
Abstract
A model has been developed that enables the viscosities of the fully liquid slag in the multi-component Al2O3-CaO-FeO-Fe2O3-MgO-Na2O-SiO2 system to be predicted within experimental uncertainties over a wide range of compositions and temperatures. The Eyring equation is used to express viscosity as a function of temperature and composition. The model links the activation and pre-exponential energy terms in the viscosity expression to the slag internal structure through the concentrations of various Si0.5O, \( {\text{Me}}^{n + }_{2/n} {\text{O}} \) , and \( {\text{Me}}^{n + }_{ 1/n} {\text{Si}}_{0. 2 5} {\text{O}} \) viscous flow structural units (SUs). The concentrations of these SUs are derived from a quasi-chemical thermodynamic model of the liquid slag using the thermodynamic computer package FactSage. The model describes a number of slag viscosity features including the charge compensation effect specific for the Al2O3-containing systems. The predictive capability of the model is enhanced by the physical aspects of the model parameters—the correlation with other physicochemical properties as well as experimental viscosity data is used to determine model parameters. The present series of two papers outlines (a) recent significant improvements introduced into the model formalism and (b) application of the model to the Al2O3-CaO-MgO-SiO2 system, review of experimental viscosity data, and optimization of the corresponding model parameters for this system.
Similar content being viewed by others


Introduction
Slag viscosity is an essential property for a number of metallurgical and power generation industrial processes. Development of a structurally based viscosity model for predictions in the multi-component slag systems valid over wide ranges of compositions and temperatures has been the focus of previous studies[1–7]; the present paper describes recent developments of the model formalism and its application for fully liquid silicate slags in the Al2O3-CaO-MgO-SiO2 system. Similar to other structurally based viscosity slag models,[8–12] the link of viscosities to the complex internal slag structure at the atomic level improves the predictive capability of slag viscosities. The overall description and application of the model to the Al2O3-CaO-MgO-SiO2 system presented in these papers is only a part of a wider development of the viscosity model for the Al2O3-CaO-FeO-Fe2O3-MgO-Na2O-SiO2 multi-component system. The systematic trends and self-consistency of the parameters across the whole multi-component system are essential features of the present development. The trend analysis was extended even further to other important slag systems, for example, to some lower-order sub-systems containing K2O and PbO. The fact of the systematic analysis of the trends across a number of key slag systems is an important feature of this work, and therefore results for some other sub-systems have been included in this manuscript.
Model
General Model Description
Frenkel’s kinetic theory[13–15] considers a liquid to have a solid-like structure with molecules, or more generally, structural units (SUs), oscillating in their energetic cells (potential wells) near average positions. Oscillations higher in magnitude than the potential barrier result in the movement of a SU into an adjacent vacant cell, or “hole,” provided the latter is vacant. These vacant cells, or “holes,” formed in the liquid as a result of fluctuations, are distributed randomly throughout the liquid. The viscosity of liquid as a reaction to the applied shear force therefore is determined by (a) the ability of a SU to jump over the potential barrier and (b) the presence of “holes” in the liquid. The following Eyring Eq. [1] for liquid viscosity was derived[16–18] using the above principles[13]:
where R (J/K/mol) and k (J/K) are the gas and the Boltzmann constants, respectively; π ≈ 3.1416; T is the absolute temperature (K); and m SU (kg) and v SU (m3) are the average mass and volume of viscous flow SUs, respectively. The activation energy \( \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{a}} \) is determined by the strength of interactions between different SUs composing the liquid. According to Eyring,[16] the energy term \( \Updelta \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{V}} \) (a product of vaporization energy and transition probability) is related to the free volume of the liquid, i.e., to the concentration of the holes in the liquid. It may be argued that \( \Updelta \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{V}} \) includes the negative of the energy of the hole formation: A higher concentration of the holes in the liquid corresponds to the higher \( \Updelta \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{V}} \) value and to the lower viscosity.
Four parameters in Eq. [1]—the average mass and volume of SUs, and the \( \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{a}} \) and \( \Updelta \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{V}} \) energies—are related to the internal structure of liquids, types, concentrations, and interactions between SUs at the atomic/cation/anion level, and require the definition of a viscous flow SU in a particular model.
A silicate slag structure is conventionally described as the silicate network of \( {\text{SiO}}_{4}^{4 - } \) tetrahedra broken by different metal cations distributed to keep the total electroneutrality.[19] A silicate slag may also be considered as a nearly close-packed arrangement of larger oxygen anions with smaller metal cations that occupy the interstices and interact with each other.[20] Fincham and Richardson[21] related properties of a silicate slag to the internal slag structure through concentrations of three different types of oxygens: “bridging” (O0—connected to two silicon cations), “non-bridging” (O−—connected to only one silicon), and “free” (O2−—associated with non-silicon cations). A simplified two-dimensional schematic picture of the internal structure of the slag illustrating these concepts is presented in Figure 1. Based on the above background, the viscous flow of the silicate slag in the present model is considered to be a movement of oxygens partly associated with metal cations under the applied shear force, so that the viscous flow SUs are defined as oxygen anions with metal cations partly associated with them (see ovals in Figure 1), including Si0.5O (=Si-O-Si=Si-Si), \( {\text{Me}}^{n + }_{ 2/n} {\text{O}} \) (=Me-O-Me=Me-Me), and \( {\text{Me}}^{n + }_{1/n} {\text{Si}}_{0.25} {\text{O}} \) (=Si-O-Me=Si-Me), where n denotes the oxidation state of a metal cation Men+. For example, for the binary MeO-SiO2 silicate slag (see Figure 1), three types of SUs can be identified, (Si-O-Si) (shaded with dark gray), (Si-O-Me) (shaded with light gray), and (Me-O-Me) (white, not shaded). Their molar fractions are indicated as X Si-Si, X Si-Me, and X Me-Me, respectively. Viscous flow SUs are specific to the present viscosity model formulation; they differ from the conventional SUs.[19–21] The viscous flow SUs in this work are the units that are assumed to move under the shear stress and include the “moving” oxygen anions with associated metal cations. For brevity, they are referred to just as “SUs.” Other models introduce SUs to describe other properties, for example, “bridging,” “non-bridging” and “free” oxygen,[21] or second-nearest-neighbor bonds.[22]

