
Abstract
Increased phosphorus availability may provoke serious eutrophication problems in wetlands. Strong evidence indicates that sulphate induced mobilization of phosphate (internal eutrophication) has been responsible for a strong decline of the biodiversity in wetlands during the last decades. It is currently underestimated, however, that the wide spread leaching of nitrate from agricultural lands can indirectly provoke strong internal phosphate eutrophication in wetlands, via its interference with sulphur and iron biogeochemistry in the subsoil. Nitrate can mobilize sulphate from geological pyrite deposits by the oxidation of FeSx in the aquifer, leading to a decrease of nitrate and an increase of groundwater sulphate concentrations. Furthermore nitrate immobilizes iron in the subsoil by oxidizing reduced (dissolved) iron. Increased sulphate concentrations may provoke strong phosphate eutrophication in wetlands fed directly or indirectly (via surface water) with groundwater as sulphate strongly interferes with iron phosphorus chemistry and stimulates anaerobic decomposition of organic matter. Management of wetlands should therefore be approached at a broader scale which includes the landscape-scale management of groundwater systems. Leaching of nitrate to the groundwater, for instance, should not only receive attention for its potential effects on drinking water quality but above all because of the resulting large scale mobilization of sulphate from geological pyrite deposits and the immobilization of ferrous iron.
Similar content being viewed by others
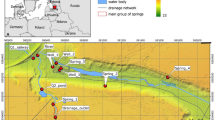

Introduction
To understand abiotic conditions of an individual wetland, understanding of landscape-scale hydrological and biogeochemical processes may be essential (e.g. Bedford, 1996). Nowadays, human activities have major impacts on landscape properties that control wetland hydrology and water chemistry. Obviously, hydrological measures may affect groundwater flows and thereby the hydrology of wetlands. Next, excessive fertilization by agricultural activities may strongly affect wetland ecosystems. Nitrogen emissions in the surroundings, for instance, are well known to affect oligotrophic systems (such as moorland pools and raised bogs) by locally causing strongly increased atmospheric deposition rates of nitrogen (e.g. Bobbink et al. 1998). Next, nitrate losses from agricultural lands are considerable and nowadays contaminate groundwater worldwide.
Although in eutrophication research nitrogen has received most attention, phosphorus availability may be a main determinant for biodiversity in groundwater fed wetlands (e.g. Wassen et al. 2005). In the soil, however, phosphorus (P) is—in contrast to the rather mobile nitrate (NO3 −)—quite immobile and vertical movement of phosphorus is assumed to be strongly restricted because of a high phosphorus fixation capacity in mineral soils (Heckrath et al. 1995; Sims et al. 1998). Although recent research revealed that leaching of phosphorus can occur from heavily fertilized agricultural lands (McDowell and Sharpley 2004), on the whole soluble P losses of <1 kg P ha−1 yr−1 are more common (Sims et al. 1998) and phosphate leaching to deeper groundwater is merely seen as a distinct possibility over a period of decades, depending on the degree of phosphate saturation of the subsoil (Behrendt and Boekhold 2006).
Although currently movement of phosphorus via aquifers does not play an important role in the eutrophication of groundwater fed surface waters, we will show that landscape-scale hydrological and biogeochemical processes can lead to increased phosphorus eutrophication of wetlands in an unexpected way. In essence, nitrate pollution of the groundwater can provoke strong phosphate eutrophication in groundwater fed wetlands, by its interference with sulphur and iron biogeochemistry. Through oxidation of iron sulphides (including pyrite), for instance, nitrate may extend its biogeochemical reach into the groundwater much further than its mere physical presence. A schematic representation of this mechanism, which is elaborated in this article, is presented in Fig. 1.

Schematic representation of nitrate mediated, sulphate-controlled P-release in wetlands
Nitrate pollution
In the past century the intensification of agricultural activities and the application of artificial fertilizers have caused a severely disturbed nitrogen balance in agricultural areas as the input of nitrogen by far exceeds the yield in these systems (e.g. Goodchild 1998; Iversen et al. 1998; Kirchmann et al. 2002; Fig. 1). Under aerobic soil conditions ammonia and ammonium ions are microbially transformed (nitrified) to nitrate. The adverse effects of high atmospheric nitrogen deposition in Western Europe (e.g. Bobbink et al. 1998) have resulted in governmental measures to decrease the loss of volatile ammonia from agricultural soils. Low emission application techniques (including manure injection) substantially reduced the ammonia emission but as a result, the leaching of nitrate to the groundwater further increased (Iversen et al. 1998).
