
Abstract
The account article treats with the characterizations of the structural and electronic transformations of Pt/C, Pt3Co/C, and Pt3Ni/C cathode electrocatalysts in polymer electrolyte fuel cells (PEFCs) involving Pt charging/discharging, Pt–O bond formation/breaking, and Pt–Pt bond breaking/reformation in cathode potential-jump processes and the rate constants of those transformations in the surface reaction sequences on the cathode electrocatalysts by in situ time-resolved X-ray absorption fine structure (XAFS) method. Spatially heterogeneous issue and mechanism for the Pt oxidation and degradation of Pt/C cathode electrocatalyst layers in PEFCs under the operating conditions, which should be uncovered for development of next-generation PEFCs, were also characterized by a scanning nano-XAFS mapping method and a laminography-XAFS method, respectively for 2D and 3D visualizations of Pt chemical species in Pt/C cathode electrocatalyst layers. These XAFS techniques provided new insights into critical issues affecting the performance and property of practical PEFCs under the operating conditions.
Graphical Abstract

Similar content being viewed by others

1 Introduction
In these decades polymer electrolyte fuel cell (PEFC) has attracted much attention as one of the most efficient clean energy generation systems, bringing low or even zero emissions into reality, which is considered to be suitable for automotive applications due to high power density at low temperatures. Indeed, PEFC vehicles have been announced to be commercialized in 2015. This is an epoch-making technology, but for widely spread commercialization of PEFC vehicles including cars for business use, further improvements of the oxygen reduction reaction (ORR) activity and durability of cathode electrocatalysts, reducing the cost of PEFC stacking, are indispensable. To solve these problems, in situ/operando techniques, which can directly characterize cathode electrocatalysts in membrane electrode assembly (MEA) of PEFC, validate catalyst fabrications, and prove fundamental issues for development of next-generation PEFCs, are mandatory. Particularly, it is necessary to elucidate key factors affecting the performance and durability of cathode electrocatalysts in PEFCs though lots of studies have been devoted to these essential problems [1, 2].
However, there are few suitable techniques for characterizing the structures and chemical states of MEA electrode catalysts under the PEFC operating conditions in the wet, heterogeneous and multiphasic reaction field. Catalyst characterizations using NMR, Raman, FT-IR/ATR-IR, TEM/SEM, XRD, neutron scattering, soft-x ray XPS, etc. have extensively been studied [3–13], but they are difficult to observe the dynamic and spatial behavior and transformation of Pt nanoparticles under PEFC operating conditions. In situ time-resolved X-ray absorption fine structure (XAFS) techniques are very powerful for in situ/operando investigation of the local coordination structures and oxidation states of supported nanoparticle catalysts under working conditions particularly in such complex systems as PEFCs, involving Pt valence change and Pt–O/Pt–Pt bonds transformation [14–20]. These key reaction processes, which regulate the durability of PEFCs as well as the ORR activity, may occur heterogeneously in the space of the cathode layer due to the spatially heterogeneous property and distribution of Pt nanoparticles and carbon supports and the microscopically non-uniform potentials loaded in the cathode layer under PEFC potential operations [21].
Since our first in situ XAFS observation of the structural change of catalyst itself in elementary reaction steps involving an intermediate structure during the course of the catalytic ethanol oxidation on a supported Mo dimer catalyst in 1985 [22] and also in situ XAFS observation of metal–metal bond breaking/reformation behavior of a supported Rh dimer catalyst during metal-assisted ethylene hydroformylation in 1990 [23], by using home-made in situ XAFS cells, nowadays in situ XAFS technique has been widely applied to structural and electronic characterization of a variety of catalysts under working conditions and more explicitly during conducting and analyzing the catalysis (often called operando) [24–26]. In situ time-resolved XAFS technique in real-time processes enables to determine reaction sequences and structural changes at active metal sites/ensembles in dynamic catalytic processes involving adsorption, diffusion and reactions as reported by our groups in 2000-2011 [27–35]. We carried out the world’s first successful quick-XAFS (QXAFS) observation and thorough analysis of structural changes of a PEFC Pt/C cathode electrocatalyst in voltage cycling processes (0.4 ⇆ 1.0 V vs RHE) and succeeded in deciding six elementary reaction steps and their rate constants in 2007 [14]. The QXAFS analysis revealed significant time lags among the changes in cell current and the structural transformations and definite hysteresis in the structural kinetics in the Pt/C cathode electrocatalyst in the voltage cycling processes. Thus, in situ real-time XAFS can provide unique and useful information on structural kinetics/dynamics of MEA Pt/C under PEFC operating conditions, which should be relevant to ORR activity and durability of the MEA catalysts.
The key reaction processes, which regulate both ORR activity and durability of PEFC Pt/C cathode electrocatalysts, may occur heterogeneously in the space of MEA cathode layer due to the spatially heterogeneous property and distribution of Pt nanoparticles and carbon supports and the microscopically non-uniform potentials loaded in the cathode layer under PEFC potential operations [21]. The cathode degradation mechanism has also been studied to improve the MEA durability, which becomes more serious issue [1, 36–39]. Therefore, the nanoscopic spatial place and mechanism of the dissolution and deterioration of the Pt/C cathode electrocatalysts in MEAs should be uncovered to develop next-generation PEFCs with high durability. Recently, 2D and 3D micro- and nano-scale observations of catalysts, batteries, etc. using X-rays have been reported [4, 5, 7, 11–13]. Some of those techniques can provide a new insight into the spatially heterogeneous issue and mechanism for the degradation of Pt/C and Pt-M alloy/C cathode electrocatalyst layers in PEFCs.
There was no synchrotron beamline capable of conducting the XAFS measurements of PEFCs with high time and spatial resolutions under operating conditions. Therefore, in 2013 we constructed a new XAFS beamline BL36XU [40] under a New Energy and Industrial Technology Development Organization (NEDO) program, which provides high time- and spatially-resolved XAFS methods along with several complementary techniques for characterizations of the cathode electrocatalysts in PEFC MEAs. A fast scanning microscopic nano-XAFS system for low concentration samples is installed at BL36XU [41]. In this method, 2D X-ray fluorescence images are measured by fast scanning of a sample at each energy point of the XAFS measurement using the X-ray beam of about 100 × 100 nm focused on samples by a Kirkpatrick-Baez mirror.
