cole J (2009) Exploitation of GFP fusion proteins and stress avoidance as a generic strategy for the production of high-quality recombinant proteins. FEMS Microbiol Lett 299:86–94
Wood TK, Peretti SW (1991) Effect of chemically-induced, cloned-gene expression on protein synthesis in E. Coli Biotechnol Bioeng 38:397–412. https://doi.org/10.1002/bit.260380410
Donovan RS, Robinson CW, Glick BR (1996) Review: Optimizing inducer and culture conditions for expression of foreign proteins under the control of thelac promoter. J Ind Microbiol 16:145–154. https://doi.org/10.1007/BF01569997
Overton TW (2014) Recombinant protein production in bacterial hosts. Drug Discov Today 19:590–601. https://doi.org/10.1016/j.drudis.2013.11.008
Morgan JLW, Strumillo J, Zimmer J (2013) Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature 493:181–186
Augimeri RV, Strap JL (2015) The phytohormone ethylene enhances cellulose production, regulates crp/fnrkx transcription and causes differential gene expression within the bacterial cellulose synthesis operon of Komagataeibacter (Gluconacetobacter) xylinus ATCC 53582. Front Microbiol 6:1459
De Ley J, Gillis M, Swings J (1984) Family VI. Acetobacteraceae. Bergey’s Maual Syst. Bacteriol,
Omadjela O, Narahari A, Strumillo J, Mélida H, Mazur O, Bulone V, Zimmer J (2013) BcsA and BcsB form the catalytically active core of bacterial cellulose synthase sufficient for in vitro cellulose synthesis. Proc Natl Acad Sci USA 110:17856–17861. https://doi.org/10.1073/pnas.1314063110
Yasutake Y, Kawano S, Tajima K, Yao M, Satoh Y, Munekata M, Tanaka I (2006) Structural characterization of the Acetobacter xylinum endo-β-1,4-glucanase CMCax required for cellulose biosynthesis. Proteins Struct Funct Bioinforma 64:1069–1077. https://doi.org/10.1002/prot.21052
Bannwarth M, Schulz GE (2003) The expression of outer membrane proteins for crystallization. Biochim Biophys Acta - Biomembr 1610:37–45. https://doi.org/10.1016/S0005-2736(02)00711-3
Korepanova A, Gao FP, Hua Y, Qin H, Nakamoto RK, Cross TA (2005) Cloning and expression of multiple integral membrane proteins from Mycobacterium tuberculosis in Escherichia coli. Protein Sci 14:148–158. https://doi.org/10.1110/ps.041022305
Tsouko E, Kourmentza C, Ladakis D, Kopsahelis N, Mandala I, Papanikolaou S, Paloukis F, Alves V, Koutinas A (2015) Bacterial cellulose production from industrial waste and by-product streams. Int J Mol Sci 16:14832–14849. https://doi.org/10.3390/ijms160714832
Dudman WF (1960) Cellulose production by Acetobacter acetigenum in defined medium. J Gen Microbiol 21:327–337
Valla S, Kjosbakken J (1982) ellulose-negative mutants of Acetobacter xylinum. J Gen Microbiol 128:1401–1408
Hestrin S, Schramm M (1954) Synthesis of cellulose by Acetobacter xylinum. 2. Preparation of freeze-dried cells capable of polymerizing glucose to cellulose. Biochem J 58:345–352
Watanabe K, Tabuchi M, Morinaga Y, Yoshinaga F (1998) Structural Features and properties of bacterial cellulose produced in agitated culture. Cellulose 5:187–200. https://doi.org/10.1023/A:1009272904582
Ross P, Mayer R, Benziman M (1991) cellulose biosynthesis and function in bacteria. Microbiol Rev 55:35–58
Haigler CH, White AR, Brown RM, Cooper KM (1982) Alteration of in vivo cellulose ribbon assembly by carboxymethylcellulose and other cellulose derivatives. J Cell Biol 94:64–69. https://doi.org/10.1083/jcb.94.1.64
Romling U, Rohde M, Olsen A, Normark S, Reinkoster J (2000) AgfD, the checkpoint of multicellular and aggregative behaviour in Salmonella typhimurium regulates at least two independent pathways. Mol Microbiol 36:10–23
Wong S-S, Kasapis S, Tan YM (2009) Bacterial and plant cellulose modification using ultrasound irradiation. Carbohydr Polym 77:280–287. https://doi.org/10.1016/j.carbpol.2008.12.038
