
Highlights
-
Edge-rich MoS2/C hollow microspheres (Edg-MoS2/C HMs) were fabricated through the simple hydrothermal treatment of MoO3–aniline nanowires and a subsequent carbonization process.
-
The Edg-MoS2/C HMs ensure the uniform deposition of Li2S on the matrix and the enhanced utilization of active edge sites.
-
The cell with an Edg-MoS2/C HM-functionalized separator displayed excellent electrochemical performance, with a high reversible discharge capacity of 478 mAh g−1 after 300 cycles at a high sulfur loading of 6.1 mg cm−2 and high rate of 0.5 C.
Abstract
As promising energy storage systems, lithium–sulfur (Li–S) batteries have attracted significant attention because of their ultra-high energy densities. However, Li–S battery suffers problems related to the complex phase conversion that occurs during the charge–discharge process, particularly the deposition of solid Li2S from the liquid-phase polysulfides, which greatly limits its practical application. In this paper, edge-rich MoS2/C hollow microspheres (Edg-MoS2/C HMs) were designed and used to functionalize separator for Li–S battery, resulting in the uniform deposition of Li2S. The microspheres were fabricated through the facile hydrothermal treatment of MoO3–aniline nanowires and a subsequent carbonization process. The obtained Edg-MoS2/C HMs have a strong chemical absorption capability and high density of Li2S binding sites, and exhibit excellent electrocatalytic performance and can effectively hinder the polysulfide shuttle effect and guide the uniform nucleation and growth of Li2S. Furthermore, we demonstrate that the Edg-MoS2/C HMs can effectively regulate the deposition of Li2S and significantly improve the reversibility of the phase conversion of the active sulfur species, especially at high sulfur loadings and high C-rates. As a result, a cell containing a separator functionalized with Edg-MoS2/C HMs exhibited an initial discharge capacity of 935 mAh g−1 at 1.0 C and maintained a capacity of 494 mAh g−1 after 1000 cycles with a sulfur loading of 1.7 mg cm−2. Impressively, at a high sulfur loading of 6.1 mg cm−2 and high rate of 0.5 C, the cell still delivered a high reversible discharge capacity of 478 mAh g−1 after 300 cycles. This work provides fresh insights into energy storage systems related to complex phase conversions.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Because of their high cost and limited capacities, conventional lithium-ion batteries cannot meet the constantly increasing demands of portable electronic devices and electrified transportation [1,2,3,4,5]. Therefore, the development of advanced battery systems with high energy densities, superior rate performances, and long cycle stabilities is increasingly urgent. Lithium–sulfur (Li–S) batteries have attracted intense interest because of their ultra-high energy densities (2600 Wh kg−1) and the low cost of the active material (sulfur), as well as its high natural abundance and environmental friendliness [6,7,8]. Despite these tremendous merits, the further development of Li–S batteries is hindered by the problems resulting from the complex phase conversion processes that occur during battery charging and discharging [7, 9,10,11].
In the first stage of the discharge process in Li–S batteries, solid S is converted into soluble polysulfides (Li2Sx, 4 ≤ x ≤ 8) in the cathode [7]. Because of osmosis, the permeation of polysulfides through the separators to the Li metal anodes occurs easily, leading to the loss of active sulfur and the corrosion of the Li metal anodes [7]. Thus, the reversible capacities of Li–S batteries fade fast and “terrible” lithium dendrites form [2]. Various strategies have been devoted to blocking the diffusion of polysulfides, thus improving the electrochemical performance of Li–S batteries, including sulfur host design [6, 12,13,14,15,16,17,18], separator functionalization [6, 19,20,21,22,23,24,25,26,27,28], and new electrolyte exploration [29,30,31]. Of these methods, separator functionalization is a promising strategy to restrict the shuttling of the polysulfides and increase the utilization of active sulfur. The materials used to functionalize separators can be divided into two main categories: first, carbon materials with large specific surface area and excellent electrical conductivity. These include graphene [19], mesoporous carbon [27], and Super P [24], which trap polysulfides through physical adsorption. However, the weak affinities of these carbon materials with polar polysulfides make it difficult to maintain the required long cycle stability of Li–S batteries, especially when using cathodes with high area sulfur loadings. Second, polar materials including metal sulfides [20, 32], metal nitrides [33, 34], and metal phosphides [35, 36] can be used. These materials exhibit excellent electrocatalytic performance for polysulfides and also strong chemical adsorption for these species. Further, they have been proven to be effective in limiting polysulfide shuttling and improving the redox reaction kinetics of polysulfides. However, to obtain high-performance Li–S batteries, the functionalized separator must be able to solve more than these two problems.