We have succeeded in mapping Pt chemical species in Pt/C cathode catalyst layers by a scanning nano-XAFS mapping method and by a nano-QXAFS method, and obtained a new insight into nano-spatial information on the site-preferential oxidation and leaching of Pt cathode nanoparticles in degraded PEFCs [42]. Spatially-resolved XAFS analysis was also successfully carried out on a single particle of a practical catalyst for the first time [43]. Significant progress has been made in visualization of the spatial distribution of metal oxidation states inside a single catalyst particle by the scanning nano-XAFS technique [44]. Three dimensional spatial visualization of PEFCs in a non-destructive manner has become a state-of-the-art topic for understanding various phenomena in MEAs relevant to the performance and property of PEFCs [45–50]. We carried out the first 4D visualization of structures and chemical states of a Pt/C cathode electrocatalyst layer in PEFC MEA by a newly developed X-ray computed laminography (XCL)–XAFS method combining 3D XCL imaging and XAFS spectroscopy [21].
In this account article we treat with the key factors affecting the performance and durability of electrocathode catalysts in PEFC MEAs by the in situ time-resolved and spatially-resolved XAFS techniques, describing our recent characterizations of elementary steps and structural kinetics of the ORR performances by in situ time-resolved XAFS method, and depicting 2D and 3D spatial mappings of the quantity and chemical species of Pt in MEAs by the scanning nano-XAFS mapping and XCL-XAFS methods [21, 42, 51].
2 Structural Kinetics/Dynamics and Reaction Mechanism for PEFC Cathode Electrocatalysis Characterized by In Situ Time-Resolved XAFS
Platinum-based bimetallic nanoparticles with 3d transition metals (e.g. Pt3Co, Pt3Ni, etc.) have been studied as one of the promising candidates for cathode electrocatalysts in PEFCs to achieve the higher cell performance and durability compared to those of Pt/C [52–57]. The promoting effects of alloying of Pt with Co, Ni, etc. have been studied by various techniques such as theoretical calculations [58–62], transmission electron microscopy [56–67], and photoelectron spectroscopy [55, 68, 69]. Although these studies have revealed relationships between the ORR enhancements and the bimetal structures, the details of dynamic surface events taking place at the cathode electrocatalyst surface remain unclear, including individual redox processes under PEFC working conditions.
In situ time-resolved XAFS spectra of Pt/C, Pt3Co/C, and Pt3Ni/C cathode electrocatalysts in MEAs were successfully measured every 100 ms for voltage cycling processes between 0.4 and 1.0 V vs RHE [51]. The size of the MEA was 3.0 × 3.0 cm2, and catalyst loadings at the cathode were 0.5 mgPtM/Pd cm−2 (M = none, Co, or Ni). The MEA was sandwiched into an in situ XAFS cell with Teflon gaskets at both cathode and anode sides. Flows of pure H2 (>99.99999 % grade) to the anode and pure N2 (>99.99995 %) or air (G1 grade) to the cathode were regulated using mass-flow controllers. The gases were bubbled through humidifiers at 351 K. The humidified gases were supplied to the in situ XAFS cell at 353 K. The back-pressures were 2 kPa at the anode and 3 kPa at the cathode. The cell voltage between the anode and the cathode was controlled using a P/G stat (VSP, BioLogic Science Instruments Co., Ltd.) with a current amplifier (VMP 3B-20, BioLogic Science Instruments). View of the BL36XU experimental hutch and an in situ time-resolved XAFS experimental setup around the sample stage are shown in Fig. 1.

a View of the BL36XU experimental hutch and b an in situ time-resolved XAFS experimental setup around the sample stage
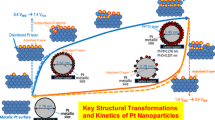
Systematic analysis of in situ time-resolved X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra in the molecular scale revealed the structural kinetics of the Pt and Pt3M (M=Co, Ni) bimetallic cathode electrocatalysts under PEFC operating conditions, and the rate constants of Pt valence change, Pt–O bond formation/breaking, and Pt–Pt bond breaking/reformation relevant to the fuel cell performance and durability were successfully determined. The results in the voltage cycling processes, that is, the potential-jump processes of 0.4 → 1.0 V and 1.0 → 0.4 V vs RHE, are summarized in Fig. 2 [51].

Time profiles of (1) electric current and charge/discharge, (2) Pt LIII-edge white line peak height, (3) CN of Pt–Pt bonds, (4) CN of Pt–M (Co, Ni) bonds, (5) R of Pt–M (Pt, Co, Ni) bonds, and (6) CN of Pt–O bonds for the voltage cycling processes (anode: H2; cathode: N2). Black solid lines represent curve-fitted data. Black dashed lines in (1) represent each component in the curve fitting. Gray markers in (2) correspond to the white line peak height of Pt foil (reference).Fig. 2 was modified from Fig. 5 of Ref. [51]
The changes in the white line heights of the in situ time-resolved QXANES spectra for the voltage cycling processes were plotted against the response time in Fig. 2 (2). It should be noted that there were significant differences in both variation range and absolute value between the white line peak heights of the Pt/C, Pt3Co/C, and Pt3Ni/C electrocatalysts. The valence level of the Pt/C electrocatalyst at 0.4 V was metallic. The white line peak heights of Pt3Co/C and Pt3Ni/C at 0.4 V were less than that for Pt/C, indicating electron transfer from Co or Ni to Pt. This is similar to our previous results for a Pt3Co/C electrocatalyst with 6 mg-PtCo cm−2 [15]. As for the structural parameters determined by the curve fitting analysis of the in situ time-resolved EXAFS Fourier transforms we found significant changes in the coordination numbers (CN) of Pt–Pt and Pt–O bonds for all catalysts [Fig. 2 (3) and (6)]. In the potential-jump process from 0.4 to 1.0 V, the formation of Pt–O bonds proceeds accompanied with partial dissociation of the Pt–Pt bonds. Note that the changes in CNs of Pt–Co and Pt–Ni were negligible [Fig. 2 (4)]. These results agree with the structural model of Pt-enriched surface and Pt-M bimetallic core. The Pt–Pt and Pt–M (M: Co or Ni) bond distances (R) remained constant in the voltage cycling processes [Fig. 2 (5)]. The four electronic and structural parameters were fitted with single or two exponential functions to estimate the rate constants (k) for each events. The determined rate constants for the Pt/C, Pt3Co/C, and Pt3Ni/C cathode electrocatalysts in the voltage cycling processes of 0.4 → 1.0 V and 1.0 → 0.4 V are shown in Fig. 3 (top) [51]. The electrochemical processes immediately proceeded and then structural changes of the Pt electrocatalysts (charging/discharging, Pt–O bond formation/breaking and Pt–Pt bond breaking/reformation) proceeded. The aspects of the rate constants for the three electrocatalysts are similar to each other, indicating the similar reaction mechanisms at the cathode electrocatalyst surfaces. It should be noted that the rates of the structural transformation for the three cathode electrocatalysts were in the order of Pt3Co/C > Pt3Ni/C > Pt/C.

