
Abstract
The elevated concentration of arsenic (As) in the groundwaters of many countries worldwide has received much attention during recent decades. This article presents an overview of the natural geochemical processes that mobilize As from aquifer sediments into groundwater and provides a concise description of the distribution of As in different global groundwater systems, with an emphasis on the highly vulnerable regions of Southeast Asia, the USA, Latin America, and Europe. Natural biogeochemical processes and anthropogenic activities may lead to the contamination of groundwaters by increased As concentrations. The primary source of As in groundwater is predominantly natural (geogenic) and mobilized through complex biogeochemical interactions within various aquifer solids and water. Sulfide minerals such as arsenopyrite and As-substituted pyrite, as well as other sulfide minerals, are susceptible to oxidation in the near-surface environment and quantitatively release significant quantities of As in the sediments. The geochemistry of As generally is a function of its multiple oxidation states, speciation, and redox transformation. The reductive dissolution of As-bearing Fe(III) oxides and sulfide oxidation are the most common and significant geochemical triggers that release As from aquifer sediments into groundwaters. The mobilization of As in groundwater is controlled by adsorption onto metal oxyhydroxides and clay minerals. According to recent estimates, more than 130 million people worldwide potentially are exposed to As in drinking water at levels above the World Health Organization’s (WHO's) guideline value of 10 μg/L. Hence, community education to strengthen public awareness, the involvement and capacity building of local stakeholders in targeting As-safe aquifers, and direct action and implementation of best practices in identifying safe groundwater sources for the installation of safe drinking water wells through action and enforcement by local governments and international water sector professionals are urgent necessities for sustainable As mitigation on a global scale.
Similar content being viewed by others

Introduction
Arsenic (As) is found naturally in the earth’s crust as the 20th most abundant element. It may exist in both soil and minerals and enter air, water, and surface environments through wind-blown dust and runoff [1–3]. The primary source of As is natural, derived mainly from the interactions between groundwater and aquifer sediments and hard rocks of minerals, particularly pyrite (FeS2), arsenopyrite (FeAsS), and other unspecified sulfide minerals [4–6], as well as secondary As-bearing minerals or mineral coatings, whereas industrial uses, agricultural activities, coal and oil exploitation and combustion, and mining of metal ores are anthropogenic sources that release As into groundwaters [7, 8]. As a result of such naturally occurring As and human-induced As concentrations, many groundwater systems greatly exceed the safe As guideline value of 10 μg/L recommended by the World Health Organization (WHO) [6, 9–13]. Thus, the elevated As concentration in groundwaters throughout the world has received much attention recently as a serious threat to public health as well as for agricultural activities [14–16].
More than 97 % of global freshwater belongs to the groundwater resources that are the main source of drinking water in many parts of the world because of its natural supply stability and good microbial quality [4, 17]. The natural release of As from river sediments under strong reducing conditions may produce elevated As levels in groundwaters [18, 19]. The naturally occurring As from geochemical processes in sedimentary aquifers has the greatest effect on the largest geographic areas. In regions where groundwater is used for drinking, many people are exposed to As contamination; the As found in the sedimentary aquifers of the Bengal Delta Plain in Bangladesh and neighboring West Bengal, India, is a good example of this type of contamination [20–22].
Naturally occurring As at exceptional concentrations has been found in potable groundwater systems of many countries, including Argentina, Chile, Mexico, China, Hungary, West Bengal (India), Bangladesh, Vietnam, and the USA. Arsenic pollution of groundwater and its deleterious effects and risks on human health have been reported in many countries [16, 23–30]. Large-scale contamination by natural As frequently appears in the groundwaters of the Bengal Basin, which are the source of drinking water for more than 40 million people [21, 26, 31–33]. Moreover, more than 10 million residents of Southeast Asia are estimated to be at risk from drinking As-contaminated groundwaters [17, 34]. Although As has not yet been observed to exist in Thailand groundwater systems, metal mining activities have resulted in significant As contamination in the surrounding environment. Analyses of surface and groundwaters in Thailand found that the concentrations of dissolved As exceed the safe threshold for human health by up to a factor of 500 [35, 36]. These high As concentrations in groundwaters pose significant risks to human health, including cancers of the skin and internal organs [15, 16, 24, 28].
A variety of natural geochemical processes play a vital role in the release, transport, and distribution of As in groundwater. The mobilization of As from the solid and/or semisolid phases to the aqueous phase is controlled by several geochemical processes, including interfacial reactions such as adsorption/desorption, ion exchange, dissolution, and oxidation/reduction [5, 11, 37]. The release of As from iron oxide deposited in aquifer sediments is thought to be the most common cause of As-rich groundwaters. Iron oxide associated with felsic volcanic rocks and alkaline aquifers has been identified as a key source of As being released into the alkaline groundwater of the western USA [38, 39]. Sulfide minerals also may act as a source of as well as a sink for As [40]. The mobility and transformation of As in groundwaters are controlled in large part by adsorption onto metal oxyhydroxides and clay minerals, depending on the interrelationships among the properties of the solid surface, pH, concentration of As and competing ions, and As speciation [10].
The main goal of this review is to provide a concise description of important naturally occurring geochemical processes involved in the mobilization and distribution of As throughout global groundwater systems. The article also provides an overview of inorganic As with an emphasis on (1) natural geochemical processes, (2) release and transport mechanisms, and (3) global distribution.
Occurrence of Arsenic in Nature
Arsenic commonly occurs in nature: in air, soil, water, rocks, plants, and animals. Natural activities such as weathering, volcanic eruption, rock erosion, and forest fires may release As into the environment. Weathering is the main naturally occurring process through which arsenic sulfides present in ores are converted into a highly mobile form of As [As(V)], which may enter the As cycle directly as a dust or via dissolution in rain, water reservoirs, or groundwater [6, 41]. As(III) also may move widely among plants and animals through the food chain. This widespread distribution of As throughout the earth’s crust, soil, sediments, water, air, and living organisms leads to serious adverse effects on the environment. The occurrence of As in certain mineral phases, soil, and water systems is linked directly to the wide variety of As(III) and As(V) species in groundwaters.
Occurrence of Arsenic in Minerals
Generally, As is a ubiquitous element in the earth’s crust, found in igneous and sedimentary rocks at an average concentration of 2 to 3 mg/kg, whereas it is present in higher levels in finer-grained argillaceous sediments, phosphorites, and reduced marine sediments [18, 41, 42]. Although As occurs in a variety of natural reservoirs, such as oceans, rocks, the atmosphere, biota, and soils, more than 99 % of As is associated with rocks and minerals [42]. It is estimated that more than 200 minerals are rich in As, 60 % as arsenates, 20 % as sulfides and sulfosalts, and the remaining 20 % as arsenides, arsenites, oxides, silicates, and elemental As. Arsenic may be coprecipitated with iron hydroxides [Fe(OH)3] and sulfides in sedimentary rocks. Iron deposits, sedimentary iron ores, and manganese nodules are considered As-rich minerals [41]. Important minerals are rich in As, including arsenolite (As2O3), olivenite (Cu2OHAsO4), cobaltite (CoAsS), proustite (Ag3AsS3), enargite (Cu3AsS4), orpiment (As2S3), realgar (As4S4), and tennantite (Cu6[Cu4(Fe,Zn)2]As4S13), and FeAsS is the most abundant As-bearing mineral species found in nature [31, 43].
Occurrence of Arsenic in Soil
The potential risk of surface and groundwater contamination by As is associated mainly with the amounts of As present in soils, because As is more highly concentrated in soils than rock [8, 33, 44]. Arsenic may be found in soil as inorganic species as well as binding to organic compounds; however, it exists predominantly as inorganic species in soil. Inorganic As compounds may undergo methylation by microorganisms, producing, under oxidizing conditions, monomethylarsonic acid, dimethylarsinic acid, and trimethylarsine oxide [2, 45, 46]. Principle factors affecting the level of As in soils include the parent rock, climate, organic and inorganic components of the soil, and redox potential status [41, 47]. Arsenate [As(V)] is the most stable form of As in soil under oxidizing conditions, and it can adsorb strongly into clays, iron and manganese oxides/hydroxides, and organic matters found in soil; however, in the presence of reducing conditions, As(III) is dominant. Arsenates of Fe and Al (AlAsO4 and FeAsO4) are the dominant phases in acid soils and are less soluble than calcium arsenate (Ca3AsO4), which is the main chemical form in alkaline and calcareous soils [41, 48]. Permissible levels of As in soils for many countries range from 0.1 to 40 mg/kg, with an average concentration of 3 to 4 mg/kg, and uncontaminated soils usually contain As at a level of 1 to 40 mg/kg [48]. However, alluvial and organic soils show much higher As concentrations, exceeding the recommended levels [41, 49]. The sorption of As in soils is influenced mainly by the bioavailability and subsequent mobility of As in soils, and the presence of inorganic PO4 3−, Ca2+, and Na+ in different types of soil, including Oxisol, Vertisol, and Alfisol, has a significant impact on the sorption of As(III) and As(V) [49–51].
Occurrence of Arsenic in Water
The occurrence of As in natural water bodies has received significant attention in recent decades. In natural waters, As may be found in low concentrations. The maximum permissible concentration of As in drinking water, which is still in force in a few countries, is 50 μg/L, and the regulatory value generally is 10 μg/L, corresponding to the WHO guidelines. In aqueous environments, inorganic As species, including As(III) and As(V), also are the most abundant species. The mobility of these species is controlled by pH, redox potential, dissolved organic matter, and the presence of adsorbents such as oxides and hydroxides of Fe(III), Al(III), Mn(III/IV), humic substances, and clay minerals [45, 52]. The presence of carbonate and bicarbonate ions in aquifer sediments directly results in a change in pH [53]. Carbonate ions can form complexes on the surface sites of iron hydroxide and substitute As from the surface of minerals and sediments, thereby releasing As into groundwater [51, 54]. The aquifers associated with As usually are linked to the input of dissolved organic matter, which also can greatly facilitate the release of As from soils and sediments to groundwaters [50, 55]. In marine ecosystems, complex organic As compounds, such as tetramethylarsonium salts, arsenocholine, arsenobetaine, dimethyl(ribosyl)arsine oxides, and As-containing lipids, have been identified. It is estimated that seawater typically contains 1 to 8 μg/L of As [2]. Only a very limited amount of the total As present in the oceans remains in solution, because a large portion of dissolved As can be adsorbed onto suspended particulate materials [41, 56].
Arsenic also may exist naturally in geothermal systems and springs as a result of the mobilization of As from rocks and mineral phases, and As-contaminated geothermal systems and springs are recognized in many areas around the world (Table 1) [3, 8, 10, 91]. The mobilization of As from rocks into geothermal fluids takes place mainly through active tectonic plate boundaries [91]. Arsenic from major geothermal reservoirs and related geothermal manifestations, such as hot springs, fumaroles, and solfataras, may directly enter cold aquifers, the vadose zone, and surface waters [8, 10]. Many volcanoes, hot springs, fumaroles, and geothermal wells may be found in the Pacific region of Latin America, and these geothermal systems are widely contaminated by As at elevated concentrations [91]. The concentration of As in the Los Humeros geothermal field in Mexico is reported to be as high as 73.6 mg/L, and the attenuation of As in the ground surface may occur via sorption and coprecipitation with iron minerals and organic matter present in sediments [91]. In deeper geothermal reservoirs, As is released predominantly from As-bearing FeS2 at temperatures in the range of 150 to 250 °C and from FeAsS at higher temperatures [10].
Natural Geochemical Processes of Arsenic
The presence of naturally occurring As in shallow reducing aquifer sediments has become a major environmental issue for millions of people worldwide because of the use of As-contaminated groundwater for drinking as well as for irrigation activities. The contamination of groundwater by naturally occurring As is much more serious; therefore, it is urgently necessary to understand and investigate the major geochemical pathways involved in the transformation and mobilization of As in aquifer sediments. The geochemistry of As is a complex phenomenon found in the environment, and it generally is a function of multiple oxidation states, speciation, and redox transformation.
Speciation of Arsenic
Eh–pH diagrams (also known as Pourbaix diagrams) conventionally are used to represent and describe the speciation of As and its environmentally significant geochemical processes [5, 46]. Figure 1 shows the Eh–pH diagram of aqueous As species in water at 25 °C. Arsenic may exist with valence states of −3, 0, +3, and +5; however, the −3 and 0 valence states occur only rarely in nature [92]. Arsenic chemistry predominantly focuses on As(V) or As(III), depending on redox potential and pH in groundwaters [93]. Both As(III) and As(V) may form protonated oxyanions in aqueous solutions [92]. The degree of protonation depends mainly on the pH of the medium. Under oxidizing conditions, As(V) can hydrolyze to four possible species depending on the range of pH encountered in surface and groundwater systems. In oxygenated water systems, arsenious acid (H3AsO4) is predominant only at extremely low pHs (<2), whereas in the range of 2 to 11 pH, As exists in the form of H2AsO4 − and HAsO4 2−. In reducing environments and at low pH, arsenious acid can convert into H2AsO3 −, and if the pH value exceeds 12, it occurs in the form of HAsO3 2− (Fig. 1) [5, 92, 94]. In the pH range of 4 to 10, As(V) species is negatively charged in water, whereas the predominant As(III) species is neutral in charge because of redox potential and the presence of complexing ions such as sulfur, iron, and calcium and microbial activity [95]. However, fully dissociated As(V) is found rarely, because very few water systems reach a pH greater than 11.5 [96]. Arsenic in its native form is rare in the environment. In reducing environments, completely protonated arsenite species becomes more predominant over a wide range of pHs without being ionized; hence, arsenite cannot adsorb as strongly as arsenate species [6].

