
Abstract
In this study H3PW12O40·9H2O and H3PMo12O40·6H2O (HPA) particles were changed into nano forms by heat-treatment in an autoclave as a simple, repaid, inexpensive and one step method. The particle size of these nanoparticles was around 25 nm. The as-synthesized nanostructures were characterized by dynamic light scattering, X-ray powder diffraction, transmission electron microscopy, Fourier transform infrared spectroscopy and inductively coupled plasma analyzer. Thermal stability of nanoparticles was surveyed by thermal gravimeter analyse. Acidity of prepared nanoparticles was investigated by pyridine adsorption method. Results showed rising acidity by declining particle size of HPA.
Similar content being viewed by others
Introduction
Heteropolyacids (HPAs), as a class of inorganic compounds, are efficient, solid and super-acid catalysts for a wide range of reactions [1–5]. 12-Tungstophosphoric acid (PWA) and 12-molybdophosphoric acid (PMA) have been chosen as catalyst because of their easy availability and extreme stability in both solution and solid state also these materials have, high proton conductivity and strong Bronsted acidity in solution form. The main disadvantage of HPAs is their very low surface area (<10 m2/g) [6]. Therefore, to address this problem, in many earlier articles these materials disperse on supports (such as silica, active carbon, acidic ion-exchange resins and etc.) which possess large surface area. Support disperses HPAs over a large surface area and increases their catalytic activity [7–13].
HPAs compounds have an impressive application in optics, electronics, catalysis and medicines [14–16]. In a review, Rhule et al. [17] described the applications of HPAs in medicines. These materials have been shown antiviral activity against several RNA viruses including orthomyxoviruses, paramyxoviruses, flaviviruses, coronaviruses, retroviruses, different influenza strains, Dengue fever virus and SARS coronavirus [18–20]. Also, HPAs have been investigated as promising anti-HIV [21] and anti-cancer agents [22, 23].
Nanoscale and nanostructure materials have potential applications in physics, biology, chemistry, catalyst, optics, magnetic data storage, ceramics, electronics and nanocomposite, due to their unique properties [24, 25]. By reducing the particle size to a few nanometers, while is kept its chemical composition, can change the fundamental properties of a material.
Several methods have been reported for the synthesis of HPAs nanoparticles; these methods include Microemulsion [26], solid state [27], hydrothermal [28] and surfactant template method [29–31]. Although these methods are valuable, however, many of these procedures have significant drawbacks such as using of surfactant and other chemical reagent, long reaction times and poly-dispersity of particle size. In this study we synthesized nano HPAs by a simple, repaid, inexpensive and one step solvothermal method. Solvothermal synthesis is generally defined as crystal synthesis or crystal growth under high temperature and high pressure solvent conditions from substances which are insoluble in ordinary temperature and pressure (<100 °C, <1 atm) in an autoclave. Thus, supercritical condition gives a favorable reaction field for particle formation, owing to the enhancement of the reaction rate and large super saturation based on the nucleation theory, due to lowering the solubility. Then as-synthesized nanostructures were characterized by several characterization methods such as dynamic light scattering (DLS), X-ray powder diffraction (XRD), transmission electron microscopy (TEM), fourier transform infrared spectroscopy (FT-IR), and inductively coupled plasma (ICP) analyzer. Thermal stability of nanoparticles was studied by thermal gravimeter analyse (TGA). For perception of particle size on acidity of the HPAs, the prepared nanoparticles were investigated by pyridine adsorption method. Results indicate that decreasing in particle size of HPAs will increase acidity.