Next, non agricultural sources, such as leaking sewage systems and home gardens (Wakida and Lerner 2005) and forests (e.g. Rothe and Mellert 2004; Gundersen et al. 2006), which catch atmospheric nitrogen deposition, may also significantly contribute to increased groundwater nitrate concentrations. In most parts of Europe, anthropogenic nitrogen deposition has led to increased nitrate leaching from forests. Pine forests have a high leaf area which is also maintained throughout the year, which leads to higher rates of N deposition from the atmosphere to the forest floor and to higher concentrations of nitrate in drainage water compared with broadleaved forests (Rothe and Mellert 2004).
World-wide, nitrate has become one of the common groundwater contaminants (e.g. Kool 1988; Postma et al. 1991; Nolan et al. 1997; Goodchild 1998; Iversen et al. 1998; Kirchmann et al. 2002; Senn and Hemond 2002; Thorburn et al. 2003; Broers et al. 2004; Surridge et al. 2007). Obviously nitrate pollution plays an important role in surface water eutrophication (Iversen et al. 1998; Kirchmann et al. 2002). However, it also poises a serious risk for drinking water quality as infants under six months of age are susceptible to nitrate poisoning (Comly 1945; Fan and Steinberg 1996). The European standard for the maximum concentrations of nitrate in potable water is 50 mg L−1 (806 μmol L−1). At present, this concentration is often exceeded in potable waters extracted from surface water or aquifers recharged by water from areas of agricultural activity.
In the Netherlands, for instance, the phreatic and shallow groundwater below infiltrating agricultural lands nowadays typically contains median nitrate concentrations as high as 2000 μmol l−1, with minimum and maximum values of 900 and 3300 μmol l−1 (Broers et al. 2004). The increase in deeper aquifers, however, is generally much less pronounced than would be expected from model studies. This phenomenon has been attributed to the consumption of nitrate due to microbially mediated nitrate reduction processes in the sub-soil.
Nitrate reduction
The reduction capacity of the aquifer soils will determine the extent to which reduction of nitrate may take place. Sedimentary organic matter (SOM) is a relevant electron donor in aquifers (reaction 1) (e.g. Hill et al. 2000; Hartog et al. 2002). The remaining reactivity of SOM in ground water systems may differ greatly, but the residual SOM tends to become more stable (less degradable) during oxidation (Cowie and Hedges 1994; Hartog et al. 2002, 2004). Sediment oxygen exposure time is probably a key factor determining the remaining reactivity of the SOM in aquifer sediments (Hartog et al. 2005). Generally, in deeper aquifers SOM is present in low concentrations (0.01–02 wt. %). Next, it is also characterized by the absence of more labile compounds such as cellulose, indicating that SOM has degraded to a considerable extent from its biomass precursors and has become relatively inert (Hartog et al. 2004).
Being a powerful electron acceptor, nitrate can also be reduced by ferrous iron and may therefore importantly influence iron cycling in natural systems. Ferrous iron (Straub et al. 1996) and ferrous iron bearing carbonates (such as siderite) and sulphides (such as pyrite) (Postma et al. 1991; Aravena and Robertson 1998; Pauwels et al. 1998; Moncaster et al. 2000; Lucassen et al. 2004; Haaijer et al. 2006, 2007; Burgin and Hamilton 2008) turn out to be important electron donors for denitrification in aquifer soils (reaction 2, 3, 4; Fig. 1). These reactions are all catalyzed by microorganisms which, withstanding a wide variety of environmental conditions, thrive in shallow and deep aquifers (e.g. Pauwels et al. 1998; Haaijer et al. 2006, 2007; Burgin and Hamilton 2008).