Simplified two-dimensional schematic diagram of internal structure and viscous flow SUs in silicate melts (O0, O−, and O2− correspond to non-bridging, bridging, and free oxygens, respectively)
The values of the \( \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{a}} \) and \( \Updelta \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{V}} \) energy terms, and the average mass and volume of SUs \( m_{\text{SU}} \) and \( v_{\text{SU}} \) in the present model, are expressed through the respective molar fractions of the various SUs X pq with Eq. [2]:
where p and q are metal cations, m pq and v pq are the masses and volumes of the respective SUs, \( \bar{E}_{{{\text{a}},pq}} \) are partial molar activation energies, \( \Updelta \bar{E}_{{{\text{V}},0}} \) is a unit constant, and \( \varepsilon_{{{\text{V}},pq}} \) are the dimensionless partial energy coefficients of the integral energy term \( \Updelta \underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E}_{\text{V}} \) of each SU. In the binary MeO-SiO2 system, partial molar activation energies are \( \bar{E}_{{{\text{a}},{\text{Si - Si}}}} \), \( \bar{E}_{{{\text{a}},{\text{Si - Me}}}} \) , and \( \overline{E}_{{{\text{a}},{\text{Me - Me}}}} \) and the integral molar activation energy is expressed as follows:
The m pq values are the weights of the corresponding molecules, such as \( {\text{Si}}_{ 1 / 2} {\text{O}} \), \( {\text{Me}}_{ 2 /n}^{n + } {\text{O}} \) , and \( {\text{Me}}_{ 1 /n}^{n + } {\text{Si}}_{ 1 / 4} {\text{O}} \). The v pq values are calculated using the effective diameters of SUs estimated from the ionic radii of various ions (O, Si, Me) composing a particular SU. The ionic radii are taken from Shannon[23]; the three-dimensional arrangements of the SUs are not taken into account in the present model at this stage. This uncertainty is “absorbed” by the model parameters later, during optimization.
In addition to one oxygen, a given SU also involves two metal cations, both of them having other neighbors and both involved in other SU(s). The partial properties \( \overline{E}_{{{\text{a}},pq}} \) and \( \varepsilon_{{{\text{V}},pq}} \) of a given SU (Me p -O-Me q ) therefore depend on the type of second nearest neighbors. For example, if a Si-Si SU has Si4+ cations with other Men+ cations as neighbors (e.g., a SU marked as “B” in Figure 1), they will have a different partial activation molar energy compared to the case when some or all other neighbors are also Si4+ cations (e.g., a SU marked as “A” in Figure 1). The effect of neighboring SUs on a given partial activation energy is expressed as a function of the concentrations of other types of SUs. The partial molar activation energy of each type of SU was previously[1–6] expressed using the following Eqs. [3] to [5]:
Note that only the effect of the second nearest neighbors was taken into account in the model. For example, \( \overline{E}_{{{\text{a}},{\text{Si - Si}}}} \) does not depend on X Me-Me and vice versa, because (Me-O-Me) SU cannot be the closest neighbor of the (Si-O-Si) SU.
A higher power term \( E_{{{\text{a}},{\text{Si - Si}}}}^{{{\text{Si - Si}},2}} \) was previously used[1–6] in the expression of \( \overline{E}_{{{\text{a}},{\text{Si - Si}}}} \) to describe experimental data in the SiO2-containing systems. More complex terms in the expression of \( \overline{E}_{{{\text{a}},{\text{Si - Si}}}} \) were later introduced[7] for the Na- and K-containing silicate slags. The composition dependencies of most of the other partial activation energies except \( \overline{E}_{{{\text{a}},{\text{Si - Si}}}} \) on the concentrations of other nearest neighbors were not taken into account due to the weak dependency on compositions and the lack or uncertainties of experimental data. The partial activation energies \( \overline{E}_{{{\text{a}},{\text{Me1 - Me}}2}} \) for the slag systems with limited experimental data available (e.g., Al2O3-“FeO” and CaO-“FeO”) were taken to be equal to 1/2 (\( \overline{E}_{{{\text{a}},{\text{Me1 - Me1}}}} \) + \( \overline{E}_{{{\text{a}},{\text{Me2 - Me2}}}} \)). The dimensionless partial energy coefficients ε v,Si-Si, ε v,Me-Si, and ε v,Me-Me were described in a similar way.
The so-called “charge compensation effect” (e.g., increase of viscosity and maximum at the Al2O3/CaO ratio close to 1 in the Al2O3-CaO-SiO2 system) was described with a specially introduced term.[1–6]
The present as well as other structurally based models[8–11] have advantages over phenomenological models that express viscosity as a function of just composition. The model parameters of the structurally based models bear physical meaning; the values of the parameters obtained from the lower-order systems can be extrapolated into multi-component and higher-order systems where no experimental data are available, enabling predictions to be performed and, in some cases, discrepancies between experimental data to be identified. Certain restrictions on the model parameters can be introduced and the trends in the values of the model parameters can be identified and used for interpolations and extrapolations in other systems in which experimental data are lacking. Such structural models are flexible enough to reflect major changes, but “rigid” for composition ranges with similar SU distributions. The models expressing viscosities as polynomial functions of compositions do not have these predictive advantages.
Improved Model
The previous formalism[1–7] had some limitations in the description of viscosities in complex silicate slag systems and therefore had been critically reviewed and subsequently revised in the present study. A number of further significant improvements introduced into the model are discussed in the following sections. As a result of the revision, the partial properties \( \overline{E}_{{{\text{a}},pq}} \) and \( \varepsilon_{{{\text{V}},pq}} \) of a given SU (Me p -O-Me q ) are described with Eqs. [6] to [8]:
where i, Ca, Mg, Al, Na, …(\( i \ne {\text{Si}} \)); j, t, Ca, Mg, …(\( j,t \ne {\text{Si}},{\text{Al}},{\text{Na}} \)); k, Si, Ca, Mg, Na, …(\( k \ne {\text{Al}} \)); l, m Ca, Mg, Na, …(\( l,m \ne {\text{Si}},{\text{Al}} \)), n, s, Si, Ca, Mg, Al,… (\( n,s \ne {\text{Na}} \)); the symbol F denotes partial molar activation energy \( \overline{E}_{\text{a}} \) or dimensionless molar energy coefficients \( \varepsilon_{\text{V}} \); the power coefficient γ Si/i in the present formalism is a system-dependent parameter; \( p_{{{\text{AlO}}_{4} }}^{ch,j} \) in Eqs. (8) and (8′) is a probability function that expresses the probability to have a tetrahedrally coordinated alumina among existing (Al-O-Me n ) and (Me j -O-Me n ) SUs; \( k_{{{\text{AlO}}_{4} }}^{ch,j} \) is an individual constant for each cation and it determines the magnitude of the probability to form a tetrahedrally coordinated alumina structure as a result of charge compensation by Me2+ cation; \( \alpha_{{{\text{Al}}/j}} \) is the power which determines the composition dependence of the probability function \( p_{{{\text{AlO}}_{4} }}^{ch,j} \) and in turn the proportion of the (Al-O-Me n ) SUs with the tetrahedrally coordinated alumina (specific for each Al2O3-MeO oxide binary aluminate system); \( \Updelta F_{\text{Si - Si}}^{{{\text{Si - Al}}(ch,j)}} \) and \( \Updelta F_{{{\text{Al}} - m}}^{ch,j} \) describe additional contributions to the partial energy coefficients \( \overline{E}_{\text{a}} \) and \( \varepsilon_{\text{V}} \) of corresponding SUs due to the presence of tetrahedrally coordinated Al-containing SUs.