Experimental
Materials and Physical Measurements
All the chemicals reagents in our experiments were of analytical grade and were used as received without further purification. Elemental analysis was carried out with an ICP analyzer (Varian, vista-pro). XRD patterns were recorded by a Rigaku D-max C III, X-ray diffractometer using Ni-filtered Cu Kα radiation. Dynamic light scattering was recorded on a HORIBA-LB550. Transmission electron microscopy images were obtained on a Philips EM208 transmission electron microscope with an accelerating voltage of 100 kV. Fourier transform infrared spectra were obtained by using a Shimadzu FT-IR 8300 spectrophotometer. TGA thermo grams were recorded on an instrument of Perkin Elmer with N2 carrier gas and the rate of temperature change of 20 °C min−1. The BET surface area was determined from nitrogen physisorption isotherms on a Micromeritics ASAP 2000 instrument at 196 °C. Before the measurement, the samples were evacuated at 250 °C for 2 h.
Synthesis of H3PW12O40 and H3PMo12O40 (HPA) Nanoparticles
H3PW12O40·nH2O and H3PMo12O40·nH2O nanoparticles (which were labeled as PWAn and PMAn respectively) were composed by heat-treatment of their bulky forms in an autoclave. HPAs are soluble in polar solvents such as methanol and water but are insoluble in non-polar solvent such as hexane. In a typical procedure, 0.3 mmol of bulk H3PW12O40·14H2O (PWAb) or H3PMo12O40·13H2O (PMAb) was dispersed in 50 ml hexane and the obtained mixture was stirred vigorously for 30 min at room temperature in order to forming a homogeneous dispersion. This dispersion was transferred into a Teflon-lined stainless autoclave and filled 80 % of its total volume. The autoclave was sealed and maintained at 150 °C for 12 h and then was cooled to room temperature. Finally, the produced powder was filtered and dried in a vacuum at 60 °C for 12 h.
For investigation of synthesis parameter on particle size, the reaction carried out in various conditions and the size of nanoparticles obtained by DLS. Table 1 shows the conditions of reactions in detail.
Characterization of H3PW12O40 and H3PMo12O40 (HPA) Nanoparticles
Elemental Analysis
The elemental analysis of PMAn and PWAn nanoparticles was estimated by an ICP analyzer and the Mo/P or W/P mole ratio of the PMAn or PWAn was investigated.
FT-IR and UV–Visible Spectroscopy
FT-IR spectroscopy is a useful tool to understand the functional group of any organic and inorganic molecule. FT-IR spectroscopy, as a means of providing the structure of the HPAs, is convenient method and widely is used for the characterization of heteropolyanions.
UV–Visible spectroscopy yield information about the electronic structure of nanoparticles.
Thermal Stability
The thermal stability of HPAs changes with heteroatom, polyatom, and polyanion structures. Thermo analysis indicates that thermolysis of H3PMo12O40 or H3PW12O40 is proceeded by the losing of crystallization water and subsequently constitutional water to form PMo12O38 or PW12O38 respectively. Above 450 °C, the Keggin structure of HPAs is completely destroyed.
X-ray Powder Diffraction (XRD)
XRD analysis, as the most useful technique for identification of crystalline structure, was employed to investigate the prepared sample.
TEM Images and DLS Analysis
Particle size, morphology and size distribution of prepared nanoparticles was performed by TEM image and DLS analysis respectively.
Acidic Properties of Heteropoly Acids
The acidity of HPAs compounds has been investigated with different methods of analysis: (i) Hammett indicators + visual inspection [32], (ii) ammonia + temperature programmed desorption (TPD) [33, 34], FT-IR spectroscopy [35], calorimetry [36, 37], (iii) pyridine + TPD, FT-IR spectroscopy [38, 39] and (iv) H1 NMR [40] and O17 NMR [41].
The interaction of pyridine with HPAs was confirmed by FT-IR spectroscopy. Pyridinium ions can be identified by the band at 1,540 cm−1 [42]. The characteristic chemisorbed pyridium ion was detected after the removal of physisorbed pyridine from HPAs by evacuation. The absorption band corresponding to the Mo=O or W=O stretch might be expected to track the extent of proton transfer upon interaction with basic molecules such as pyridine. We measured the IR intensity of pyridium ion at 1,540 cm−1 of the pyridine-exposed PMA and PWA and calculated the intensity ratio of I(1,540 cm−1)/I(Mo=O or W=O) bands in an attempt to track the acidic properties of PMA and PWA.