Oh SY, Yoo DI, Shin Y, Kim HC, Kim HY, Chung YS, Park WH, Youk JH (2005) Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr Res 340:2376–2391. https://doi.org/10.1016/j.carres.2005.08.007
Carrillo F, Colom X, Suñol JJ, Saurina J (2004) Structural FTIR analysis and thermal characterisation of lyocell and viscose-type fibres. Eur Polym J 40:2229–2234. https://doi.org/10.1016/j.eurpolymj.2004.05.003
Kacurakova M, Smith AC, Gidley MJ, Wilson RH (2002) Molecular interactions in bacterial cellulose composites studied by 1D FT-IR and dynamic 2D FT-IR spectroscopy. Carbohydr Res 337:1145–1153
Huang HC, Chen LC, Lin SB, Hsu CP, Chen HH (2010) In situ modification of bacterial cellulose network structure by adding interfering substances during fermentation. Bioresour Technol 101:6084–6091. https://doi.org/10.1016/j.biortech.2010.03.031
Łojewska J, Miśkowiec P, Łojewski T, Proniewicz LM (2005) Cellulose oxidative and hydrolytic degradation: In situ FTIR approach. Polym Degrad Stab 88:512–520. https://doi.org/10.1016/j.polymdegradstab.2004.12.012
Zogaj X, Nimtz M, Rohde M, Bokranz W, Romling U (2001) The multicellular morphotypes of Salmonella typhimurium and Escherichia coli produce cellulose as the second component of the extracellular matrix. Mol Microbiol 39:1452–1463
Romling U (2002) Molecular biology of cellulose production in bacteria. Res Microbiol 153:205–212. https://doi.org/10.1016/s0923-2508(02)01316-5
Da Re S, Ghigo JM (2006) A CsgD-independent pathway for cellulose production and biofilm formation in Escherichia coli. J Bacteriol 188:3073–3087. https://doi.org/10.1128/jb.188.8.3073-3087.2006
Jiao Y, Cody GD, Harding AK, Wilmes P, Schrenk M, Wheeler KE, Banfield JF, Thelen MP (2010) Characterization of extracellular polymeric substances from acidophilic microbial biofilms. Appl Environ Microbiol 76:2916–2922. https://doi.org/10.1128/AEM.02289-09
Poletto M, Pistor V, Zattera AJ (2013) Structural characteristics and thermal properties of native cellulose. InTech, Rijeka, Ch. 0
Ullah H, Wahid F, Santos HA, Khan T (2016) Advances in biomedical and pharmaceutical applications of functional bacterial cellulose-based nanocomposites. Carbohydr Polym 150:330–352. https://doi.org/10.1016/j.carbpol.2016.05.029
Shezad O, Khan S, Khan T, Park JK (2010) Physicochemical and mechanical characterization of bacterial cellulose produced with an excellent productivity in static conditions using a simple fed-batch cultivation strategy. Carbohydr Polym 82:173–180. https://doi.org/10.1016/j.carbpol.2010.04.052
Ji K, Wang W, Zeng B, Chen S, Zhao Q, Chen Y, Li G, Ma T (2016) Bacterial cellulose synthesis mechanism of facultative anaerobe Enterobacter sp. FY-07. Sci Rep 6:21863
Mangayil R, Rajala S, Pammo A, Sarlin E, Luo J, Santala V, Karp M, Tuukkanen S (2017) Engineering and characterization of bacterial nanocellulose films as low cost and flexible sensor material. ACS Appl Mater Interfaces 9:19048–19056. https://doi.org/10.1021/acsami.7b04927
Florea M, Hagemann H, Santosa G, Abbott J, Micklem CN, Spencer-Milnes X, de Arroyo Garcia L, Paschou D, Lazenbatt C, Kong D, Chughtai H, Jensen K, Freemont PS, Kitney R, Reeve B, Ellis T (2016) Engineering control of bacterial cellulose production using a genetic toolkit and a new cellulose-producing strain. Proc Natl Acad Sci 113:E3431–E3440. https://doi.org/10.1073/pnas.1522985113
Imai T, Sun SJ, Horikawa Y, Wada M, Sugiyama J (2014) Functional reconstitution of cellulose synthase in Escherichia coli. Biomacromolecules 15:4206–4213. https://doi.org/10.1021/bm501217g