The concentrations of SUs in the present study are determined using the quasi-chemical thermodynamic model of the liquid slag[22,24,25] that takes into account short-range ordering of second-nearest-neighbor cations in the ionic melt. For a binary MeO-SiO2 slag, the quasi-chemical thermodynamic model considers the formation of two nearest-neighbor pairs (Si-Me) from (Me-Me) and (Si-Si) pairs, referred to as “second nearest neighbour bonds” (SNNB).[22,24,25] The concentrations of the various viscous flow SUs are equal to the concentrations of the corresponding second-nearest-neighbor bonds of the quasi-chemical thermodynamic model (see Figure 1). The quasi-chemical thermodynamic model as part of the FactSage computer package[26] has been successfully applied to describe experimental phase equilibria, thermodynamic and other types of data in many slag systems from binary to multi-component systems, and the SNNB concentrations calculated with the quasi-chemical thermodynamic model were taken as a reasonable approximation of the slag internal structure. The important point here is that valuable information on the structure of the liquid slag at the atomic level can be obtained from the quasi-chemical thermodynamic model of the liquid phase developed by experimental data on phase equilibria and thermodynamic properties. This information can be used as a basis for the description of other physicochemical properties, e.g., viscosity in this study. The SNNB concentrations were calculated using the thermodynamic computer package FactSage[26] with the latest thermodynamic database[27–29] and a specially developed software tool.
Key Principles in Developing Model Formalism and Model Parameters
Key principles in developing model formalism and model parameters include (a) critical review of experimental data, (b) determination of and fitting into the experimental values for \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) where possible, (c) application of restrictions to parameters derived from the physical basis of the model, and (d) systematic analysis of the trends of the model parameters’ correlations with available structural, physicochemical, and thermochemical properties.
Critical review of experimental data
All existing experimental viscosity data were first carefully analyzed with particular attention to the experimental procedures. Only the viscosity data measured at temperatures higher than liquidus were taken into account for viscosity modeling (FactSage was used to predict the liquidus). The contamination of the melt by the dissolution of container or sensor materials is one of major sources of uncertainties in high-temperature viscosity measurements affecting slag composition and container or sensor geometry. In this study, experimental results which include post-chemical analyses were preferred and those which contain the possibility of the contamination were given low weights. Temperature control is also essential—experimental results where temperature was measured by thermocouples close to the sample were preferred rather than those where an optical pyrometer was used. The rotating bob/crucible method was regarded as a more accurate and reliable technique for the high-temperature viscosity measurements compared to other methods including vibration viscometer.
Derivation of the experimental \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) values
Experimental values of the integral \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies were derived using Eq. [1] from the gradient and intercept, respectively, of the relationship of \( \ln \left( {\frac{\eta }{{T^{3/2} }}} \right) \) vs inverse temperature 1/T for a given composition (see Kondratiev and Jak[1] for details). These \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) values were then used along with the viscosity experimental data for optimizations.
Physical meaning of the model parameters and (d) systematic analysis of the trends of the model parameters
Present model parameters (partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies) have physical basis, and they are directly related to the internal slag structure and the physics of interactions at atomic scale. Strong, mostly covalent bonds linking silicate tetrahedrons in the melt result in high activation energy and high viscosities in the silica-rich slags. Addition of basic metal oxides (e.g., CaO, MgO, …) strongly affects the bonds between silicate tetrahedrons and therefore has a strong effect on the slag properties, e.g., decrease viscosity of the high-silicate slags significantly. Some cations (e.g., Al3+) behave in a different way depending on the chemical environment. The individual effect of various metal oxides on viscosity as well as on other properties is determined by a series of factors including atomic structure, cation size, inner and outer electronic arrangement, the “ease” to donate valent electrons to the oxygen anions in the oxide melt, etc.
The physical basis of the parameters in the present viscosity model is a foundation (i) to introduce particular restrictions (e.g., integral \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies should be positive over the whole composition range) and (ii) to relate these parameters to other known metal cation characteristics (e.g., cation size, ionic potential) or other physicochemical properties (e.g., heat of vaporization). The model formulation was revised to facilitate the linkage of the model parameters to other physical properties and metal cation characteristics. The optimization procedures involved active assessments and estimates of physicochemical trends and the use of correlations with other properties. The present viscosity model parameters can also help to “de-convolute” the relative physicochemical behavior of different cations in the oxide melt. These considerations were used in the revision of the model formalism and optimization of the model parameters. It is an essential feature that the model for the Al2O3-CaO-MgO-SiO2 system presented in these papers is only a part of a wider development of the viscosity model for the Al2O3-CaO-FeO-Fe2O3-MgO-Na2O-SiO2 multi-component system. The systematic analysis of the trends and self-consistency of the parameters across the whole multi-component system extending even further to other important slag systems, for example, to some lower-order sub-systems containing K2O and PbO, is an important factor of the present development.
Model Parameters
Systematic optimization of the systems was carried out using the above principles in cycles from lower-order systems to the multi-component systems and back until satisfactory agreement with all accepted experimental data was achieved. Table 1 reports the present model parameters.
Agreement with the experimental data using the modified model compared to the previous version[1–7] has been improved. In particular, the description of viscosities at high SiO2 concentrations within experimental uncertainties is demonstrated in the relevant figures below. Note that the present parameters for the Al2O3-CaO-MgO-SiO2 system are self-consistent with and derived using trends of the broader range of important slag systems; therefore, some results for other sub-systems containing Fe, Na, K, and Pb oxides are also presented below.
Details of Model Development
Pure Oxide Properties
The partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies of pure SiO2, Al2O3, and PbO liquids have been determined first using available experimental data for pure oxides.[30–40] The calculated and experimental viscosities of pure oxide melts are shown in Figure 2, demonstrating good agreement. Experimental data for the SiO2 melt by Bockris et al.[31] appear to be inconsistent with the others. Experimental data by Iida et al.[40] for the PbO melt were given a low weight during optimization due to a possible contamination of the melt by the dissolution of crucible material. The partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies for pure CaO, MgO, Na2O, and K2O melts were derived from not only (i) the data in the close composition ranges in the corresponding binary and ternary silicate systems but also (ii) using analysis of the \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) trends as functions of available structural and other physicochemical data. Various types of data and trends were analyzed, and ionic potentials and enthalpy of vaporization were selected to assist in evaluation of the partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies (see Figure 3).