As long as groundwater nitrate concentrations are high, ferrous iron levels tend to remain low because nitrate acts as a redox buffer, preventing the reduction of iron(III)oxides (Lucassen et al. 2004; Smolders et al. 2006a). Additionally, ferrous iron may be microbially oxidized by nitrate (Straub et al. 1996; Senn and Hemond 2002). High nitrate loads therefore strongly decrease groundwater iron concentrations (Fig. 2). An advantage of this process might be that P entering aquifers along with nitrate, can be bound to oxidized iron in aquifers, potentially reducing the supply of P to wetlands via groundwater.

Relationship between nitrate and iron concentrations in groundwater derived from a total of 1012 analyses of phreatic groundwater samples from the Netherlands. The horizontal line indicates the median iron concentrations for the nitrate range and the vertical line the range of values measures. The inset presents the relationship between the mean iron and the mean nitrate concentration for each nitrate range. Data are derived from the authors’ personal data archive
The chemoautolithotrophic oxidation of iron sulphide deposits by nitrate, frequently leads to a concomitant increase of groundwater sulphate concentrations (Postma et al. 1991, 1998; Aravena and Robertson 1998; Moncaster et al. 2000; Broers and Van der Grift 2004; Broers et al. 2004; Van Beek et al. 2006). Although the reduction of nitrate by organic matter is thermodynamically favourable, iron sulphide oxidation appears to be kinetically favoured in deeper aquifers, probably because of the different microbiological availability of iron sulphides (including amorphic pyrite) and organic matter (Postma et al. 1991; Wriedt and Rode 2006).
As a result of nitrate leaching, groundwater chemistry has changed profoundly in many parts of the world and is nowadays characterized by lower iron and much higher sulphate concentrations (Postma et al. 1991; Aravena and Robertson 1998; Hoffmann et al. 1998; Pauwels et al. 1998; Moncaster et al. 2000; Molenat et al. 2002; Broers and Van der Grift 2004). In the Pleistocene regions of the Netherlands, for instance, increased agricultural activities have led to a strongly increased leaching of nitrate and to a concomitant increase of the groundwater sulphate concentrations. Near the pumping station of Vierlingsbeek (in the province of Limburg, the Netherlands), sulphate concentrations have increased from 400 to 1450 μmol l−1 between the 1960s and 2000, of which atmospheric sulphur deposition only contributes +200 μmol l−1. In the same time span groundwater nitrate concentrations have increased from hardly detectable to +500 μmol l−1 (Van Beek et al. 2006). Estimations reveal that at present up to 70% of the sulphate present in groundwater in the Netherlands is derived from nitrate induced pyrite oxidation resulting in mean groundwater sulphate concentrations of 833 μmol l−1 (Van Beek et al. 2006). Before the 1950’s, groundwater sulphate concentrations in these regions were typically lower than 200 μmol l−1 (STOM 1983).
Internal eutrophication
Under anaerobic conditions, the availability of alternative electron acceptors strongly affects the breakdown of organic matter. In addition to the availability of electron acceptors, (anaerobic) decomposition is obviously strongly regulated by the availability of degradable organic matter (Drever 1997). In wetlands the availability of reactive SOM will be much higher compared to (deeper) aquifers due to the constant input of plant litter. This means that nitrate and sulphate, supplied by groundwater enriched with these compounds, can strongly stimulate the decomposition of organic matter (e.g. Roden and Edmonds 1997; Holmer and Storkholm 2001; Smolders et al. 2006a).
Next, the increased input of sulphate and decreased input of iron in groundwater fed (semi-)aquatic ecosystems may also cause phosphate release due to the fact that sulphide (produced by sulphate reduction) interferes with the iron-phosphorus cycle (Fig. 1). The main product of sulphate reduction is dissolved sulphide. Sulphide may react with dissolved reduced iron and particulate iron (hydr)oxides. The products of the reaction between sulphide and iron are highly insoluble iron sulphide minerals while phosphate adsorbed to the iron (hydr)oxides or present as iron phosphates becomes mobilized (Sperber 1958; Caraco et al. 1989; Søndergaard et al. 1993; Roden and Edmonds 1997; Holmer and Storkholm 2001; Smolders et al. 2001; Lamers et al. 2002; Smolders et al. 2006a).