Eh–pH diagram illustrating the stability fields of the oxidized and reduced species of As in water at 25 °C [77]
Mobilization and Redox Transformation of Arsenic
Natural geochemical and biologic processes play a vital role in controlling the mobilization and transformation of As in the surface and subsurface environment [8, 37, 40]. In groundwater, mobilization of As takes place in the range of pH 6.5 to 8.5 under both oxidizing and reducing conditions [17]. Both abiotic and biotic processes favor the natural redox transformation and mobilization of As in humid as well as arid environments. Figure 2 illustrates the process of mobilization and redox transformation of As in aquifer sediments.

Illustration of the mechanisms of mobilization and redox transformation of As in aquifer sediments
Redox Transformation of Arsenic
Under reducing conditions in natural environments, deltaic and alluvial sediments usually are associated with the mobilization of As [17, 93]. Aquifer sediments, along with a layer of clay or silt, may act as a cap, which effectively restricts the penetration of atmospheric oxygen to the aquifers, thereby creating an anaerobic environment. Highly reducing conditions (anaerobic) also may be developed in the presence of natural organic matter deposited with sediments. These highly reducing conditions facilitate the release of As adsorbed on amorphous Fe oxides commonly occurring in the aquifer sediments. Reductive dissolution of As-bearing amorphous Fe(III) oxides plays a key role in the mobilization of As from aquifer sediments to groundwaters. Dissolved organic carbon (DOC) deposited in sediments and anaerobic metal-reducing bacteria such as Shewanella algae, Shewanella oneidensis, and Geobacter metallireducens also may play a vital role in As mobilization [38, 97]. The existence of As in groundwaters to a large extent depends on several factors, including areal and vertical distributions of peat deposits, the degradation of which is the major redox controller; the redox driver in the groundwater system; groundwater movement; pH; HCO3 −; Fe, Mn, and Al oxides; and DOC concentrations of sediments [17]. Therefore, it is clear that reducing reactions under anaerobic conditions in aquifer sediments undoubtedly may result in As concentrations in groundwaters as high as several hundred micrograms per liter [98].
Oxidation of Arsenite
The form of As(III) is thermodynamically unstable in aerobic conditions; hence, it oxidizes easily to the less mobile form of As(V). However, this reaction is a slow process, when oxygen becomes the only oxidant. The presence of some other redox-sensitive species, such as ferric iron [Fe(III)], manganese oxides (MnO2), clay minerals, and some microorganisms, can intensively increase the rate of As(III) oxidation converting into the less toxic As(V) form. These oxidation reactions are highly favorable in arid and semiarid areas as a result of extensive mineral reactions and evaporation. In the presence of Fe(III) in aqueous solutions, the rate of As(III) oxidation is accelerated below pH 7. Manganese oxides commonly associated with aquifer solids have been recognized as an important oxidant to oxidize As(III) to As(V), and this reaction is thermodynamically feasible over a wide range of pH values [99]. Reactions of As(III) and As(V) with synthetic birnessite (MnO2) have been studied in which As(III) is oxidized by MnO2, followed by the adsorption of the As(V) reaction product on the MnO2 solid phase [100]. Clay minerals also can oxidize As(III). Three types of clay minerals have been used successfully to oxidize As(III), suggesting that the oxidation and adsorption of produced As(V) occur on the surface of clay minerals [101]. Moreover, the oxidation of trivalent As is found to be catalyzed by some microorganisms, including Pseudomonas arsenitoxidans, Alcaligenes faecalis, Cenibacterium arsenoxidans, Thermus sp., Thermus thermophilus, and Agrobacterium tumefaciens, using oxygen as the terminal electron acceptor [31, 94, 102, 103]. Bacteria species attached to submerged macrophytes in a stream near geothermal sources are reported to be capable of mediating the rapid oxidation of As(III) [104], and the oxidation reaction may be expressed as follows [95]:
Mechanisms of Arsenic Release to Groundwater
The As release mechanisms in aquifer rock or sediment obviously are linked to the extent of As contamination in groundwater systems. Public health risks usually are associated with the consumption of ground and surface water with As concentrations above WHO standard levels (As >10 μg/L). In general, the average concentration of As in the continental crust is substantially low, and its concentration varies between 1 and 2 mg/kg [105]; consequently, only low levels are expected in groundwater. However, under critical geochemical environments, processes including adsorption/desorption, oxidation/reduction, and oxidation of As-bearing sulfide minerals may release high concentrations of As in groundwater [8, 18]. Hence, it is these geochemical triggers, more than the As content in aquifer media, that create the risk of drinking water contamination with elevated concentrations of As. Four major natural geochemical processes initiate the natural release of As from aquifer materials into groundwater: (i) reductive dissolution, (ii) alkali desorption, (iii) sulfide oxidation, and (iv) geothermal processes (Fig. 3).

Natural geochemical processes that release As into groundwater in different parts of the world
Arsenic release processes may occur in a variety of geochemical environments; however, the most serious As occurrences are located in the young alluvial basins adjacent to active mountain belts. Therefore, many people living in or near these areas have been greatly affected by As-contaminated groundwaters [106]. All these processes, except sulfide oxidation, are virtually independent from human-induced activities in the subsurface environment. However, the pumping of groundwater, particularly by using deep tube wells, leads to widespread distribution of dissolved As in the subsurface [107]. The reductive dissolution of As-bearing Fe(III) oxides and sulfide oxidation in aquifer sediments are thought to be the most significant geochemical triggers that release As into groundwaters to a large extent [50].
Reductive Dissolution of Iron Oxide
Iron hydroxides Fe(OH)3 are one of the most common phases associated with aquifer sediments. The desorption mechanisms of As from Fe(OH)3 are directly responsible for the existing high concentrations of mobile As(III) in groundwaters [40, 106, 108]. Biologically mediated reactions in the presence of organic carbon generated from sedimentary organic matter and anthropogenic organic compounds are capable of releasing As from Fe(OH)3, a process referred to as dissimilatory iron reduction [37]. The dissimilatory iron-reducing bacterium S. algae promotes As(V) release from the crystalline mineral scorodite (FeAsO4·2H2O) via dissimilatory reduction of Fe(III) to Fe(II).
The primary mechanism for As release from soil and sediments is the competition between As and organic anions present at sorption sites, whereas redox reactions likely play a minor role [50]. In aqueous medium, the reaction of Fe(OH)3 is stimulatory, depending on the pH; at high pH values, desorption processes become stronger, leading to high levels of As in groundwaters [37]. Sulfidization of ferrihydrite [Fe(III)OAs(OH)2] is the major process by which As mobilization takes place via reductive dissolution [109]. Sulfides, particularly H2S, accumulated in sediments can reductively dissolve As from Fe(III)OAs(OH)2, releasing As(III) and thereby increasing the dissolved As concentration in groundwaters [71]. High concentrations of aqueous Fe(II) and the presence of more ordered phases, such as hematite, may result in chemical conditions in which sulfate reduction is thermodynamically favorable. Moreover, sulfide may be transported or diffused from zones of sulfate reduction into Fe(OH)3-rich environments (Reaction 2). Hence, As bound to Fe(OH)3 may be subject to transformation upon reaction with dissolved sulfide:
Sulfide then may eliminate sorbed As(III) from the surface of Fe(III)OAs(OH)2 via a ligand exchange reaction (Reaction 3), or As(III) may be mobilized via the reductive dissolution of Fe(III)OAs(OH)2 by dissolved sulfide (Reaction 4) [110]:
Furthermore, phosphate ions present in water also may react with As-bearing Fe(III) minerals, resulting in much greater As concentrations in groundwaters [111]. Application of some phosphate-containing fertilizers is the major cause of high As concentrations in groundwaters in these areas.
Oxidation of Sulfide Minerals
The primary source of As in the environment is the oxidation of As sulfides, such as FeAsS and FeS2 minerals. Oxidation of As-bearing sulfides is recognized as an important cause of As contamination of groundwater due to production of acid drainage containing toxic inorganic pollutants at higher levels. In oxygen-rich waters, As is released predominantly through pyrite and FeAsS minerals. Arsenopyrite is the most common As-bearing mineral, and FeS2 is found commonly in gangue sulfide minerals associated with ore deposits, which have a serious impact on mine drainage [1, 37, 71].
Arsenic Release from Pyrite
FeS2 oxidation takes place via several reaction pathways; the first step involves the chemical oxidation of FeS2 in the presence of dissolved oxygen (DO), resulting in a certain amount of As in groundwaters. This reaction may be expressed as follows [37]:
The second possible pathway is the oxidation of FeS2 by aqueous nitrate at pH >5 [20, 37]. In groundwater supplies affected by nitrogen as the result of human-induced activities, nitrates may act as an oxidant for the oxidation of sulfide minerals deposited in aquifer sediments (Reaction 6):
The Fe(III) ions that have accumulated in aquifers also can oxidize FeS2 (Reaction 7), and Fe(II) produced in this reaction may be oxidized further to Fe(III) (Reaction 8):
Reaction 8 is a slow process under abiotic conditions; nevertheless, extremophilic bacteria such as Acidithiobacillus ferrooxidans and Leptospirillum ferrooxidans can concentrate Fe(III) ions by complexation through uronic acids or other residues present at the mineral surface, allowing an oxidative attack on the sulfide and thereby enhancing the dissolution of the metal sulfide [112]. The oxidation of Fe(II) to Fe(III) and As adsorption on Fe(OH)3 also depend on the DOC content and pH in groundwater [113]. In water with neutral pH, most Fe(III) can exist in the solid or colloidal phase, which leads to the release and transport of As in drinking water aquifers. At near-neutral pH, dissolved Fe(III) would not be present at high concentrations unless it were complexed with DOC and, consequently, would be a less important oxidant than DO [5]. Thus, it is suggested that when Fe(OH)3 is present as a coating on aquifer materials, As may be immobilized via adsorption, whereas if it is present as colloidal-size Fe(OH)3, As may undergo colloidal transport.
Arsenic Release from Arsenopyrite
Dissolution of FeAsS also is of environmental concern and therefore has received much attention in recent years. FeAsS can be oxidized by Fe(III), a process more than 10 times faster than oxidation of FeS2 [114, 115]. This oxidation reaction is catalyzed in the presence of microorganisms, including T. ferrooxidans, and its role in releasing As from FeAsS present in leachate has been documented [114]. The first step in this process involves a slow but strong adhesion of bacteria onto mineral surfaces; the appearance of a surface phase of elemental sulfur; the weak solubilization of Fe(II), As(III), and As(V); and the presence of the first corrosion patterns, which follow the fragility zones and the crystallographic orientation of mineral grains [114, 115]. After this short step, growth of the unattached bacteria begins, while ferrous ions in solution are oxidized by them. The Fe(III) ions produced by the bacteria can oxidize the sulfide directly and are then regenerated by Fe(II) bacterial oxidation. At that point, a bioleaching cycle occurs and a coarse surface phase of ferric arsenate (FeAsO4·2H2O) and deep ovoid pores appear. At the end of the bioleaching cycle, the high concentration of Fe(III) and As(V) in the solution promotes the precipitation of a second phase of amorphous FeAsO4·4H2O in the leachate. Then, the bio-oxidation process tends to cease. The bacteria attached to the mineral surfaces are coated by the ferric arsenates, and the concentration of Fe(III) on the leachate tends to decrease greatly [107, 114].
The release of As from FeAsS is a highly pH-dependent process, and the dissolution of As is favorable in the pH range of 1.8 to 6.4, whereas it is reduced to a minimum in the range of 7 to 8. This may be the result of oxidation of Fe2+ to hydrous ferric oxide with attenuation of dissolved As onto the freshly precipitated hydrous ferric oxide, which can strongly adsorb both As(V) and As(III) species [107, 115]. Thus, in the presence of excess hydrous ferric oxide, reductive dissolution of iron may be the dominant mechanism by which As is released into the solution. Dissolved As(III) may be reversed into a solid phase by adsorption of As(III), by forming an iron precipitate: Fe(OH)3. Under high-pH conditions, this Fe(OH)3 is the most dominant species and can adsorb or coprecipitate the As. The overall reaction of oxidation may be expressed as follows [37]:
Both As and sulfate may be released from FeAsS in an approximate 1:1 molar ratio, suggesting that the oxidative dissolution by oxygen under circumneutral pH (6.3–6.7) is congruent [116]. The reduction of As(V) increases its solubility at circumneutral pH. The rate of FeAsS oxidation reaction was determined to be 10−10.14 ± 0.03 mol m−2 s−1, which essentially is independent of DO over the geologically significant range of 0.3 to 17 mg/L. Kinetics of the FeAsS oxidation process indicate that the rate-determining step in FeAsS oxidation is the reduction of water at the anodic site rather than the transfer of electrons from the cathodic site to oxygen, as observed in other sulfide minerals, such as FeS2 [116].
