
Abstract
Gene expression analyses using native populations can contribute to the understanding of plant development and adaptation in multiple ways. These include the identification of candidate genes and genetic polymorphisms affecting expression and phenotypic traits and characterization of transcriptional networks. We analyzed the expression of 111 genes with probable roles in xylem/wood development in a population of loblolly pine (Pinus taeda L.) covering much of the natural range. Loblolly pine is one of the most commercially important forest tree species in the United States, and the discovery of genes and alleles contributing to desirable wood properties would be valuable. Of the 111 genes analyzed using quantitative reverse transcription–polymerase chain reaction, there were significant differences in gene expression between clones for 106 genes. Genes encoding lignin biosynthetic enzymes and arabinogalactan proteins were more variable than those encoding cellulose synthases or those involved in signal transduction. Several groups of genes with related functions form clusters. A network analysis identified transcription factors that may be key regulators of xylem development in pine. Secondary wall-associated NAC domain protein 1 (SND1) in particular appears to be involved in the regulation of many other genes. The cluster analysis using clones did not result in discrete populations but did identify some expression differences between regions. In the future, the gene expression data will be used for association analyses and promoter studies to understand how these gene expression differences are associated with specific genetic polymorphisms in other genes and promoters.
Similar content being viewed by others


Introduction
Differences in gene expression play a significant role in phenotypic variation within and among species. Within a species, expression levels vary not only among cell types within an individual but also among individuals (Storey et al. 2007). Natural variation is caused by spontaneous mutations that have been maintained by selection (Alonso-Blanco et al. 2009). Intraspecific variation in expression may be due to mutations in promoter or enhancer regions or in transcription factors or other genes in the signal transduction cascade. Expression differences between individuals can be particularly interesting when looking at a species found in its native habitat and adapted to a variety of environmental conditions. The study of gene expression in natural populations also has a great potential for understanding molecular population genetics and evolution (Townsend et al. 2003). The analyses of natural variation in crop plants and Arabidopsis thaliana have provided information on the genetic and molecular mechanisms that determine intraspecific variation and help us to understand the molecular bases of phenotypic differences that help in their adaptation (Mitchell-Olds et al. 2007).
Loblolly pine (Pinus taeda L.) is a species native to the southeastern United States and has considerable variation in traits of economic importance, including those involved in wood properties. Wood properties are determined by the activity of the genes and proteins expressed during xylogenesis, and variation in wood properties is partially due to the regulation of these genes in response to developmental and environmental cues (Whetten et al. 2001). There is a great deal of interest in the identification of genes or alleles controlling wood/xylem development as wood is a major source of terrestrial biomass and is an economically important plant tissue (Plomion et al. 2001). Genes that are of particular interest are those that affect wood properties such as cell wall thickness, wood-specific gravity, microfibril angle, fiber length, lumen diameter, and chemical composition of major cell wall components such as cellulose, lignin, and hemicelluloses. These genes are potential targets for modification of wood properties through breeding or genetic engineering (Yang and Loopstra 2005).
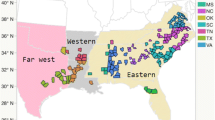
We are using gene expression analyses to try to identify genes and alleles controlling xylem development and to better understand the natural genetic variation in wood characteristics. There is abundant evidence for differential expression of genes involved in wood/xylem development among tissues (Loopstra and Sederoff 1995; Allona et al. 1998; Zhang et al. 2000; Yang et al. 2004; 2005). However, very little work has been done to examine differential expression among individuals (Yang and Loopstra 2005). In this article, we present our work to determine how gene expression differs between genotypes in a natural population of loblolly pine using a population of 400 clones (unique genotypes) representing much of the natural range (Fig. 1). Loblolly pine is commercially the most important forest tree species in the southern United States, growing on approximately 29 million acres through 14 states from southern New Jersey south to central Florida and west to Texas. It is a model system to study xylogenesis in a gymnosperm (Sederoff et al. 1994). To better understand the molecular basis of xylogenesis and variation in gene expression, quantitative reverse transcription–polymerase chain reaction (qRT-PCR) analysis was performed on 111 genes with probable roles in xylem development. To the best of our knowledge, there is no comparable data set for any other plant species.

The natural range of loblolly pine (http://esp.cr.usgs.gov/data/atlas/little/; Little 1971). Tree markers indicate the counties from which the parent trees were collected for the population. The natural range was divided into five regions based on distance from the Atlantic and Gulf Coasts and direction from the Mississippi River
Extensive research has been done to infer gene regulatory networks (GRNs) from expression data obtained from microarrays (Friedman 2004; Nachman et al. 2004; Basso et al. 2005; Bansal et al. 2007). Bansal et al. (2007) reviewed various approaches to infer GRNs using gene expression data through reverse engineering networks, including Bayesian networks. We used Bayesian networks to infer GRNs from our qRT-PCR gene expression data. Genes interconnected in GRNs suggest that one gene regulates the transcription of another directly or indirectly. Therefore, GRNs can be used to suggest functional and regulatory roles to poorly characterized genes (Needham et al. 2009). Association studies and promoter cloning are being conducted to understand how these gene expression differences are associated with specific genetic polymorphisms. The gene expression and association studies will contribute to our understanding of the molecular mechanisms that control formation of wood.
Results
Variation in gene expression
The genes analyzed in this project were primarily selected based on a review of the literature related to xylem development in woody and nonwoody species. Additional genes were included based on prior results from our laboratory. The genes selected for expression analyses are listed in Table 1. The justification for the selection of these genes is given in the supplemental materials (Supplemental text, Supplemental Table 1).