Results and Discussion
As is shown in Fig. 1, PMA with Keggin structure consisted of one PO4 tetrahedron surrounded by four Mo3O13 sets formed by three edge-sharing octahedra. There are four types of oxygen atoms in H3PMo12O40; O* (the central oxygen atoms in PO4 tetrahedron), Oc (the corner-sharing oxygen between different Mo3O13 sets), Oe (the edge-sharing oxygen-bridge within Mo3O13 sets), and Ot (the terminal oxygen atoms).

Keggin structure of H3PW12O40 and color of PMA bulk and nano
The elemental analysis of these nanoparticles was estimated by ICP. The Mo/P or W/P mole ratio of the PMAn or PWAn was 12.2 and 12.5 respectively. These nanoparticles have Keggin unit cell structure because Keggin units have one central atom (P), 12 transition metal atoms (W or Mo) and an appropriate number of charge balancing protons or cations.
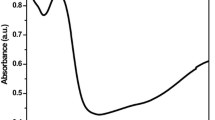
Optical properties are commonly characterized using spectroscopic techniques including UV–Visible spectroscopy, which yield information about the electronic structure of nanoparticles. The UV–Visible absorption measured as a function of wavelength reflects the strength of the electronic transition between the valence (VB) and conduction bands (CB). The bandgap energy increases with decreasing particle size, resulting in a blue-shift of the absorption peak. This behavior is due to what is termed quantum confinement. For spherical particles, a quantification of quantum confinement is embodied in Eq. 1 [43]:
where Eg(b) is the bulk band gap, me and mh are the effective masses of the electron and hole, R is particle size and e is the bulk optical dielectric constant. The second term on the right hand side shows that the effective band gap is inversely proportional to R2 and increases as size decreases. On the other hand, the third term shows that the band gap energy decreases with decreasing R due to increased Columbic interaction. However, since the second term becomes dominant with small R, the effective band gap is expected to increase with decreasing R, especially when R is small.
The UV–Vis absorption spectrum of the PMAb and PMAn were recorded as shown in Fig. 2. In the UV, the Keggin heteropolyanions (PMAb) show two absorption bands, one around 235 nm and another around 315 nm. The bands are assigned respectively to the vibrations of terminal Mo=Ot and bridging bonds (Mo–Oe). As can be seen, the absorption peak blue shifts with decreasing the particle size (225 and 290 nm for PMAn). Values of absorption bands (λmax) and the band gap energy are summarized in Table 2.

UV–Vis absorption spectrum of the PMAb and PMAn
The band gap energy of PMAb and PMAn were 5.3 and 5.5 eV respectively which calculated according to Eq. 2 [44]:
where Eg is the band-gap energy and λmax is the wavelength (nm).
Nanoparticles of the same material with different sizes exhibit various colors from blue to red, due to the quantum confinement effect. Smaller particles have a bigger band gap then larger particles and therefore have absorption smaller wavelength. The photograph of PMAb and PMAn are shown in Fig. 1. As mentioned above PMAn nanoparticles have larger band gap then PMAb. While PMAb is yellow color, PMAn nanoparticles is green. PWAn and PWAb are white color.
FT-IR spectroscopy, as a means of providing the structure of the HPAs, is convenient and widely is used for the characterization of heteropolyacid [45, 46]. FT-IR spectra of PMAn, PMAb, PWAn and PWAb are shown in Fig. 3. The FT-IR spectrum for PMAn shows four bands in the range 500–1,250 cm−1 that these bands are assigned at 754, 837, 954 and 1,062 cm−1, which are correspond to ν as(Mo–Oe–Mo), ν as(Mo–Oc–Mo), ν as(Mo–Ot) and ν as(P–O*), respectively [4–8]. It also shows a weak band in the region of 592 cm−1 that corresponds to the δ(P–O) vibration. Also, IR spectra of PWAn can display specific absorption bands correspond to the Keggin structure at 763, 871, 972 and 1,080 cm−1, can be assigned to the stretching vibrations ν as(W–Oe–W), ν as(W–Oc–W), ν as(W–Ot) and ν as(P–O*), respectively.