Geochemical Processes in Controlling Arsenic Mobility and Transformation
Adsorption is the most prominent geochemical process that controls the mobility and transport of As in groundwaters. Adsorption reactions between As and mineral surfaces generally are the most critical phenomena in controlling the dissolved concentrations of As in groundwaters. Adsorption of As depends on several external factors, such as solid surface, pH, Eh, concentration of As and competing ions, and As speciation [5]. Redox-active oxide surfaces of iron, aluminum, and manganese minerals are potentially the most important sources of As in aquifer sediments [107]. The adsorption properties of these oxides generally are a result of the presence of surface functional groups containing OH groups. When metal ions on the oxide surface are exposed to bound water, they can complete their coordination shells with OH groups, and depending on pH, these OH groups may bind or release, developing a surface charge that often is associated with adsorption properties governing a particular adsorption mechanism [6, 39]. It is reported that As(V) adsorption takes place in the net release of OH− at a pH range of 4.6 to 9.2, whereas As(III) adsorption results in net OH− release at pH 9.2 and net H+ release at pH 4.6 [2, 39].
Arsenic Adsorption on Iron Oxides
The solubility of As in groundwaters is strongly influenced by adsorption at the Fe(OH)3 surfaces, which exist as discrete particles or as coatings on other mineral surfaces. Both As(V) and As(III) may be adsorbed and coprecipitated on Fe(OH)3, depending on several important factors, including pH, the amount of Fe(OH)3 deposited, and the concentration of other competing ions found in the medium. The adsorption of As onto Fe(OH)3 occurs mainly at the oxic/anoxic boundary, referred to as ferrihydrite (Fe2O3·2H2O), which has a large specific area resulting in an increased adsorption capacity. Ferrihydrite plays a significant role in controlling dissolved As concentrations and limits the mobility and bioavailability of As(III) and As(V). Adsorption occurs via ligand exchange of the As species for OH2 and OH− in the coordination spheres of the surface structural Fe atoms [39, 117].
One of the most important key parameters that may affect the adsorption of As(III) and As(V) is pH. The greatest adsorption of As(V) usually occurs at low pH values (pH 3–6) and decreases with increasing pH (pH >6) [118, 119]. Greater adsorption of As(V) at low pH is attributable to more favorable adsorption energies between the more positively charged surface and negatively charged H2AsO4 −, which is the predominant As(V) species between pH 2 and 6. Because HAsO4 − becomes the predominant aqueous species at pH >6, the surface charge tends to be less positive; therefore, causing adsorption is less favorable. However, even at pH 10, some surface functional groups can exchange As(V) [5]. The kinetics of As(V) adsorption by ferrihydrite was investigated at a pH range of 7.5 to 9.0 [120]. The study revealed that a period of rapid (<5 min) As(V) adsorption from solution is followed by continued adsorption for at least 8 days as As(V) diffuses to adsorption sites on ferrihydrite surfaces within aggregates of colloidal particles [120]. The adsorption of As(III) with the ferrihydrite is a fast process compared with that of As(V) [119, 121].
Arsenic Adsorption on Aluminum Oxides
Oxides and hydroxides of Al also have significant adsorption capacity for As. Because the Al(III) atom has the same charge and a nearly identical radius as the Fe(III) atom, the common hydrous Al oxide phases are structurally similar to hydrous ferric oxide [5]. As(V) species such as CH3AsO(OH)2 and (CH3)2AsOOH can be absorbed by amorphous Al(OH)3, crystalline Al(OH)3 (gibbsite), α-Al2O3, and β-Al2O3 up to 7.0 pH, and adsorption decreases significantly at higher pH values, whereas As(III) adsorption increases with increasing pH. The adsorption of As(III) onto α-Al2O3 increases with increasing pH from 3 to 4.5, whereas adsorption decreases with increases between pH 4.5 and 9.0 [122]. However, As(V) adsorption decreases with increasing pH from 3.5 to 10. The As(III) inner- and outer-sphere adsorption coexists, whereas for As(V), inner-sphere complexes are predominant [123]. Chemical models also confirmed that arsenate adsorption occurs over a pH range of 4.5 to 9 on gibbsite and amorphous Al hydroxide [51, 118]. The degree of crystallinity of Al oxides and hydroxides also is important for As adsorption. Under comparable experimental conditions, x-ray amorphous material tends to absorb more As(V) per gram than crystalline and gibbsite [5, 124].
Arsenic Adsorption on Manganese Oxide
The oxidation of As(III) to As(V) by manganese oxide is an important process in the natural cycling of As, as well as in reducing the concentration of dissolved As(III) in groundwater systems. As(III) is oxidized by MnO2 followed by the adsorption of the As(V) reaction product on the MnO2 solid phase. The As(V)–MnO2 complex likely is a bidentate binuclear corner-sharing complex occurring at MnO2 crystallite edges and interlayer domains [100, 125]. Manganese oxides may act as an electron acceptor in the oxidation process and therefore can adsorb significant amounts of As. The birnessite groups are the most common Mn oxides derived from weathering processes, and they possess a net negative surface charge at pH values common in groundwater [5, 126]. As(III) oxidation occurs directly via a surface mechanism, with no effect on DO concentration, and the oxidation product, As(V), is adsorbed almost as quickly, whereas release of the reduction product, Mn(II), is slightly slower [20]. Reductive dissolution of synthetic birnessite by arsenious acid (H3AsO3) proceeds in two steps [127]. The first step is associated with the reduction of Mn(IV) to Mn(III), producing an intermediate reaction product, MnOOH* (Reaction 10), which is a combination of Mn(III), hydroxyl, and H2O, according to x-ray photoelectron spectroscopic data:
In the second step, MnOOH* may react with H3AsO3 to release Mn(II) to the solution (Reaction 11):
This oxidation process transforms a toxic species of As [As(III)] to a less toxic aqueous species, which further precipitates with Mn2+ as a mixed As–Mn solid characterized by a low-solubility product. The ion activity product of Mn(II) and As(V) is the solubility product of a protonated manganese arsenate, and this precipitation reaction may be expressed as follows. The manganese arsenate precipitate formed in the reaction may aggregate and adsorb on the edges of H-birnessite layers (Reaction 12) [128]:
The oxidation of As(III) and adsorption of As(V) may be catalyzed by specific microorganisms present in the medium. The oxidation of As(III) and adsorption of As(III/V) may be induced by biogenic manganese oxide formed by a manganese oxide-depositing fungus [6, 129]. Biogenic manganese oxide with a low proportion of Mn(II) may increase the oxidation rates of As(III) significantly, resulting in great adsorption capacity; hence, this study provides insights into the applicability of Mn-depositing microorganisms in the biologic treatment of As-contaminated waters.
Arsenic Adsorption on Clay Minerals
Adsorption and oxidation/reduction reactions of As at clay mineral surfaces also play a crucial role in the natural attenuation and transformation of As in groundwater systems. Aluminosilicate clay minerals are composed of alternating layers of silica oxide and aluminum oxide, providing several types of binding sites to adsorb a variety of metal ions [130]. The OH groups associated with Al ions bound on the surface of clay particles act as proton acceptors, forming anionic species of As. Three types of clay minerals have been used successfully to oxidize As(III), suggesting that the oxidation and adsorption of As(V) product take place on the surface of clay minerals and that the oxidation rate and adsorption capacity are affected by the type of clay mineral [101]. Adsorption of As(III) on several clay minerals, such as kaolinite, illite, montmorillonite, and amorphous aluminum hydroxide [am-Al(OH)3], is a function of pH as well as ionic strength, and the oxidation of As(III) to As(V) is enhanced by heterogeneous oxidation on kaolinite and illite surfaces [131, 132]. Maximum adsorption of As(V) by kaolinite, montmorillonite, illite, halloysite, and chlorite may occur up to pH values near 7 and decrease with further increases in pH. The adsorption process also may be affected by competing anions, such as phosphate (P), chromate, sulfate, and molybdate, present in the medium. Maximum adsorption of arsenate on kaolinite, montmorillonite, and illite occurs at pH 5, 6, and 6.5, respectively, and the adsorption rate decreases in the presence of competing anions, such as phosphates [133, 134]. Surface area is another important key factor affecting the adsorption of As by clay minerals. It is reported that montmorillonite can adsorb approximately twice as much As(III) and As(V) as kaolinite, because the surface area of montmorillonite is just over two times greater than that of kaolinite [135].
Worldwide Distribution of Arsenic
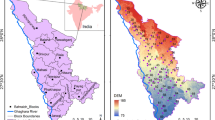
The occurrence and distribution of elevated As concentrations in groundwaters have received much attention in the recent decade, because most groundwater systems are considered an essential source of drinking and irrigation water in rural areas throughout the world. Concentrations of naturally occurring As in groundwater systems in different parts of the world may fluctuate according to climate and geologic conditions. During the past decade, As-rich groundwaters have appeared to be developing at an alarming frequency, and contamination of groundwaters with geogenic As poses a major health risk for many people worldwide. The occurrence and distribution of As globally have been predicted by using a variety of model parameters. Thus far, more than 70 countries with a population of more than 140 million have been affected by As-contaminated groundwaters. Hence, understanding the global occurrence and distribution of As in groundwaters would pave the way to controlling and mitigating the serious problems associated with As pollution. Figure 4 and Table 1 demonstrate highly vulnerable regions with groundwaters containing As at elevated concentrations distributed worldwide.

Global occurrence of arsenic in groundwater and the most vulnerable regions/countries of the world
Southeast Asia
The occurrence and distribution of high concentrations of naturally occurring As in the groundwaters of the Southeast Asia region, particularly in the densely populated river deltas, have received much attention in recent years [22, 136]. Countries of particular concern in this region include Bangladesh, West Bengal (India), China, Vietnam, Taiwan, Thailand, and Nepal because of widespread problems associated with As within their populations [1, 13, 137, 138]. Arsenic found in the Ganges–Meghna–Brahmaputra basin in southern Asia is an important example of natural contamination. In Southeast Asia, groundwater is a significant source of daily inorganic As intake in humans. It is estimated that more than 100 million people in this region are at risk from groundwater As contamination, and so far, more than 700,000 people have been affected by As-related diseases [17]. Most groundwaters used for drinking water in rural areas of this region have been found to be contaminated with As at levels exceeding the WHO drinking water guideline of 10 μg/L [21, 22].
Bangladesh and West Bengal (India)
The original source of groundwater As in the Ganges–Meghna–Brahmaputra basin (India and Bangladesh) may have been the formation of the Himalayan orogenic belt by deep-seated tectono-magmatism resulting in the introduction of As to the surficial system by unearthing, after which it was transported by sedimentary processes to the peripheral foreland basin, where solid-phase As is released to the circulating groundwater by biogeochemical processes [139]. The ingestion of contaminated As (directly and/or indirectly through the food chain; i.e., arsenite and arsenate) was reported to induce carcinogenesis and cardiovascular disorders in Bangladesh and West Bengal, India [23–26] (http://www.sos-arsenic.net/). In Bangladesh, at least 25 million to 40 million people recently were exposed to As concentrations above the national limit of 50 μg/L [14]. It is estimated that more than 5.6 million people currently are consuming drinking water containing more than 200 μg/L of As, a level more than 20-fold greater than the WHO-recommended value [14]. Shallow hand tube wells are the main source of drinking water in many rural areas of Bangladesh. An analysis of 521 groundwater samples collected from 144 wells from 2009 to 2011 found that high As concentrations in groundwater correspond to black sediments with an average concentration of 239 μg/L, which by far exceeds the safe drinking water standard of Bangladesh (50 μg/L) [58]. However, average As concentrations in 39 tube wells in which red sediments were observed were lower than the WHO guideline value of 10 μg/L, implying safe water with regard to the Bangladesh drinking water standard for As (50 μg/L) [58, 140]. Consuming high levels of As indeed is the main cause of the arsenicosis symptoms reported among at least 40,000 people living in the most vulnerable locations of Bangladesh. Moreover, this study identified 492 As-affected villages in 141 police stations of 42 As-affected districts in Bangladesh. Among the water samples collected, about 59 % contained As levels greater 50 μg/L, with 93 % of those samples, on average, containing As in concentrations above the toxic level. Six districts of West Bengal, covering an area of 34,000 km2 with a population of 30 million, have As groundwater concentrations exceeding the permissible limit [141]. At present, 37 areas of these six districts near the Ganges River are highly affected, and more than 800,000 people from 312 villages use As-contaminated water for drinking; approximately 175,000 of them have developed arsenical skin lesions [141, 142].
Groundwater is used extensively for drinking in Bangladesh, and about 6 million to 11 million tube wells currently are in use, about a quarter of which have As at concentrations exceeding the national drinking water standard (50 μg/ L) [143]. Recent reports estimate that as many as 35 million people are drinking As-contaminated groundwater in Bangladesh. Southeast Bangladesh is recognized as the region most affected; here, the sediments are of the Holocene age and As concentrations in groundwater frequently exceed 200 μg/L [143]. In Bangladesh, As is predominantly of natural origin, and problems may arise with even minute quantities. The mobilization and transfer of As in groundwater systems do not occur via the oxidation of FeS2, as some articles suggest [108, 143]. However, the aquifers overlain by finer sediments contain greater concentrations of As in groundwater, whereas As concentrations are remarkably low in aquifers with permeable sandy materials or a thinner silt/clay layer at the surface. This near-surface lithology controls spatial distributions of groundwater As within very shallow depths (<15 m). Shallow alluvial aquifers may provide drinking water with low As concentrations in many areas of Bangladesh [143].