Gene expression values (Cycle threshold, or Ct) for 111 genes known or hypothesized to be involved in wood development were collected from 400 clones of loblolly pine using qRT-PCR (Table 2). Of the 111 genes analyzed, statistically significant differences among clones were observed for 106 genes. The differences between the clone with the lowest expression and that with the highest expression ranged from 2.1 cycles (4.3-fold) for Hap5a to 8.5 cycles (362-fold) for XET3. The average difference between low- and high-expressing clones for all genes was 4.4 cycles (20.8-fold). The genes showed normal distributions in their expression patterns among the clones. Figure 2 shows the distribution and range of ΔΔCt values for different clones across the population. As we expected, most of the clones fall in a narrower range of expression. Eighty-five of 111 genes had at least 75% of clones falling within one cycle higher or lower than the average ΔΔCt value (a 4-fold range). However, there were two genes, XET2 and MADS, where less than half of the clones had expression values within this window. We also observed differences between categories of genes. The average difference between the lowest and highest expressing clones for the 19 lignin biosynthesis genes (including laccases) was 5.3 cycles (38.6-fold); for the 14 cellulose synthase and related genes, it was 3.4 cycles (10.6-fold); for the cell wall protein (AGPs/PRP/GRP) genes, it was 5.2 cycles (35.8-fold); and for 29 genes involved in signal transduction, it was 3.8 cycles (13.9-fold). On average, 24% and 27% of the 400 clones were not within a two-cycle (4-fold) window for genes encoding proteins involved in lignin biosynthesis and cell wall proteins, respectively. Only 7.4% and 14.5% of clones were not within the two-cycle window for cellulose synthase genes and genes involved in signal transduction. Therefore, it appears that there is greater variation between clones for lignin biosynthesis and cell wall structural proteins than for genes involved in signal transduction or cellulose biosynthesis.

Normal distribution plots and barplots showing the range of ΔΔCt values for the CCR and CCoAMT genes among different clones in the population. The mean is zero for the normal distribution plots. The ΔΔCt of the clone with the highest expression was considered zero when constructing the barplots
Primer-binding and amplification efficiency
The observed differences between clones could be due to true differences in RNA levels present in the tissues or inefficient primer binding resulting from polymorphisms in primer-binding sites. The regions amplified by RT-PCR were sequenced to determine if single nucleotide polymorphisms (SNPs) in the primer-binding sites were responsible for differences in gene expression values. SNPs were observed in the primer-binding sites for several of the primers. All of the SNPs were in the middle or 5′ end of the primer sequence, except for SAM, which had a pair of SNPs at the 3′ end of the primer-binding site. When the same SNPs were present in both high- and low-expressing clones, we decided that the expression value differences were not due to the SNPs. We redesigned primers for six genes and performed qRT-PCR to determine if the SNPs were responsible for the expression differences due to improper primer binding. The gene expression values with the new primer pairs were identical with those from old primer pairs (±0.05 cycles), suggesting that the SNPs did not have much impact on primer binding. This might be due to the fact that the SNPs were mostly present toward the 5′ end of the primers. Boyle et al. (2009) have shown that SNPs present at the 5′ end of the primer do not affect the binding efficiency of the primer, and our results are in agreement with that observation.
Correlation of gene expression values
To determine if there were correlations between pairs of genes based on their expression, Pearson correlation in SPSS (Levesque 2007) was used. Significant correlations (r 2 > 0.66) were observed between 145 pairs of genes based on their gene expression (ΔΔCt) values (Supplemental Table 3). Expression of the PtMYB1 gene has significant positive correlations with all of the analyzed lignin biosynthesis genes (Table 3), in accordance with the hypothesis by Bomal et al. (2008) that MYB1 might be involved in transcriptional activation of genes involved in the phenylpropanoid pathway. Expression of the SND1 gene showed significant positive correlations with the expression of several other transcription factors involved in wood development as well as genes encoding AGPs, enzymes involved in lignin biosynthesis, and other proteins involved in xylogenesis (Table 3). No strong correlations were observed between the gene expression data and the geographical location of the trees in the population or the average precipitation of the counties from which the trees in the population were initially collected.
Hierarchical clustering of the gene expression profiles
Gene clustering
In order to get a general description of how the expression of genes covaried, autoscaled data were analyzed using a hierarchical Ward-linkage clustering with Euclidean distance as a similarity metric (Fig. 3). All the lignin biosynthesis and laccase genes except LAC1 clustered together with a bootstrap probability (BP) value of 63%. Eight of the nine genes encoding AGPs clustered together along with seven other genes with a BP value of 62%. The BP values of the other gene clusters were usually less than 30%, suggesting the weak nature of these clusters. However, eight of the ten cellulose synthase genes analyzed as well as one cellulose synthase-like gene and one calose synthase gene clustered together. The four tubulin genes also clustered together.