Drew D, Froderberg L, Baars L, de Gier JW (2003) Assembly and overexpression of membrane proteins in Escherichia coli. Biochim Biophys Acta 1610:3–10
Sleight SC, Bartley BA, Lieviant JA, Sauro HM (2010) Designing and engineering evolutionary robust genetic circuits. J Biol Eng 4:12. https://doi.org/10.1186/1754-1611-4-12
Striedner G, Pfaffenzeller I, Markus L, Nemecek S, Grabherr R, Bayer K (2010) Plasmid-free T7-based Escherichia coli expression systems. Biotechnol Bioeng 105:786–794. https://doi.org/10.1002/bit.22598
Heap JT, Ehsaan M, Cooksley CM, Ng Y-K, Cartman ST, Winzer K, Minton NP (2012) Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res 40:e59–e59. https://doi.org/10.1093/nar/gkr1321
Dormiani K, Khazaie Y, Sadeghi HM, Rabbani M, Moazen F (2007) Cloning and expression of a human tissue plasminogen activator variant: K2S in Escherichia coli. Pak J Biol Sci 10:946–949
Vera A, Gonzalez-Montalban N, Aris A, Villaverde A (2007) The conformational quality of insoluble recombinant proteins is enhanced at low growth temperatures. Biotechnol Bioeng 96:1101–1106. https://doi.org/10.1002/bit.21218
Silva F, Passarinha L, Sousa F, Queiroz JA, Domingues FC (2009) Influence of growth conditions on plasmid DNA production. J Microbiol Biotechnol 19:1408–1414
Eichhorn SJ, Dufresne A, Aranguren M, Marcovich NE, Capadona JR, Rowan SJ, Weder C, Thielemans W, Roman M, Renneckar S, Gindl W, Veigel S, Keckes J, Yano H, Abe K, Nogi M, Nakagaito AN, Mangalam A et al (2010) Review: current international research into cellulose nanofibres and nanocomposites. J Mater Sci 45:1–33. https://doi.org/10.1007/s10853-009-3874-0
Kuboki T, Lee YH, Park CB, Sain M (2009) Mechanical properties and foaming behavior of cellulose fiber reinforced high-density polyethylene composites. Polym Eng Sci 49:2179–2188. https://doi.org/10.1002/pen.21464
Pickering KL, Efendy MGA, Le TM (2016) A review of recent developments in natural fibre composites and their mechanical performance. Compos Part A Appl Sci Manuf 83:98–112. https://doi.org/10.1016/j.compositesa.2015.08.038
Czaja W, Romanovicz D, Brown R malcolm (2004) Structural investigations of microbial cellulose produced in stationary and agitated culture. Cellulose 11:403–411. https://doi.org/10.1023/B:CELL.0000046412.11983.61
Fang L, Catchmark JM (2015) Characterization of cellulose and other exopolysaccharides produced from Gluconacetobacter strains. Carbohydr Polym 115:663–669. https://doi.org/10.1016/j.carbpol.2014.09.028
Ishihara M, Matsunaga M, Hayashi N, Tišler V (2002) Utilization of d-xylose as carbon source for production of bacterial cellulose. Enzyme Microb Technol 31:986–991. https://doi.org/10.1016/S0141-0229(02)00215-6
Yang YK, Park SH, Hwang JW, Pyun YR, Kim YS (1998) Cellulose production by Acetobacter xylinum BRC5 under agitated condition. J Ferment Bioeng 85:312–317. https://doi.org/10.1016/S0922-338X(97)85681-4
Hwang JW, Yang YK, Hwang JK, Pyun YR, Kim YS (1999) Effects of pH and dissolved oxygen on cellulose production by Acetobacter xylinum BRC5 in agitated culture. J Biosci Bioeng 88:183–188. https://doi.org/10.1016/s1389-1723(99)80199-6
Acknowledgements
The authors gratefully acknowledge the financial support of the Turkey National Ministry of Education for funding Gizem Buldum.
Author information
Authors and Affiliations
Contributions
GB designed and performed the experiments. AB designed the BC analysis experiments. AM designed the experiments. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Buldum, G., Bismarck, A. & Mantalaris, A. Recombinant biosynthesis of bacterial cellulose in genetically modified Escherichia coli . Bioprocess Biosyst Eng 41, 265–279 (2018). https://doi.org/10.1007/s00449-017-1864-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-017-1864-1