The high As concentrations in Bangladesh groundwaters may be the result of a combination of factors, such as (i) younger Holocene sediments, which can undergo rapid change from an oxidizing to a reducing environment following sediment burial; (ii) the desorption and dissolution of As from iron oxides found in sediments; and (iii) the very low hydraulic gradients throughout much of Bangladesh, meaning that groundwater flow is very slow, which combined with the “young” age of many of the sediments, reduces the natural flushing of the shallow aquifer, allowing any mobilized As to accumulate in the groundwater [14]. The high rate of sediment deposition in Bangladesh and the Indian part of the Bengal Basin suggests that the chance of a well receiving As-rich water likely is relatively high compared with smaller deltas and other alluvial environments where the sedimentation rate is much lower [143].
High levels of As concentrations (>10 μg/L, the WHO-recommended guideline) have been reported in the groundwaters of 14 (of 19) districts in West Bengal, including Kolkata metropolitan city (India) [144]. More than 9.2 million inhabitants in West Bengal are drinking As-contaminated groundwater (>10 μg/L) [144]. According to a previous report, in the combined regions of West Bengal (India) and Bangladesh, about 50 million inhabitants are potentially at risk from As-enriched groundwater [4, 12, 59, 60, 104, 144–148]. In North 24 Parganas district (India), one of the worst As-affected districts in West Bengal (India), 50 % of the tube wells are contaminated with As concentrations greater than the WHO limit (10 μg/L) and 2.0 million inhabitants (one third of the total population of the district) are exposed to As-contaminated drinking groundwater [60].
The Moyna and Ardebok villages (peri-urban villages of North 24 Parganas) are severely contaminated with groundwater As [15]. A recent report indicates that of the 2467 residents of Moyna and Ardebok villages, 17.3 % are using safe drinking water (>10 μg/L of As), whereas 57.7 % are using As-contaminated (>50 μg/L) drinking water [15]. The district of Nadia is another region in West Bengal (India) where groundwater is highly contaminated with As [15, 59, 60]. In 2014, Rahman et al. [60] reported that 51.4 and 17.3 % of the tube wells had As concentrations above 10 and 50 μg/L, respectively. In this article, the authors also reported that the groundwater of all 17 blocks of Nadia shows As concentrations above 50 μg/L (maximum, 3200 μg/L) [60]. Furthermore, their research indicates that about 2.1 million and 0.6 million people are using water contaminated with As at concentrations greater than 10 and 50 μg/L, respectively, for drinking; moreover, 0.048 million people are living with high health risks from drinking water contaminated with As concentrations greater than 300 μg/L, the level at which arsenical skin lesions are predicted to occur [60]. Besides the state of West Bengal, some parts of the states of Bihar (the middle Ganga plain, Bihar, India; e.g., Semria Ojha Patti village) [62] and Uttar Pradesh (upper and middle Ganga plain, India) are severely contaminated with As-rich groundwater [63]. One study of 4780 tube well water samples from three districts (Ballia, Varanasi, and Ghazipur in Uttar Pradesh in the upper and middle Ganga plain, India) revealed that As concentrations exceeded 10 μg/L in 46.5 %, 50 μg/L in 26.7 %, and 300 μg/L in 10 % of the samples (maximum As concentration level, 3192 μg//L). Furthermore, the report indicated that of the 989 villagers (691 adults, and 298 children) screened, 137 (19.8 %) of the adults and 17 (5.7 %) of the children had arsenical skin lesions, arsenical neuropathy, or adverse obstetric outcomes [28].
The individuals in the aforementioned studies were exposed to high As concentrations mainly through different direct and indirect exposure pathways, such as the drinking of As-enriched groundwater [47] and ingestion of As-contaminated food (food grains and vegetables) [149–151]. Ingested As accumulates in the hair, nails, and other tissues of the human body, with the excess partly released through urine, which is why hair, nail, and urine are the primary bio-indicators of As contamination. The research reported from the peri-urban villages of Moyna and Ardebok (North 24 Parganas, India) indicates that the As concentrations in hair, nail, and urine samples increased with age, peaking at 29 to 56 years of age [15]. Subjects younger than 14 years had the lowest As concentrations [15]. Biswas et al. [151] also reported lower As concentrations in hair and nail samples from children compared with adults in the Samta village of Bangladesh. Nath et al. [152] reported similar findings in their study population of Baruipur, South 24 Parganas district, West Bengal. Medical screening in the Bangladesh region [153] showed that most of the study populations were affected by keratosis and melanosis, whereas a few subjects were affected by chronic bronchitis and whole-body infection. In the West Bengal region, the individuals studied exhibited a full range of arsenical skin lesions, including hyperkeratosis, Bowen’s disease (suspected), and non-healing ulcers (suspected cancer) [15, 23, 154]. Furthermore, a significant number of people in the Bhagirathi (India) and Padma–Meghna (Bangladesh) sub-basins are affected by As-contaminated groundwater, and the number is increasing daily.
Nepal
Elevated concentrations of As have been found in the groundwaters of Nepal, used as a major source of drinking water. Since the pioneering studies by Tandukar [155], As-rich groundwater has been recognized as a serious public health concern in Nepal. Arsenic concentrations are particularly high in the Terai alluvial plain (TAP), comprising 20 districts and home to approximately 47 % of the total population, 90 % of whom are using groundwater for drinking [156]. It is estimated that about 0.5 million people are at risk for As poisoning, and 3.5 million people are being exposed to As levels between 10 and 50 μg/L [156–158]. Recent studies reported a 1.3 to 5.1 % prevalence of arsenicosis-related dermatosis, and the amounts of As accumulated in hair and nail samples are much higher than the acceptable level [158].
Hydrochemical data from shallow wells ranging in depth from 7 to 55 m in the southwestern part of Nawalparasi district in the Terai belt suggest a predominant reducing character with high HCO3 − and low SO4 2 − and NO3 − concentrations [157, 159, 160]. Elevated HCO3 − levels primarily are a result of the oxidation of organic matter associated with the sediments. Low levels of NO3 − coupled with elevated NH4 + concentrations in well water samples suggest dissimilatory nitrate reduction in the TAP aquifers. The redox levels (Eh < − 0.2 V) measured at the well heads are low, triggering the reduction of Fe(III) and Mn(IV) oxides in the aquifer sediments of the region. The sequence of redox reactions or terminal electron-accepting processes prevalent in the TAP aquifers plays a critical role in mobilizing As through the natural interaction between the aqueous phases and the aquifer sediments under anoxic conditions in the groundwater environment of the Terai aquifers [156–160].
China
China also is recognized as a highly As-affected region. An analysis of groundwaters from 20 of 34 provinces revealed high levels of As contamination [161–163], and most of the As-contaminated groundwater systems are located in the arid to semiarid inland basins as well as river deltas [161, 164–166]. Shallow groundwaters, distributed mostly in northwestern China, contain high As concentrations in alluvial–lacustrine or lacustrine sediment aquifers while the fluvial–marine sedimentary aquifers in the river deltas mostly are responsible for the highest As contamination of groundwaters in China [48, 165–171]. The use of As-contaminated groundwaters for drinking was first recognized in the 1960s [172]. Recently, it was documented that 19.6 million people in China are at risk of being affected by consuming naturally occurring high As concentrations in groundwater [172]. As a result, many people in rural areas have been exposed to chronic As poisoning, and more than 20,000 cases of arsenicosis recently were identified and confirmed [172]. The provinces of Xinjiang and Shanxi, as well as parts of Inner Mongolia, have been identified as the main locations in which problems related to As contamination are severe. Some regions of Inner Mongolia, such as Keshenketeng county, the Hetao Basin, and the Huhhot Basin, covering an area of approximately 3000 km2 with more than one million people, are highly affected by excessive As concentrations in groundwater (>50 μ/L) [164]. It also is estimated that at least 3000 people living in 776 villages have arsenicosis symptoms [161–164, 172, 173].
Arsenic contamination in the Huhhot alluvial basin is of particular concern because of the existence of As at elevated concentrations in groundwaters under metal-reducing redox conditions [48, 164, 168]. Groundwater systems in the areas of the Huhhot Basin have been greatly affected by weathering of feldspars and equilibrium with some minerals, such as gibbsite and anorthite [48]. The mobilization of As and its transport into the groundwater possibly occur as the result of recharging water flows via the various types of rock minerals found in the surrounding mountain areas of the basin. The major mechanism of As mobilization in the Huhhot Basin is microbially mediated reductive dissolution of metal oxides and hydroxides [48]. The median As concentration in 37 of 45 groundwater samples collected from the basin was found to be 0.7 μM, more than fivefold greater than the WHO maximum contaminant level (10 μg/L). It also was reported that the total As concentration at shallow depths of the basin is ≥19 μM [48, 164–170]. These high As levels in shallow aquifers of the basin likely are the result of many biogeochemical and redox transformation processes triggered by the recharged waters associated with other redox-sensitive species, including Fe and Mn [48, 169–171]. In the Huhhot Basin, redox conditions are attributed mainly to the SO4 2− reduction based on high HS− concentrations [48, 169, 171].
In the Ba Men region of Inner Mongolia, As is contaminating groundwaters at a maximum concentration of 1200 μg/L, which is far higher than China’s national standard for drinking water (50 μg/L) [172, 173]. Individual drinking water wells in this region have been found containing As in the range of 20 to 300 μg/L; hence, areas of chronic arsenism are widely here [173]. Many groundwaters distributed in the Hetao Basin of Inner Mongolia, northern China, also have high As concentrations, ranging from 76 to 1093 μg/L [167–169]. Based on the geochemical analysis of aquifer sediments of the northwestern Hetao Basin, total As concentrations are reported to be in the range of 6.8 to 58.5 mg/kg, with a median of 14.4 mg/kg [168]. As mobilization from aquifer sediments into groundwater systems of the Hetao Basin is governed predominantly by the reductive dissolution of Fe oxide minerals, and this process may be enhanced by indigenous microbes, such as Pseudomonas, Dietzia, and Rhodococcus, associated with the sediments [171].
Vietnam and Cambodia
In Vietnam also, groundwater contamination by As poses a serious health threat to millions of people [174]. Arsenic levels are elevated in groundwaters of the Holocene and Pleistocene aquifers as well as in the Red River Delta in the city of Hanoi [174, 175]. The groundwaters tend to be anoxic because of naturally occurring organic matter in the river sediments and are rich in iron minerals. With an average As concentration of 159 μg/L, the contamination levels vary from 1 to 3050 μg/L in rural groundwater samples from private small-scale tube wells. In one highly affected rural area, groundwater used directly for drinking has an average As concentration of 430 μg/L [175]. Most family-based tube wells tap the Holocene aquifer, whereas the Hanoi waterworks extract more than 600,000 m3/day of groundwater from the Pleistocene aquifer [175]. High and low levels of dissolved As may be seen in average concentrations of 121 and 21 μg/L, respectively, at the river bank, and medium levels of dissolved As, averaging 60 μg/L, may be found in an area of buried peat and excessive groundwater abstraction [175]. Reductive dissolution of iron oxyhydroxides and oxidation of sulfide phases associated with sediments likely are the major mechanisms responsible for the mobilization of As into groundwaters [69, 175]. The high As concentrations found in tube wells in the Mekong and Red River Deltas in Vietnam and Cambodia present a considerable risk of chronic As poisoning to several million people who consume untreated groundwater for drinking [69].
Taiwan
Elevated As levels in groundwater were found in two neighboring catchments including the Chianan Plain and the southern Choushui River alluvial fan in Taiwan [21, 67, 137, 162, 176]. The reduction potential in the Chianan Plain is stronger than that of the Choushui River alluvial fan, and the difference in reduction potentials between these two vicinal areas is believed to be the cause of the exceptional concentrations of As in groundwaters [177, 178]. The reductive dissolution of As-rich Fe-oxyhydroxide is the major mechanism responsible for the release of As into the groundwater in the Chianan Plain and the Choushui River alluvial fan [67, 177, 178]. Groundwater wells in southwest Taiwan also show considerable As contamination, on the average of 672 μg/L of total dissolved As, whereas interestingly, the wells of Hsinchu in northwest Taiwan show levels less than 0.7 μg/L. It is reported that the dominant As species of these wells is As(III), with an average As(III)/As(V) ratio of 2.6 [21, 179].
High As concentrations also have been reported in both aquifers and aquitards in the southern Choushui River alluvial fan, and these elevated levels have a direct impact on the contamination of shallow groundwaters (Table 1) [67, 177, 178]. Most groundwater As associated with the river originates from the aquitard of marine sequence; hence, the high As levels in marine formations with a high clay content may lead to the bioaccumulation of As in sea organisms. Some geochemical models have demonstrated that As(III) is the dominant As species in groundwater in the Chianan Plain of southwestern Taiwan, and the presence of Fe-bearing carbonates, sulfides, and oxide phases may act locally as potential sinks for As [138]. A significant number of bladder cancers due to ingestion of As have been reported in an arseniasis-endemic area in southwestern Taiwan, where many households in a village share only a few wells for drinking. However, in some arseniasis-endemic areas in northeastern Taiwan, each household tends to have its own well for drinking water [177–179]. This likely is the main reason for the significantly increased incidence of As toxicity reported in the general population of Taiwan.