Hierarchical clustering of genes using qRT-PCR expression data. Pvclust in R was used to produce the dendrogram. Ward's hierarchical clustering, with Euclidean distance as the similarity metric, was used for the analyses. Two types of p-values are shown here: the approximately unbiased (AU) p-value and the bootstrap probability (BP) value. Cluster 1 contains all the lignin biosynthesis genes analyzed. Cluster 2 includes all the AGPs except AGP5D. Cluster 3 contains all the tubulins analyzed. Cluster 4 contains eight of ten CesAs, along with one CaS and one CslA gene
Clone clustering
To determine if expression patterns are different across the range of loblolly pine, autoscaled gene expression data were used to perform cluster analyses on the 400 clones in the population (Fig. 4). The clustering analysis was done using hierarchical Ward-linkage clustering with Euclidean distance as the similarity metric. The dendrogram grouped individuals based on similar patterns of expression for the 111 genes. Most clones (50 of 55) from west of the Mississippi River (Fig. 1, region 5) formed two distinct clusters that contained only 5 other clones. Thirteen of the 26 clones from the region along the Gulf Coast (Fig. 1, region 4) formed a cluster, and almost half (16 of 33) of the clones from region closest to the Atlantic coast (Fig. 1, region 2) formed a cluster. A large number of the clones (69%) come from areas we have indicated as regions 1 and 3 (Fig. 1). This includes the parts of Mississippi, Alabama, Georgia, South Carolina, North Carolina, and Virginia that are not close to the Gulf of Mexico or the Atlantic Ocean. We did not observe strong clustering of clones within these regions.

Hierarchical clustering of clones using qRT-PCR expression data. Ward's linkage algorithm was used with Euclidean distances as the similarity metric for the clustering analysis. Clones from the Atlantic Coast (AC), the Gulf Coast region (GC), and from counties west of the Mississippi River (WMR) formed distinct clusters. The clones from counties west of the Mississippi River formed two distinct clusters
Inference of a GRN
Correlations between gene expression patterns can be used to infer GRNs (Ma and Chan 2008). We employed steady-state Bayesian network inference (BANJO) of interactions between genes involved in wood development (Fig. 5). In an inferred gene network, an interaction between genes does not necessarily imply a physical interaction. It can refer to an indirect regulation by proteins or metabolites (Bansal et al. 2007). If two genes are joined by an edge (arrow), it can be hypothesized that the expression pattern of these two genes is highly correlated and the expression of the source gene might affect the expression of the target gene. The edge connecting a gene encoding a transcription factor and a target nontranscription factor gene suggests the transcriptional regulation of the target by the transcription factor. The edge between MYB1 and LAC7, LAC8 and EndChi genes suggests that MYB1 transcriptionally regulates expression of these three genes. If two transcription factors are joined by an edge, then such an edge can be an indication that the two transcription factors act as coregulators of the expression of other genes or one of the two transcription factors is a transcriptional regulator of the other. The transcription factors SND1 and MOR1 may jointly regulate the target gene HSP82 or SND1 may regulate MOR1, which further regulates the expression of HSP82. The network analysis indicates that the SND1 gene may be involved in the regulation of many of the genes we analyzed. We analyzed 111 genes, which is only a fraction of the total genes involved in xylem development. Therefore, critical links in the network may be missing.

Inferred gene network from the qRT-PCR data using BANJO at the MARIMBA Web site. Red circles represent genes involved in lignin biosynthesis; blue circles represent transcription factors; yellow circles represent no-hit genes; green circles represent AGPs
Discussion
Considerable natural variation exists within most forest tree species, some of which reflects adaptations to different environments (Linhart and Grant 1996). This natural variation is the result of the interaction of multiple genes and environmental factors (Keurentjes and Sulpice 2009). In order to understand the genetic basis and molecular mechanisms behind this naturally occurring developmental variation, genome-wide or candidate gene-based approaches can be used to identify the genes and nucleotide polymorphisms causing the observed diversity. Thus, the analysis of natural intraspecific variation helps us to discover the genes involved in plant adaptation to different environments through developmental modifications (Alonso-Blanco et al. 2005).
Genetic polymorphisms affecting plant development or adaptation may affect protein structure or gene expression. Studies investigating natural variation in gene expression have been carried out in several species including humans (Cheung et al. 2003), yeast (Steinmetz et al. 2002), fish (Oleksiak et al. 2002), Arabidopsis (Vuylsteke et al. 2005), rice (Liu et al. 2010), and maize (Auger et al. 2005). Cheung et al. (2003) examined the transcript levels of five genes in human lymphoblastoid cells among unrelated individuals, related individuals, and monozygotic twins. They found that the genes showed less variability in expression level in more closely related individuals; i.e., expression levels varied the least in monozygotic twins, with intermediate variability in siblings from the same family (2–5 times greater) and greatest variability in unrelated individuals (3–11 times greater). Oleksiak et al. (2002) used microarray technology to study the variation in gene expression within and between natural populations of teleost fish of the genus Fundulus and observed statistically significant differences in expression for approximately 18% of 907 genes. Liu et al. (2010) have shown that in two different rice cultivars, the expression of four phenylpropanoid pathway-related genes [C3H, CCR1, CCR10, and CHS8 (Chalcone synthase8] differs 3- to 500-fold under normal conditions and 85- to 1,150-fold during oxidative stress. We analyzed the expression profiles of 111 genes, hypothesized to be involved in xylem development, in a population of 400 loblolly pine plants. Out of these 111 genes, 106 genes showed statistically significant differences (ranging from 4.3- to 362-fold) in their gene expression among the clones. The large amounts of variation in expression we observed support the idea that expression differences may be important factors responsible for evolutionary changes.
