
Abstract
The binary Al-Mn system has been critically evaluated based upon available phase equilibrium and thermodynamic data, and optimized model parameters have been obtained giving the Gibbs energies of all phases as functions of temperature and composition. The liquid solution has been modeled with the modified quasichemical model to account for short-range ordering. The results have been combined with those of our previous optimizations of the Al-Mg and Mg-Mn systems to evaluate and optimize the Mg-Al-Mn system. All available data for the ternary system are reproduced with only one small ternary model parameter for the liquid phase.
Similar content being viewed by others
Introduction
Although magnesium-based materials have a long history of important commercial applications, including automotive, there remains much to be learned about the basic properties of the metal and its alloys. With the recent renewed interest in lightweight wrought materials, including both sheet and tube applications, there has been an increased focus on developing a better understanding of novel magnesium alloys, including those that incorporate additions of Mn and Al. These alloy systems, along with other potential candidates, are being actively pursued as possible routes to develop magnesium materials with improved ductility, or even practical room temperature formability.
The properties of cast or wrought material depend first and foremost upon the phases and microstructural constituents (eutectics, precipitates, solid solutions, etc.) which are present. In an alloy with several alloying elements, the phase relationships are very complex. In order to investigate and understand these complex phase relationships effectively, it is very useful to develop thermodynamic databases containing model parameters giving the thermodynamic properties of all phases as functions of temperature and composition. Using Gibbs free energy minimization software such as FactSage,[1,2] the automotive and aeronautical industries and their suppliers will be able to access the databases to calculate the amounts and compositions of all phases at equilibrium at any temperature and composition in multicomponent alloys, to follow the course of equilibrium or non-equilibrium cooling, to calculate corresponding heat effects, etc.
Such thermodynamic databases are prepared by critical evaluation, modeling, and optimization. In a thermodynamic “optimization,” adjustable model parameters are calculated using, simultaneously, all available thermodynamic and phase-equilibrium data in order to obtain one set of model equations as functions of temperature and composition. Thermodynamic data, such as activities, can aid in the evaluation of the phase diagrams, and information on phase equilibria can be used to deduce thermodynamic properties. Thus, it is frequently possible to resolve discrepancies in the available data. From the model equations, all of the thermodynamic properties and phase diagrams can be back-calculated, and interpolations and extrapolations can be made in a thermodynamically correct manner. The data are thereby rendered self-consistent and consistent with thermodynamic principles, and the available data are distilled into a small set of model parameters, ideal for computer storage.
As part of a broader research project to develop a thermodynamic database for Mg-alloys containing up to 25 potential alloying elements, the present study reports on evaluations and optimizations of the Al-Mn and Mg-Al-Mn systems. Previous optimizations[3-5] were based upon a Bragg-Williams (BW) random-mixing model for the liquid phase. However, the liquid phase in the Al-Mn binary system is expected to exhibit short-range ordering (SRO) as evidenced by the relatively large negative enthalpy of mixing.[6] As has been shown by the present authors,[7] the use of the BW model in liquids with a high degree of SRO generally results in unsatisfactory results and in poor predictions of ternary properties from binary model parameters. Hence the Al-Mn system was reoptimized with the modified quasichemical model (MQM) for the liquid phase; the present optimization reproduces all available data in the ternary Mg-Al-Mn system with only one very small ternary model parameter for the liquid solution. Care was taken to ensure that all optimized properties, such as the entropies of formation of intermetallic compounds, have physically reasonable values.