FT-IR spectra of PMAb, PMAn, PWAb and PWAn
In the case of nanoparticles, quantum confinement effect, size effect, dipolar interactions, interfacial effects, surface amorphousness, high internal stress etc. play some important role in the observed changes in vibrational frequencies. According to FT-IR spectroscopy data that are shown in Table 3, the characteristic bands of Keggin structure for PMAn and PWAn in comparison with PMAb and PWAb, are shifted to higher wave numbers (blue shift).
Due to the small size of grains and the large surface to volume ratio of nanoparticles (PMAn and PWAn), the atomic arrangements on the boundaries differ greatly from that of bulk particles (PMAb and PWAb). This surface amorphousness results in the shifting of IR peaks. The variation in the band position may be due to variation in the cation–oxygen bond length resulting from the change in particle size.
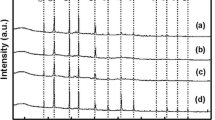
Thermal stability of HPAs was investigated by TGA. Figure 4 shows the TGA curves for pure PWAb and PMAb. For pure PMAb (Fig. 3a), a small weight loss region was found at 110 °C, and larger ones were found from 137 to 400 °C and at 430 to 567 °C. Similarly, for pure PWAb (Fig. 3c), three weight loss regions could be observed at 134, 340, and 500 °C. The HPAs are usually obtained with large amounts of crystallization water, and most of these water molecules are released around 100 °C. Two mass loss signals between 100 and 140 °C corresponding to the departure of water of crystallization or hydration. A second mass loss at temperatures between 250 and 350 °C, was attributed to water content resulting from combination of H+ ions and network oxygen [47].

Thermogravimetric patterns of PMAb (a), PMAn (b), PWAb (c) and PWAn (d)
Decomposition of HPAs, which takes place at 350–600 °C, is believed to occur according to [47]:
TG studies of the thermal transformation of bulk and nano HPAs, indicate that for PMAb, PWAb, PMAn and PWAn “n” is 13, 14, 6 and 9 respectively.
X-ray powder diffraction patterns of bulk and nano PMA and PWA are shown in Fig. 5. The strong characteristic peaks at 2θ = 9.05°, 23.7°, 26.35°, 28.25° and 32.4°, and 2θ = 10.55°, 21.10°, 25.95°, 30.33° and 35.30° are assigned to Keggin structure of MPA [JCPDS File 1-32] and PWA [JCPDS File 50-0657] respectively [48, 49]. These characteristic peaks are observed in PMAn and PWAn. As mentioned due to the small size of grains and the large surface–volume ratio of PMAn and PWAn, the atomic arrangements on the boundaries differ greatly from that of PMAb and PWAb. This surface amorphousness also results in the intensity and position of XRD peaks.

XRD pattern of PMAb, PMAn, PWAb and PWAn
From XRD data, the crystallite size (D c ) of the as-prepared PMAn and PWAn, were calculated by using the Debey–Scherrer equation [14],
where \([ \beta ]\) is the FWHM (full width at half maximum), K is the so-called shape factor, which usually takes a value of about 0.9, and λ is the wavelength of X-ray source used in XRD. The average size of the PMAn and PWAn, was calculated to be 25 and 27 nm respectively, which is to some extent in agreement with that observed from TEM and DLS analysis.