Thailand
In Thailand, As so far has not been determined to be mobilized naturally from geogenic sources. Instead, anthropogenic sources, such as tin mining, farming, and ore dressing plant wastes, have contributed greatly to the significant emission of As, resulting in large-scale As contamination of groundwater systems there [36]. More recently, As concentrations >700 μg/L in groundwater in shallow wells have been reported, exceeding the WHO guideline for drinking water by a factor of 70 [17]. The Ron Phibun district in Nakhon Sri Thammarat province on Thailand’s southern peninsula is a well-known As-affected area. In 1987, skin manifestations due to chronic As poisoning were first diagnosed among the district’s population [34, 36]. In this region, the main source of high As levels in the groundwater is FeAsS, likely the result of mining processes during the past 10 decades. High rainfall tends to leach As from mining sites, leading to the contamination of soil, groundwater, and surface water over an area greater than 500 km2 in the district. Moreover, the Mae Moh open-pit lignite mine and associated power stations in Lampang province in northern Thailand are considered major As environmental fluxes. Bashkin and Wongyai [36] estimated the average As concentrations in borehole lignite, fly ash, reservoir bottom sediment, mine sump water, and Mae Moh reservoir water to be 515 mg/kg, 312 mg/kg, 5213 mg/kg, 325 mg/kg, and 8 μg/L, respectively.
The USA
Naturally occurring As in groundwaters at moderate or high concentrations has been documented in several parts of the USA, including the western states and parts of Maine, Michigan, Minnesota, South Dakota, Oklahoma, and Wisconsin. Widespread As concentrations in the western USA generally are associated with specific geochemical environments, such as basin-fill deposits of alluvial–lacustrine origin, particularly in semiarid areas, volcanic deposits, geothermal systems, thermal springs, and uranium and gold mining areas [3, 37, 91]. Alluvial and lacustrine sedimentary deposits are considered an important source of As in volcanic areas, such as Lane County, Oregon, and in areas underlain by basin-fill deposits, such as the Carson Desert in Nevada and the Tulare Lake basin in California. Arsenic in sedimentary aquifers is mobilized partially by changes in the geochemical environment due to agricultural irrigation. On the other hand, the compaction caused by groundwater withdrawal may result in high As concentrations in the deeper subsurface. Geothermal water and high evaporation rates also are associated with As concentrations greater than 10 mg/L in groundwater in the western USA.
Concentrations of dissolved As in groundwaters in alluvial basins of Arizona have been recorded as high as 1300 μg/L, far exceeding the WHO standard limit (10 μg/L) [180]. In this area, As occurs mainly in the fully oxidized form of As(V), most likely as HAsO4 2 –, under oxidizing and specific pH conditions [180]. The sources of As in the basins are the sulfide and arsenide deposits found in mineralized areas of the surrounding mountains, where As is oxidized to soluble forms before being transported into the basins. Under oxidizing conditions, As in the Arizona groundwater is controlled in part by sorption or desorption of HAsO4 2 – on active ferric oxyhydroxide surfaces [180]. The concentration and speciation of As exhibit systematic changes along the Carrizo Sand aquifer flow path, depending on changes in dissolved Fe(II) and Fe(III) concentrations, dissolved sulfide concentrations, pH, and alkalinity [181]. The speciation, mobility, and transformation of As in the Carrizo Sand aquifer are governed by certain mechanisms, such as ferric iron reduction, adsorption, and pH-related desorption [181].
Europe
Many countries in Europe, including Greece, Hungary, Romania, Croatia, Serbia, Turkey, Italy, and Spain, are significantly affected by elevated As concentrations in groundwater [2, 3, 7, 8, 85–88]. Among these nations, Greece is of particular concern because many of its groundwater resources contain large amounts of As. Geothermal-affected waters, the alluvial deposits of rivers, and aquifers have been identified as major sources that directly contribute to As release into groundwater [87, 88]. Arsenic concentrations in geothermal waters vary from 30 to 4500 μg/L; in the regions close to alluvial deposits, they vary from 15 to 100 μg/L and in areas affected by mining activities from 20 to 60 μg/L [7]. Arsenic concentrations in groundwater in the volcanic regions of central and southern Italy are widely variable, ranging from <0.1 to 6940 μg/L [87] with variable hydrochemical characteristics [87].
Other areas of Europe, such as the Pannonian Basin of Hungary and Romania, the Kütahya plain in Turkey, and the Chalkidiki region of Central Macedonia in northern Greece, are known for high As concentrations in the groundwater. Nearly 600,000 people living around the Pannonian Basin frequently are subjected to the danger of As toxicity [85, 88]. In Hungary, approximately 500,000 people are drinking water with As concentrations exceeding 10 μg/L. Arsenic dissolution in groundwater at an increased level is controlled by the mechanism of reductive dissolution of iron oxyhydroxides. In Romania, As is found mainly in the western plains, which belong to the Pannonian Basin, which is shared between Hungary and Romania, and where about 500,000 people are exposed to high As concentrations. In eastern Croatia, more than 200,000 people are affected by As concentrations of up to 610 μg/L in drinking water; here, reductive dissolution of iron oxyhydroxides is the main mechanism that releases As to groundwater. The Vojvodina region of northern Serbia belongs to the South Pannonian Basin, where As is present in average concentrations of 150 μg/L in groundwater, and more than 600,000 people—or 40 % of the population of the South Pannonian Basin—use drinking water with As concentrations higher than 10 μg/L [85, 88].
Latin America
Although high As concentrations have a long history in Latin American countries, As levels in water resources increased sharply during the past few decades. Today, high As levels may be detected in ground and surface water in 15 of the 20 Latin American countries [30]. A population of more than 1.2 million in rural Argentina depends on groundwater where As concentration exceeds the WHO guideline as well as the national drinking water standard of 10 μg/L [77, 78]. The most affected regions are extensive areas of the Pampean Plain, some parts of the Chaco Plain, and some small areas of the Andean range, where drinking water wells contain 50 to 15,000 μg/L of As [30, 46, 70, 76–83]. The sedimentary aquifers in this region comprise Tertiary aeolian loess-type sediments in the Pampean Plain and predominantly fluvial sediments of Tertiary and Quaternary age in the Chaco region. Drinking water for the rural populations is supplied by the shallow aquifers and contains around 200 μg/L of As (nearly 30 % as As) as well as high concentrations of fluoride (2.1 mg/L) [30, 46, 70, 76–83].
Approximately 400,000 residents of northern Chile drink water from public supplies diverted from rivers in the Andes Mountains to the arid regions. However, many of these rivers have high levels of natural As, which often end up in northern Chile’s drinking water [30, 182]. In the past, the public water system of Antofagasta (the largest city in this region) contained approximately 800 to 1000 μg/L of As, whereas other water supplies in northern Chile reportedly have As levels as high as 5000 μg/L [30].
In groundwaters of the Zimapan valley and several surrounding towns in Mexico, high As concentrations are attributed to multiple sources which includes both natural and anthropogenic activities related to mining in the region. Arsenic levels in groundwater are recorded as high as 1100 μg/L [73, 74], and shallow wells contaminated by mine tailings and fumes emanating from the smelters reportedly have As concentrations as high as 530 μg/L [75]. Elevated levels of natural As are also reported in groundwater from the unconfined alluvial aquifers in the region around Lagunera, causing adverse health effects in both people and livestocks [75]. Arsenic concentrations in the groundwater vary in the range of 5–750 μg/L, and extensive areas in the region are significantly higher than both the WHO drinking water guideline as well as the Mexican standard for drinking water (25 μg/L).
As in other volcanic regions where natural As contamination has been reported, the southern area of Lake Poopó in the Bolivian highlands, or the Altiplano, has elevated amounts of As in the water [3, 183, 184]. Samples of ground and surface water collected from 24 different sites, including drinking water wells and rivers, in the southern Poopó basin revealed various levels of As in shallow drinking water wells, from below detection level to 207 μg/L [3, 183, 184]. Many other occurrences of high As levels found in ground and surface waters in Central America, the Caribbean, and South American regions were reviewed extensively by Bundschuh et al. [30].
Conclusions
The primary source of groundwater As is natural and derived predominantly from interactions between groundwater and aquifer sediments of minerals, including FeS2, FeAsS, and other sulfide minerals. However, mining is highly responsible as an anthropogenic mobilization for As contamination of groundwaters. The geochemistry of As is a complex phenomenon in the environment; it generally is a function of multiple oxidation states, speciation, and redox transformation. Mobilization of As from aquifer sediments into the groundwater is governed by four main natural geochemical processes: reductive dissolution, alkali desorption, sulfide oxidation, and geothermal activities. The mobility and transformation of As in groundwaters may be controlled by adsorption onto metal oxyhydroxides and clay minerals. The occurrence and distribution of high concentrations of naturally occurring As in groundwaters of the Southeast Asia region, the USA, and some European countries have received much attention in recent years. Countries in Southeast Asia, including Bangladesh, India (West Bengal), China, Vietnam, Taiwan, Thailand, and Nepal, are of particular concern because of widespread As-related health problems within these populations. The western USA, parts of Maine, Michigan, Minnesota, South Dakota, Oklahoma, and Wisconsin; Greece, Hungary, Romania, Croatia, Serbia, Turkey, and Spain in Europe; and many Latin American countries, including Mexico, Argentina, Chile, Bolivia, and El Salvador also are exposed to elevated concentrations of As in groundwaters. This worldwide distribution of As in groundwaters poses a significant risk to human health, irrigation, and agricultural activities. Hence, raising awareness through public education as well as direct action and enforcement by the national governments is an urgent necessity toward achieving sustainable As mitigation worldwide.
References
Bhattacharya P, Ahmed KM, Hasan MA, Broms S, Fogelström J, Jacks G, et al. Mobility of arsenic in groundwater in a part of Brahmanbaria district, NE Bangladesh. In: Naidu R, Smith E, Owens G, Bhattacharya P, Nadebaum P, editors. Managing arsenic in the environment: from soil to human health. Melbourne: CSIRO Publishing; 2006. p. 95–115.
Bhattacharya P, Welch AH, Stollenwerk KG, McLaughlin MJ, Bundschuh J, Panaullah G. Arsenic in the environment: biology and chemistry. Sci Total Environ. 2007;379:109–20.
Ormachea Muñoz M, Bhattacharya P, Sracek O, Ramos Ramos O, Aguirre JQ, Bundschuh J, et al. Arsenic and other trace elements in thermal springs and in cold waters from drinking water wells on the Bolivian Altiplano. J S Am Earth Sci. 2015;60:10–20.
Ahmed KM, Bhattacharya P, Hasan MA, Akhter SH, Alam SM, Bhuyian MH, et al. Arsenic enrichment in groundwater of the alluvial aquifers in Bangladesh: an overview. Appl Geochem. 2004;19:181–200.
Welch AH, Stollenwerk KG. Arsenic in ground water: geochemistry and occurrence. New York: Springer Science & Business Media; 2002.
Maity JP, Kar S, Liu J-H, Jean J-S, Chen C-Y, Bundschuh J, et al. The potential for reductive mobilization of arsenic [As(V) to As(III)] by OSBH2 (Pseudomonas stutzeri) and OSBH5 (Bacillus cereus) in an oil-contaminated site. J Environ Sci Health. 2011;46:1239–46.
Katsoyiannis IA, Mitrakas M, Zouboulis AI. Arsenic occurrence in Europe: emphasis in Greece and description of the applied full-scale treatment plants. Desalin Water Treat. 2014;54(1–8):2100–7.
Bundschuh J, Maity JP, Nath B, Baba A, Gunduz O, Kulp TR, et al. Naturally occurring arsenic in terrestrial geothermal systems of western Anatolia, Turkey: potential role in contamination of freshwater resources. J Hazard Mater. 2013;262:951–9.
Vithanage M, Dabrowska BB, Mukherjee AB, Sandhi A, Bhattacharya P. Arsenic uptake by plants and possible phytoremediation applications: a brief overview. Environ Chem Lett. 2012;10:217–24.
Bundschuh J, Maity JP. Geothermal arsenic: occurrence, mobility and environmental implications. Renew Sust Energ Rev. 2015;42:1214–22.
Liu C-C, Kar S, Jean J-S, Wang C-H, Lee Y-C, Sracek O, et al. Linking geochemical processes in mud volcanoes with arsenic mobilization driven by organic matter. J Hazard Mater. 2013;262:980–8.
Nath B, Maity JP, Jean J-S, Birch G, Kar S, Yang H-J, et al. Geochemical characterization of arsenic-affected alluvial aquifers of the Bengal Delta (West Bengal and Bangladesh) and Chianan Plains (SW Taiwan): implications for human health. Appl Geochem. 2011;26:705–13.
Liu C-C, Maity JP, Jean J-S, Reza AS, Li Z, Nath B, et al. Geochemical characteristics of the mud volcano fluids in southwestern Taiwan and their possible linkage to elevated arsenic concentration in Chianan plain groundwater. Environ Earth Sci. 2012;66:1513–23.
Bhattacharya P, Jacks G, Ahmed KM, Routh J, Khan AA. Arsenic in groundwater of the Bengal Delta Plain aquifers in Bangladesh. Bull Environ Contam Toxicol. 2002;69:538–45.