In the second stage of the discharge process, soluble polysulfides are further transformed into solid Li2S [7]. This electrochemical process contributes to three-quarters of the theoretical capacity of Li–S batteries [11]. However, the insulation and insolubility of the discharge product, Li2S, usually result in its aggregation [7, 11]. The cores of these Li2S aggregates become “dead sulfur” in the charging process because of the loss of electrical contact with the conductive network, leading to sluggish Li2S oxidation kinetics and the irreversible loss of active sulfur [7, 10, 11]. Therefore, regulating the uniform deposition of Li2S in the discharge process is crucial to improving the reversible capacity and cycling stability of Li–S batteries. Generally, the distribution of Li2S precipitates deposited on the matrix depends on the electron transfer capability of the matrix, the binding energy between Li2S and the matrix, and the distribution of active Li2S binding sites [7, 10,11,12, 15, 16, 25, 35,36,37,38,39,40,41,42,43,44]. Therefore, it is expected that Li–S batteries with separators functionalized with a material that strongly chemisorbs polysulfides, and exhibits excellent electrocatalytic performance for polysulfides and the capability to regulate the uniform nucleation and growth of Li2S could achieve excellent electrochemical performance.
In this study, we first designed and fabricated hollow, edge-rich MoS2/C microsphere (Edg-MoS2/C HMs) functionalized separators to regulate the uniform deposition of Li2S in Li–S batteries. The Edg-MoS2/C HMs were obtained by the facile hydrothermal treatment of MoO3–aniline (MoO3–AN) nanowires with thiourea and sucrose and a subsequent carbonization process. These microspheres consist of uniformly distributed MoS2/C nanosheets that are rich in edge sites and a carbon network. The liquid-phase lithium polysulfides are effectively entrapped by MoS2 (Fig. 1a). Furthermore, the outstanding electrocatalytic performance of Edg-MoS2/C HMs accelerates the conversion kinetics of the polysulfides. Additionally, the carbon network facilitates electron transfer, and the many edge sites of the uniformly distributed MoS2/C nanosheets provide abundant, strong Li2S binding sites. As a result, the uniform nucleation and growth of Li2S on the matrix was realized (Fig. 1b). Moreover, the hollow structure and the ultrathin edge-rich MoS2/C nanosheets of the Edg-MoS2/C HMs increase the utilization of active edge sites and, thus, reduce the weight of the interlayers and guarantee the excellent performance of the corresponding Li–S batteries. Thanks to these advantages, the Edg-MoS2/C HMs-functionalized separators consequently improve the specific capacity, rate performance, and cyclic stability of Li–S batteries, especially with high sulfur contents and high areal sulfur loadings of cathodes.

Schematic illustration of the synthesis and application of the Edg-MoS2/C@PP separators in Li–S batteries. a Schematic of polysulfides entrapped in the Li–S battery using the Edg-MoS2/C@PP separator. b Schematic of Li2S deposition in the Edg-MoS2/C@PP and PP cells. c Synthesis route of the Edg-MoS2/C HMs and schematic of the edge and terrace sites of the Edg-MoS2/C HMs
2 Experimental Section
2.1 Preparation of Edg-MoS2/C HMs and Carbon Network
The MoO3–AN nanowires were prepared according to a previously reported method [45]. In a typical procedure, 100 mg of as-obtained MoO3–AN, 1.0 g thiourea, and 300 mg sucrose were added to 60 mL deionized water. Then, the mixture was ultrasonicated for 1 h. Subsequently, the obtained suspension was transferred to a 100-mL Teflon-lined autoclave, which was heated to 200 °C for 12 h in an oven. The resultant black products were collected by vacuum filtration, washed with deionized water and ethanol several times, and then dried in a vacuum oven at 60 °C for 12 h. The obtained product was the Edg-MoS2/partially carbonized carbon hollow microspheres (Edg-MoS2/PC HMs). The Edg-MoS2/C HMs were obtained after annealing in a tube furnace at 700 °C for 2 h under N2 flow. Edg-MoS2/C400 HMs and MoS2 microflowers (MFs) were obtained using the same procedures with or without the addition of 400 mg sucrose, respectively. In addition, the MoS2 in the Edg-MoS2/C HMs was removed with aqua regia, leaving only the carbon network (CN).