TEM images and DLS analysis of the PMAn and PWAn have been given in Fig. 6. Figure 6a and c are shown TEM images of PMAn and PWAn respectively, The size of nanoparticles obtained from the TEM images which shows sizes of 20 and 25 nm for PMA and PWA nanoparticles respectively, and their shapes are nearly spherical. To investigate the size distribution of the nanoparticles, size histograms were prepared for PMAn and PWAn (Fig. 6b and d respectively) by DLS analysis. This size distribution is centered at a value of 27 nm for PMAn and 29 nm for PWAn.

TEM image of a PMAn and c PWAn, and DLS pattern of b PMAn and d PWAn
The surface areas of bulk and nano HPAs were measured by N2 absorption. While the BET surface area of bulk HPAs was 10.0 and 12.0 m2/g for PMAb and PWAb respectively, nano HPAs have far higher surface area. Reducing the particle size of HPAs from bulk powder to nanometer scale, was increased the surface area from 10 to 270 m2/g for PMA and from 12 to 242 m2/g for PWA.
The interaction of pyridine with HPAs which confirmed by FT-IR, as was shown in Fig. 7. The P–O*, W=Ot (terminal oxygen), and W–O–W (corner sharing and edge-sharing) bands of fresh PWAn appeared at 1,080, 972, 872 and 764 cm−1, respectively. Pyridinium ions can be identified by the band at 1,540 cm−1 [24]. The characteristic chemisorbed pyridium ion was detected after the removal of physisorbed pyridine from HPAs by evacuation. The P–O*, W=Ot (terminal oxygen), and W–O–W (corner sharing and edge-sharing) bands of pyridine-exposed PWAn (PWAn-Py) appeared at 1,034, 942, 839 and 737 cm−1, respectively. Shifting of IR bands of pyridine-exposed HPAs were attributed to the interaction of the cationic species, pyridinium ions, with the heteropolyanions. Also, the FT-IR analyses of pyridine-exposed PWAb, PMAb and PMAn are shown in Fig. 7 and Table 3. The quantification of bridging oxygen bands was not simple. Thus, the absorption band correspond to the Mo=Ot or W=Ot stretch might be expected to track the extent of proton transfer upon interaction with basic molecules such as pyridine. The acidic properties of PMA and PWA are measured by the IR intensity of pyridium ion at 1,540 cm−1 of the pyridine-exposed PMA and PWA and calculated the intensity ratio of I(1,540 cm−1) to I(Mo=Ot or W=Ot) bands. As shown in Table 3 the racidity value for nanoparticles are larger than bulk particles.

Pyridine (A a, B a), PWAn (A b), PWAn-Pyridine (A c), PWAb (A d), PWAb-Pyridine (A e), PMAn (B b), PMAn-Pyridine (B c), PMAb (B d), PMAb-Pyridine (B e)
In addition, some other conditions such as concentration of PMA, reaction time and temperature were examined to investigate the size of products, if any, and compare them with each other. The results are shown in Table 1.
For investigating the concentration of PMAb on the size of PMAn, the reaction carried out with 2, 6 and 8 mM concentration and results are shown in Table 1. In a series of experiments, the temperature was kept at 150 °C and the reaction time was 12 h (sample no. 2, 5 and 8). With increasing of reactant concentration (from 2 to 8 mM) the size of nanoparticles became bigger (from 28 to 45 nm), due to the rate of particles growth will increase [50].
The effect of reaction time on the particle sized of the PMAn was investigated. There is generally growth in particle size with increase in time, which is consistent with Ostwald ripening process [50]. When the reaction time was prolonged (upper 12 h), at constant reactant concentration and temperature, the size of products became bigger (sample nos. 5 and 6). But at lower time at 12 h, reaction time for conversion of bulk particles to nanoparticles does not sufficient; therefore the products are larger than 33 nm (sample nos. 4 and 5). This illustrates that the particle size can be effectively controlled by setting the growth time.