Maity JP, Nath B, Kar S, Chen C-Y, Banerjee S, Jean J-S, et al. Arsenic-induced health crisis in peri-urban Moyna and Ardebok villages, West Bengal, India: an exposure assessment study. Environ Geochem Health. 2012;34:563–74.
Kar S, Maity JP, Jean J-S, Liu C-C, Liu C-W, Bundschuh J, et al. Health risks for human intake of aquacultural fish: arsenic bioaccumulation and contamination. J Environ Sci Health. 2011;46:1266–73.
Kim K-W, Chanpiwat P, Hanh H, Phan K, Sthiannopkao S. Arsenic geochemistry of groundwater in Southeast Asia. Front Med. 2011;5:420–33.
Bhattacharya P, Frisbie SH, Smith E, Naidu R, Jacks G, Sarkar B. Arsenic in the environment: a global perspective. In: Sarkar B, editor. Handbook of Heavy Metals in the Environment. New York: Marcell Dekker; 2002. p. 147–215.
Rango T, Vengosh A, Dwyer G, Bianchini G. Mobilization of arsenic and other naturally occurring contaminants in groundwater of the Main Ethiopian Rift aquifers. Water Res. 2013;47:5801–18.
Bhattacharya P, Welch AH, Ahmed KM, Jacks G, Naidu R. Arsenic in groundwater of sedimentary aquifers. Appl Geochem. 2004;19:163–7.
Maity JP, Nath B, Chen C-Y, Bhattacharya P, Sracek O, Bundschuh J, et al. Arsenic-enriched groundwaters of India, Bangladesh and Taiwan—comparison of hydrochemical characteristics and mobility constraints. J Environ Sci Health. 2011;46:1163–76.
Chakraborty M, Mukherjee A, Ahmed KM. A review of groundwater arsenic in the Bengal Basin, Bangladesh and India: from source to sink. Curr Pollut Rep. 2015;1:220–47.
Guha Majumder DN, Chakraborty AK, Ghosh A, Chakraborty DP, Dey SB, Chattopadhyay N. Chronic As toxicity from drinking tube well water in rural West Bengal. WHO Bull. 1988;66:499–506.
Joseph T, Dubey B, McBean EA. Human health risk assessment from arsenic exposures in Bangladesh. Sci Total Environ. 2015;527–528:552–60.
Rahman MM, Dong Z, Naidu R. Concentrations of arsenic and other elements in groundwater of Bangladesh and West Bengal, India: potential cancer risk. Chemosphere. 2015;139:54–64.
SOS arsenic poisoning in Bangladesh/India. http://www.sos-arsenic.net/. Accessed 24 Nov 2015.
Hu J, Liu Y-F, Wu C-F, Xu F, Shen Z-X, Zhu Y-M, et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2009;106:3342–7.
Samal AC, Kar S, Maity JP, Santra SC. Arsenicosis and its relationship with nutritional status in two arsenic affected areas of West Bengal, India. J Asian Earth Sci. 2013;77:303–10.
Schreiber ME, Simo JA, Freiberg PG. Stratigraphic and geochemical controls on naturally occurring arsenic in groundwater, eastern Wisconsin, USA. Hydrogeol J. 2000;8:161–76.
Bundschuh J, Litter MI, Parvez F, Román-Ross G, Nicolli HB, Jean J-S, et al. One century of arsenic exposure in Latin America: a review of history and occurrence from 14 countries. Sci Total Environ. 2012;429:2–35.
Smedley P, Kinniburgh D. A review of the source, behaviour and distribution of arsenic in natural waters. Appl Geochem. 2002;17:517–68.
Stollenwerk KG, Breit GN, Welch AH, Yount JC, Whitney JW, Foster AL, et al. Arsenic attenuation by oxidized aquifer sediments in Bangladesh. Sci Total Environ. 2007;379:133–50.
Bhattacharyya R, Jana J, Nath B, Sahu S, Chatterjee D, Jacks G. Groundwater As mobilization in the Bengal Delta Plain, the use of ferralite as a possible remedial measure—a case study. Appl Geochem. 2003;18:1435–51.
Mukherjee A, Sengupta MK, Hossain MA, Ahamed S, Das B, Nayak B, et al. Arsenic contamination in groundwater: a global perspective with emphasis on the Asian scenario. J Health Popul Nutr. 2006;24:142–63.
Williams M, Fordyce F, Paijitprapapon A, Charoenchaisri P. Arsenic contamination in surface drainage and groundwater in part of the southeast Asian tin belt, Nakhon Si Thammarat Province, southern Thailand. Env Geol. 1996;27:16–33.
Bashkin V, Wongyai K. Environmental fluxes of arsenic from lignite mining and power generation in northern Thailand. Environ Geol. 2002;41:883–8.
Welch AH, Westjohn D, Helsel DR, Wanty RB. Arsenic in ground water of the United States: occurrence and geochemistry. Groundwater. 2000;38:589–604.
Cummings DE, Caccavo F, Fendorf S, Rosenzweig RF. Arsenic mobilization by the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY. Environ Sci Technol. 1999;33:723–9.
Jain A, Raven KP, Loeppert RH. Arsenite and arsenate adsorption on ferrihydrite: surface charge reduction and net OH- release stoichiometry. Environ Sci Technol. 1999;33:1179–84.
Islam A, Maity JP, Bundschuh J, Chen C-Y, Bhowmik BK, Tazaki K. Arsenic mineral dissolution and possible mobilization in mineral–microbe–groundwater environment. J Hazard Mater. 2013;262:989–96.
Mandal BK, Suzuki KT. Arsenic round the world: a review. Talanta. 2002;58:201–35.
Francesconi KA, Kuehnelt D. Arsenic compounds in the environment. Environ Chem Arsenic. 2002:51–94.
Al-Abed SR, Jegadeesan G, Purandare J, Allen D. Arsenic release from iron rich mineral processing waste: influence of pH and redox potential. Chemosphere. 2007;66:775–82.
Meharg AA, Rahman MM. Arsenic contamination of Bangladesh paddy field soils: implications for rice contribution to arsenic consumption. Environ Sci Technol. 2003;37:229–34.
Bissen M, Frimmel FH. Arsenic—a review. Part I: occurrence, toxicity, speciation, mobility. Acta Hydrochim Hydrobiol. 2003;31:9–18.
Nicolli HB, Bundschuh J, Blanco MC, Tujchneider OC, Panarello HO, Dapena C, et al. Arsenic and associated trace-elements in groundwater from the Chaco-Pampean plain, Argentina: results from 100 years of research. Sci Total Environ. 2012;429:36–56.
Kar S, Maity JP, Jean J-S, Liu C-C, Nath B, Yang H-J, et al. Arsenic-enriched aquifers: occurrences and mobilization of arsenic in groundwater of Ganges Delta Plain, Barasat, West Bengal, India. Appl Geochem. 2010;25:1805–14.
Mukherjee A, Bhattacharya P, Shi F, Fryar AE, Mukherjee AB, Xie ZM, et al. Chemical evolution in the high arsenic groundwater of the Huhhot basin (Inner Mongolia, PR China) and its difference from the western Bengal basin (India). Appl Geochem. 2009;24:1835–51.
Smith E, Naidu R, Alston AM. Chemistry of inorganic arsenic in soils. J Environ Qual. 2002;31:557–63.
Bauer M, Blodau C. Mobilization of arsenic by dissolved organic matter from iron oxides, soils and sediments. Sci Total Environ. 2006;354:179–90.
Wang S, Mulligan CN. Effect of natural organic matter on arsenic release from soils and sediments into groundwater. Environ Geochem Health. 2006;28:197–214.
Klump S, Kipfer R, Cirpka OA, Harvey CF, Brennwald MS, Ashfaque KN, et al. Groundwater dynamics and arsenic mobilization in Bangladesh assessed using noble gases and tritium. Environ Sci Technol. 2006;40:243–50.
Anawar HM, Akai J, Sakugawa H. Mobilization of arsenic from subsurface sediments by effect of bicarbonate ions in groundwater. Chemosphere. 2004;54:753–62.
Biswas A, Nath B, Bhattacharya P, Halder D, Kundu AK, Mandal U, et al. Hydrogeochemical contrast between brown and grey sand aquifers in shallow depth of Bengal Basin: consequences for sustainable drinking water supply. Sci Total Environ. 2012;431:402–12.
Kar S, Maity JP, Jean J-S, Liu C-C, Nath B, Lee Y-C, et al. Role of organic matter and humic substances in the binding and mobility of arsenic in a Gangetic aquifer. J Environ Sci Health. 2011;46:1231–8.
Kohlmeyer U, Kuballa J, Jantzen E. Simultaneous separation of 17 inorganic and organic arsenic compounds in marine biota by means of high‐performance liquid chromatography/inductively coupled plasma mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:965–74.
Chakraborti D, Rahman MM, Das B, Murrill M, Dey S, Mukherjee SC, et al. Status of groundwater arsenic contamination in Bangladesh: a 14-year study report. Water Res. 2010;44:5789–802.
Hossain M, Bhattacharya P, Frape SK, Jacks G, Islam MM, Rahman MM, et al. Sediment color tool for targeting arsenic-safe aquifers for the installation of shallow drinking water tubewells. Sci Total Environ. 2014;493:615–25.
Rahman MM, Mandal BK, Roychowdhury T, Sengupta MK, Chowdhury UK, Lodh D, et al. Arsenic groundwater contamination and sufferings of people in North 24-Parganas, one of the nine arsenic affected districts of West Bengal, India. J Environ Sci Health Part A Environ Sci. 2003;38:25–59.
Rahman MM, Mondal D, Das B, Sengupta MK, Ahamed S, Hossain MA, et al. Status of groundwater arsenic contamination in all 17 blocks of Nadia district in the state of West Bengal, India: a 23-year study report. J Hydrol. 2014;518:363–72.
Shankar S, Shanker U, Shikha. Arsenic contamination of groundwater: a review of sources, prevalence, health risks, and strategies for mitigation. Sci World J. 2014.
Chakraborti D, Mukherjee SC, Pati S, Sengupta MK, Rahman MM, Chowdhury UK, et al. Arsenic groundwater contamination in Middle Ganga Plain, Bihar, India: a future danger? Environ Health Perspect. 2003;111(9):1194–201.
Ahamed S, Sengupta MK, Mukherjee A, Amir Hossain M, Das B, Nayak B, et al. Arsenic groundwater contamination and its health effects in the state of Uttar Pradesh (UP) in upper and middle Ganga plain, India: a severe danger. Sci Total Environ. 2006;370(2–3):310–22.
Shukla DP, Dubey C, Singh NP, Tajbakhsh M, Chaudhry M. Sources and controls of arsenic contamination in groundwater of Rajnandgaon and Kanker District, Chattisgarh Central India. J Hydrol. 2010;395:49–66.
Das N, Patel AK, Deka G, Das A, Sarma, KP, Kumar M. Geochemical controls and future perspective of arsenic mobilization for sustainable groundwater management: A study from Northeast India. Groundwater for Sustainable Development. 2015;1:92–104.
Nordstrom DK. Worldwide occurrences of arsenic in ground water. Science. 2002;296:2143–5.
Liu C-C, Maity JP, Jean J-S, Li Z, Kar S, Sracek O, et al. The geochemical characteristics of the mud liquids in the Wushanting and Hsiaokunshui mud volcano region in southern Taiwan: implications of humic substances for binding and mobilization of arsenic. J Geochem Explor. 2013;128:62–71.
Baig JA, Kazi TG, Arain MB, Afridi HI, Kandhro GA, Sarfraz RA, et al. Evaluation of arsenic and other physico-chemical parameters of surface and ground water of Jamshoro, Pakistan. J Hazard Mater. 2009;166:662–9.
Berg M, Stengel C, Trang PTK, Hung Viet P, Sampson ML, Leng M, et al. Magnitude of arsenic pollution in the Mekong and Red River Deltas—Cambodia and Vietnam. Sci Total Environ. 2007;372:413–25.
Smedley PL, Nicolli HB, Macdonald DMJ, Kinniburgh DG. Arsenic in groundwater and sediments from La Pampa Province, Argentina. In: Bundschuh J, Armienta MA, Birkle P, Bhattacharya P, Matschullat J, Mukherjee AB, editors. Natural arsenic in groundwaters of Latin America. Boca Raton: Taylor & Francis; 2008. p. 35–45.
Nriagu J, Bhattacharya P, Mukherjee A, Bundschuh J, Zevenhoven R, Loeppert R. Arsenic in soil and groundwater: an overview. Trace Met Contam Environ. 2007;9:3–60.
Donahue R, Hendry M. Geochemistry of arsenic in uranium mine mill tailings, Saskatchewan, Canada. Appl Geochem. 2003;18:1733–50.
Armienta MA, Rodriguez R, Aguayo A, Ceniceros N, Villaseñor G, Cruz O. Arsenic contamination of groundwater at Zimapán, Mexico. Hydrogeol J. 1997;5:39–46.
Rodriguez R, Ramos JA, Armienta A. Groundwater arsenic variations: the role of local geology and rainfall. Appl Geochem. 2004;19(2):245–50.