Modified Quasichemical Model
The MQM in the pair approximation[8] was used to model the liquid Al-Mn alloys. The liquid phases in the Mg-Al and Mg-Mn sub-systems of the Mg-Al-Mn system were also modeled previously with the MQM.[9,10] This model, which takes SRO into account, has been used extensively for molten salts,[11-13] slags[14] and sulfides.[15-17] All details of the model and notation have been described previously[8] and only a brief summary is given here.
In the MQM in the pair approximation, the following pair exchange reaction between atoms A and B on neighboring lattice sites is considered:
where (i − j) represents a first-nearest-neighbor pair. The non-configurational Gibbs energy change for the formation of two moles of (A − B) pairs is \( \Updelta g_{AB} \).
Let n A and n B be the number of moles of A and B, \( n_{ij} \) be the number of moles of (i − j) pairs, and Z A and Z B be the coordination numbers of A and B. The pair fractions, mole fractions, and “coordination-equivalent” fractions are defined respectively as:
The following equations may be written:
The Gibbs energy of the solution is given by:
where \( g_{A}^{ \circ } \) and \( g_{B}^{ \circ } \) are the molar Gibbs energies of the pure components, and \( \Updelta S^{\text{config}} \) is the configurational entropy of mixing given by randomly distributing the (A − A), (B − B) and (A − B) pairs in the one-dimensional Ising approximation:[8]
\( \Updelta g_{AB} \) is expanded in terms of the pair fractions:
where \( \Updelta g_{AB}^{ \circ } \), \( g_{AB}^{i0} \) and \( g_{AB}^{0j} \) are the parameters of the model which can be functions of temperature.
The equilibrium pair distribution is calculated by setting
This gives the “equilibrium constant” for the “quasichemical reaction” of Eq 1:
As \( \Updelta g_{AB} \) becomes progressively more negative, the reaction (Eq 1) is shifted progressively to the right, and the calculated enthalpy and configurational entropy of mixing assume, respectively, the negative “V” and “m” shapes characteristic of SRO.
The composition of maximum SRO is determined by the ratio of the coordination numbers Z B /Z A , as given by the following equations:[8]
where \( Z_{AA}^{A} \) and \( Z_{AB}^{A} \) are the values of Z A respectively when all the nearest neighbors of an A are A’s, and when all nearest neighbors of an A are B’s, and where \( Z_{BB}^{B} \) and \( Z_{BA}^{B} \) are defined similarly. (Note that \( Z_{AB}^{A} \) and \( Z_{BA}^{A} \) represent the same quantity and can be used interchangeably.) In order to set the composition of maximum SRO at X Mn = 0.5 in the binary systems we set the \( Z_{ij}^{i} /Z_{ij}^{j} = 1 \) so that the composition of maximum SRO occurs at the equimolar composition. Although the model is sensitive to the ratio of the coordination numbers, it is less sensitive to their absolute values. The use of the one-dimensional Ising model in Eq 8 introduces a mathematical approximation into the model which we have found, by experience, can be partially compensated by selecting values of Z B and Z A which are smaller than the actual values. The values of the coordination numbers selected in the present study are listed in Table 1. The liquid phase in the Al-Mg and the Mg-Mn systems show maximum SRO near the equimolar composition[9,10]; hence \( Z_{AB}^{A} = Z_{BA}^{B} \)in all cases.
From the MQM model parameters for the binary liquid phases, the thermodynamic properties of a ternary liquid phase may be estimated as discussed previously.[18] If ternary experimental data are available, additional ternary model parameters may be added if required.
The Al-Mn System
All calculations and optimizations in the present study were performed with the FactSage thermochemical software.[1,2]
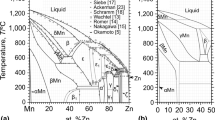
The optimized model parameters for the binary phases are reported in Table 1. Gibbs energies of all stable and metastable phases of the elements were taken from Dinsdale.[19] Crystallographic data[20-22] for the phases are listed in Table 2. The optimized phase diagram of this system is shown in Fig. 1.