Top Structural kinetics of the surface events for the voltage-cycling processes between 0.4 and 1.0 V vs RHE in anode: H2 and cathode: N2. Bottom (A) Variation of Pt LIII-edge XANES white line peak heights at 0.4 V with the specific power density. B, C Enhancements by Co and Ni (relative rate constants) vs. specific power density. Figure 3 was revised by merging Figs. 6 and 7 of Ref. [51]
Figure 3 (bottom A) shows the variation of the Pt LIII-edge XANES white line peak heights at 0.4 V with the specific power density [51], which is in agreement with the linear relationship between ORR activity and d-band center shift for Pt3Co, Pt3Ni, and Pt [70–72]. Although we did not investigate the particle size effect on the structural kinetics, the ORR kinetics may not be affected by the particle size of Pt cathode electrocatalysts (below 5 nm size) [73, 74]. The enhancements of the rate constants by the Co and Ni addition to Pt/C are shown in Fig. 3 (bottom) [51]. The increments compared to Pt/C were larger for the potential-jump process of 1.0 → 0.4 V than the 0.4 → 1.0 V jump process. Figure 3 (bottom C) revealed that the rate enhancement of the structural kinetics of the Pt electrocatalysts has a positive correlation with the cell performance (regarded as catalyst activity approximately in this condition). The Pt–O bond breaking at the electrocatalyst surface as well as the reduction of Pt valence is known to be a key step in the steady-state ORR [58–62, 70–72, 75]. The present results of the in situ time-resolved XAFS analysis demonstrate a nearly linear relationship between the increase of the rate constants k′Pt–O and k′Pt and the improvement of PEFC performances. The rate constant (k′Pt–Pt) for Pt–Pt reformation was also promoted by the Co and Ni addition, but the difference in the enhancement between Co and Ni was negligible. In summary, the overall reaction sequences and the structural and electronic transformations at the surface of a typical Pt/C cathode electrocatalyst under the potential-jump processes (0.4 ⇆ 1.4 V vs RHE) elucidated by the in situ time-resolved XAFS analysis are illustrated in Fig. 4.

Reaction mechanism and structural kinetics for Pt surface events of an MEA Pt/C cathode catalyst in the potential-jump transient-response processes between 0.4 ⇆ 1.4 V vs RHE. The rate constants k elec(1), k elec(2), k′ elec(1), and k′ elec(2) were determined by electrochemical current changes, and the rate constants k Pt–O, k Pt, k Pt–Pt, k′ Pt–O, k′ Pt, and k′ Pt–Pt were determined by time-resolved XAFS
3 Spatially-Resolved XAFS Imaging of Degraded PEFC Pt/C Cathode Electrocatalysts
3.1 Mapping of Pt Chemical Species in PEFC MEAs by a Scanning Nano-XAFS Mapping Method and a Nano-QXAFS Method
The new beamline BL36XU that we designed and constructed with JASRI/SPring-8 under a NEDO program in 2013 [40] and experimental setup for nano-XAFS measurements are shown in Fig. 5. The beamline BL36XU is the worldwide only-one and high-performance beamline for real-time and spatially-resolved XAFS for PEFCs under the operating conditions.

To measure the scanning nano-XAFS and nano-QXAFS spectra, we prepared two types of MEAs as typical examples: (a) MEA with a flat interface and few micro-crack (denoted as MEA-A) and (b) MEA with micro-cracks (denoted as MEA-B). These MEAs were electrochemically treated with aging by I–V load cycles and accelerated durability test (ADT) by rectangle cycles between 0.60 and 1.0 V vs RHE. We applied the ADT cycles to both MEA-A and MEA-B until the maximum power density decreased by 25 % from those of the aging MEAs. The Pt nanoparticles (2.5 nm) at the cathode grew to 8.5 nm on average by the ADT cycles. Sliced MEA samples were used for the nano-XAFS measurements. For slicing the sample pieces, a cryo-type Ultra microtome (Leica EM UC7) was used at sample temperature 298 K, blowing humid N2. After slicing the sample, we put the sample to the humid N2-filled glove-bag again, then put the sliced sample on a SiN membrane substrate (1 μm thick). The sliced sample-membrane was put in a designed XAFS cell and nano-XAFS spectra were measured. The resultant nano-XAFS spectra are regarded to be equivalent to the nano-XAFS spectra measured in situ after the aging and ADT cycles because the spatial aspect of the degradation is irreversible in the time scale under the present experimental conditions. For the nano-XAFS measurements, nano-beams of 570 × 540 nm2 and 228 × 225 nm2 were used. We performed the scanning nano-XAFS mappings every 1 μm step in the 45 × 45 μm2 or 125 × 95 μm2 regions. We also carried out nano-XAFS measurements by the nano-QXAFS method. The acquisition time for the scanning nano-XAFS and nano-QXAFS measurements was 1.6 s or 15 min for each spectrum, respectively. We used the scanning nano-XAFS for XANES analysis and the nano-QXAFS for EXAFS analysis.