Boochs PW, Billib M, Gutiérrez C, Aparicio J. Groundwater contamination with arsenic, Región Lagunera, México. In: M.I. Litter, H.B. Nicolli, M. Meichtry, N. Quici, J. Bundschuh, P. Bhattacharya & R. Naidu (eds.) Once Century of the Discovery of Arsenicosis in Latin America (1914-2014) As2014. Interdisciplinary book series: Arsenic in the Environment - Proceedings. Series Editors: J. Bundschuh & P. Bhattacharya, CRC Press/Taylor and Francis (ISBN 978-1-138-00141-1), 2014; pp. 132–134.
Bejarano Sifuentes G, Nordberg E. Mobilisation of arsenic in the Rio Dulce alluvial cone, Santiago del Estero province, Argentina. TRITA-LWR Master Thesis: 2003; Department of Land and Water Resources Engineering, KTH Royal Institute of Technology, Stockholm Sweden. 75p.
Bundschuh J, Farias B, Martin R, Storniolo A, Bhattacharya P, Cortes J, et al. Grounwater arsenic in the Chaco-Pampean Plain, Argentina: case study from Robles County, Santiago del Estero Province. Appl Geochem. 2004;19(2):231–43.
Smedley PL, Nicolli HB, Macdonald DMJ, Barros AJ, Tullio JO. Hydrogeochemistry of arsenic and other inorganic constituents in groundwaters from La Pampa, Argentina. Appl Geochem. 2002;17:259–84.
Smedley PL, Kinniburgh DG, Macdonald DMJ, Nicolli HB, Barros AJ, Tullio JO, et al. Arsenic associations in sediments from the loess aquifer of La Pampa, Argentina. Appl Geochem. 2005;20:989–1016.
Bhattacharya P, Claesson M, Bundschuh J, Sracek O, Fagerberg J, Jacks G, et al. Distribution and mobility of arsenic in the Río Dulce Alluvial aquifers in Santiago del Estero Province, Argentina. Sci Total Environ. 2006;358(1–3):97–120.
Nicolli HB, García JW, Falcón CM, Smedley PL. Mobilization of arsenic and other trace elements of health concern in groundwater from the Salí River Basin, Tucumán Province, Argentina. Environ Geochem Health. 2012;34:251–62.
Aullón Alcaine A, Sandhi A, Bhattacharya P, Jacks,G, Bundschuh J, Thunvik R, Schulz C, Mörth CM. Distribution and mobility of geogenic arsenic in the shallow aquifers of the northeast of La Pampa, Argentina. In: J.C. Ng, B.N. Noller, R. Naidu, J. Bundschuh & P. Bhattacharya (eds.) Understanding the Geological and Medical Interface of Arsenic As 2012, Interdisciplinary book series: Arsenic in the Environment - Proceedings. Series Editors: J. Bundschuh and P. Bhattacharya, CRC Press/Taylor and Francis (ISBN-13: 978-0-415-63763-3), 2012; pp. 132–134.
Panigatti MC, Boglione RM, Griffa CA, Schierano MC. Groundwater arsenic in the central-west of the Santa Fe Province, Argentine. In: M.I. Litter, H.B. Nicolli, M. Meichtry, N. Quici, J. Bundschuh, P. Bhattacharya & R. Naidu (eds.) Once Century of the Discovery of Arsenicosis in Latin America (1914-2014) As2014. Interdisciplinary book series: Arsenic in the Environment - Proceedings. Series Editors: J. Bundschuh & P. Bhattacharya, CRC Press/Taylor and Francis (ISBN 978-1-138-00141-1), 2014; pp. 159–161.
Robinson B, Kim N, Marchetti M, Moni C, Schroeter L, van den Dijssel C, et al. Arsenic hyperaccumulation by aquatic macrophytes in the Taupo Volcanic Zone, New Zealand. Environ Exp Bot. 2006;58:206–15.
Leonardi G, Vahter M, Clemens F, Goessler W, Gurzau E, Hemminki K, et al. Inorganic arsenic and basal cell carcinoma in areas of Hungary, Romania, and Slovakia: a case–control study. Environ Health Perspect. 2012;120:721–6.
Romić Ž, Habuda-Stanić M, Kalajdžić B, Kuleš M. Arsenic distribution, concentration and speciation in groundwater of the Osijek area, eastern Croatia. Appl Geochem. 2011;26:37–44.
Aiuppa A, D’Alessandro W, Federico C, Palumbo B, Valenza M. The aquatic geochemistry of arsenic in volcanic groundwaters from southern Italy. Appl Geochem. 2003;18:1283–96.
Rowland HA, Omoregie EO, Millot R, Jimenez C, Mertens J, Baciu C, et al. Geochemistry and arsenic behaviour in groundwater resources of the Pannonian Basin (Hungary and Romania). Appl Geochem. 2011;26:1–17.
Appleyard SJ, Angeloni J, Watkins R. Arsenic-rich groundwater in an urban area experiencing drought and increasing population density, Perth, Australia. Appl Geochem. 2006;21:83–97.
Smith JVS, Jankowski J, Sammut J. Vertical distribution of As(III) and As(V) in a coastal sandy aquifer: factors controlling the concentration and speciation of arsenic in the Stuarts Point groundwater system, northern New South Wales, Australia. Appl Geochem. 2003;18:1479–96.
López DL, Bundschuh J, Birkle P, Armienta MA, Cumbal L, Sracek O, et al. Arsenic in volcanic geothermal fluids of Latin America. Sci Total Environ. 2012;429:57–75.
Panagiotaras D, Papoulis D, Panagopoulos G, Avramidis P. Arsenic geochemistry in groundwater system. Intech Open Access Publisher; 2012.
Sracek O, Bhattacharya P, Jacks G, Gustafsson JP, von Brömssen M. Behavior of arsenic and geochemical modeling of arsenic contamination. Appl Geochem. 2004;19(2):169–80.
Gong Z, Lu X, Watt C, Wen B, He B, Mumford J, et al. Speciation analysis of arsenic in groundwater from Inner Mongolia with an emphasis on acid-leachable particulate arsenic. Anal Chim Acta. 2006;555:181–7.
Katsoyiannis IA, Zouboulis AI. Application of biological processes for the removal of arsenic from groundwaters. Water Res. 2004;38:17–26.
Brookins DG. Eh-pH diagrams for geochemistry. Berlin Heidelberg: Springer; 2012.
Islam FS, Gault AG, Boothman C, Polya DA, Charnock JM, Chatterjee D, et al. Role of metal-reducing bacteria in arsenic release from Bengal delta sediments. Nature. 2004;430:68–71.
Routh J, Bhattacharya P, Jacks G, Ahmed K, Khan A, Rahman M. Arsenic geochemistry of Tala groundwater and sediments from Satkhira District, Bangladesh. EOS Trans Am Geophys Union. 2000;81:550.
Scott MJ, Morgan JJ. Reactions at oxide surfaces. 1. Oxidation of As(III) by synthetic birnessite. Environ Sci Technol. 1995;29:1898–905.
Manning BA, Fendorf SE, Bostick B, Suarez DL. Arsenic(III) oxidation and arsenic(V) adsorption reactions on synthetic birnessite. Environ Sci Technol. 2002;36:976–81.
Lin Z, Puls RW. Adsorption, desorption and oxidation of arsenic affected by clay minerals and aging process. Env Geol. 2000;39:753–9.
Santini JM, Sly LI, Wen A, Comrie D, Wulf-Durand PD, Macy JM. New arsenite-oxidizing bacteria isolated from Australian Gold Mining Environments—phylogenetic relationships. Geomicrobiol J. 2002;19:67–76.
Valenzuela C, Campos V, Yañez J, Zaror C, Mondaca M. Isolation of arsenite-oxidizing bacteria from arsenic-enriched sediments from Camarones River, Northern Chile. Bull Environ Contam Toxicol. 2009;82:593–6.
Wilkie JA, Hering JG. Rapid oxidation of geothermal arsenic(III) in streamwaters of the Eastern Sierra Nevada. Environ Sci Technol. 1998;32:657–62.
Taylor SR, McLennan SM. The continental crust: its composition and evolution. London: Blackwell Scientific; 1985. 365 pp.
Bhattacharya P, Chatterjee D, Jacks G. Occurrence of arsenic-contaminated groundwater in alluvial aquifers from delta plains, Eastern India: options for safe drinking water supply. Int J Water Resour Dev. 1997;13:79–92.
Bhattacharya P, Hasan MA, Sracek O, Smith E, Ahmed KM, von Brömssen M, et al. Groundwater chemistry and arsenic mobilization in the Holocene flood plains in south-central Bangladesh. Environ Geochem Health. 2009;31:23–43.
Welch AH, Lico MS, Hughes JL. Arsenic in ground water of the Western United States. Ground Water. 1988;26:333–47.
O’Day PA, Vlassopoulos D, Root R, Rivera N. The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions. Proc Natl Acad Sci U S A. 2004;101:13703–8.
Kocar BD. Soil-sediment processes perpetuating history’s largest mass poisoning through release of arsenic to Asian groundwaters. ProQuest; 2008.
Sracek O, Bhattacharya P, Jacks G, Gustafsson J-P, Von Brömssen M. Behavior of arsenic and geochemical modeling of arsenic enrichment in aqueous environments. Appl Geochem. 2004;19:169–80.
Sand W, Gehrke T. Extracellular polymeric substances mediate bioleaching/biocorrosion via interfacial processes involving iron(III) ions and acidophilic bacteria. Res Microbiol. 2006;157:49–56.
Bhattacharya P, Jacks G, Jana J, Sracek A, Gustafsson J, Chatterjee D. Geochemistry of the holocene alluvial sediments of Bengal Delta Plain from West Bengal, India: implications on arsenic contamination in groundwater. Groundwater Arsenic Contam Bengal Delta Plain Bangladesh. 2001;3084:21–40.
Fernandez MGM, Mustin C, de Donato P, Barres O, Marion P, Berthelin J. Occurrences at mineral–bacteria interface during oxidation of arsenopyrite by Thiobacillus ferrooxidans. Biotechnol Bioeng. 1995;46:13–21.
Yu Y, Zhu Y, Gao Z, Gammons CH, Li D. Rates of arsenopyrite oxidation by oxygen and Fe(III) at pH 1.8–12.6 and 15–45 °C. Environ Sci Technol. 2007;41:6460–4.
Walker FP, Schreiber ME, Rimstidt JD. Kinetics of arsenopyrite oxidative dissolution by oxygen. Geochim Cosmochim Acta. 2006;70:1668–76.
Youngran J, Fan M, Van Leeuwen J, Belczyk JF. Effect of competing solutes on arsenic(V) adsorption using iron and aluminum oxides. J Environ Sci. 2007;19:910–9.
Masue Y, Loeppert RH, Kramer TA. Arsenate and arsenite adsorption and desorption behavior on coprecipitated aluminum:iron hydroxides. Environ Sci Technol. 2007;41:837–42.
Raven KP, Jain A, Loeppert RH. Arsenite and arsenate adsorption on ferrihydrite: kinetics, equilibrium, and adsorption envelopes. Environ Sci Technol. 1998;32:344–9.
Fuller CC, Davis JA, Waychunas GA. Surface chemistry of ferrihydrite: part 2. Kinetics of arsenate adsorption and coprecipitation. Geochim Cosmochim Acta. 1993;57:2271–82.
Grafe M, Eick MJ, Grossl PR, Saunders AM. Adsorption of arsenate and arsenite on ferrihydrite in the presence and absence of dissolved organic carbon. J Environ Qual. 2002;31:1115–23.
Wang Y, Deng Y. Environmental geochemistry of high-arsenic aquifer systems. Wang, Lawrence K, Paul Chen, Jiaping, Hung, Yung-Tse (Eds) Heavy metals in the environment. CRC Press, Taylor & Francis Group, Boca Raton, London. 2009:123–54.
Arai Y, Elzinga EJ, Sparks DL. X-ray absorption spectroscopic investigation of arsenite and arsenate adsorption at the aluminum oxide–water interface. J Colloid Interface Sci. 2001;235:80–8.
Paktunc D, Foster A, Heald S, Laflamme G. Speciation and characterization of arsenic in gold ores and cyanidation tailings using X-ray absorption spectroscopy. Geochim Cosmochim Acta. 2004;68:969–83.
Tufano KJ, Reyes C, Saltikov CW, Fendorf S. Reductive processes controlling arsenic retention: revealing the relative importance of iron and arsenic reduction. Environ Sci Technol. 2008;42:8283–9.
Borch T, Kretzschmar R, Kappler A, Cappellen PV, Ginder-Vogel M, Voegelin A, et al. Biogeochemical redox processes and their impact on contaminant dynamics. Environ Sci Technol. 2009;44:15–23.
Nesbitt HW, Canning GW, Bancroft GM. XPS study of reductive dissolution of 7Å-birnessite by H3AsO3, with constraints on reaction mechanism. Geochim Cosmochim Acta. 1998;62:2097–110.
Tournassat C, Charlet L, Bosbach D, Manceau A. Arsenic(III) oxidation by birnessite and precipitation of manganese(II) arsenate. Environ Sci Technol. 2002;36:493–500.
Tani Y, Miyata N, Ohashi M, Ohnuki T, Seyama H, Iwahori K, et al. Interaction of inorganic arsenic with biogenic manganese oxide produced by a Mn-oxidizing fungus, strain KR21-2. Environ Sci Technol. 2004;38:6618–24.