Prolonged high sulphate loads to such sediments will ultimately result in sediments in which up to 80% of the iron can be bound to reduced sulphur (FeSx) (Smolders et al. 1995). As a consequence, the capacity of the sediment to retain P will greatly decrease (FeSx has far fewer sorption sites for P than have iron(hydr)oxides), which may result in a high mobility of P in the soils and thus in a strong eutrophication of such systems (Caraco et al. 1989; Søndergaard et al. 1993; Roden and Edmonds 1997; Lamers et al. 1998; Wetzel 2001; Lamers et al. 2002; Smolders et al. 2006a). This may be enhanced by the decreased input of ferrous iron via the groundwater due to nitrate induced immobilization of iron. If sulphate reduction continues, toxic concentrations of hydrogen sulphide may accumulate in the sediment pore water and cause serious problems for rooted aquatic macrophytes by inducing sulphide toxicity and iron deficiency (Smolders and Roelofs 1996; Lamers et al. 2002; Smolders et al. 2006a). In extremely dry years desiccation of such sediments can lead to a strong acidification and mobilization of toxic metals, due to the oxidation of accumulated reduced sulphur to sulphuric acid (Lucassen et al. 2002; Smolders et al. 2006b).
In general, the classic iron cycle can explain the actual release of P from the sediment. In the oxygenated boundary layer between sediment and water layer, dissolved iron becomes oxidized and phosphate is effectively bound by iron(III)(hydr)oxides (Wetzel 2001; Smolders et al. 2006a). This mechanism explains the frequently found positive relation between the phosphate release to the water layer and the dissolved-P: dissolved-Fe ratios in sediment pore water (Caraco et al. 1989; Wetzel 2001; Smolders et al. 2001). Under sulphur-rich reducing conditions, however, this mechanism will no longer function as all dissolved ferrous iron is precipitated with sulphide as FeSx, (Caraco et al. 1989; Smolders and Roelofs 1996; Wetzel 2001; Smolders et al. 2001). Furthermore, dissolved sulphide (and other reduced compounds) consume oxygen in the top sediment layer, thus decreasing the thickness of the oxidized boundary layer. This may greatly boost the release of dissolved phosphate from the sediment. Increased methane production rates under highly reductive conditions may further stimulate P release by ebullition, causing mixing of anaerobic phosphate rich sediment pore water with surface water, especially in organic sediments (Søndergaard et al. 1993; Wetzel 2001).
Geurts et al. (2008) recorded the aquatic vegetation and collected surface water, sediment pore water and sediment samples in 145 fen waters in the Netherlands, Ireland and Poland. In halve of these waters they found a decreased biodiversity and increased phosphate concentrations in the water layer which appeared to be SO4 2−-induced and especially occurred below certain threshold values for total sediment Fe:P (10 mol mol−1). This suggests that especially sediments with a relatively low iron content and/or a high phosphorus content are prone to sulphate induced eutrophication.
Conclusion
Leaching of nitrate to the groundwater should receive much more attention for its potentially large scale mobilization of sulphate from geological pyrite deposits and the immobilization of ferrous iron. As nitrate is concomitantly stripped (denitrified to nitrogen gas and dinitrogen gas) from the groundwater, pyrite oxidation has been wrongfully promoted to be beneficial since it was assumed to act as a natural cleaning mechanism. It is still underestimated that during the last decades, sulphate induced eutrophication and sulphide toxicity have been responsible for the strong decline of the biodiversity in wetlands which are directly or indirectly (via surface water) fed by sulphate rich groundwater (e.g. Søndergaard et al. 1993; Roden and Edmonds 1997; Lamers et al. 2002; Smolders et al. 2006a).
Under low nutrient availability, plants will compete mainly for nutrients (Grime 1979) while under high nutrient availability, competition for light becomes the main factor, resulting in the dominance of a few high-yield species. Therefore, low concentrations of nutrients, P in particular, seem to be a prerequisite for long-term co-existence of plant species. Being a key nutrient limiting autotrophic growth in most wetlands, phosphorus availability has been proposed to be a main determinant for biodiversity (e.g. Janssens et al. 1998; Wassen et al. 2005) and increased phosphorus availability indeed provokes serious eutrophication problems in many wetlands. However, the reduction of nitrogen loads to the groundwater should be a major objective to tackle this problem. In this respect, measures to reduce the leaching of nitrate from agricultural soils are urgently needed. Such measures might include environmental indexing of fields, reduction of nitrogen gifts to soils to levels slightly below those expected to give optimum yield and a range of counter measures (such as catch crops) (see Kirchmann et al. 2002).