Calculated viscosities of pure oxide liquids as functions of temperature. Results for “FeO” and Fe2O3 were taken from Ref. [42]

(a) Relationship between the partial activation energy of pure oxide MeO liquid and ionic potential for the corresponding cation Men+. (b) Relationship between the partial energy term \( \Updelta \overline{E}_{\text{V}} \) for pure oxide Me2/n O and the enthalpy of decomposing oxide component in elemental gas species, calculated using the FactSage computer package. Data for “FeO” and Fe2/3O were taken from Ref. [42]. Blank squares critically evaluated directly from experimental results for pure oxide melts, filled circles determined from experimental results for higher-order systems. Dashed lines are drawn to indicate a trend for all the oxides except Si1/2O
Figure 3(a) shows the relationship between the partial molar activation energies for pure oxides and the ionic potentials of the corresponding cations (ratio of the charge to the radius of a cation)—the activation energy is higher for the cations with higher ionic potentials, indicating stronger bonds with oxygen anions. Figure 3(b) shows the comparison between the partial energy terms \( \Updelta \overline{E}_{\text{V}} \) for pure oxides and the enthalpies of vaporizing liquid oxides in elemental gas species calculated using the latest thermodynamic databases of the FactSage thermodynamic computation package[26–29,41] on the basis of the following reaction:
where n denotes the oxidation state of a cation Men+. Figure 3(b) indicates that \( \Updelta \overline{E}_{\text{V}} \) for pure SiO2 is much higher than the others, which may be attributed to the strong covalent components of the bonds involving silicon cations. The \( \Updelta \overline{E}_{\text{V}} \) values of the other pure oxide liquids are approximately in the range of values of the corresponding enthalpies of vaporization of pure liquid oxides in elemental gas species. It should be noted that \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) values for SiO2, Al2O3, and PbO (described by squares) in Figure 3 have been derived from experimental values available for the corresponding pure oxides; those for the others (described by circles) have no experimental data for pure oxides and were derived from experimental data in the composition areas close to the corresponding oxides in binary and higher-order systems. The trends given in Figure 3 were also used as a guide to determine corresponding molar energies of the pure oxide liquids in addition to the experimental data in the corresponding unary, binary, and ternary systems.
Binary Silicate Systems
Figure 4 presents the fractions of the (Si-O-Si), (Si-O-Me), and (Me-O-Me) SUs in various Si1/2O-Me2/n O systems, calculated using the quasi-chemical thermodynamic model[22,24,25] and the FactSage computer package[26]—the so-called ordering tendency (preference to form Si-Me over Me-Me and Si-Si SNNB) decreases from the Na2O-, K2O-, and CaO- to MgO-, “FeO”-, and Al2O3-silicate system. An important point is that at high SiO2 concentrations, the Si-Me and Si-Si are predominant SUs and therefore determine the viscosity in the melts. This internal structural information was carefully analyzed and used in optimization of the model parameters, and is essential for understanding of the detailed trends in the developed model.

The concentrations of the SUs in the Si1/2O-Me2/n O systems at 1800 K (Me = K, Na, Pb, Ca, Mg, Al, n ionic valence of the Men+ cation)
Figures 5 and 6 show the calculated and experimental slag viscosities of binary silicate systems indicating that the addition of other metal oxides into the silica-rich slag significantly (by several orders of magnitude) decreases viscosity. This significant viscosity decrease is commonly attributed to the disturbance of the strong covalent bonds in the silicate tetrahedral network structures by basic cations. The degree of this viscosity decrease therefore should be related to such characteristics as cation size, valence, inner and outer electronic arrangement, the “ease” for cations to donate valent electrons to the oxygen anions in the melt, and the tendency toward more basic or acidic chemical behavior, and therefore should be specific for a given metal oxide. The use of a second power term \( E_{\text{Si - Si}}^{{{\text{Si - Si}},2}} X_{\text{Si - Si}}^{2} \) for all binary silicate systems in the previous QCV model[1–6] resulted in systematic discrepancies—predicted viscosities were higher than experimental data at high SiO2 concentrations. The revised model has the (Si-O-Si) partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies in different SiO2-MeO systems expressed by a power function of the concentrations of the (Si-O-Me) SUs (Eq. [6] reduces to Eq. [9] in the binary system):
where the parameters \( \gamma_{{{\text{Si}}/{\text{Me}}}} ,\;E_{{{\text{a}},{\text{Si - Si}}}}^{\text{Si - Me}} \), and \( \varepsilon_{{{\text{V}},{\text{Si - Si}}}}^{\text{Si - Me}} \) (the latter two with negative values) describe the degree of viscosity decrease with the increase of the (Si-O-Me) SU concentrations as a result of complex interactions in the silicate melt. The power parameter \( \gamma_{{{\text{Si}}/{\text{Me}}}} \) may be related to the apparent number of the (Si-O-Si) SUs which are affected by one given (Si-O-Me) SU. These parameters are system specific, dependent on how basic the metal cation Men+ behavior is in affecting the strength of the predominantly covalently bonded silicate tetrahedral structures, and are expected to correlate with the ionic potential I Me—defined by the ratio of the charge to the size of a cation. These parameters therefore may be derived from correlations with other properties if direct experimental viscosity data are not sufficient. Alternatively, if viscosity data exist, the trend of the correlation between the parameters and other properties may be established.

Comparison of calculated viscosities of fully liquid slag for binary silicate systems: (a) CaO-SiO2 at 1873 K, (b) Na2O-SiO2 at 1573 K, (c) K2O-SiO2 at 1373 K

Calculated viscosities of fully liquid slag for binary silicate systems: (a) Al2O3-SiO2, (b) PbO-SiO2, (c) MgO-SiO2
The correlations among the parameters \( \gamma_{{{\text{Si}}/{\text{Me}}}} ,\;E_{{a,{\text{Si - Si}}}}^{\text{Si - Me}} ,\;\varepsilon_{{{\text{V}},{\text{Si - Si}}}}^{\text{Si - Me}} \), and ionic potentials of corresponding cations are shown in Figure 7. Figure 7(a) indicates that the value of the power parameter \( \gamma_{{{\text{Si}}/{\text{Me}}}} \) increases proportional to the ratio of ionic potentials I Me/I Si. Figure 7(b) indicates that the value of the activation energy coefficient for the Al2O3-SiO2 system \( E_{{{\text{a}},{\text{Si - Si}}}}^{\text{Si - Al}} \) is higher than those for other binary silicate systems, but besides this there is no clear trend between the activation energy coefficient \( E_{{a,{\text{Si - Si}}}}^{\text{Si - Me}} \) and the ratio of ionic potentials I Me/I Si. It may be argued that a Men+ cation with larger size and lower ionic potential compared to Si4+ cation has the ability to affect more surrounding Si-Si SUs and to reduce their partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies more significantly, which is expressed by lower \( \gamma_{{{\text{Si}}/{\text{Me}}}} \) values, and therefore more significantly decrease the viscosity of the SiO2-Me2/n O silicate melt with the addition of Me2/n O. Figure 8 demonstrates that for a number of binary silicate systems, the integral molar activation energy at high SiO2 concentrations shows significant decrease with the \( \gamma_{{{\text{Si}}/{\text{Me}}}} \) decrease.