Variation in expression of a particular gene may be due to the environment, developmental stage, mutations in promoter or enhancer regions of the gene, or to mutations in transcription factors or other genes in the signal transduction cascade. The additive and epistatic effects of the genes can result in large numbers of individuals with phenotypes (in our case, expression levels) close to the mean, with fewer having extreme phenotypes (expression levels; Benfey and Mitchell-Olds 2008). While in some cases, we observed very large differences between high- and low-expressing clones, we did find that for over three-fourths of our genes, less than 25% of the clones had expression values more than two-fold higher or lower than the population average. Growth conditions are not the primary reason for the observed gene expression differences as growth conditions were as uniform as possible. We feel that the differences in expression are primarily due to genetic polymorphisms. Since expression appears to be quantitative with a continuous distribution between the low- and high-expressing individuals, this suggests that multiple genetic polymorphisms are involved. Gene expression profiles help us identify genes with highly variable expression, but the reasons for this variation cannot be determined easily.
Natural variation in a population provides a resource to discover novel gene functions (Benfey and Mitchell-Olds 2008). Theoretically, genes in the same expression cluster must share some common function or regulatory elements. It might be possible to hypothesize the function of an unknown gene by looking at the other genes with which it clusters (Hruschka et al. 2006). Alternatively, the known and unknown genes may be coregulated or one could regulate the other. We used Ward’s linkage hierarchical clustering algorithm to group genes according to similar expression patterns. Euclidean distance was used as a nonparametric distance function. In the analysis with our data set, the lignin biosynthetic genes and AGPs formed separate clusters with significant bootstrapping values. All laccases clustered closely together and close to the lignin biosynthesis genes, supporting studies that indicated that the activities of laccases are closely correlated with lignin deposition in developing xylem (Bao et al. 1993; Dean and Eriksson 1994). PtMYB1, which has been hypothesized to regulate lignin biosynthesis in differentiating xylem (Patzlaff et al. 2003a), clustered with the lignin biosynthesis genes.
The KORRIGAN (KORRI) gene encodes a plasma membrane-bound member of the endo-1,4-β-d-glucanase family and has been shown to be involved in rapid cell elongation in Arabidopsis (Nicol et al. 1998). COBRA (COB), a regulator of oriented cell expansion (Schindelman et al. 2001) and KORRI clustered together with the lignin biosynthesis genes and laccases. All CeSAs, CaSs, and Csl genes clustered together, except CeSA2, CeSA9, and CaS3, which formed a cluster with UDP-glucosyl transferase, adenolyte kinase, prxC2 (horseradish peroxidase), and transcription factor LIM1. Kaothien et al. (2002) showed that LIM1 is a transcription factor binding to a PAL-box motif of the horseradish C2 peroxidase (prxC2) promoter in tobacco plants, which is responsible for the wound-induced expression of plant peroxidase genes. The similar expression pattern of these two genes in our analysis suggests that this relationship is also true in loblolly pine. The CslA1 gene formed a cluster with MYB8 and PIN1, suggesting that it might be regulated by these two transcription factors, either directly or indirectly. Of the seven no-hit genes (genes with no significant matches in other plants but selected due to preferential expression in loblolly pine xylem; Yang et al. 2004) included in our project, five of them clustered together with the following genes: LP-6 (a chitinase homolog), PutAMS (a putative S-adoMet synthetase), translation initiation factor eIF-4A, and transcription factor Hap5A. One of the no-hit genes, NH-9, formed a cluster with 1,4-benzoquinone reductase (BQR), pinoresinol-lariciresinol reductase (PLR), phenylcoumaran benzylic ether reductase (PCBER), and β-ketoacyl-ACP synthetase I-2 (BKACPS) genes. BQR is shown to be up-regulated in cotton during the fiber initiation stage and is suggested to be involved in cell elongation and secondary cell wall synthesis (Turley and Taliercio 2008). PCBER and PLR are involved in the biosynthesis of important phenylpropanoid-derived plant defense compounds, and PCBER is considered to be the progenitor of PLR (Gang et al. 1999). These correlations and the inferred network analyses described below help us to interpret the function of the no-hit genes. The no-hit genes may have functions similar to or be coregulated with the genes with which they cluster. Although these predictions are not certain, they at least provide a point from which one can start to interpret the function of these genes.
Continuous distribution across large geographical expanses makes the identification of genetic clusters difficult or inappropriate for species such as loblolly pine. However, based on the results from principal component analyses (PCAs; Jolliffe 2002) and STRUCTURE (Pritchard et al. 2000; Falush et al. 2003), a program for detecting population structure, Eckert et al. (2009),have shown that patterns of population structure for loblolly pine do exist in natural populations. PCA of SNP and SSR marker data revealed the presence of seven significant PCs defining eight genetic clusters, of which three were clearly differentiated clusters. The remaining five significant clusters lacked a strong geographical basis. One of the strong clusters is separated from the other two by the Mississippi River Valley, with a further division of the eastern cluster into Gulf and Atlantic Coast clusters. The clusters from the gene expression analyses are in partial agreement with the results of the population structure analyses. Of the 55 clones from the region west of the Mississippi River, 50 of them formed a distinct cluster, in agreement with the results of Eckert et al. (2009). However, we did not find that most clones from the regions east of the Mississippi River Valley formed clusters resembling those determined by PCA.