Optimized phase diagram of the Al-Mn system
McAlister and Murray[23] presented an extensive literature review of the system up to 1987. Jansson[3] performed the first thermodynamic optimization of the system, treating the liquid phase with a BW random-mixing model. Liu et al.[4] re-optimized the system in the light of their new data[24] for the HCP phase. Du et al.[5] optimized the Al-Mn system as a first step in their assessment of the Mg-Al-Mn system.
The solid solution phases CBCC, CUB, FCC, BCC, γ (BCC) and ε (HCP) (Fig. 1) were modeled by a single-sublattice substitutional model. There are numerous data[25-34] for the solubility of Mn in FCC-Al obtained by various techniques (electrical resistivity (ER), optical microscopy (OM), lattice parameter (LP), hardness measurements (HD), electron probe microanalysis (EPMA)). Figure 2 compares the present optimization with these data.

Optimized solubility of Mn in the FCC phase
The optimized phase diagram for X Mn ≤ 0.2 is compared with experimental data in Fig. 3. Schaefer et al.,[35] by X-ray diffraction (XRD) and metallography, identified Al12Mn as a stable phase. They reported the pertectoid decomposition of Al12Mn into Al and Al6Mn between 504 and 521 °C. The present calculated temperature for this reaction is 511 °C.

Optimized phase diagram of the Al-Mn system for X Mn ≤ 0.2
Dix et al.[34] and Phillips[36] studied the system by metallography and thermal analysis. They reported the intermetallic compounds Al6Mn and Al4Mn (μ-Al4Mn in Fig. 1). Godecke and Koster[37] studied the system by the same techniques. They confirmed the presence of Al11Mn4 which was also noted by Philips.[36] They reported high- and low-temperature allotropes of Al11Mn4, the high-temperature form with a single-phase composition range of approximately 4 at.%. As the exact nature of the phase boundaries of the high-temperature form are unknown, this compound is treated as two stoichiometric phases Al11Mn4 in the present calculations with a transition temperature of 916 °C.[37] In the absence of any thermodynamic data, the Gibbs energy of the transformation was assumed to be zero. That is, the parameters for this compound listed in Table 1 apply to both the phases.
Taylor[38] by XRD and thermal analysis, and Murray et al.[39] by thermal analysis, reported the existence of a second phase close to μ-Al4Mn. Du et al.[5] by XRD and differential thermal analysis (DTA) confirmed the presence of two distinct phases: μ-Al4Mn at X Mn = 0.2 and λ-Al4Mn at X Mn = 0.186. They modeled the phase λ-Al4Mn as stoichiometric Al461Mn107 based on the crystallographic data of Kreiner and Franzen.[21]
Koch et al.[40] studied the system by thermal analysis in the range 25-100 at.% Mn. Koster and Wachtel[41] studied the system in the range 30-100 at.% Mn by thermal and magnetic analysis, microhardness, and XRD. Later, Godecke and Koster,[37] by metallography and thermal analysis, reported three phases in the region from 30 to 50 at.% Mn: γ, γ1, and γ2 (In Fig. 1, γ is denoted γ (BCC), while γ1 and γ2 are the Al8Mn5 phase). Ellner,[20] using high temperature XRD, showed that the γ phase has a BCC structure. As very little information is available about the γ1 and γ2 phases, they were modeled as a single phase “Al8Mn5” (Fig. 1), as was also done in previous optimizations.[4,5]. Following the suggestion, based on crystallographic data,[20] of Du et al.,[5] the Al8Mn5 phase was modeled by the compound energy formalism[42,43] as Al12Mn5(Al,Mn)9 (the first sublattice containing only Al, the second only Mn and the third a random mixture of Al and Mn).
The optimized phase diagram for the region from 0.2 ≤ X Mn ≤ 0.5 is compared with experimental data in Fig. 4. In the absence of any further experimental evidence, the order-disorder transformation in the γ (BCC) phase suggested by Liu et al.[4] based upon preliminary differential scanning calorimetry (DSC) results[44] was ignored. For modeling purpose, γ (BCC) was formally treated as the same phase as the terminal BCC solid solution of Al and Mn, but for clarity of representation, this region has been denoted as γ (BCC) in the figures.

Optimized phase diagram of the Al-Mn system for 0.2 ≤ X Mn ≤ 0.5
The optimized phase diagram in the region from 0.5 ≤ X Mn ≤ 0.1 is compared with the experimental data in Fig. 5. The phase equilibria for the ε (HCP) phase were first studied by XRD and specific heat measurements by Kono.[45] Koster and Wachtel[41] studied the boundaries of the phase by magnetic analysis, micro-hardness, XRD, and thermal analysis, and denoted the phase as ε. Muller et al.[46] established phase equilibria for this phase by DTA. Liu et al.[24] investigated the phase mainly by a diffusion couple technique, and also by metallography, XRD, DSC and transmission electron microscopy (TEM). They reported a wider single-phase region than Koster and Wachtel,[41] attributing the difference to the transformation of ε (HCP) at compositions richer in Mn than 58 at.% into the CUB phase during the quenching experiments.

Optimized phase diagram of the Al-Mn system for 0.5 ≤ X Mn ≤ 1.0
Meschel and Kleppa,[47] by direct synthesis calorimetry, reported the enthalpy of formation at 25 °C for alloys at 60 and 80 at.% Mn. Kubaschewski and Heymer,[48] by high temperature reaction calorimetery, reported enthalpies of formation for four compositions: Al6Mn, Al4Mn, Al11Mn4 and AlMn. The optimized standard enthalpy of formation of the intermediate compounds is compared with the experimental data and the previous optimizations in Fig. 6.

Optimized standard enthalpies of formation of solid Al-Mn alloys
Partial enthalpies of mixing in the liquid phase at 1353 °C were measured by high-temperature vacuum isothermal calorimetry by Esin et al.[6] who reported only smoothed data. The present optimized enthalpy of mixing is compared with these data and with previous optimizations in Fig. 7.

Optimized partial enthalpies of mixing in liquid Al-Mn alloys at 1353 °C
Batalin et al.[49] performed electromotive force (EMF) measurements in the liquid phase at 1297 °C, reporting activities of Mn, while Kematick and Myers[50] measured Al and Mn activities at 902 °C by Knudsen cell/mass spectrometry in the range 42-62 at.% Mn. These data are inconsistent with the other data for the system and were ignored. Chastel et al.[51] determined activities of Mn and Al in the melt in the range from 0 to 50 at.% Mn at 1247 °C by Knudsen cell/mass spectrometry. The optimized activities are compared with the experimental data and previous optimizations in Fig. 8.

Optimized activity of Al and Mn in liquid Al-Mn alloys at 1247 °C
The optimized entropy of mixing in the liquid phase at 1400 °C is compared with the previous optimizations in Fig. 9. The present positive entropy of mixing is physically more probable than the negative values of the previous optimizations. The optimized standard entropies of formation of the solid alloys from the elements at 25 °C are compared with previous optimizations in Fig. 10 (see also Table 1). Generally, such entropies of formation are expected to be small, as in the case in the present study. It is not possible to obtain a closer fit to the liquid activity data in Fig. 8, simultaneously with all the other data for the system, without introducing a relatively large negative non-configurational entropy term for the liquid phase as well as significantly larger entropies of formation of the solid phases. Since such large entropies are physically improbable, we believe it to be more likely that the activity data are in error.