Comparisons between the activation energy coefficients for binary silicate systems and the ratio of ionic potential of Si4+ cation to that of basic cation Men+ I Si /I Me: (a) \( \gamma_{{{\text{Si}}/{\text{Me}}}} \)—I Me/I Si, (b) \( E_{\text{a,Si - Si}}^{\text{Si - Me}} \)—I Me/I Si. Data for the SiO2-“FeO” system are taken from Ref. [42]. Dashed lines are drawn to follow a tendency for all the binary silicate systems

Comparison between the calculated integral activation energies in the Si1/2O-Me2/n O systems. (Me = K, Na, Pb, Ca, Mg, Al, n ionic valence of the Men+ cation). (a) overall compositions, (b) limited to high SiO2 concentrations
The individual contributions of partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies to the corresponding integral molar energies in the present model can accurately describe all available experimental integral \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies derived from reliable experimental viscosity data at different temperatures.[30–33,42,43] Details are given in Figure 9 for the CaO-SiO2 system as an example.

Calculated results of (a) the activation energy, (b) the energy term \( \Updelta \underline{E}_{\text{V}} \) of the CaO-SiO2 system
The revised QCV model describes experimental viscosity data within experimental uncertainties, as illustrated in Figures 5 and 6 for the CaO, MgO, PbO, Al2O3, Na2O, and K2O-SiO2 systems. Comparison with the viscosity model developed by Decterov et al.[11] incorporated into the FactSage version 6.3[22,24–26] given in Figures 5 and 6 shows close viscosity description by both models in the composition ranges where experimental data are available.
Extrapolation of Binary Parameters into Multi-Component Silicate Systems
Analysis of the trends in the ternary and multi-component silicate systems indicates that the integral \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies vary as follows:
-
(i).
as a function of SiO2 concentrations along the constant ratio of two metal oxides (section A in Figure 10), similar to those in the binary silicate systems, and
Fig. 10 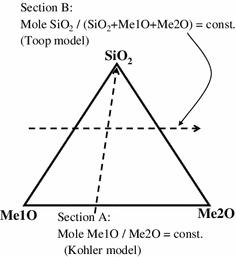
Schematic diagram showing extrapolation of binary parameters
-
(ii).
as a function of the basic metal oxide ratios along the constant SiO2 concentrations (section B in Figure 10), approximately linearly.

To describe these trends, the parameters determined for binary systems in the present viscosity model were extrapolated into the multi-component systems using the following expression:
Note that the right-hand side of Eq. [10] reduces to the left-hand side in binary silicate systems. This extrapolation of the binary parameters into ternary or multi-component silicate systems is similar to the Kohler/Toop “asymmetric” model[44] used in many other models for various properties of complex solutions (for example, Chartland and Pelton[45] in the quasi-chemical thermodynamic model).
Figure 11 shows the sections in the CaO-MgO-SiO2 ternary system used for comparison of predictions with experimental results[30,31,37,46–52] which are represented with thick solid lines. Good agreements of predicted and experimental values of the \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies and the corresponding viscosities are illustrated in Figures 12 and 13 for the CaO-MgO-SiO2 system. Figure 12(a) shows that the concentrations of the Si-Si, Si-Ca, and Si-Mg SUs as functions of SiO2 concentration at a fixed molar ratio of MgO to CaO in the CaO-MgO-SiO2 system vary similarly to those in the binary silicate systems [Figure 12(b)]—both are calculated with FactSage.[22,24–26] The trends of the calculated integral \( \underline{E}_{\text{a}} \)and \( \Updelta \underline{E}_{\text{V}} \) energies [Figure 12(c)] and viscosities [Figure 12(d)] are similar in the binaries and ternary systems.

Compositions of selected viscosity measurements in the CaO-MgO-SiO2 system along with the liquidus isotherms predicted by FactSage[26]

Calculated results of (a) SU concentrations in the CaO-MgO-SiO2 ternary system, (b) SU concentrations in the CaO-SiO2 and MgO-SiO2 binary systems, (c) integral \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies, (d) slag viscosity in the CaO-MgO-SiO2 system at 1773 K (1550 °C), where molar ratio MgO/CaO is kept as 45/55. \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies and viscosities in the CaO-SiO2 and MgO-SiO2 binary slags are also shown in Figs. 12(c) and (d)

(a) Proportions of SUs, (b) integral \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies, (c) slag viscosity in the CaO-MgO-SiO2 system at 50 mol pct SiO2
Figure 13(a) shows the fractions of SUs in the CaO-MgO-SiO2 system at a fixed SiO2 concentration of 50 mol pct. It can be seen that the proportions of the Si-Si SUs do not change significantly, those of the Ca-Ca and Mg-Mg SUs are almost zero, while those of the Si-Ca and Si-Mg SUs change nearly linearly with the MgO/(CaO+MgO) molar ratio. Figure 13(b) and (c) shows the \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies, and the viscosities as a function of the MgO/(CaO + MgO) molar ratio, indicating that these properties change nearly linearly with the MgO/(CaO+MgO) ratio.
Experimental viscosity data by Licko et al.[46] at 50 SiO2-50 mol pct MgO binary composition were not taken for optimizing partial energy coefficients \( \overline{E}_{\text{a}} \) and \( \varepsilon_{\text{V}} \) because viscosities were partially measured below liquidus temperatures.
Description of the Charge Compensation Effect
Experimental results indicate that the viscosities in the Al2O3-containing silicate slag systems Al2O3-(Me+, Me2+) x O-SiO2 are at a maximum at intermediate (Me+, Me2+)O/Al2O3 ratios (for example, in the systems Al-Ca-O-Si,[30,53,54] Al-Mg-O-Si,[54,55] and Al-Na-O-Si,[56,57] see Figures 14 and 15). This viscosity maximum is commonly attributed[20] to the so-called “charge compensation effect” explained as the Al3+ cation’s ability to take the tetrahedral interstitial between the oxygen anions if the excess negative charge for Al3+ is compensated by the alkali or alkaline earth cations; this is sometimes described as Me+AlO2 or Me2+Al2O4 associates in the melt. It is argued that the positioning of the Al3+ cations into the tetrahedral interstitials increases viscosity due to (i) the formation of stronger covalent (Al-O-Al) and (Al-O-Si) bonding in place of weaker ionic bonds and (ii) reduction of the weakening effect of the octahedral Al3+ on the strength of the nearest (Si-O-Si) bonds.