Using BANJO, we inferred a gene network from our expression data. The inferred network supported the previous assumptions of genes with known functions involved in certain metabolic pathways. This inferred gene network might also help to shed some light on the regulatory interactions among genes and identify genes that regulate each other. Zhong et al. (2007) have shown that simultaneous RNA interference (RNAi) inhibition of both the Secondary wall-associated NAC domain protein 1 (SND1) and NAC secondary wall-thickening promoter factor 1 (NST1) genes results in loss of secondary wall formation in fibers of Arabidopsis stems and also down-regulation of several fiber-associated transcription factor genes. Overexpression of SND1 activates the expression of secondary wall biosynthetic genes and results in ectopic secondary wall deposition (Zhong et al. 2006). Expression of several transcription factors, including MYB85, KNAT4 (a Knotted1-like homeodomain protein), and KNAT7, are regulated by SND1 (Zhong et al. 2006, 2007). Secondary wall defects were observed in Arabidopsis plants with repressed expression of MYB85 and KNAT7 (Zhong et al. 2008). PtMYB8 is a close homolog of the Arabidopsis MYB61 whose overexpression could cause ectopic lignin deposition (Zhong and Ye 2009). Our inferred gene network has edges between SND1 and NST1, KNAT7, MOR1, PtMYB8, MYB85, XET2, and lignin biosynthetic genes. This inferred network is in accordance with the results of Zhong et al. (2006), suggesting that SND1 is indeed a master transcriptional switch activating the developmental program of secondary wall biosynthesis in gymnosperms as well as angiosperms. Zhong and Ye (2009) have shown that the biosynthesis of other secondary wall components, including cellulose and xylan, are under the control of the same transcriptional network as lignin. Our analyses indicate that regulation of secondary cell wall synthesis in pines is similar to that in Arabidopsis. As pointed out by Zhong and Ye (2009), identification of these transcription factors may provide tools valuable for manipulating wood properties.
PtMYB1, PtMYB2, and PtMYB4 are preferentially expressed in developing xylem tissues (Patzlaff et al. 2003a, b). These MYBs bind AC elements and activate transcription from lignin biosynthetic gene promoters in plant cells (Patzlaff et al. 2003a, b). Our inferred gene network shows edges connecting MYB1 with PAL, Endo chitinase, COMT, and most of the laccases, supporting the previous observations by Patzlaff et al. (2003a, b) and Bao et al. (1993). In Arabidopsis, KORRI and CTL1, a chitinase-like gene implicated in cellulose deposition during primary cell wall formation, were highly correlated with the primary cell wall cellulose complex (Persson et al. 2005). In the inferred gene network, KORRI is connected to Endchi (P. taeda homolog of CTL1) through PAL and PtMYB1. KORRI is connected to the lignin biosynthetic genes in the inferred network, suggesting that it might be coordinately regulated along with those genes. We analyzed only a fraction of the total genes involved in xylem development, and therefore, critical links in the network are likely missing. Incorporation of more genes into the analyses will help us better understand the loblolly pine xylem GRN.
Conclusion
Due to loblolly pine being a native species with most of the natural variation still intact, we were able to analyze gene expression in a large number of individuals from across the natural range and found considerable variation in expression of xylem-related genes. This leads us to believe that further investigations into the role that regulatory variation plays during adaptive evolution are warranted. The amount of variation for some genes was somewhat surprising to us as was the difference in variation between different classes of genes. This variation allowed us to determine how gene expression values are correlated and to start development of a regulatory network that will help us determine genes, such as SND1, that are key regulators of xylogenesis in loblolly pine. In the future, we hope that with the help of association mapping, we can identify molecular markers associated with expression phenotypes and that they will aid in marker-assisted selection and breeding practices.
Materials and methods
Plant material
A population of loblolly pine-rooted cuttings was created at North Carolina State University from 600 independent seed lots obtained from the three southern pine breeding cooperatives (Murthy and Goldfarb 2001; Rowe et al. 2002; LeBude et al. 2004). It is composed of more than 500 loblolly pine clones (unique genotypes) that represent most of the natural range of loblolly pine and has no mating design (Fig. 1). Three rooted cuttings from each of 475 clones were transplanted into pots, all containing the same potting mixture and were grown for four additional months (April–August 2006) in a common greenhouse environment with evaporative cooling in College Station, TX. Conditions were as uniform as possible, although there could be small differences in light or temperature in different parts of the greenhouse, and there may be variability between bags of potting mixture. The stems, needles, and roots were collected from each plant, frozen in liquid nitrogen, and stored at −80°C.
RNA extraction and cDNA synthesis
Total RNA was extracted from the stems of two ramets (biological replicates) of each clone using the method of Chang et al. (1993), except for an additional chloroform extraction. Residual DNA was removed using DNA-free (Ambion Inc., Austin, TX). The first-strand cDNAs for each sample were synthesized using 5 μg of total RNA, random hexamers, and a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), following the manufacturer’s recommendations.
Gene selection
Genes shown or hypothesized to be involved in xylem development were selected for the expression studies. Genes were selected based on reviews of the current literature and prior research in our laboratory. The selected genes include those involved in cell wall formation, lignin biosynthesis, transcription factors, and genes of unknown function that are preferentially expressed in loblolly pine xylem tissue. The genes selected and reasons for selecting particular genes are given in the supplemental material.
Primer design and testing the efficiency of amplification
Putative orthologs of the selected genes were identified in loblolly pine using the NCBI EST database and BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi; Altschul et al. 1990) and the loblolly EST database at the University of Georgia (http://fungen.org/Projects/Pine/Pine.htm). Contigs were assembled from these EST sequences, and gene-specific primers were designed for qRT-PCR using Primer Express (Applied Biosystems). The primers used for qRT-PCR are listed in Supplemental Table 2. The primers were tested on a panel of 12 clones to see if there were significant differences in expression among the clones, and melting curve analyses were performed to check if the primers were amplifying a single product.