Figure 6 shows the scanning nano (570 × 540 nm2 (A, a, B, b) Pt LIII-edge XANES mapping for MEA-A before (A–C) and after (a–c) the durability test; A and a: Pt quantity mapping, B and b: Pt valence mapping. Figure 6 corresponds to 2D depth mapping of the MEAs. In the aging MEA- A the Pt valence mapping (B) in the observed whole cathode area did not show any change significantly. The line profiles of the Pt quantity (blue) and Pt valence (red) along the red arrow of Fig. 6 B are shown in Fig. 6 C, which elucidates that the Pt valence is metallic in the whole region. In contrast, the distribution of Pt quantity in MEA-A after the ADT cycles was heterogeneous (Fig. 6 a). Interestingly, the Pt valence mapping (Fig. 6 b) characterized the unique region between the cathode layer edge and Pt band. The line profiles of the Pt quantity (blue) and Pt valence (red) along the red arrow of Fig. 6 b are shown in Fig. 6 c. It is to be noted that the Pt valence in the cathode layer began to increase from the place at the distance of roughly 3.2 μm far from the cathode layer edge and showed a maximum (0.33+) at d of the line profile c in the electrolyte membrane toward the Pt band as shown Fig. 6. Thus, the Pt valence mapping of Fig. 6 b visualizes the existing area of oxidized Pt species in the degraded MEA-A. At the other parts than the boundary areas in the cathode layer no oxidized Pt species were observed. The resultant oxidized Pt species is suggested to be produced first in the boundary of the cathode electrocatalyst layer during the operating process involving repeated potential loads, while keeping the Pt nanoparticles at the other parts of the cathode electrocatalyst layer to be metallic.

A and a: Pt LIII-edge jump mapping (Pt quantity mapping), B and b: : Pt valence mapping, for MEA-A after the aging (A, B) and ADT cycles (a, b), respectively by the scanning nano (570 × 540 nm2) XAFS mapping method. C and c: Line profiles of the absorbance (μ) at 11.600 keV (blue) and Pt valence (red) in the scanning nano-XANES spectra along the red arrows in B and b, respectively. The cathode boundary with the polymer electrolyte membrane in the line profile c was enlarged to exhibit the preferentially oxidized region. D and E: Pt LIII-edge jump mapping (Pt quantity mapping) and Pt valence mapping, respectively for another MEA-A piece after the aging by the scanning nano (228 nm x 225 nm) XAFS mapping method. The quantity scale in D is not equivalent to that in A. Figure 6 was revised from Fig. 2 of Ref. [42]
Figure 7 exhibits the Pt LIII-edge jump mapping (A), normalized white line peak area (WLPA) mapping (B) and Pt valence mapping (C) around the Pt/C cathode layer with a micro-crack, in MEA-B after the ADT cycles. It is to be noted that the Pt valence in most parts of the micro-crack region was calculated as 2+ by the linear relationship between the normalized WLPA and Pt valence [17, 76, 77], whereas Pt nanoparticles in the other cathode areas were metallic as shown in Fig. 7C. The aging MEA-B exhibited the metallic Pt0 valence similar to the aging MEA-A in Fig. 6B. Figure 7D shows the line-scan profiles of the Pt LIII-edge jump (Pt quantity) and Pt valence in the scanning nano-XAFS spectra along the red arrow of Fig. 7B. It is notable that the Pt valence in the cathode electrocatalyst layer began to increase from the places at the distances of roughly 1.2 and 2.4 μm on both cathode sides of the micro-crack far from the boundary edge to show the maximum Pt valence (Pt2+) in the micro-crack area as shown in Fig. 7. It was found that the micro-crack areas 1, 5 and 6 accommodated Pt2+ species and that the CN of Pt–O at 0.200 nm was 4.0 (± 0.4) but no Pt–Pt bonding was observed. This means that the Pt2+ species does not have any Pt–Pt bonds but have a four-coordinated Pt2+–O4 structure. The presence of the Pt2+–O4 coordination species was also suggested with the degraded MEA-A in the region between the cathode layer edge and the Pt band. There are Nafion ionomers with sulfonic group and water in the micro-crack region and the Pt2+ species may be stabilized by coordination with the sulfonic group (Nf-SO3) as well as H2O to form [Pt(Nf-SO3)x(H2O)y]2+. During the ADT cycles, Pt nanoparticles at the boundary of the Pt/C cathode layer toward the micro-crack are regarded to be first oxidized, then dissolved into the ionomers with moisture in the micro-crack region. The possibility that the oxidized Pt species might have formed elsewhere in the cathode electrocatalyst layer in the MEA-B and migrated toward the micro-crack during the ADT cycles may be excluded because the metallic Pt nanoparticles in the other parts of the cathode layer than the 1.2/2.4 μm areas around the micro-crack were not oxidized to dissolve significantly by the ADT cycles at the early stage of the degradation. As for the other micro-crack areas 2–4 of Fig. 7B, the CNs of Pt–Pt and Pt–O were 1.0–1.8 and 2.7–3.1, respectively, where the Pt valences are estimated as 1.8–1.9+. It is probable that both small Pt0 clusters and Pt2+–O4 species coexist in the micro-crack areas 2–4. Thus, the micro-cracks larger than about 2 μm as well as about 3 μm boundary are suggested to promote the MEA degradation by oxidizing and leaching Pt nanoparticles as Pt2+–O4 coordination species. These results and information on the degradation mechanism could be used to design MEA cathodes with better durability.