Calculated slag viscosities of the Al-Ca-O-Si and Al-Mg-O-Si systems where SiO2 = 67 mol pct

Calculated slag viscosities of the Al-Me-O-Si systems at 1773 K (Me = Na, Ca, Mg, Fe2+) where SiO2 = 50 mol pct. Results for the Al-Fe2+-O-Si systems were taken from Ref. [40]
The “charge compensation effect” in the Ca-, Fe-, Mg-, and K-containing aluminate systems is not specifically described in the latest quasi-chemical thermodynamic model by a corresponding Al-Me-O associate,[8] which is different from the AlNaO2 associate introduced into the quasi-chemical thermodynamic model for the Al-Na-containing systems. Therefore, a special treatment is required to describe the charge compensation effect on the viscosity of the corresponding melts.
A special charge compensation term added to the activation energy of viscous flow in the initial model[1–6] was based on the assumption[20] that the “charge compensation effect” appears when the Al3+ replaces Si4+ in the tetrahedral coordination, thus keeping the silicate network structure instead of breaking it. This mechanism related the “charge compensation effect” to the presence of the silicate networks, and therefore the “charge compensation term” in the previous formulation[1–6] was made proportional to the concentrations of the Si-Si SUs. However, that description had subsequently been revised[7] due to the following reasons. It did not reflect the fact that the viscosity maximum was observed also in the SiO2-free systems (e.g., CaO-Al2O3[58,59]). More importantly, it was argued[7] that the slag viscous flow is the movement of individual Si-Si and other SUs due to the breaks of the individual bonds (rather than long networks). The networks in slags are not permanent and individual (Si-O-Si) bonds do break during liquid flow. The viscous flow depends on the strength of the individual (Si-O-Si) bonds rather than on the network length (on the contrary, the length of the network depends on the strength of the individual bonds). Acceptance of the suggestion that formation of tetrahedral Al3+ influences the strength of the individual bonds in the long networks would also accept significance of the long-range (more than several atomic distances) interactions; the latter concept is believed to be incorrect. Instead, the strength of the individual (Si-O-Si) bonds is taken to be independent of the formation of the tetrahedral Al3+ at a distant atomic position (only the second-nearest-neighbor interaction is taken to be significant). The presence of a network is not taken to justify high viscosity. The increase of viscosity and the presence of networks are taken to be (i) the result of the strength of the individual interactions involving tetrahedral Al3+ with higher proportion of covalent bonding and (ii) the reduced concentrations of octahedral Al3+ and therefore their reduced effect on the interactions between Si-Si SUs. This assumption means that the increase of viscosity associated to the charge compensation effect is due to (i) stronger (Al-O-Al) and (Al-O-Si) bonds for the tetrahedral Al3+ and (ii) smaller effect of Al3+ cations on the predominantly covalently bonded (Si-O-Si) SU. These effects do not necessarily depend on the (Si-O-Si) SU concentrations, which agree with experimental evidence.[58,59] The Al3+ cations taking tetrahedral coordination as a result of charge compensation form strong covalent bonds and reduce the proportion of Al3+ in the octahedral coordination.
In the present model, the probability of Al3+ to take a tetrahedral coordination \( p_{{{\text{AlO}}_{4} }}^{ch,j} \) has been introduced, and the additional contributions of the charge-compensated Al3+ to the \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies of the Al2O3-containing SUs are described to be proportional to the \( p_{{{\text{AlO}}_{4} }}^{ch,j} \) value. Moreover, the reduction of the effect of Al3+ cations on decreasing the partial molar energies of the (Si-O-Si) SUs, due to the fact that a proportion of Al3+ is not in the octahedral but in the tetrahedral coordination, is described by the addition of special term with positive coefficients \( \Updelta F_{\text{Si - Si}}^{{{\text{Si - Al}}(ch,j)}} \) to the partial \( \overline{E}_{\text{a}} \) and \( \Updelta \overline{E}_{\text{V}} \) energies of (Si-O-Si) SU, which is proportional to the concentration of tetrahedral (Al-O-Si) SUs, which affects one (Si-O-Si) SU. The above improvements are shown in Eqs. [6] to [8].
Similar to Al2O3-containing systems, tetrahedrally coordinated Fe3+-containing SUs should be taken into account in Fe2O3-containing systems because there are some lines of analytical evidence indicating that tetrahedrally coordinated Fe3+ ions exist in the oxide melts or glasses due to the charge compensation effect by basic cations.[49,60] However, previous viscosity-measuring studies have indicated that the charge compensation effect of Fe3+ ions on slag viscosity is not high compared to that of Al3+ ions.[61] In this case, the model formalism in Fe2O3-containing systems can be described by adjusting parameters in the partial molar activation energies of Fe3+-containing SUs instead of introducing the special treatment used in Al2O3-containing systems.
Figure 16 shows the calculated result of the proportion of tetrahedrally coordinated Al3+ ions among total Al3+ ions in the Al2O3-CaO-SiO2 system and it depends on \( \alpha_{{{\text{Al}}/{\text{Ca}}}} \) and \( k_{{{\text{AlO}}_{4} }}^{ch,Ca} \) values in probability function \( p_{{{\text{AlO}}_{4} }}^{ch,Ca} \). The probability function has a maximum at intermediate Al2O3/CaO ratio, and the maximum positions are shifted to the higher Al2O3 concentrations for the higher \( \alpha_{{{\text{Al}}/{\text{Ca}}}} \) values. The maximum value of the probability function is proportional to \( k_{{{\text{AlO}}_{4} }}^{ch,Ca} \) values. The experimental result by Neuville et al.[62] obtained by MAS NMR analysis and Raman spectroscopy for corresponding glass compositions indicates that almost all Al3+ in the system are charge compensated by Ca2+ and thus tetrahedrally coordinated. This experimental structural information was used to determine the values as \( \alpha_{{{\text{Al}}/{\text{Ca}}}} = 2.5 \) and \( k_{{{\text{AlO}}_{4} }}^{ch,Ca} = 14.0 \) for the Al2O3-CaO and Al2O3-CaO-SiO2 systems. For other Al2O3-MeO and Al2O3-MeO-SiO2 systems, the \( \alpha_{{{\text{Al}}/{\text{Me}}}} \) and \( k_{{{\text{AlO}}_{4} }}^{{ch,{\text{Me}}}} \) values were assumed to be equal to \( \alpha_{{{\text{Al}}/{\text{Ca}}}} \) and \( k_{{{\text{AlO}}_{4} }}^{{ch,{\text{Ca}}}} \) , respectively, at this stage as the initial guess, due to the lack of reliable experimental structural data on the concentration of tetrahedrally coordinated Al3+.