The reaction temperature was the key parameters which were changed for more investigation. With increasing of reactant temperature upper than 150 °C the size of nanoparticles became bigger (sample nos. 5 and 13), due to the rate of particles growth will increase. But at lower temperature at 150 °C, temperature for conversion of bulk particles to nanoparticles does not sufficient; therefore the products are larger than 33 nm (sample nos. 5 and 12).
Conclusions
A novel method for the synthesis of HPAs nanoparticles from bulk HPAs was developed by hydrothermal treatment method. The PMA and PWA nanoparticles had a uniform spherical structure with a diameter of 20–25 nm.
References
M. N. Timofeeva (2003). Appl. Catal. A: Gen. 256, 19.
A. M. Alsalme, P. V. Wiper, Y. Z. Khimyak, E. F. Kozhevnikova, and I. V. Kozhevnikov (2010). J. Catal. 276, 181.
M. M. Heravi, Kh Bakhtiari, and F. F. Bamoharram (2006). Catal. Commun. 7, 499.
M. Mohammadpour Amini, A. Shaabani, and A. Bazgir (2006). Catal. Commun. 7, 843.
F. E. Celik, H. Lawrence, and A. T. Bell (2008). J. Mol. Catal. A: Chem. 288, 87.
G. S. Kumar, M. Vishnuvarthan, M. Palanichamy, and V. Murugesan (2006). J. Mol. Catal A: Chem. 260, 49.
G. Sartori and R. Maggi (2006). Chem. Rev. 106, 1077.
H. Li, H. Yin, T. Jiang, T. Hu, J. Wu, and Y. Wada (2006). Catal. Commun. 7, 778.
L. Cheng and X. P. Ye (2009). Catal. Lett. 130, 100.
G. D. K. Kumar and S. Baskaran (2005). J. Org. Chem. 70, 4520.
Z. Zhang, F. Zhang, Q. Zhu, W. Zhao, B. Ma, and Y. Ding (2011). J. Colloid Interface Sci. 360, 189.
H. J. Kim, Y. G. Shul, and H. Han (2006). J. Power Sources 158, 137.
H. J. Kim, Y. G. Shul, and H. Han (2006). Appl. Catal. A: Gen. 299, 46.
M. N. Timofeeva (2003). Appl. Catal. A: Gen. 256, 19.
T. Yamase (1998). Chem. Rev. 98, 307.
D. E. Katsoulis (1998). Chem. Rev. 98, 359.
J. T. Rhule, C. L. Hill, and D. A. Judd (1998). Chem. Rev. 98, 327.
S. Shigeta, S. Mori, T. Yamase, N. Yamamoto, and N. Yamamoto (2006). Biomed. Pharmacother. 60, 211.
B. Hasenknopf (2005). Front. Biosci. 10, 275.
H. U. V. Gerth, A. Rompel, B. Krebs, J. Boos, and C. Lanvers-Kaminsky (2005). Anti-Cancer Drug 16, 101.
A. Flütsch, T. Schroeder, M. G. Grütter, and G. R. Patzke (2011). Bioorg. Med. Chem. Lett. 21, 1162.
D. Menon, R. T. Thomas, S. Narayanan, S. Maya, R. Jayakumar, F. Hussain, V. K. Lakshmanan, and S. V. Nair (2011). Carbohydr. Polym. 84, 887.