a Pt LIII-edge jump mapping (Pt quantity mapping), b normalized WLPA mapping, and c Pt valence mapping for MEA-B after the ADT cycles by the scanning nano-XAFS mapping method. d Line profiles of the absorbance (μ) at 11.600 keV (blue) and Pt valence (red) in the scanning nano XANES spectra along the red arrow in b. The micro-crack boundary of the line profile d was enlarged to exhibit the preferentially oxidized region. e Nano-QEXAFS analysis by the curve-fittings (CN(Pt–Pt): blue, CN(Pt–O): red) and Pt valence (green) for each micro-crack area (1–6) in b. Figure 7 was revised from Fig. 3 of Ref. [42]
4 Spatial Visualization of a Cathode Pt/C Layer in PEFC MEA by a 3D XCL–XAFS Method
XCT can be applied to a sample smaller than observation field area in all projected directions of XCT, but in general it cannot analyze a flat sample. On the other hand, X-ray computed laminography (XCL) is applicable to partial 3D imaging of a such-shaped sample [78–80]. X-rays from an undulator were monochromatized by a Si(111) double-crystal monochromator to irradiate an MEA sample through a rotating beam diffuser to reduce speckle noises coming from optical components in the X-ray beam path. A high spatial resolution X-ray image detector consisted of a visible light conversion unit (BM3, AA50, Hamamatsu Photonics) and a cooled charge coupled device (CCD) camera (C-4880-41S, Hamamatsu Photonics) were used. A field of view of the sectional image (X–Y section) obtained from laminography reconstruction was 400 × 400 μm2. The rotational axis of the MEA sample was inclined from the vertical direction to 30° as shown in Fig. 8 [21]. The whole sample was fixed on the acrylic sample holder using metallic retainer plates. Sectional images were reconstructed using a filtered back projection method, considering the inclination of the rotational axis. The 3D image was constructed by stacking X–Y sectional images along Z axis. In the laminography reconstruction, however, it is impossible to reconstruct a complete 3D image because of the inclination of the rotational axis. Therefore, there is a difference of spatial resolution depending on the direction of the reconstructed 3D image. The spatial resolution in the reconstructed plane (X–Y plane) is 1.5 μm, while the spatial resolution in the depth direction (Z direction) is approximately 5 μm [21].

We succeeded in reconstructing 3D images of the distributions of Pt nanoparticles in the cathode catalyst layer in the fresh and degraded MEAs by the XCL-XAFS technique as shown in Fig. 9 [21]. Bright and dark areas indicate high and low Pt concentrations, respectively. In the fresh MEA, Pt nanoparticle catalysts were almost fully and mostly uniformly loaded throughout the cathode electrocatalyst layer [Fig. 9 (A-1)]. Note that the distribution of Pt nanoparticles changed markedly in the three dimensions for the degraded MEA [Fig. 9 (B-1)]. There were several aggregated Pt spots colored with red heterogeneously, indicating sintering and breakdown/dissolution of Pt nanoparticles in MEA during ADT cycles. Besides, large cavities were observed, indicating carbon corrosion. These results suggest that Pt dissolution, breakdown, migration and aggregation as well as carbon corrosion spread throughout the cathode electrocatalyst layer in the degraded MEA. It has been reported that Pt nanoparticles on carbon supports migrate with ease under the repeated durability testing conditions, resulting in dissolution, detachment, and agglomeration [81, 82].

(A-1 & B-1) The distribution of Pt catalysts in the cathode Pt/C catalyst layers of fresh MEA (A-1) and degraded MEA (B-1) as observed by 3D XCL–XANES. The change in intensity (colored scale bar), which reflects Pt quantity, was calculated from the difference between the intensity of X-ray absorption at 11.572 keV and that at 11.496 keV. (A-2 & B-2) Series of depth-resolved Pt LIII-edge XCL–XANES spectra along the Z axis for the cathode catalyst layers in the fresh MEA (A-2) and degraded MEA (B-2). XANES spectra are presented at each depth every 12 μm. (B-3) Cross-sectional image of Pt quantity observed by 3D-Laminography XANES; b1 and b2: measurement areas of DX = 27.6 μm and DY = 32.4 μm, respectively. c XANES spectrum of Pt foil. (B-4) Spatially resolved Pt LIII-edge Laminography–XANES spectra at a depth Z of 113.2 μm for the degraded MEA. Vertical axis (intensity) was normalized by Pt LIII-edge jump. Figure 9 was revised by merging Figs. 1, 2, and 3 of Ref. [21]
Depth (Z)-resolved XANES spectra were successfully obtained by integrating 3D XCL–XANES spectra over the X–Y plane at each depth in the X-ray energy range of 11.439–11.608 keV [Fig. 9 (A-2 and B-2)]. A difference in the edge intensity of the Pt LIII-edge XANES spectra indicates difference in the Pt quantity in the MEAs along the Z depth. In the fresh MEA, the Pt quantity gradually increased with depth and reached a maximum at about 170 μm deep [Fig. 9 (A-2)]. Pt LIII-edge 3D XCL–XANES spectra for different parts of the electrocatalyst layer were similar to each other, indicating that the Pt catalysts in those areas were in similar chemical states. In contrast, a series of depth-resolved XCL–XANES spectra of the degraded MEA were wavy in the Z direction, which probably reflects the degradation of the MEA [Fig. 9 (B-2)] [21].
Figure 9 B3 and B4 shows a typical cross-sectional image of the Pt quantity in the degraded MEA observed by the 3D-laminography–XANES technique and 3D-laminography–XANES spectra for particular areas at a depth Z of 113.2 μm, respectively. In the fresh MEA, no significant aggregation of the Pt catalysts was observed, and the Pt catalysts in the whole area were in similar chemical states (not shown here). In the degraded MEA, the chemical states of the Pt catalysts changed, depending on the area/position in the cathode layer. The white-line intensity had a lower maximum intensity at the Pt LIII-edge in the area where aggregation occurred (b1) than the area where Pt was evenly distributed (b2). The 3D XCL–XANES spectra mapping visualized the heterogeneous chemical states of the Pt nanoparticle electrocatalysts in the cathode layer in the degraded MEA, which cannot be monitored by conventional XAFS method which averages 3D structural information with different chemical states in the MEA. Indeed, the 3D XCL–XANES spectra integrated for the entire cathode electrocatalyst layer agreed with the XANES spectra measured by the conventional XANES technique [21]. The dissolution, breakdown, detachment, migration, and agglomeration of Pt nanoparticles may be promoted by the carbon corrosion to make cavities and cracks.