Calculated result of the proportion of tetrahedrally coordinated Al3+ ions among total Al3+ ions in the Al2O3-CaO-SiO2 system at different SiO2 concentrations as a function of Al2O3/CaO ratio
Unlike the Ca-, Fe-, and Mg-containing aluminate systems, the “charge compensation effect” in the Na-containing aluminate systems is described in the latest quasi-chemical thermodynamic model by the formation of the NaAlO2 associates[26,27] and in the present viscosity model by formalism for the \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies of the corresponding SUs without the above special adjustments.
Currently, the viscosity model including the charge compensation effect has been developed up to Al2O3-(Na+, Ca2+, Mg2+, Fe2+) x O-SiO2 systems. Figure 14 shows the calculated slag viscosities of the Al2O3-CaO-SiO2 and Al2O3-MgO-SiO2 systems at constant SiO2 concentration of 67 mol pct. The predicted viscosities satisfactorily reproduce available experimental data. It was indicated that the use of the present viscosity model is valid to reasonably describe slag viscosities of aluminosilicate systems over wide composition ranges. Figure 15 provides the comparison of the viscosities at SiO2 = 50 mol pct for different ternary aluminosilicate melts. The predicted viscosities show upward curvatures due to the contribution of tetrahedral Al3+ coordination, which agree with the corresponding experimental data. It was found that the viscosity maximum decreases in the series of (Na-Al-Si-O) > (Ca-Al-Si-O) > (Mg-Al-Si-O) > (Fe2+-Al-Si-O) systems.
Conclusions
The quasi-chemical viscosity model has been revised, which makes it possible to predict the viscosities of fully liquid multi-component silicate slags over wide compositions and temperature ranges on the basis of physicochemical characteristics of the oxide melts.
The present viscosity model uses information from a structurally based (e.g., quasi-chemical) thermodynamic model for phase equilibria and thermodynamic properties. There are a number of important advantageous features in the present viscosity model. The values of the molar partial \( \overline{E}_{\text{a}} \) and \( \varepsilon_{\text{V}} \) energy coefficients obtained using the experimental data in the binary and ternary systems can be extrapolated into higher-order systems where no experimental data are available. This treatment enables the viscosity predictions to be performed and, in some cases, discrepancies between experimental data to be identified. The model is flexible enough to reflect major structural changes, but “rigid” for composition ranges with similar SUs distributions. The integral \( \underline{E}_{\text{a}} \) and \( \Updelta \underline{E}_{\text{V}} \) energies can be “de-convoluted” to partial contributions from a given SU and can be systematically analyzed. It enables various aspects which affect the viscosity of molten slag to be evaluated.
Some relationships have been found between model parameters and other physicochemical properties of oxide components, such as ionic potentials and enthalpy of vaporization. These correlations have significantly assisted in the selection of model parameters for the systems in which experimental data are lacking and can be further used in other chemical systems. The parameters for the Al2O3-CaO-MgO-SiO2 system presented in these papers are only a part of a wider development of the viscosity model for the Al2O3-CaO-FeO-Fe2O3-MgO-Na2O-SiO2 multi-component system and therefore are based on the analysis of the systematic trends and self-consistency across whole multi-component system—this is an essential feature of the present development.
Agreement with the experimental data using the modified model compared to the previous version has been improved. For example, viscosities at high SiO2 concentrations are reproduced within experimental uncertainties. A comparison with the viscosity model incorporated in FactSage presented in some figures shows good description of experimental viscosities by both models.
References
A. Kondratiev and E. Jak, Metall. Mater. Trans. B, 2005, vol. 36B, pp 623–38.
A. Kondratiev, P.C. Hayes, and E. Jak: 150 th ISIJ Int. Meeting, Hiroshima, Japan, Sept 2005, CAMP-ISIJ, vol. 18(4), pp. 821–25.
A. Kondratiev, P.C. Hayes, and E. Jak: ISIJ Int., 2006, vol. 46(3), pp. 359–67.
A. Kondratiev, P.C. Hayes, and E. Jak: ISIJ Int., 2006, vol. 46(3), pp. 368–74.
A. Kondratiev, P.C. Hayes, and E. Jak: ISIJ Int., 2006, vol. 46(3), pp. 375–84.
A. Kondratiev, P.C. Hayes, and E. Jak: ISIJ Int., 2008, vol. 48(1), pp. 7–16.
E. Jak: Proc. 8 th Int. Conf. Molten Slags Fluxes Salts, Santiago, Chile, 2009, pp. 433–48.
A.N. Grundy, H. Liu, I.H. Jung, S.A. Decterov, and A.D. Pelton: Int. J. Mat. Res. (formerly: Z. Metallkunde), 2008, vol. 99(11), pp. 1185–94. ibid pp. 1195–1209.
L. Zhang and S. Jahanshahi: Metall. Mater. Trans. B, 1998, vol. 29B, pp. 177–86. ibid. pp. 187–95.
T. Iida, H. Sakai, Y. Kita, and K. Shigeno: ISIJ Int., 2000, vol. 40, pp. S110–14.
M. Nakamoto, J. Lee, and T. Tanaka: ISIJ Int., 2003, vol. 45(5), pp. 651–56.
W.Y. Kim, A.D. Pelton, and S.A. Decterov: ISIJ Int., 2012, vol. 103(3), pp. 313–28.
J. Frenkel: Z. Phys., 1926, vol. 35, pp. 652–69.
J. Frenkel: Trans. Faraday Soc., 1937, vol. 33, pp. 58–65.
J. Frenkel: Kinetic Theory of Liquids, Oxford University Press, Oxford, 1946.
H. Eyring: J. Chem. Phys., 1936, vol. 4, pp. 283–91.
R. Ewell and H. Eyring: J. Chem. Phys., 1937, vol. 5, pp. 726–36.
S. Glasstone, K.J. Laidler, and H. Eyring: Theory of the Rate Processes, McGraw-Hill, New York, 1941.