A template titration assay was done using a dilution series of cDNA templates (1,000, 250, 62.5, 15.625, and 3.90 ng) and two control samples: a no template control (NTC) and a no reverse transcriptase control (−RT). The slope can be affected by template quality, pipetting errors, etc. β-Actin was also run on the same plate to normalize the expression data. All lignin genes were evaluated in the standard curve trials to ensure that they gave efficient amplification, and the efficiency of amplification was calculated from a plot of ΔCt versus the template concentration.
Melting curve analyses were done to ensure product specificity and to differentiate between the true product and primer dimers. Four primer pairs gave more than one peak. These primers were discarded, and new primers were designed and tested. These redesigned primer pairs gave single peaks, suggesting the amplification of one product. All the valid primer sets had a slope of approximately −3.3 and a correlation coefficient (R 2-value) >0.95 for the standard curve. These standard curve analyses provided evidence for the efficiency of the amplification reactions.
Relative gene expression analysis
Transcript levels of the genes of interest were determined using qRT-PCR. The technical variability of the PCR reaction was standardized by inclusion of a template normalization step using stably expressed reference genes, 18 S rRNA and β-actin. An NTC and a−RT control were included on some plates. Amplification of the NTC sample indicates the presence of primer–dimer formed during the reaction. The−RT sample is included to confirm the absence of genomic amplification. Samples were run in duplicate on each plate using SYBR-Green PCR Master Mix (Applied Biosystems) on a GeneAmp 7900HT Sequence Detection System (Applied Biosystems), following the manufacturer’s recommendations. Real-time RT-PCR was performed in an 8-μl reaction containing 2.5 μl ddH2O, 4 μl SYBR-Green PCR Master Mix, 0.5 μl forward primer (1 mM), 0.5 μl reverse primer (1 mM), and 0.5 μl of template cDNA (10 ng/μl). The PCR conditions were 2 min of preincubation at 50°C, 10 min of predenaturation at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C, followed by steps for dissociation curve generation (15 s at 95°C, 15 s at 60°C, and 15 s at 95°C).
Analysis of the qRT-PCR data
Relative transcript levels for each sample were obtained using the “relative standard curve method” (see User Bulletin #20 ABI PRISM 7900 Sequence Detection System for details) and were normalized to the transcript level of 18 S rRNA or β-actin of each sample to get ΔCt values. The clone with the closest expression values for all the genes between the ramets was selected as a calibrator, and SDS 2.3 software (Applied Biosystems) was used to collect the ΔΔCt values of all the genes for all the clones. The selective amplification of individual gene family members was judged based on dissociation curves. These experiments were conducted for 111 genes ×400 clones ×2 ramets/clone ×2 reps/ramet. A paired t-test and an analysis of variance (ANOVA), using a p-value of 0.01, were used on normalized and calibrated transcript levels to test for variation in gene expression among clones.
Sequencing of primer-binding sites
In order to rule out low primer-binding efficiency as a factor responsible for low expression, new primers were designed for most genes outside of the initial set of primers used for qRT-PCR, and PCR was performed in low- and high-expressing clones. These PCR transcripts were sequenced to check for the presence of SNPs in the primer-binding sites. If SNPs were seen only in the primer-binding sites of clones with low expression, then qRT-PCR was performed using a different set of primers to check if SNPs affected primer-binding efficiency and expression values.
Correlations and clustering analyses
The gene expression data (ΔΔCt values) was autoscaled as described in Stahlberg et al. (2008) so that the average expression of each gene in all clones is zero and its standard deviation is one. This allows equal weights to all genes in clustering analyses. Pearson correlation in SPSS was used to determine if there were correlations between pairs of genes based on their ΔΔCt values. We applied Ward’s linkage hierarchical clustering algorithm (Ward 1963) to group genes according to similar expression patterns using Euclidean distances. Clone clustering was also done using Ward’s linkage hierarchical clustering algorithm. We used bootstrapping (10,000 replicates) to obtain estimates for the reliability of the groupings using the pvclust (Suzuki and Shimodaira 2006) package as part of the R computing environment (R Core Development Team 2007).
Gene network inference
Bayesian Network inference with Java Objects (BANJO, http://www.cs.duke.edu/~amink/software/banjo/) was used to infer a gene network from the expression data. BANJO can infer gene networks from gene expression data (Hartemink 2005; Yu et al. 2004). Results for BANJO were obtained using the default parameters at the MARIMBA Web site (http://marimba.hegroup.org/index.php). The gene expression data were changed from continuous to discrete using their q3 discretization function.