Optimized entropy of mixing in liquid Al-Mn alloys at 1400 °C

Optimized standard entropies of formation at 25 °C of solid Al-Mn alloys from the elements
The Mg-Al-Mn System
The previously optimized phase diagrams of the Al-Mg[9] and Mg-Mn[10] systems are shown in Fig. 11 and 12 respectively. The parameters optimized by Chartrand[9] for the phases in the Al-Mg system pertinent to the present work are given in Table 3. Crystallographic data of all the solid phases appearing in the Mg-Al-Mn system are in Table 2. It may be noted that the calculated consolute temperature of the miscibility gap in the Mg-Mn system, Fig. 9, is about 1500-2000 °C lower than in the previous optimizations[52,53] of this binary system.

Previously optimized phase diagram of the Al-Mg system[9]

Previously optimized phase diagram of the Mg-Mn system[10]
Our previous optimizations[9,10] of the Al-Mg and Mg-Mn systems were combined with the present optimization of the Al-Mn system in order to calculate the polythermal projection of the liquidus of the Mg-Al-Mn system shown in Fig. 13. The thermodynamic properties of the ternary liquid phase were calculated by the MQM from the binary model parameters. The “asymmetric approximation”[18,54] with Al as “asymmetric component” was used, since the Mg-Mn liquid exhibits positive deviations from ideality, whereas the Al-Mg and Al-Mn liquids exhibit negative deviations. A small ternary interaction parameter (Table 1) was included for the liquid phase.

Predicted polythermal projection of the liquidus in the Mg-Al-Mn system. Calculated invariant temperatures are shown (°C)
The HCP phase in the Al-Mg and Mg-Mn systems[9,10] and the FCC phase in the Al-Mg system were modeled with single-sublattice substitutional models. For modeling purposes, the Mg-rich HCP phase in the Al-Mg and Mg-Mn systems and the ε (HCP) phase in the Al-Mn system were formally treated as the same phase. The thermodynamic properties of the ternary HCP and FCC phases were estimated from the binary model parameters. The “symmetric” (Kohler) approximation[54] was used with no ternary interaction parameters. The predicted stability of the ε (HCP) phase at 1200 °C is shown in Fig. 14.

Calculated isothermal section of the Mg-Al-Mn phase diagram at 1200 °C
γ-AlMg has the same structure as the CBCC-Mn phase (Table 2). A small solubility of Mn in this compound or combined solubility of Al and Mg in CBCC-Mn might therefore be expected. No data for these solubilities could be found. Pending further experimental work, the binary phase γ-AlMg and CBCC-Mn were treated as separate phases. Possible mutual solubilities between any other intermetallic phases were assumed to be negligible in the absence of any experimental evidence and since they all have different structures and stoichiometries.
Mg-Rich Alloys
The solubilities of Mn in liquid Mg reported by Hanawalt et al.[55] are significantly lower than later findings[56,57] and have been rejected. Beerwald[56] and Nelson[57] used a settling technique to determine solubilities. Oberländer et al.,[58] and later Simensen et al.[59] from the same laboratory, identified the composition of precipitated solids around 700-750 °C by a centrifuging technique supplemented with XRD and metallography. They concluded that at 700-750 °C, CUB and Al8Mn5 are the equilibrium phases at compositions 0 ≤ wt.% Mn ≤ 3 and 0 ≤ wt.% Al ≤ 15. The present calculations agree well with these data. In another work, Simensen et al.,[60] reported solubilities at 750, 710 and 670 °C by the same technique. Thorvaldsen and Aliravci[61] measured the solubility of Mn in the liquid phase by settling and decantation followed by emission spectrometry and inductively coupled plasma (ICP) measurements.
The data of Nelson,[57] Beerwald,[56] Simensen et al.[60] and Thorvaldsen and Aliravci[61] are compared with the present calculations in Fig. 15. All data except those of Simensen et al.[60] are reasonably well reproduced below 780 °C. The solubilities reported by Simensen et al.[60] are lower than the present calculations and the disagreement increases with increasing temperature. This same trend was noted by Ohno and Schmid-Fetzer[62] in their assessment. Thorvaldsen and Aliravci[61] reported that the results of Simensen et al.[60] may have been influenced by iron contamination. A calculated isopleth at 5.05 wt.% Al is compared with the data of Thorvaldsen and Aliravci[61] in Fig. 16.

Optimized liquidus surface in Mg-rich solutions at different temperatures T: (a) T < 700 °C, (b) 700 < T ≤ 750 °C, and (c) T > 750 °C

Calculated section of the Mg-Al-Mn phase diagram at constant 5.05 wt.% Al
Mirgalovskaya et al.,[63] by microstructural and microhardness tests, studied liquid-solid and solid-solid phase equilibria in Mg-rich alloys. Their data are compared with the present calculations in Fig. 17. Their results at 850 °C are inconsistent with the measurements of other authors as can be seen by comparing Fig. 17(b) and 15(c). Other measurements of Mirgalovskaya et al.[63] and Ageev et al.[64] in Mg-rich alloys at temperatures below 400 °C were rejected because they report large solubilities of Mn and Al in Mg which are inconsistent with the other data.