At present, nitrate pollution of groundwater might be one of the most serious environmental threats for wetlands. Understanding the spatial scales at which wetland processes operate, including hydrological and biogeochemical elements, are of utmost importance. Management of wetlands should therefore be approached at a broader scale which includes the landscape-scale management of groundwater systems, including aquifer geology. Knowledge on aquifer geology is highly important in order to be able to estimate the potential effects of nitrate leaching in different parts of the world. For instance, it is important to estimate how long it will take to oxidize the reduced sulphur present in the subsoil, given a certain nitrate load.
References
Aravena R, Robertson WD (1998) Use of multiple tracers to evaluate denitrification in ground water: study of nitrate from a large-flux septic system plume. Ground Water 36:975–982
Bedford BL (1996) The need to define hydrologic equivalence at the landscape scale for freshwater wetland mitigation. Ecol Appl 6:57–68
Behrendt H, Boekhold A (2006) Phosphorus saturation in soils and groundwaters. Land Degrad Dev 4:233–243
Bobbink R, Hornung M, Roelofs JGM (1998) The effects of air-borne nitrogen pollutants on species diversity in natural and semi-natural European vegetation. J Ecol 86:717–738
Broers HP, Van der Grift B (2004) Regional monitoring of temporal changes in groundwater quality. J Hydrol 296:192–220
Broers HP, Griffioen J, Willems WJ, Fraters B (2004) Naar een andere toetsdiepte voor nitraat in grondwater? Achtergronddocument voor de Evaluatie Meststoffenwet. TNO Utrecht, The Netherlands, NITG 04-066-A., 66 pp
Burgin AJ, Hamilton SK (2008) NO3 − driven SO4 2− production in freshwater ecosystems: implications for N and S cycling. Ecosystems 11:908–922
Caraco NF, Cole JJ, Lickens GE (1989) Evidence for sulphate-controlled phosphorus release from sediment of aquatic systems. Nature 341:316–318
Comly HH (1945) Cyanosis in infants caused by nitrates in well water. J Am Med Assoc 129:112–116 (Reprinted in 1987 in the same Journal 257:2788–2792)
Cowie GL, Hedges JI (1994) Biochemical indicators of diagenetic alteration in natural organic matter mixtures. Nature 369:304–307
Drever JI (1997) The geochemistry of natural waters, surface and ground water environments, 3th edn. Prentice Hall, Upper Saddle River, NJ 07458
Fan AM, Steinberg VE (1996) Health implications of nitrate and nitrite in drinking water: an update on methemoglobinemia occurrence and reproductive and developmental toxicity. Regul Toxicol Pharmacol 23:35–43
Geurts JJM, Smolders AJP, Verhoeven JTA, Roelofs JGM, Lamers LPM (2008) Sediment Fe:PO4 ratio as a diagnostic and prognostic tool for the restoration of aquatic biodiversity in fen waters. Freshw Biol 53:2101–2116
Goodchild RG (1998) EU politics for the reduction of nitrogen in water: the example of the nitrates directive. Environ Pollut 102:737–740
Grime JP (1979) Plant strategies and vegetation processes. Wiley, Chichester, 222 pp
Gundersen P, Schmidt IK, Raulund-Rasmussen K (2006) Leaching of nitrate from temperate forests—effects of air pollution and forest management. Environ Rev 14:1–57
Haaijer SCM, van der Welle MEW, Schmid MC, Lamers LPM, Jetten MSM, Op den Camp HJM (2006) Evidence for the involvement of betaproteobacterial thiobacilli in the nitrate-dependent oxidation of iron sulfide minerals. FEMS Microbiol Ecol 58:439–448
Haaijer SCM, Lamers LPM, Smolders AJP, Jetten MSM, Op den Camp HJM (2007) Iron sulphide and pyrite as potential electron donors for microbial nitrate reduction in freshwater wetlands. Geomicrobiol J 24:391–401