References
Allona I, Quinn M, Shoop E, Swope K, St. Cyr S, Carlis J, Riedl J, Retzel E, Campbell MM, Sederoff R, Whetten R (1998) Analysis of xylem formation in pine by cDNA sequencing. Proc Natl Acad Sci USA 95:9693–9698
Alonso-Blanco C, Mendez-Vigo B, Koornneef M (2005) From phenotypic to molecular polymorphisms involved in naturally occurring variation of plant development. Int J Dev Biol 49:717–732
Alonso-Blanco C, Arts MGM, Bentsink L, Keurentjes JJB, Reymond M, Vreugdenhil D, Koornneef M (2009) What has natural variation taught us about plant development, physiology, and adaptation. Plant Cell 21:1877–1896
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Auger DL, Gray AD, Ream TS, Kato A et al (2005) Nonadditive gene expression in diploid and triploid hybrids in maize. Genetics 169:389–397
Bansal M, Belcastro V, Ambesi-Impiombato A, di Bernardo D (2007) How to infer gene networks from expression profiles. Mol Syst Biol 3:78–87
Bao W, O’Malley DM, Whetten R, Sederoff RR (1993) A laccase associated with lignification in Loblolly pine xylem. Science 260:672–674
Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A (2005) Reverse engineering of regulatory networks in human B cells. Nat Genet 37:382–390
Benfey PN, Mitchell-Olds T (2008) From clone to phenotype: systems biology meets natural variation. Science 320:495–497
Bomal C, Bedon F, Caron S, Mansfield SD, Levasseur C, Cooke JE, Blais S, Tremblay L, Morency MJ, Pavy N, Grima-Pettenati J, Séguin A, Mackay J (2008) Involvement of Pinus taeda MYB1 and MYB8 in phenylpropanoid metabolism and secondary cell wall biogenesis: a comparative in planta analysis. J Exp Bot 59:3925–39
Boyle B, Dallaire N, MacKay J (2009) Evaluation of the impact of single nucleotide polymorphisms and primer mismatches on quantitative PCR. BMC Biotech 9:75–90
Chang S, Puryear J, Cairney JA (1993) Simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Rep 11:114–117
Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS (2003) Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Gen 33:422–425
Dean JFD, Eriksson K-EL (1994) Laccase and the deposition of lignin in vascular plants. Holzforschung 48:21–33
Eckert AJ, van Heerwaarden J, Wegrzyn JL, Nelson CD, Ross-Ibarra J, González-Martínez SC, Neale DB (2009) Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., Pinaceae). Genetics (submitted).
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164:1567–1587
Friedman N (2004) Inferring cellular networks using probabilistic graphical models. Science 303:799–805
Gang DR, Kasahara H, Xia ZQ, Mijnsbrugge KV, Bauw G, Boerjan W, Van Montagu M, Davin LB, Lewis NG (1999) Evolution of plant defense mechanisms: relationships of phenylcoumaran benzylic ether reductases to pinoresinol-lariciresinol and isoflavone reductases. J Biol Chem 274:7516–7527
Hartemink A (2005) Reverse engineering gene regulatory networks. Nat Biotech 23:554–555
Hruschka ER, Campello RJGB, de Castro LN (2006) Evolving clusters in gene-expression data. Inf Sci 176:1898–1927
Jolliffe IT (2002) Principal component analysis. Springer Series in Statistics, 2nd ed., Springer, NY, XXIX, p 487
Kaothien P, Kawaoka A, Ebinuma H, Yoshida K, Shinmyo A (2002) Ntlim1, a PAL-box binding factor, controls promoter activity of the horseradish wound-inducible peroxidase gene. Plant Mol Biol 49:591–599
Keurentjes JJB, Sulpice R (2009) The role of natural variation in dissecting genetic regulation of primary metabolism. Plant Signal Behav 4:244–246
LeBude AV, Goldfarb B, Blazich FA, Wise FC, Frampton LJ Jr (2004) Mist, medium water potential, and cutting water potential influence rooting of stem cuttings of loblolly pine. Tree Physiol 24:823–831
Levesque R (2007) SPSS programming and data management: a guide for SPSS and SAS users, 4th edn. SPSS, Chicago
Linhart YB, Grant MC (1996) Evolutionary significance of local genetic differentiation in plants. Annu Rev Ecol Syst 27:237–277
Little EL Jr (1971) Atlas of United States trees, volume 1, conifers and important hardwoods: U.S. Department of Agriculture Miscellaneous Publication 1146, 9 p., 200 maps
Liu F, Xu W, Wei Q, Zhang Z, Xing Z et al (2010) Gene expression profiles deciphering rice phenotypic variation between Nipponbare (Japonica) and 93-11 (Indica) during oxidative stress. PLoS ONE 5(1):e8632. doi:10.1371/journal.pone.0008632
Loopstra CA, Sederoff RR (1995) Xylem-specific gene expression in loblolly pine. Plant Mol Biol 27:277–291
Ma PCH, Chan KCC (2008) Inferring gene regulatory networks from expression data by discovering fuzzy dependency relationships. IEEE Trans Fuzzy Syst 16:455–465
Mitchell-Olds T, Willis JH, Goldstein DB (2007) Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat Rev Genet 8:845–856
Murthy R, Goldfarb B (2001) Effect of handling and water stress on water status and rooting of loblolly pine stem cuttings. New For 21:217–230
Nachman I, Regev A, Friedman N (2004) Inferring quantitative models of regulatory networks from expression data. Bioinformatics 20:i248–256
Needham CJ, Manfield IW, Bulpitt AJ, Gilmartin PM, Westhead DR (2009) From gene expression to gene regulatory networks in Arabidopsis thaliana. BMC Systems Biol 3:85–93
Nicol F, His I, Jauneau A, Vernhettes S, Canut H, Höfte H (1998) A plasma membrane-bound putative endo-1, 4-β-D-glucanase is required for normal wall assembly and cell elongation in Arabidopsis. EMBO J 17:5563–5576
Oleksiak MF, Churchill GA, Crawford DL (2002) Variation in gene expression within and among natural populations. Nat Genet 32:261–266