Calculated liquidus surface in the Mg-Al-Mn system: (a) 700 °C and (b) 850 °C
The solidus measurements of Nelson[57] are compared with the calculations in Fig. 18. The disagreement is due to the fact that these measurements are inconsistent with other data in the binary Al-Mg system (wt.% Mn = 0 in Fig. 18) which were used in the optimization of this binary system.

Calculated solidus curves in the Mg-Al-Mn system
In the present work, for all practical purposes the solubility data up to 760 °C can be reproduced without any ternary interaction parameters. The small ternary term shown in Table 1 is only required to refine the optimization at the higher temperatures.
Al-Rich Alloys
Leemann and Hanemann[65] studied Al-rich alloys by metallography and thermal analysis. Wakeman and Raynor[66] doubted the attainment of equilibrium in Leemann and Hanemann’s work[65] and carried out microstructural observations of alloys annealed at 400 °C. These authors[66] reported a ternary compound by XRD and tentatively reported its composition to be MnMg2Al10. Later, Fun et al.[67] determined the crystal structure of this phase by XRD and reported its composition to be Mn2Mg3Al18. This phase is denoted as T in the present work. Du et al.[5] reported the enthalpy of formation of T as −10.2 kJ/(mol of atoms) by first principles calculations and as −8.7 kJ/(mol of atoms) by a CALPHAD-type assessment. The present optimization gives the enthalpy of formation as −9.9 kJ/(mol of atoms).
Barlock and Mondolfo[68] reported a eutectic invariant reaction L = (Al) + β-AlMg + T at 447 °C. The present computed temperature for this reaction is 451 °C. According to the present calculations, the T phase should melt peritectically near 471 °C. The primary crystallization field for this ternary phase is extremely small and is very close to the Mg-Al binary edge of the composition triangle. It is not visible on the scale of Fig. 13.
Ohnishi et al.[69] studied Al-rich alloys at 400 and 450 °C by metallography and XRD. Isothermal sections at 400 and 450 °C are compared with the experimental data in Fig. 19 and 20. Ohnishi et al.[69] also reported two-phase (FCC + Al6Mn) regions (not shown here) at very low Mg and Mn contents at 400 and 450 °C which are inconsistent with the optimized Al-Mn binary phase diagram. In a different work, Ohnishi et al.[70] studied six Al-rich alloys, showing the two-phase FCC + Al6Mn region to be stable for 1 ≤ wt.% Mn ≤ 2 and 0 ≤ wt.% Mg ≤ 4, in agreement with the present calculations.

Calculated isothermal sections of the Mg-Al-Mn phase diagram: (a) 400 °C and (b) 450 °C

Calculated liquidus surface in the Mg-Al-Mn system
Butchers et al.,[71] from cooling curves, reported smoothed liquidus curves between 630 and 650 °C. The data at 650 °C are compared with the present calculations in Fig. 20.
Little et al.[72] by microstructure observations, and Fahrenhorst and Hoffman[25] by electrical resistance measurements, reported solubilities of Mn and Mg at 500 °C in the FCC phase. These data are compared with the present calculations in Fig. 21.