The nano-focused XAFS and XCL–XAFS methods are very unique and powerful in study on such complex functional materials and systems as PEFC MEAs. Nevertheless, in situ nano-XAFS and XCL–XAFS techniques for time-dependent mapping of the quantity and chemical species of Pt or active metals in PEFC MEAs under various PEFC operating conditions will provide new impacts to further understanding and more detailed discussion of the MEA degradation mechanism and performance property for development of next-generation PEFCs.
5 Conclusions
Key factors affecting the performance and durability of cathode electrocatalysts in PEFC MEAs were characterized and discussed by in situ real-time and spatially resolved XAFS techniques. Systematic analysis of the in situ time-resolved QXAFS spectra provided the reaction sequences and rate constants of the dynamic surface events on the Pt/C, Pt3Co/C, and Pt3Ni/C cathode electrocatalysts in the potential-jump processes from 0.4 to 1.0 V and from 1.0 to 0.4 V vs RHE. The redox reactions on the Pt-based bimetallic electrocatalysts with Pt-enriched surfaces and Pt–Co/Ni bimetallic cores between 0.4 and 1.0 V took place on the Pt-enriched surfaces at the similar mechanisms for all three electrocatalysts. We found notable increase in the rate constants of the structural kinetics, in particular, in the rate constants k′Pt-O (Pt-O dissociation) and k′Pt (Pt discharging) for the reductive processes of the Pt cathode electrocatalysts, and the extent of the enhancement was larger with Co than with Ni.
The present spatially-resolved nano-XAFS study on the MEAs enabled to map the Pt valence and to identify the major places for the oxidation and dissolution of Pt nanoparticles by the ADT cycles. This technique gained new insights into the 2D distribution of metallic and oxidized Pt species and the site-preferential mechanism for Pt oxidation and dissolution to form the Pt2+ monomeric species with a Pt–O4 coordination structure in the degradation process. The 3D XCL–XAFS technique also provided a new way of the 4D visualization of the structure/morphology, Pt distribution, and chemical states of the cathode electrocatalyst layer in PEFC MEA in a non-destructive manner. The spatially-resolved 3D XCL–XANES revealed the 3D spatially heterogeneous presence of aggregation and chemical states of the Pt nanoparticle electrocatalyst in the degraded MEA.
In situ time-resolved and spatially-resolved XAFS techniques are promising techniques, which can address critical issues and mechanisms for the ORR performance and degradation of the MEA cathode electrocatalysts and also aid the further development of next-generation electrocatalysts, by presenting real-time structural kinetics/dynamics of potential-dependent surface reactions on cathode electrocatalysts and mapping time-dependent chemical changes of Pt nanoparticles in PEFC MEAs under various electrochemical conditions in a non-destructive manner. The techniques can be consorted with other new techniques such as in situ 3D XCT, real-time XRD, ambient-pressure HAXPES, etc. The developed XAFS techniques may also be applied to a variety of other functional materials such as catalysts, solid-oxide FCs, batteries, devices, biomaterials, etc., particularly under in situ/operando conditions.
References
Debe MK (2012) Nature 486:43–51
Houchins C, Kleen GJ, Spendelow JS, Kopasz J, Peterson D, Garland NL, Ho DL, Marcinkoski J, Martin KE, Tyler R, Papageorgopoulos DC (2012) Membranes 2:855–878
Promfret MB, Owrutsky JC, Walker RA (2007) Anal Chem 79:2367–2372
Choi K-S, Kim B-G, Park K, Kim H-M (2012) Fuel Cells 12:908–938
Rice C, Tong Y, Oldfield E, Wieckowski A, Hahn F, Gloaguen F, Léger J-M, Lamy C (2000) J. Phys. Chem. B 104:5803–5807
Healy J, Hayden C, Xie T, Olson K, Waldo R, Brundage M, Gasteiger H, Abbott J (2005) Fuel Cells 5:302–308
Manke I, Hartnig Ch, Grünerbel M, Kaczerowski J, Lehnert W, Kardjilov N, Hilger A, Banhart J, Treimer W, Strobl M (2007) Appl Phys Lett 90(184101):1–3
Kramer D, Zhang J, Shimoi R, Lehmann E, Wokaun A, Shinohara K, Scherer G (2005) Electrochim Acta 50:2603–2614
Feindel KW, LaRocque LP-A, Starke D, Bergens SH, Wasylishen RE (2004) J Am Chem Soc 126:11436–11437
Lamy C, Rousseau S, Belgsir EM, Coutanceau C, Léger J-M (2004) Electrochim Acta 49:3901–3908
Owejan JP, Trabold TA, Jacobson DL, Arif M, Kandlikar SG (2007) Int J Hydrog Energy 32:4489–4502