Patzlaff A, Newman LJ, Dubos C, Whetten RW, Smith C, McInnis S, Bevan MW, Sederoff RR, Campbell MM (2003a) Characterisation of PtMYB1, an R2R3-MYB from pine xylem. Plant Mol Biol 53:597–608
Patzlaff A, McInnis S, Courtenay A, Surman C, Newman LJ, Smith C, Bevan MW, Mansfield S, Whetten RW, Sederoff RR, Campbell MM (2003b) Characterisation of a pine MYB that regulates lignification. Plant J 36(6):743–754
Persson S, Hairong W, Milne J, Grier O, Somerville C (2005) Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. PNAS 102:8633–8638
Plomion C, Leprovost G, Stokes A (2001) Wood formation in trees. Plant Physiol 127:1513–1523
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rowe DB, Blazich FA, Goldfarb B, Wise FC (2002) Nitrogen nutrition of hedged stock plants of loblolly pine. II. Influence of carbohydrate and nitrogen status on adventitious rooting of stem cuttings. New For 24:53–65
Schindelman G, Morikami A, Jung J, Baskin TI, Carpita NC, Derbyshire P, McCann MC, Benfey PN (2001) COBRA encodes a putative GPI-anchored protein, which is polarly localized and necessary for oriented cell expansion in Arabidopsis. Genes Dev 15:1115–1117
Sederoff RR, Campbell M, O’Malley D, Whetten R (1994) Genetic regulation of lignin biosynthesis and the potential modification of wood by genetic engineering in loblolly pine. Rec Adv Phytochem 28:313–355
Stahlberg A, Elbing K, Andrade-Garda JM, Sjogreen B, Forootan A, Kubista M (2008) Multiway real-time PCR gene expression profiling in yeast Saccharomyces cerevisiae reveals altered transcriptional response of ADH-genes to glucose stimuli. BMC Genomics 9:170–184
Steinmetz LM, Sinha H, Richards DR, Spiegelman JI, Oefner PJ, McCusker JH, Davis RW (2002) Dissecting the architecture of a quantitative trait locus in yeast. Nature 416:326–330
Storey JD, Madeoy J, Strout JL, Wurfel M, Ronald J, Akey JM (2007) Gene-expression variation within and among human populations. Am J Hum Genet 80:502–509
Suzuki R, Shimodaira H (2006) Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22:1540–1542
Townsend JP, Cavalieri D, Hartl DL (2003) Population genetic variation in genome-wide gene expression. Mol Biol Evol 20:955–963
Turley RB, Taliercio E (2008) Cotton benzoquinone reductase: up-regulation during early fiber development and heterologous expression and characterization in Pichia pastoris. Plant Physiol Biochem 46:780–785
Vuylsteke M, van Eeuwijk F, Van Hummelen P, Kuiper M, Zabeau M (2005) Genetic analysis of variation in gene expression in Arabidopsis thaliana. Genetics 171(3):1267–75
Ward JH (1963) Hierarchical grouping to optimize an objective function. J Am Statistical Asscn 58:236–244
Whetten RW, Sun Y-H, Zhang Y, Sederoff RR (2001) Functional genomics and cell wall biosynthesis in loblolly pine. Plant Mol Biol 47:275–291
Yang S-H, van Zyl L, No E-G, Loopstra CA (2004) Microarray analysis of genes preferentially expressed in differentiating xylem of loblolly pine (Pinus taeda). Plant Sci 166:1185–1195
Yang S-H, Loopstra CA (2005) Seasonal variation in gene expression for loblolly pines (Pinus taeda) from different geographical regions. Tree Physiol 25:1063–73
Yang S-H, Wang H, Sathyan P, Stasolla C, Loopstra CA (2005) Real-time RT-PCR analysis of loblolly pine (Pinus taeda) arabinogalactan-protein and arabinogalactan-protein-like genes. Physiol Plant 124:91–106
Yu J, Smith VA, Wang PP, Hartemink AJ, Jarvis ED (2004) Advances to bayesian network inference for generating causal networks from observational biological data. Bioinformatics 20:3594–3603
Zhang Y, Sederoff RR, Allona I (2000) Differential expression of genes encoding cell wall proteins in vascular tissues from vertical and bent loblolly pine trees. Tree Physiol 20:457–466
Zhong R, Demura T, Yea ZH (2006) SND1, a NAC domain transcription factor, is a key regulator of secondary wall synthesis in fibers of Arabidopsis. Plant Cell 18:3158–3170
Zhong R, Richardson EA, Ye ZH (2007) Two NAC domain transcription factors, SND1 and NST1, function redundantly in regulation of secondary wall synthesis in fibers of Arabidopsis. Planta 225:1603–1611
Zhong R, Lee C, Zhou J, McCarthy RL, Ye ZH (2008) A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell 20:2763–2782
Zhong R, Ye ZH (2009) Transcriptional regulation of lignin biosynthesis. Plant Signal Behav 4:1028–1034
Acknowledgements
We would like to thank David Neale for his valuable support and suggestions regarding the manuscript. We would also like to thank Jeffrey Puryear for helping with plant propagation and for sequencing and Dana Nelson for microsatellite analyses to verify plant identification. Funding for this project was provided by the National Science Foundation (Grant number DBI-0501763).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Dean
Electronic supplementary material
Below is the link to the electronic supplementary material.
Esm 1
Gene selection—Reasons for selecting particular genes for the expression analysis (DOC 64 kb)
Table S1
Genes analyzed with their GenBank accession numbers (XLSX 17 kb)
Table S2
Primers used in qRT-PCR analysis (XLSX 19 kb)
Table S3
Correlations among the genes based on their expression data (XLSX 96 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Palle, S.R., Seeve, C.M., Eckert, A.J. et al. Natural variation in expression of genes involved in xylem development in loblolly pine (Pinus taeda L.). Tree Genetics & Genomes 7, 193–206 (2011). https://doi.org/10.1007/s11295-010-0325-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-010-0325-7