Goldberg S. Competitive adsorption of arsenate and arsenite on oxides and clay minerals. Soil Sci Soc Am J. 2002;66:413–21.
Manning BA, Goldberg S. Adsorption and stability of arsenic(III) at the clay mineral–water interface. Environ Sci Technol. 1997;31:2005–11.
Mohapatra D, Mishra D, Chaudhury GR, Das RP. Arsenic adsorption mechanism on clay minerals and its dependence on temperature. Korean J Chem Eng. 2007;24:426–30.
Manning BA, Goldberg S. Modeling arsenate competitive adsorption on kaolinite, montmorillonite and illite. Clay Clay Miner. 1996;44:609–23.
Wang S, Mulligan CN. Natural attenuation processes for remediation of arsenic contaminated soils and groundwater. J Hazard Mater. 2006;138:459–70.
Frost RR, Griffin RA. Effect of pH on adsorption of arsenic and selenium from landfill leachate by clay minerals. Soil Sci Soc Am J. 1977;41:53–7.
Samal AC, Kar S, Bhattacharya P, Santra SC. Human exposure to arsenic through foodstuffs cultivated using arsenic contaminated groundwater in areas of West Bengal, India. J Environ Sci Health. 2011;46:1259–65.
Maity JP, Liu C-C, Nath B, Bundschuh J, Kar S, Jean J-S, et al. Biogeochemical characteristics of Kuan-Tzu-Ling, Chung-Lun and Bao-Lai hot springs in southern Taiwan. J Environ Sci Health Part A. 2011;46:1207–17.
Nath B, Jean J-S, Lee M-K, Yang H-J, Liu C-C. Geochemistry of high arsenic groundwater in Chia-Nan plain, Southwestern Taiwan: possible sources and reactive transport of arsenic. J Contam Hydrol. 2008;99:85–96.
Chakraborty M, Mukherjee A, Ahmed KM. A review of groundwater arsenic in the Bengal Basin, Bangladesh and India: from source to sink. Curr Pollut Rep. 2015. doi:10.1007/s40726-015-0022-0.101.
Bhattacharya P, Samal A, Majumdar J, Santra S. Arsenic contamination in rice, wheat, pulses, and vegetables: a study in an arsenic affected area of West Bengal, India. Water Air Soil Pollut. 2010;213:3–13.
Roy Chowdhury T. Arsenic in ground water in six districts of West Bengal, India: the biggest arsenic calamity in the world. Part I. Arsenic species in drinking water and urine of the affected people. Analyst. 1995;120:643–50.
Kinniburgh D, Smedley P, Davies J, Milne C, Gaus I, Trafford J, et al. The scale and causes of the groundwater arsenic problem in Bangladesh. In: Welch A, Stollenwerk K, editors. Arsenic in ground water. US: Springer; 2003. p. 211–57.
von Brömssen M, Jakariya M, Bhattacharya P, Ahmed KM, Hasan MA, Sracek O, et al. Targeting low-arsenic aquifers in Matlab Upazila, southeastern Bangladesh. Sci Total Environ. 2007;379:121–32.
Chakraborti D, Das B, Rahman MM, Chowdhury UK, Biswas B, Goswami AB, et al. Status of groundwater arsenic contamination in the state of West Bengal, India: a 20-year study report. Mol Nutr Food Res. 2009;53:542–51.
Nickson RT, McArthur JM, Ravenscroft P, Burgess WB, Ahmed KM. Mechanism of As poisoning of groundwater in Bangladesh and West Bengal. Appl Geochem. 2000;15:403–13.
Mukherjee AB, Bhattacharya P. Arsenic in groundwater in the Bengal Delta Plain: slow poisoning in Bangladesh. Environ Rev. 2001;9:189–220.
Bhattacharya R, Chatterjee D, Nath B, Jana J, Jacks G, Vahter M. High arsenic groundwater: mobilization, metabolism and mitigation—an overview in the Bengal Delta Plain. Mol Cell Biochem. 2003;253:347–55.
Chatterjee D, Halder D, Majumder S, Biswas A, Nath B, Bhattacharya P, et al. Assessment of arsenic exposure from groundwater and rice in Bengal Delta Region, West Bengal, India. Water Res. 2010;44:5803–12.
Roychowdhury T, Uchino T, Tokunaga H, Ando M. Survey of As in food composites from an As-affected area of West Bengal, India. Food Chem Toxicol. 2002;40:1611–21.
Talukdar T, Ghosh AK, Srivastava KK. Arsenic in ground water of North 24 Pargasnas district, West Bengal. Bhu-jal News. 2009;24:95–106.
Biswas BK, Dhar RK, Samanta G, Mandal BK, Chakraborti D, Faruk I, et al. Detailed study report of Samta, one of the arsenic-affected villages of Jessore District, Bangladesh. Curr Sci. 1998;74:134–45.
Nath B, Sahu SJ, Jana J, Mukherjee-Goswami A, Roy S, Sarkar MJ, et al. Hydrochemistry of arsenic enriched aquifer from rural West Bengal, India: a study of the arsenic exposure and mitigation option. Water Air Soil Pollut. 2008;190:95–113.
Kadono T, Inaoka T, Murayama N, Ushijima K, Nagano M, Nakamura S, et al. Skin manifestations of arsenicosis in two villages in Bangladesh. Int J Dermatol. 2002;41:841–6.
Rahman MM, Sengupta MK, Ahamed S, Chowdhury UK, Lodh D, Hossain A, et al. Arsenic contamination of groundwater and its health impact on residents in a village in West Bengal, India. WHO Bull. 2005;83:49–57.
Tandukar N. Arsenic contamination in groundwater in Rautahat District of Nepal—an assessment and treatment. Unpublished M. Sc. Thesis, Institute of Engineering, Lalitpur, Nepal. (2000).
Tandukar N, Bhattacharya P, Mukherjee A B. Preliminary assessment of arsenic contamination in groundwater in Nepal. Book of Abstracts, Arsenic in the Asia-Pacific Region Workshop, Adelaide (2001) pp. 103–105.
Bhattacharya P, Tandukar N, Neku A, Valero AA, Mukherjee AB, Jacks G. Geogenic arsenic in groundwaters from Terai alluvial plain of Nepal. J Phys IV France. 2003;107:173–6.
Pokhrel D, Bhandari B, Viraraghavan T. Arsenic contamination of groundwater in the Terai region of Nepal: an overview of health concerns and treatment options. Environ Int. 2009;35:157–61.
Gurung J, Ishiga H, Khadka M. Geological and geochemical examination of arsenic contamination in groundwater in the Holocene Terai Basin, Nepal. Environ Geol. 2005;49:98–113.
Brikowski TH, Neku A, Shrestha SD, Smith LS. Hydrologic control of temporal variability in groundwater arsenic on the Ganges floodplain of Nepal. J Hydrol. 2014;518:342–53.
Rodríguez LL, Sun G, Berg M, Zhang Q, Xue H, Zheng Q, et al. Groundwater arsenic contamination throughout China. Science. 2013;341:866–8.
Guo H, Wen D, Liu Z, Jia Y, Guo Q. A review of high arsenic groundwater in Mainland and Taiwan, China: distribution, characteristics and geochemical processes. Appl Geochem. 2014;41:196–217.
Wen D, Zhang F, Zhang E, Wang C, Han S, Zheng Y. Arsenic, fluoride and iodine in groundwater of China. J Geochem Explor. 2013;135:1–21.
Shi F. Arsenic in groundwater in Huhhot alluvial basin in Inner Mongolia, People’s Republic of China. Master Thesis, Department of Land and Water Resources Engineering, KTH Royal Institute of Technology, Stockholm, Sweden. 2004. TRITA-LWR Master Thesis 04–04. 37p
Guo H, Wang Y. Geochemical characteristics of shallow groundwater in Datong basin, northwestern China. J Geochem Explor. 2005;87:109–20.
Xie ZM, Zhang YM, Naidu R. Extent and severity of arsenic poisoning in China. In: Naidu R, Smith E, Owens G, Bhattacharya P, Nadebaum P, editors. Managing arsenic in the environment: from soils to human health. Collingswood: CSIRO Publishing; 2006. p. 541–52.
Guo H, Yang S, Tang X, Li Y, Shen Z. Groundwater geochemistry and its implications for arsenic mobilization in shallow aquifers of the Hetao Basin, Inner Mongolia. Sci Total Environ. 2008;393:131–144r.
Deng Y, Wang Y, Ma T, Gan Y. Speciation and enrichment of arsenic in strongly reducing shallow aquifers at western Hetao Plain, northern China. Environ Geol. 2009;56:1467–77.
Guo H, Zhang B, Wang G, Shen Z. Geochemical controls on arsenic and rare earth elements approximately along a groundwater flow path in the shallow aquifer of the Hetao Basin, Inner Mongolia. Chem Geol. 2010;270:117–25.
Deng Y, Wang Y, Ma T, Yang H, He J. Arsenic associations in sediments from shallow aquifers of northwestern Hetao Basin, Inner Mongolia. Environ Earth Sci. 2011;64:2001–11.
Li Y, Guo H, Hao C. Arsenic release from shallow aquifers of the Hetao basin, Inner Mongolia: evidence from bacterial community in aquifer sediments and groundwater. Ecotoxicology. 2014;23:1900–14.
Guo X, Fujino Y, Kaneko S, Wu K, Xia Y, Yoshimura T. Arsenic contamination of groundwater and prevalence of arsenical dermatosis in the Hetao plain area, Inner Mongolia, China. In: Shi X, Castranova V, Vallyathan V, Perry W, editors. Molecular mechanisms of metal toxicity and carcinogenesis. Developments in molecular and cellular biochemistry. US: Springer; 2001. p. 137–40.
Ning Z, Lobdell DT, Kwok RK, Liu Z, Zhang S, Ma C, et al. Residential exposure to drinking water arsenic in Inner Mongolia, China. Toxicol Appl Pharmacol. 2007;222:351–6.
Berg M, Tran HC, Nguyen TC, Pham HV, Schertenleib R, Giger W. Arsenic contamination of groundwater and drinking water in Vietnam: a human health threat. Environ Sci Technol. 2001;35:2621–6.
Berg M, Trang PTK, Stengel C, Buschmann J, Viet PH, Van Dan N, et al. Hydrological and sedimentary controls leading to arsenic contamination of groundwater in the Hanoi area, Vietnam: the impact of iron-arsenic ratios, peat, river bank deposits, and excessive groundwater abstraction. Chem Geol. 2008;249:91–112.
Wang S-W, Liu C-W, Jang C-S. Factors responsible for high arsenic concentrations in two groundwater catchments in Taiwan. Appl Geochem. 2007;22:460–76.
Liu C-W, Wang S-W, Jang C-S, Lin K-H. Occurrence of arsenic in ground water in the Choushui River alluvial fan, Taiwan. J Environ Qual. 2006;35:68–75.
Chen S-L, Dzeng SR, Yang M-H, Chiu K-H, Shieh G-M, Wai CM. Arsenic species in groundwaters of the blackfoot disease area, Taiwan. Environ Sci Technol. 1994;28:877–81.
Chiou H-Y, Chiou S-T, Hsu Y-H, Chou Y-L, Tseng C-H, Wei M-L, et al. Incidence of transitional cell carcinoma and arsenic in drinking water: a follow-up study of 8,102 residents in an arseniasis-endemic area in Northeastern Taiwan. Am J Epidemiol. 2001;153:411–8.
Robertson FN. Arsenic in ground-water under oxidizing conditions, south-west United States. Environ Geochem Health. 1989;3–4:171–85. doi:10.1007/BF01758668.
Haque S, Johannesson KH. Arsenic concentrations and speciation along a groundwater flow path: the Carrizo Sand aquifer, Texas, USA. Chem Geol. 2006;228:57–71.
Ferreccio C, Sancha AM. Arsenic exposure and its impact on health in Chile. J Health Popul Nutr. 2006;24:164–75.
Ormachea Muñoz M, Wern H, Johnsson F, Bhattacharya P, Sracek O, Thunvik R, et al. Geogenic arsenic and other trace elements in the shallow hydrogeologic system of Southern Poopó Basin, Bolivian Altiplano. J Hazard Mater. 2013;262:924–40.
Ormachea Muñoz, M., Huallpara, L., Coariti, E.B., Aróstegui, J.L.G., Kohfahl, C., Estévez M.C.H. & Bhattacharya, P. Natural arsenic occurrence in drinking water and assessment of water quality in the southern part of the Poopó lake basin, Bolivian Altiplano. In: M.I. Litter, H.B. Nicolli, M. Meichtry, N. Quici, J. Bundschuh, P. Bhattacharya & R. Naidu (eds.) Once Century of the Discovery of Arsenicosis in Latin America (1914-2014) As2014. Interdisciplinary book series: Arsenic in the Environment - Proceedings. Series editors: J. Bundschuh & P. Bhattacharya, CRC Press/Taylor and Francis (ISBN 978-1-138-00141-1). 2014; pp. 154–156.
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is part of the Topical Collection on Water Pollution
Rights and permissions
About this article

Cite this article
Herath, I., Vithanage, M., Bundschuh, J. et al. Natural Arsenic in Global Groundwaters: Distribution and Geochemical Triggers for Mobilization. Curr Pollution Rep 2, 68–89 (2016). https://doi.org/10.1007/s40726-016-0028-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40726-016-0028-2