Hartog N, Griffioen J, Van der Weijden CH (2002) Distribution and reactivity of O2-reducing components in sediments from a layered aquifer. Environ Sci Technol 36:2436–2442
Hartog N, van Bergen PF, de Leeuw JW, Griffioen J (2004) Reactivity of organic matter in aquifer sediments: geological and Geochemical controls. Geochim Cosmochim Acta 68:1281–1292
Hartog N, Griffioen J, van Bergen PF (2005) Depositional and paleohydrogeological controls on the distribution of organic matter and other reactive reductants in aquifer sediments. Chem Geol 216:113–131
Heckrath N, Brookes PC, Poulton PR, Goulding KWT (1995) Phosphorous leaching from soils containing different phosphorous cocnetrations in the Broadbalk experiment. J Environ Qual 24:904–910
Hill AR, Devito KJ, Campagnolo S, Sanmugadas K (2000) Subsurface denitrification in a forest riparian zone: interactions between hydrology and supplies of nitrate and organic carbon. Biogeochemistry 51:193–223
Hoffmann CC, Pedersen ML, Krontvang B, Ovig L (1998) Restoration of the Rivers Brede, Cole and Skerne: a joint Danish ad British EU-LIFE demonstration project, IV-implications for nitrate and iron transformations. Aquat Conserv Freshw Ecosyst 8:223–240
Holmer M, Storkholm P (2001) Sulphate reduction and sulphur cycling in lake sediments: a review. Freshw Biol 46:431–451
Iversen TM, Grant K, Nielsen K (1998) Nitrogen enrichment of European inland and marine waters with special attention to Danish policy measures. Environ Pollut 102:771–780
Janssens F, Peeters A, Tallowin JRB, Bakker JP, Bekker RM, Fillat F, Oomes MJM (1998) Relationship between soil chemical factors and grassland diversity. Plant Soil 202:69–78
Kirchmann H, Johnston AEJ, Bergstrøm LF (2002) Possibilities for reducing nitrate leaching from agricultural lands. Ambio 31:404–408
Kool HJ (1988) Health risks in relation to drinking water treatments. In: Larson RA (ed) Biohazards of drinking water treatment. Taylor & Francis Ltd, New York
Lamers LPM, Tomassen HBM, Roelofs JGM (1998) Sulfate induced eutrophication and phytotoxicity in freshwater wetlands. Environ Sci Technol 32:199–205
Lamers LPM, Falla S-J, Samborska EM, van Dulken IAR, van Hengstum G, Roelofs JGM (2002) Factors controlling the extent of eutrophication and toxicity in sulfate-polluted freshwater wetlands. Limnol Oceanogr 47:585–593
Lucassen ECHET, Smolders AJP, Roelofs JGM (2002) Potential sensitivity of mires to drought, acidification and mobilization of heavy metals: the sediment S/(Ca + Mg) ratio as diagnostic tool. Environ Pollut 120:635–646
Lucassen ECHET, Smolders AJP, Van der Salm AL, Roelofs JGM (2004) High groundwater nitrate concentrations inhibit eutrophication of sulphate-rich freshwater wetlands. Biogeochemistry 67:249–267
McDowell RW, Sharpley AN (2004) Variation of phosphorus leached from Pennsylvanian soils amended with manures, composts or inorganic fertilizer. Agric Ecosyst Environ 102:17–27
Molenat J, Durand P, Odoux C, Gacuel P, Davy P, Gruau G (2002) Mechanisms of nitrate transfer from soil to stream in an agricultural watershed of French Brittany. Water Air Soil Pollut 133:161–183
Moncaster SJ, Bottrell SH, Tellam JH, Lloyd JW, Konhauser KO (2000) Migration and attenuation of agrochemical pollutant: insights from isotopic analysis of groundwater sulphate. J Contam Hydrol 43:147–163
Nolan BT, Ruddy BC, Hitt KH, Helsel DR (1997) Risks of nitrate in ground waters of the United States—national perspective. Environ Sci Technol 31:2229–2236
Pauwels H, Kloppmann W, Foucher JC, Martelat A, Fritsche V (1998) Field tracer test for denitrification in a pyrite-bearing aquifer. Appl Geochem 13:767–778
Postma D, Boesen C, Kristiansen H, Larsen F (1991) Nitrate reduction in an unconfined aquifer: water chemistry, reduction processes an geochemical modeling. Water Resour Res 27:2027–2045