Calculated solubility of Mn and Mg in FCC-Al
Conclusions
Gibbs energy functions for all phases in the Al-Mn system have been obtained. All available thermodynamic and phase equilibrium data have been critically evaluated in order to obtain one set of optimized model parameters of the Gibbs energies of all phases which can reproduce the experimental data within experimental error limits. Tentative calculated phase diagrams of the Mg-Al-Mn system have been given. For all practical purposes, the available data below 760 °C in the Mg-Al-Mn system can be reproduced solely from the optimized binary model parameters. A small ternary parameter has been included for the liquid phase to refine the optimization at higher temperatures.
The use of the MQM for the liquid phase has permitted SRO to be taken into account. Use of this model results in a better fitting of the data for the liquid phase than is the case when a Bragg-Williams random-mixing model is used, as well as a better representations of the partial properties of solutes in dilute solution in magnesium, the activities of solutes in dilute solution being of much practical importance. As shown by the present authors,[7] the use of the MQM generally also results in better estimations of the properties of ternary and higher-order liquid alloys. These estimations of phase equilibria in magnesium alloys will aid in the design of novel magnesium alloys.
References
C. W. Bale, P. Chartrand, S. A. Degterov, G. Eriksson, K. Hack, R. Ben Mahfoud, J. Melançon, A. D. Pelton and S. Petersen (2002) FactSage Thermochemical Software and Databases, Calphad, 26(2), 189–228.
C.W. Bale, A.D. Pelton, and W. Thompson, FactSage Thermochemcial Software and Databases, http://www.crct.polymtl.ca (2008)
A. Jansson 1992 Thermodynamic Evaluation of the Al-Mn System, Metall. Mater. Trans. A, 23A, 2953–2962.
X. J. Liu, I. Ohnuma, R. Kalnuma, and K. Ishida (1999) Thermodynamic Assessment of the Al-Mn Binary Phase Diagram, J. Phase Equilib., 20(1), 45–56.
Y. Du, J. Wang, J. Zhao, J. C. Schuster, F. Weitzer, R. Schmid-Fetzer, M Ohno, H. Xu, Z. Liu, S. Shang and W. Zhang (2007) Reassessment of the Al-Mn System and a Thermodynamic Description of the Al-Mg-Mn System, Int. J. Mat. Res. 98, 855–871.
Y. O. Esin, N. T. Bobrov, M. S. Petrushevskii and P. V. Geld 1973 Concentration Variation of the Enthalpies of Formation of Mn-Al Melts at 1626 K, Russ. J. Phys. Chem. 47, 1103–1105.
A. D. Pelton and Y. -B. Kang 2007 Modeling Short-range Ordering in Solutions. Int. J. Mat. Res. 10, 907–917.
A. D. Pelton, S. A. Degterov, G. Eriksson, C. Robelin and Y. Dessureault 2000 The Modified Quasichemical Model I – Binary Solutions. Metall. Mater. Trans. B. 31B (6), 651–659.
P. Chartrand, CRCT, Ecole Polytechnique, Montreal, 2006 (unpublished work)
Y.-B. Kang, A. D. Pelton, P. Chartrand, P. Spencer and C. D. Fuerst (2007) Critical Evaluation and Thermodynamic Optimization of the Binary Systems in the Mg-Ce-Mn-Y System. J. Phase Equilib. Diffus. 28 (4), 342–354.
P. Chartrand and A. D. Pelton 2001 Thermodynamic Evaluation and Optimization of the LiCl-NaCl-KCl-RbCl-CsCl-MgCl2-CaCl2 System Using the Modified Quasichemical Model, Metall. Mater. Trans. A. 32A (6), 1361–1383.
P. Chartrand and A. D. Pelton 2001 Thermodynamic Evaluation and Optimization of the LiF-NaF-KF-MgF2-CaF2 System Using the Modified Quasichemical Model. Metall Mater Trans A 32A (6), 1385–1396.
P. Chartrand and A. D. Pelton 2001 Thermodynamic Evaluation and Optimization of the Li, Na, K, Mg, Ca//F, Cl Reciprocal System Using the Modified Quasichemical Model. Metall. Mater. Trans. A. 32A (6), 1417–1430.
S.A. Decterov, I.-H. Jung, E. Jak, Y.-B. Kang, P. Hayes, and A.D. Pelton, Thermodynamic Modelling of the Al2O3-CaO-CoO-CrO-Cr2O3-FeO-Fe2O3-MgO-MnO-NiO-SiO2-S System and Application in Ferrous Process Metallurgy, Proceedings of the VII International Conference on Molten Slags, Fluxes and Salts, C. Pistorius, Ed. (Johannesburg, South Africa), The South African Institute of Mining and Metallurgy, 2004, p 839-850
P. Waldner, A. D. Pelton 2004 Thermodynamic Modeling of the Ni-S System. Z. Metallkunde 95, 672–681.
P. Waldner and A. D. Pelton 2004 Critical Thermodynamic Assessment and Modeling of the Fe-Ni-S System. Metall. Mater. Trans. B. 35B (5), 897–907.