Casalongue HS, Kaya S, Viswanathan V, Miller DJ, Friebel D, Hansen HA, Nøskov JK, Nilsson A, Ogasawara H (2013) Nature Commun. doi:10.1038/ncomms3817
Arlt T, Manke I, Wippermann K, Riesemeier H, Mergel J, Banhart J (2013) J Power Sources 221:210–216
Tada M, Murata S, Asakoka T, Hiroshima K, Okumura K, Tanida H, Uruga T, Nakanishi H, Matsumoto S, Inada Y, Nomura M, Iwasawa Y (2007) Angew Chem Int Ed 46:4310–4315
Ishiguro N, Saida T, Uruga T, Nagamatsu S, Sekizawa O, Nitta K, Yamamoto T, Ohkoshi S, Iwasawa Y, Yokoyama T, Tada M (2012) ACS Catal. 2:1319–1330
Ishiguro N, Saida T, Uruga T, Sekizawa O, Nagasawa K, Nitta K, Yamamoto T, Ohkoshi S, Yokoyama T, Tada M (2013) Phys Chem Chem Phys 15:18827–18834
Nagamatsu S, Arai T, Yamamoto M, Ohkura T, Oyanagi H, Ishizaka T, Kawanami H, Uruga T, Tada M, Iwasawa Y (2013) J Phys Chem C 117:13094–13107
Russell AE, Rose A (2004) Chem Rev 104:4613–4636
Friebel D, Miller DJ, O’Grady CP, Anniyev T, Barger J, Bergmann U, Ogasawara H, Wikfeldt KT, Pettersson LGM, Nilsson A (2011) Phys Chem Chem Phys 13:262–266
Kityakarn S, Saida T, Sode A, Ishiguro N, Sekizawa O, Uruga T, Nagasawa K, Yamamoto T, Yokoyama T, Tada M (2014) Top Catal 57:903–910
Saida T, Sekizawa O, Ishiguro N, Hoshino M, Uesugi K, Uruga T, Ohkoshi S, Yokoyama T, Tada M (2012) Angew Chem Int Ed 124:10457–10460
Iwasawa Y, Asakura K, Ishii H, Kuroda H (1985) Z Phys. Chem. N. F. 144:105–115
Asakura K, Bando KK, Isobe K, Arakawa H, Iwasawa Y (1990) J Am Chem Soc 112(3242–3244):9096–9104
Iwasawa Y (ed) (1996) X-Ray absorption fine structure for catalysts and surfaces. World Scientific, Singapore
Weckhuysen BM (2003) Phys Chem Chem Phys 5:4351–4360
Singh J, Lamberti C, van Bokhoven JA (2010) Chem Soc Rev 39:4754–4766
Yamaguchi A, Shido T, Inada Y, Kogure T, Asakura K, Nomura M, Iwasawa Y (2000) Catal Lett 68:139–145
Yamaguchi A, Suzuki A, Shido T, Inada Y, Asakura K, Nomura M, Iwasawa Y (2001) Catal Lett 71:203–208
Iwasawa Y (2003) J Catal 216:165–177
Suzuki A, Inada Y, Yamaguchi A, Chihara T, Yuasa M, Nomura M, Iwasawa Y (2003) Angew Chem Int Ed 42:4795–4799
Suzuki A, Yamaguchi A, Chihara T, Inada Y, Yuasa M, Abe M, Nomura M, Iwasawa Y (2004) J. Phys. Chem. B 108:5609–5616
Yamamoto T, Suzuki A, Nagai Y, Tanabe T, Dong F, Inada Y, Nomura M, Tada M, Iwasawa Y (2007) Angew Chem Int Ed 46:9253–9256
Tada M, Uemura Y, Bal R, Inada Y, Nomura M, Iwasawa Y (2010) Phys Chem Chem Phys 12:5701–5706
Uemura Y, Inada Y, Bando KK, Sasaki T, Kamiuchi N, Eguchi K, Yagishita A, Nomura M, Tada M, Iwasawa Y (2011) J Phys Chem C 115:5823–5833
Uemura Y, Inada Y, Bando KK, Sasaki T, Kamiuchi N, Eguchi K, Yagishita A, Nomura M, Tada M, Iwasawa Y (2011) Phys Chem Chem Phys 13:15833–15844
Yu Y, Li H, Wang H, Yuan X-Z, Wang G, Pan M (2012) J. Power Sources 205:10–23 and references there in
Burlatsky SF, Gummalla M, Atrazhev VV, Dmitriev DV, Kuzminyh NY, Erikhman NS (2011) J Electrochem Soc 158:B322–B330
Bi W, Gray GE, Fuller TF (2007) Electrochem Solid-State Lett 10:B101–B104
Zhang J, Litteer BA, Gu W, Liu H, Gasteiger HA (2007) J Electrochem Soc 154:B1006–B1011
Sekizawa O, Uruga T, Tada M, Nitta K, Kato K, Tanida H, Takeshita K, Takahashi S, Sano M, Aoyagi H, Watanabe A, Nariyama N, Ohashi H, Yumoto H, Koyama T, Senba Y, Takeuchi T, Furukawa Y, Ohata T, Matsushita T, Ishizawa Y, Kudo T, Kimura H, Yamazaki H, Tanaka T, Bizen T, Seike T, Goto S, Ohno H, Takata M, Kitamura H, Ishikawa T, Yokoyama T, Iwasawa Y (2013) J. Phys. Conf. Ser. 430:012020-1–012020-4
Tsuji T, Uruga T, Nitta K, Kawamura N, Mizumaki M, Suzuki M, Sekizawa O, Ishiguro N, Tada M, Ohashi H, Yamazaki H, Yumoto H, Koyama T, Senba Y, Takeuchi T, Terada Y, Nariyama N, Takeshita K, Fujiwara A, Goto S, Yamamoto M, Takata M, Ishikawa T (2013) J. Phys: Conf. Ser. 430:012019-1–012019-4
Takao S, Sekizawa O, Nagamatsu S, Kaneko T, Yamamoto T, Samjeske G, Higashi K, Nagasawa K, Tsuji T, Suzuki M, Kawamura N, Mizumaki M, Uruga T, Iwasawa Y (2014) Angew. Chem. Int. Ed. doi:10.1002/anie.201408845
Tada M, Ishiguro N, Uruga T, Tanida H, Terada Y, Nagamatsu S, Iwasawa Y, Ohkoshi S (2011) Phys Chem Chem Phys 13:14910–14913
Ishiguro N, Uruga T, Sekizawa O, Tsuji T, Suzuki M, Kawamura N, Mizumaki M, Nitta K, Yokoyama T, Tada M (2014) ChemPhysChem 15:1563–1568
Inukai J, Miyatake K, Takada K, Watanabe M, Hyakutake T, Nishide H, Nagumo Y, Watanabe M, Aoki M, Takano H (2008) Angew Chem Int Ed 47:2792–2795