Roden EE, Edmonds JW (1997) Phosphate mobilization in iron-rich anaerobic sediments: microbial Fe(III) oxide reduction versus iron-sulfide formation. Archiv für Hydrobiologie 139:347–378
Rothe A, Mellert K (2004) Effects of forest management on nitrate concentrations in seepage water of forests in Southern Bavaria, Germany. Water Air Soil Pollut 156:337–355
Senn DB, Hemond HF (2002) Nitrate controls on iron and arsenic in an urban lake. Science 296:2373–2376
Sims JT, Simard RR, Joern BC (1998) Phosphorus loss in agricultural drainage: historical perspective and current research. J Environ Qual 29:111–116
Smolders AJP, Roelofs JGM (1996) The roles of internal iron hydroxide precipitation, sulphide toxicity and oxidizing ability in the survival of Stratiotes aloides roots at different iron concentrations in sediment pore water. New Phytol 133:253–260
Smolders AJP, Nijboer RC, Roelofs JGM (1995) Prevention of sulphide accumulation and phosphate mobilization by the addition of iron(II) chloride to a reduced sediment: an enclosure experiment. Freshw Biol 34:559–568
Smolders AJP, Lamers LPM, Moonen M, Zwaga K, Roelofs JGM (2001) Controlling phosphate release from phosphate-enriched sediments by adding various iron compounds. Biogeochemistry 54:219–228
Smolders AJP, Lamers LPM, Lucassen ECHET, Van der Velde G, Roelofs JGM (2006a) Internal eutrophication: ‘How it works and what to do about it’, a review. Chem Ecol 22:93–111
Smolders AJP, Moonen M, Zwaga K, Lucassen ECHET, Lamers LPM, Roelofs JGM (2006b) Changes in pore water chemistry of desiccating freshwater sediments with different sulphur contents. Geoderma 132:372–383
Søndergaard M, Kristensen P, Jeppesen E (1993) Eight years of internal phosphorus loading and changes in the sediment phosphorus profile of lake Søbygaard, Denmark. Hydrobiologia 253:345–356
Sperber JI (1958) Release of phosphate from soil minerals by hydrogen sulphide. Nature 181:934
STOM (1983) Lange termijn ontwikkelingen van voedselarme milieus en grondwater van de Pleistocene zandgronden. Een verkenning van de periode 1900-2025. University of Utrecht, the Netherlands, 289 pp
Straub KL, Benz M, Schink B, Widdel F (1996) Anaerobic nitrate dependent microbial oxidation of ferrous iron. Appl Environ Microbiol 62:1458–1460
Surridge BWJ, Heathwaite AL, Baird AJ (2007) The release of phosphorus to porewater and surface water from river riparian sediments. J Environ Qual 36:1534–1544
Thorburn PJ, Biggs JS, Weier KL, Keating BA (2003) Nitrate in groundwater of intensive agricultural areas in coastal Northeastern Australia. Agric Ecosyst Environ 94:49–58
Van Beek K, Hesen P, Kappelhof J, Vink K (2006) Consequenties huidige vermesting voor de waterleidingbedrijven. H2O 39(11):25–28
Wakida FT, Lerner DN (2005) Non-agricultural sources of groundwater nitrate: a review and case study. Water Res 39:3–16
Wassen MJ, Venterink HO, Lapshina ED, Tanneberger F (2005) Endangered plants persist under phosphorus limitation. Nature 437:547–550
Wetzel RG (2001) Limnology. W.B. Saunders Company, Philadelphia, 1006 pp
Wriedt G, Rode M (2006) Modeling nitrate transport and turnover in a lowland catchment system. J Hydrol 328:157–176
Acknowledgements
The authors wish to thank Prof. Joshua Schimel, Prof. Rudy van Diggelen and two anonymous reviewers for their valuable comments which significantly improved the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Smolders, A.J.P., Lucassen, E.C.H.E.T., Bobbink, R. et al. How nitrate leaching from agricultural lands provokes phosphate eutrophication in groundwater fed wetlands: the sulphur bridge. Biogeochemistry 98, 1–7 (2010). https://doi.org/10.1007/s10533-009-9387-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-009-9387-8