P. Waldner and A. D. Pelton, 2005, Thermodynamic Modeling of the Fe-S System, J. Phase Equilib. Diffus., 26 (1), 23–38.
A. D. Pelton and P. Chartrand 2001 The Modified Quasichemical Model: Part II. Multicomponent solutions. Metall. Mater. Trans. A. 32A (6), 1355–1360.
A.T. Dinsdale, SGTE Data for Pure Elements, CALPHAD, 1991, 15(4), p 317-425 plus updates (private communication), 2000, http://www.sgte.org
M. Ellner (1990) The Stucture of the High-Temperature Phase MnAl (h) and the Displacive Transformation from MnAl (h) into Mn5Al8. Metall. Mater. Trans. A 21A, 1669–1672.
G. Kreiner, H. F. Franzen (1997) The Crystal Structure of λ-Al4Mn. J. Alloys Compd. 261, 83–104.
P. Villars, L. D. Calvert (1991) Pearson’s Handbook of Crystallographic Data for Intermetallic phases 2nd edition. ASM, Materials Park, Ohio, USA.
A. J. McAlister, J. L. Murry (1987) The Al-Mn System. Bulletin of Alloy Phase Diagrams 8(5), 438–446.
X. J. Liu, I. Ohnuma, R. Kalnuma, K. Ishida 1996 Phase Equilibria in the Mn-Rich Portion of the Binary System Mn-A1. J. Alloys Compd. 235, 256–261.
E. Fahrenhorst, W. Hoffman 1940 The Solubility of Manganese in Aluminum Containing up to 2 Percent of Magnesium. Metallwirtschaft, 19, 891–893.
E. Butchers, W. Hume-Rothery 1945 The Solubility of Manganese in Aluminum. J. Inst. Met. 71, 87–91.
I. Obinata, E. Hata, K. Yamaji 1953 Chiefly on the Sub-Cooled Al-Mn Alloys. J. Inst. Met. 17, 496–501.
G. M. Kuznetsov, A. D. Barsukov and M. I. Abas 1983 Study of Manganese, Chromium, Titanium, and Zirconium Solubility in Solid Aluminum, Sov. Non Ferrous Met. Res. 11:47–51.
Y. Minamino, T. Yamane, H. Araki, N. Takeuchi, Y.-S. Kang, Y. Miyamoto and T. Okamoto 1991 Solid Solubilities of Mn and Ti in Aluminum at 01 MPa and 21 GPa. Metall. Mater. Trans. A 22A, 783–786.
V. A. Livanov and V. M. Vozdvizhenskii 1958 Recrystallization of Aluminum Alloys with Manganese. Trudy Moskov. Aviatsion, Tekhnol. Inst. 31, 65–83.
E.H. Dix and W.D. Keith, Equilibrium Relations in Al-Mn Alloys of High Purity, Proc. AIME, Inst. Metals Div., 1927, p 315-335
M. E. Drits, E. S. Kadaner, E. M. Padzhnova and N. R. Bochvar 1964 Determination of the Boundaries of Common Solubility of Mn and Cd in Solid Aluminum. Zh. Neorg. Khim. 9(6), 1397–1402.
C. Sigli, CALPHAD XXIV Conference, Kyoto, Japan, 1995, quoted by Du et al. [5]
E. H. Dix, W. L. Fink and L. A. Willey 1933 Equilibrium Relations in Al-Mn Alloys of High Purity II. Trans. AIME 104, 335–352.
R. J. Schaefer, F.S. Biancaniello and J. W. Cahn 1986 Formation and Stability Range of the G Phase in the Al-Mn System. Scr. Metall 20(10), 1439–44.
H.W.L. Phillips 1942 The Constitution of Alloys of Aluminium with Manganese Silicon and Iron. J. Inst. Met 69, 275–316.
T. Godecke and W. Koster 1971 A Supplement to the Constitution of the Al-Mn System. Z. MetaIlkd. 62(10), 727–32.
M. A. Taylor 1960 Intermetallic Phases in the Al-Mn Binary System. Acta. Metall. 8, 256–262.
J. L. Murray, A. J. McAlister, R. J. Schaefer, L. A. Bendersky, F. S. Biancaniella and D. L. Moffatt 1987 Stable and Metastable Phase Equilibria in the Al-Mn System. Metall. Trans. A 18A, 385–392.
A. J. J. Koch, P. Hokkeling, M.G.v.d. Steeg, and K. J. Devos 1960 New Material for Permanent Magnets on a Base of Mn and Al. J.Appl. Phys., 31(5), 75S–77S.
W. Koster and E. Wachtel 1960 Magnetic Investigation of Al-Mn Alloys Containing More Than 25 at. % Mn, Z. Metallkd. 51, 271–280.
M. Hillert and L. -I. Staffansson 1970 Regular Solution Model for Stoichiometric Phases and Ionic Melts, Acta Chem. Scand. 24(10), 3618–26.
J. O. Andersson, A. F. Guillermet, M. Hillert, B. Jansson, B. Sundman 1986 A Compound-Energy Model of Ordering in a Phase with Sites of Different Coordination Numbers. Acta Metall. 34(3), 437–45.
X.J. Liu, Ph.D. Thesis, Tohoku University, Japan, 1998
H. Kono 1958 On the Ferromagnetic Phase in Mn-Al System. J. Phys. Soc. Jpn. 13, 1444–1451.
C. Muller, H. Stadelmaier. B. Reinsch and G. Petzow 1996 Metallurgy of the Magnetic τ-Phase in Mn-Al and Mn-Al-C. Zeitschrift fuer Metallkunde 87(7), 594–597.
S. V. Meschel and O. J. Kleppa 1994 The Standard Enthalpies of Formation of Some 3d Transition Metal Aluminides by High-Temperature Direct Synthesis Calorimetery. NATO ASI Series, Ser.E 256, 103–112.