F. Zhai, D. Li, C. Zhang, X. Wang, and R. Li (2008). Eur. J. Med. Chem. 43, 1911.
M. Salavati-Niasari, J. Javidi, and F. Davar (2010). Ultrason. Sonochem. 17, 870.
M. Salavati-Niasaria, J. Javidi, F. Davar, and A. Amini.Fazl (2010). J. Alloy Compd. 503, 500.
G. Maayan, R. Popovitz-Biro, and R. Neumann (2006). J. Am. Chem. Soc. 128, 4968.
Z. Kang, Y. wang, E. wang, S. Lian, L. Gao, W. You, C. Hu, and L. Xu (2004). Solid State Commun. 129, 559.
S. Farhadi, R. Zareisahamieh, and M. Zaidi (2011). J. Braz. Chem. Soc. 22, 1323.
T. Zhang, C. Spitz, M. Antonietti, and C. F. J. Faul (2005). Chem. Eur. J. 11, 1001.
G. Maayan, R. Popovitz-Biro, and R. Neumann (2006). J. Am. Chem. Soc. 128, 4968.
A. Stein, M. Fendorf, T. P. Jarvie, K. T. Mueller, A. J. Benesi, and T. E. Mallouk (1995). Chem. Mater. 7, 304.
T. Okuhara, T. Nishimura, H. Watanabe, and M. Misono (1992). J. Mol. Catal. 74, 247.
L. C. Jozefowicz, H. G. Karge, E. Vasilyeva, and J. B. Moffat (1993). Microporous Mesoporous Mater. 1, 313.
N. Essayem, R. Frety, G. Coudurier, and J. C. Vedrine (1997). J. Chem. Soc. Faraday Trans. 93, 3243.
A. Biela´nski, A. Małelcka, and L. Kubelkova (1989). J. Chem. Soc. Faraday Trans. 85, 2847.
F. Lefebvre, F. X. L. Cai, and A. Auroux (1994). J. Mater. Chem. 4, 125.
G. I. Kapustin, T. R. Brueva, A. L. Klyachko, M. N. Timofeeva, S. M. Kulikov, and I. V. Kozhevnikov (1990). Kinet. Katal. 31, 896.
M. Misono, N. Mizuno, K. Katamura, A. Kasai, Y. Konishi, K. Sakata, T. Okuhara, and Y. Yoneda (1982). Bull. Chem. Soc. Jpn. 55, 400.
E. M. Serwicka, K. Bruckman, J. Haber, E. A. Paukshtis, and E. N. Yurchenko (1991). Appl. Catal. 73, 153.
G. M. Maksimov, M. N. Timofeeva, and V. A. Likholobov (2001). Russ. Chem. Bull. Int. Ed. 50, 1529.
I. V. Kozhevnikov, A. Sinnema, and H. van Bekkum (1995). Catal. Lett. 34, 213.
S. Mahmoud Kaba, A. Mark Barteau, Wha Young Lee, and In. Kyu Song (2000). Appl. Catal. A: Gen. 194, 129.
L. E. Brus (1984). J. Chem. Phys. 80, 4403.
S. B. Khan, M. Faisa, M. M. Rahman, K. Akhtar, A. M. Asiri, A. Khan, and K. A. Alamry (2013). Int. J. Electrochem. Sci. 8, 7284.
T. Uma and M. Nogami (2007). Chem. Mater. 19, 3604.
K. T. Venkateswara Rao, P. S. N. Rao, P. Nagaraju, P. S. Sai Prasad, and N. Lingaiah (2009). J. Mol. Catal. A: Chem. 303, 84.
G. Lakshminarayana and M. Nogami (2009). J. Phys. Chem. C. 113, 14540.
P. A. Jalil, M. Faiz, N. Tabet, N. M. Hamdan, and Z. Hussain (2003). J. Catal. 217, 292.
H. G. Manyar, G. S. Chaure, and A. Kumar (2006). J. Mol. Catal. A: Chem. 243, 244.
M. Salavati-Niasaria and D. Ghanbari (2012). Particuology 10, 628.
Acknowledgments
Authors are grateful to the council of Iran National Science Foundation and University of Shiraz for their unending effort to provide financial support to undertake this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Javidi, J., Esmaeilpour, M., Rahiminezhad, Z. et al. Synthesis and Characterization of H3PW12O40 and H3PMo12O40 Nanoparticles by a Simple Method. J Clust Sci 25, 1511–1524 (2014). https://doi.org/10.1007/s10876-014-0745-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-014-0745-x