Y. Waseda and J.M. Toguri: The Structure and Properties of Oxide Melts, 1998, World Scientific, Singapore, 1998.
B.O. Mysen: Dev. Geochem., 1988, vol. 4, pp. 1–354.
J.B. Fincham and F.D. Richardson: Proc. R. Soc. (Lond.), 1954, vol. 223, pp. 40–62.
M. Blander and A.D. Pelton: 2 nd Int. Symp. Metal. Slags Fluxes, TMS-AIME, Warrendale, PA, 1986, pp. 295–304.
R.D. Shannon: Acta Crystallogr., 1976, vol. A32, pp. 751–67.
A.D. Pelton, S.A. Decterov, G. Eriksson, C. Robelin, and Y. Dessureault: Metall. Mater. Trans. B, 2000, vol. 31B, pp. 651–59.
A.D. Pelton and P. Chartrand: Metall. Mater. Trans. A, 2001, vol. 32A, pp. 1355–60.
FactSage, Ecole Polytechnique, Montréal, 2013, http://www.factsage.com/, Accessed 18 Feb 2013.
S.A. Decterov, I.H. Jung, E. Jak, Y.B. Kang, P.C. Hayes, and A.D. Pelton: Proc. 7 th Int Conf Molten Slags Fluxes Salts, Cape Town, South Africa, publ. SAIMM, Johannesburg, SA, 2004, pp. 839–50.
E. Jak and P.C. Hayes: Final report, “Prediction of liquidus temperatures and high temperature phase equilibria for the system SiO2-Al2O3-FeO-Fe2O3-CaO with addition of Na2O-K2O-MgO”, The Univerisity of Queensland, Centre for Coal in Sustainable Development (CCSD), Brisbane, Australia, www.ccsd.biz, 2004.
E. Jak, P.C. Hayes, A.D. Pelton, and S.A. Decterov: Proc. 8 th Int. Conf. Molten Slags Fluxes Salts, Santiago, Chile, 2009, pp. 473–90.
G. Urbain, Y. Bottinga, and P. Richet: Geochim. Cosmochim. Acta, 1962, vol. 46, pp. 1061–72.
J.O’M. Bockris, J.D. MacKenzie, and J.A. Kitchener: Trans. Faraday Soc., 1955, vol. 51, pp. 1734–48.
G. Hofmaier and G. Urbain: Sci. Ceram., 1968, vol. 4, pp. 25–32.
V.K. Leko, E.V. Meshcheryakova, N.K. Gusakova, and R.B. Lebedeva: Sov. J. Opt. Technol., 1974, vol. 41, pp. 600–03.
D.W. Bowen and R.W. Taylor: Ceram. Bull., 1978, vol. 57, pp. 818–19.
R.A. Blomquest, J.K. Fink, and L. Leibowitz: Ceram. Bull., 1978, vol. 57, p. 522.
V.P. Elyutin, B.C. Mitin, and Y.A. Nagibin: Fiz. Aerodispersnykh Sist., 1972, vol. 7, pp. 129–35.
G. Hofmaier: Berg- und Huttenmannische Monatshefte, 1968, vol. 113, pp. 270–81.
G. Urbain: Rev. Int. Hautes Temper. Refract., 1982, vol. 19, pp. 55–7.
E. Sinn: Kristall und Technik., 1979, vol. 14, pp. 117–27.
T. Iida, Z. Morita, and T. Rokutanda: 3rd Int. Conf. Molten Slags Fluxes, Glasgow, UK, 1989, pp. 195–98.
ChemApp, GTT Technologies, Germany, 2013, http://www.gtt-technologies.de/, Accessed 18 Feb 2013.
E. Jak, M. Suzuki, and M. Chen: Private communication, 2011.
H. Ito and T. Yanagase: Trans. Jpn Inst. Met., 1986, vol. 1, pp. 115–20.
G.W. Toop: Trans-AIME, 1965, vol. 233, pp. 850–54.
P. Chartland and A.D. Pelton: J. Phase Equil., 2000, vol. 21, pp. 141–47.
T. Licko and V. Danek: Phys. Chem. Glasses, 1986, vol. 27, pp. 22–6.
J.S. Machin and T.B. Yee: J. Am. Ceram. Soc., 1948, vol. 31, pp. 200–04.
C.M. Scarfe, D.J. Cronin, J.T. Wenzel, and D.A. Kauffman: Am. Min., 1983, vol. 68, pp. 1083–88.
K. Morinaga, Y. Suginohara, and T. Yanagase: J. Jpn. Inst. Met., 1976, vol. 40, pp. 480–86.
J.S. Machin, T.B. Yee, and D.C. Hanna: J. Am. Ceram. Soc., 1952, vol. 35, pp. 322–25.
J.S. Machin and T.B. Yee: J. Am. Ceram. Soc., 1954, vol. 37, pp. 177–86.
C.M. Scarfe and D.J. Cronin: Am. Miner., 1986, vol. 71, pp. 767–71.
P. Kozakevitch: Rev. Metall., 1960, vol. 57, pp. 149–60.
M.J. Toplis and D.B. Dingwell: Geochim. Cosmochim. Acta., 2004, vol. 68, pp. 5169–88.
K.C. Mills: Slag Atlas, 2nd edn, Verlag Stahleisen GmbH, Düsseldorf, 1995.
M.J. Toplis, D.B. Dingwell, and T. Lenci: Geochim. Cosmochim. Acta., 1997, vol. 61, pp. 2605–12.
E.F. Riebling: J. Chem. Phys., 1966, vol. 44, pp. 2857–65.
G. Hofmaier: Berg und Huttenm. Monatsh. Montan. Hochschule Loeben, 1968, vol. 113, pp. 270–81.
G. Urbain: Rev. Int. Haut. Temp. Refract., 1983, vol. 20, pp. 135–39.
B.O. Mysen: Geochim. Cosmochim. Acta., 2006, vol. 70, pp. 2337–53.
V. Danek, T. Lioko, and Z. Panek: Silikaty, 1985, vol. 29, pp. 291–99.
D.R. Neuville, L. Cormier, and D. Massiot: Chem. Geol., 2006, vol. 229, pp. 173–85.
R. Knoche, D.B. Dingwell, F.A. Seifert, and S.L. Webb: Chem. Geol., 1994, vol. 116, pp. 1–16.
E. Eipeltauer, G. Jangg: Kolloid Z. Z. Polymere, 1955, vol. 142, pp. 77–84.
T.P. Shvaiko-Shvaikovskaya, O.V. Mazurin, and Z.S. Bashun: Izv. Akademii Nauk SSSR. Neorg. Mater., 1971, vol. 7(1), pp. 143–47.
L. Sasek: Silikaty., 1977, vol. 21, pp. 291–305.
L. Shartsis, S. Spinner, and W. Capps: J. Am. Ceram. Soc., 1952, vol. 35, pp. 155–60.
V.P. Elyutin, V.I. Kostikov, B.C. Mitin, and Y.A. Nagibin: Rus. J. Phys. Chem., 1969, vol. 43, pp. 316–20.
S.K. Gupta: Metall. Mater. Trans. B, 1995, vol. 26B, pp. 281–87.
G. Urbain: Rev. Int. Hautes Temper. Refract., 1984, vol. 21, pp. 107–111.
T. Yasukouchi, K. Nakashima, and K. Mori: Tetsu-to-Hagane, 1999, vol. 85, pp. 571–77.
F. Johannsen and H. Brunion: Z. Erzbergbau Metall., 1959, vol. 12, pp. 211–19. ibid, vol. 12, pp. 272–79.
Acknowledgments
The authors are grateful to Prof. Peter Hayes of the PYROSEARCH, The University of Queensland, for useful critical discussions, suggestions on model development, support, and review of the paper. The authors would like to acknowledge financial support from the Australian Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted April 22, 2013.
Rights and permissions
About this article
Cite this article
Suzuki, M., Jak, E. Quasi-Chemical Viscosity Model for Fully Liquid Slag in the Al2O3-CaO-MgO-SiO2 System—Part I: Revision of the Model. Metall Mater Trans B 44, 1435–1450 (2013). https://doi.org/10.1007/s11663-013-9928-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-013-9928-3