O. Kubaschewski and G. Heymer 1960 Heats of Formation of Transition-Metal Aluminides, Trans. Faraday Soc. 56, 473–478.
G. I. Batalin, E. A. Beloborodova, V. A. Stukalo and A. A. Chekhovskii 1972 Thermodynamic Properties of Molten Alloys of Aluminum with Manganese. Ukr. Khim Zh. 38(8), 825–827.
R. J. Kematick, C. E. Myers 1992 Thermodynamics and Phase Equilibria in the Al-Mn System. J. Alloys and Comp 178, 343–349.
R. Chastel, M. Saito and C. Bergman 1994 Thermodynamic Investigation on Al1-xMnx Melts by Knudsen Cell Mass Spectrometry. J. Alloys and Comp. 205, 39.
J. Tibbals, Mg-Mn System, COST 507 – Thermochemical Databases for Light Metal Alloys, I. Ansara, A.T. Dinsdale, and M.H. Rand, Eds., Vol. 2, EUR 18499, 1998, p 215-217
J. Gröbner, D. Mirkovic, M. Ohno and R. Schmid-Fetzer 2005 Experimental Investigation and Thermodynamic Calculation of Binary Mg-Mn Phase Equilibria. J. Phase Equilib. Diffus. 26(3) 234–239.
A. D. Pelton 2001 A General “Geometric” Thermodynamic Model for Multicomonent Solutions, Calphad 25(2), 319–328.
J.D. Hanawalt, C.E. Nelson, and G.E. Holdeman, Removal of Iron from Mg-Base Alloys, US Patent No. 2267862, December 30, 1941
A. Beerwald 1944 On the Solubility of Iron and Manganese in Magnesium and in Magnesium-Aluminium Alloys. Metallwirtschaft 23, 404–407.
B. J. Nelson 1951 Equilibrium Relations in Mg-Al-Mn Alloys. J. Metals 3, 797–799.
B.C. Oberländer, C.J. Simensen, J. Svalestuen, and A. Thorvaldsen, Phase Diagram of Liquid Magnesium-Aluminium-Manganese Alloys, Magnesium Technology, Pros. Conf., London, 1986, p 133-137
C. J. Simensen, B. C. Oberländer, J. Svalestuen and A. Thorvaldsen 1988 Determination of the Equilibrium Phases in Molten Mg–4 wt. % Al-Mn Alloys. Z. Metallkd. 79, 537–540.
C. J. Simensen, B. C. Oberländer, J. Svalestuen and A. Thorvaldsen 1988 The Phase Diagram for Magnesium-Aluminium-Manganese above 650 °C. Z. Metallkd. 79, 696–699.
A. Thorvaldsen and C.A. Aliravci, Solubility of Mn in Liquid Mg-Al Alloys, Proc. Int. Symp. Adv. Prod. Fabr. Light Met. Met. Matrix Comp., 1992, p 277-288
M. Ohno and R. Schmid-Fetzer 2005 Thermodynamic Assessment of Mg-Al-Mn Phase Equilibira on Mg-Rich Alloys. Z. Metallkd. 96(8), 857–869.
M. S. Mirgalovskaya, L. N. Matkova and E. M. Komova 1957 The System Mg-Al-Mn. Trudy Inst. Met. Im. A.A. Baikova, Akad. Nauk 2, 139–148.
N. V. Ageev, I. I. Kornilov, A. N. Khlapova 1948 Magnesium-Rich Alloys of the System Magnesium-Aluminium-Manganese. Izv. Inst. Fiz.-Khim. Anal., Inst. Obshcheii Neorg. Khim., Akad. Nauk SSSR 14, 130–143.
W. G. Leemann and H. Hanemann 1938 The Ternary System Aluminium-Magnesium-Manganese. Aluminium Arch. 9, 6–17.
D. W. Wakeman and G. V. Raynor 1948 The Constitution of Aluminium-Manganese-Magnesium and Aluminium-Manganese-Silver Alloys, with Special Reference to Ternary Compound Formation. J. Inst. Met. 75, 131–150.
H. -K. Fun, H. -C. Lin, T. -J Lee, B. -C. Yipp 1994 T-Phase Al18Mg3Mn2. Acta Crystallogr. C50, 661–663.
J. G. Barlock and L. F. Mondolfo 1975 Structure of Some Aluminum-Iron-Magnesium-Manganese-Silicon Alloys. Z Metallkd 66(10), 605–611.
T. Ohnishi, Y. Nakatani and K. Shimizu 1973 Phase Diagrams and Ternary Compounds of the Al-Mg-Cr and the Al-Mg-Mn Systems in Al-Rich Side. Light Metals Tokyo 23, 202–209.
T. Ohnishi, Y. Nakatani and K. Shimizu 1973 Phase Diagram in the Al-Rich Side of the Al-Mg-Mn-Cr Quaternary System. Light Metals Tokyo , 23, 437–443.
E. Butchers, G. V. Raynor and W. Hume-Rothery 1943 The Constitution of Magnesium-Manganese-Zinc-Aluminium Alloys in the Range 0–5 % Magnesium, 0–2 % Manganese, 0–8 % Zinc, I-The Liquidus. J. Inst. Met. 69, 209–228.
A. T. Little, G. V. Raynor, W. Hume-Rothery 1943 The Constitution of Magnesium-Manganese - Zinc - Aluminium Alloys in the Range 0–5 % Magnesium, 0–2 % Manganese and 0–8 % Zinc, III-The 500 °C and 400 °C Isothermals. J Inst Met 69, 423–440.
Acknowledgments
Financial support from General Motors of Canada Ltd. and the Natural Sciences and Engineering Research Council of Canada through the CRD grants program is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Shukla, A., Pelton, A.D. Thermodynamic Assessment of the Al-Mn and Mg-Al-Mn Systems. J. Phase Equilib. Diffus. 30, 28–39 (2009). https://doi.org/10.1007/s11669-008-9426-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11669-008-9426-5