Lee SJ, Lim N-Y, Kim S, Park G-G, Kim C-S (2008) J Power Sources 185:867–870
Sinha PK, Mukherjee PP, Wang C-Y (2007) J Mater Chem 17:3089–3103
Manke I, Hartnig C, Kardjilov N, Riesemeier H, Goebbels J, Kuhn R, Kruger P, Banhart J (2010) Fuel Cells 10:26–34
Sasabe T, Deevanhxay P, Tsushima S, Hirai S (2011) Electrochem Commun 13:638–641
Flückiger R, Marone F, Stampanoni M, Wokaun A, Büchi FN (2011) Electrochim Acta 56:2254–2262
Ishiguro N, Kityakarn S, Sekizawa O, Uruga Y, Sasabe T, Nagasawa K, Yokoyama Y, Tada M (2014) J. Phys. Chem. C 118:15874–15883
Markovic NM, Schmidt TJ, Stamenkovic V, Ross PN (2001) Fuel Cells 1:105–116
Paulus UA, Wokaun A, Scherer GG, Schmidt TJ, Stamenkovic V, Radmilovic V, Markovic NM, Ross PN (2002) J. Phys. Chem. B 106:4181–4191
Mukerjee S, Srinivasan S (1993) J Electroanal Chem 357:201–224
Koh S, Toney MF, Strasser P (2007) Electrochim Acta 52:2765–2774
Yu P, Pemberton M, Plasse P (2005) J Power Sources 144:11–20
Chen S, Sheng W, Yabuchi N, Ferreira PJ, Allard LF, Shao-Horn Y (2009) J Phys Chem C 113:1109–1125
Kitchin JR, Nørskov JK, Barteau MA, Chen JG (2004) J. Chem. Phys. 120:10240–10246
Xu Y, Ruban AV, Mavrikakis M (2004) J Am Chem Soc 126:4717–4725
Stamenkovic V, Mun BS, Mayrhofer KJJ, Ross PN, Markovic NM, Rossmeisl J, Greeley J, Nørskov JK (2006) Angew. Chem. Int. Ed. 45:2897–2901
Callejas-Tovar R, Balbuena PB (2008) Surf Sci 602:3531–3539
Matanovic I, Garzon FH, Henson NJ (2011) J Phys Chem C 115:10640–10650
Xin HL, Mundy JA, Liu Z, Cabezas R, Hoven R, Kourkoutis LF, Zhang J, Subramanian NP, Makharia R, Wagner FT, Muller DA (2012) Nano Lett 12:490–497
Carlton CE, Chen S, Ferreira PJ, Allard LF, Shao-Horn Y (2012) J. Phys. Chem. Lett. 3:161–166
Durst J, Lopez-Haro M, Dubau L, Chatenet M, Soldo-Olivier Y, Guétaz L, Bayle-Guillemaud P, Maillard F (2014) J. Phys. Chem. Lett. 5:434–439
Yu Y, Xin HL, Hovden R, Wang D, Rus ED, Mundy JA, Muller DA, Abruña H (2012) Nano Lett 12:4417–4423
Heggen M, Oezaslan M, Houben L, Strasser P (2012) J Phys Chem C 116:19073–19083
Menning CA, Hwu HH, Chen JG (2006) J. Phys. Chem. B 110:15471–15477
Yu X, Ye S (2007) J Power Sources 172:145–154
Stamenkovic VR, Mun BS, Mayhofer KJJ, Ross PN, Markovic NM (2006) J Am Chem Soc 128:8813–8819
Stamenkovic VR, Fowler B, Mun BS, Wang G, Ross PN, Lucas CA, Marcovic NM (2007) Science 315:493–497
Stamenkovic VR, Mun BS, Arenz M, Mayrhofer KJJ, Lucas CA, Wang G, Ross PN, Markovic NM (2007) Nat Mater 6:241–247
Nesselberger M, Ashton S, Meier JC, Katsounaros I, Mayrhofer KJJ, Arenz M (2011) J Am Chem Soc 133:17428–17433
Perez-Alonso FJ, McCarthy DN, Nierhoff A, Hernandez-Fernandez P, Strebel C, Steohens IEL, Nielsen JH, Chorkenfdorff I (2012) Angew. Chem. Int. Ed. 51:4641–4643
Mun BS, Watanabe M, Rossi M, Stamenkovic V, Markovic NM, Ross PN (2005) J. Chem. Phys. 123:204717
Nagasawa K, Takao S, Higashi K, Nagamatsu S, Samjeské G, Imaizumi Y, Sekizawa O, Yamamoto T, Uruga T, Iwasawa Y (2014) Phys Chem Chem Phys 16:10075–10087
Christensen ST, Elam JW, Rabuffetti FA, Ma Q, Weigand SJ, Lee B, Seifert S, Stair PC, Poeppelmeiter KR, Hersam MC, Bedzyk MJ (2009) Small 5:750–757
Helfen L, Baumbach T, Mikul P, Kiel D, Pernot P, Cloetens P, Baruchel J (2005) Appl Phys Lett 86(071915):1–3
Harasse S, Yashiro W, Momose A (2011) Opt Express 19:16560
Helfen L, Myagotin A, Mikul P, Pernot P, Voropaev A, Elyyan M, Michiel MD, Baruchel J, Baumbach T (2011) Rev Sci Instrum 82:063702
Mayrhofer KJJ, Ashton SJ, Meier JC, Wiberg GKH, Hanzlik M, Arenz M (2008) J Power Sources 185:734–739
Perez-Alonso FJ, Elkjar CF, Shima SS, Abrams BL, Stephens IEL, Chorkendorff I (2011) J Power Sources 196:6085–6091
Acknowledgments
This work has been performed under NEDO program. The work was performed with the approval of SPring-8 subject numbers 2010B1015, 2011A1031, 2011A1032, 2011B1018, 2011B1039, 2011B1040, 2012A1015, 2012A1024, 2012A1026, 2012B1024, 2012B1008, 2013A7803, 2013A7820, 2013B7803, 2013B7820, and 2014A7806.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Tada, M., Uruga, T. & Iwasawa, Y. Key Factors Affecting the Performance and Durability of Cathode Electrocatalysts in Polymer Electrolyte Fuel Cells Characterized by In Situ Real Time and Spatially Resolved XAFS Techniques. Catal Lett 145, 58–70 (2015). https://doi.org/10.1007/s10562-014-1428-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1428-9