2.2 Preparation of Edg-MoS2/C@PP and CN@PP Separators
The Edg-MoS2/C HMs (or CN), Super P (super conductive carbon black), and poly(vinylidene fluoride) binder (6:3:1 in mass ratio) were stirred in N-methyl-2-pyrrolidone (NMP) to form a uniform slurry, which was then coated on a Celgard 2400 polypropylene (PP) membrane. After drying in a vacuum oven at 60 °C overnight, the Edg-MoS2/C@PP and CN@PP separators were obtained and punched into disks with diameters of 19 mm. The mass loadings of the coating materials on the Edg-MoS2/C@PP and CN@PP separators were controlled to be approximately 0.34 mg cm−2.
2.3 Preparation of the Sulfur Cathode
Typically, sublimed sulfur and carbon nanotubes (CNTs, 4:1 by mass) were ground uniformly together and sealed in a 50-mL reaction kettle. The mixture was then heated at 155 °C for 10 h. The obtained product, CNT/S (approximately 80 wt%), carbon black (10 wt%), and LA133 binder (10 wt%) were stirred in an aqueous n-propanol solution. The slurry was coated onto carbon-coated aluminum foil and dried at 60 °C overnight. Then, the sulfur electrode was obtained and punched into disks with diameters of 12 mm. The average sulfur loading mass on the electrodes was controlled to be 1.7 mg cm−2. Higher sulfur loadings of 3.5 and 6.1 mg cm−2 were also prepared.
2.4 Lithium Polysulfide Adsorption Tests
The lithium polysulfide adsorption tests were conducted by adding an equivalent amount (10 mg) of Edg-MoS2/C HMs and CN to the lithium polysulfide (Li2S6) solution and holding for 6 h. The lithium polysulfide (Li2S6) solution was prepared by dissolving stoichiometric amounts of sulfur and lithium sulfide (Li2S) in a molar ratio of 5:1 in a 1,2-dimethoxyethane/1,3-dioxolane (DME/DOL) mixture (1:1 by volume).
2.5 Assembly of Symmetric Cells and Kinetic Evaluation of Polysulfide Conversion
Corresponding electrodes were fabricated without sulfur. The active material (Edg-MoS2/C HMs and CN) and poly(vinylidene fluoride) (PVDF) in a mass ratio of 6:1 were dispersed in NMP by vigorous stirring to form uniform slurry, which was subsequently overlaid on Al foils with an areal loading of approximately 0.31 mg cm−2. Two identical electrodes were used as the working and the counter electrodes. They were assembled into typical 2032-type coin cells with a PP membrane as the separator and 40.0 μL Li2S6 electrolyte (containing 0.5 mol L−1 Li2S6 and 1.0 mol L−1 lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in DOL/DME solution with a volume ratio of 1:1). The cyclic voltammetry (CV) curves of the symmetric cells were measured at scan rates of 50–2000 mV s−1 with a voltage range between − 1.0 and 1.0 V.
2.6 Fabrication of Li–S Cells and Electrochemical Measurements
The electrochemical performances of the Li–S cells were determined in CR2032 coin cells with CNT/S electrodes as the cathodes, functionalized PP films as the separators, and lithium foil as the anodes. The electrolyte contained 1 M LiTFSI in DOL and DME binary solvents (1:1 by volume) with 2 wt% LiNO3. The ratio of added electrolyte to sulfur in the coin cells was 12 µL mg−1 sulfur. The amounts of electrolyte added to the cell with sulfur areal loadings of 1.7, 3.5, and 6.1 mg cm−2 were 23, 47, and 83 μL, respectively. The galvanostatic charge–discharge tests were carried out using a battery test system (CT2001C, LAND) in a voltage range of 1.8–2.7 V. CV measurements and electrochemical impedance spectroscopy (EIS) were conducted using an electrochemical workstation (Chenhua CHI 600). CV measurements were carried out from 1.7 to 2.8 V, and all electrochemical tests were performed at room temperature.
2.7 Characterization
The crystal structures of the samples were determined using powder X-ray diffractometry (XRD) measurements on a Bruker/D8 Focus diffractometer (Germany). Cu Kα X-rays (λ = 1.5405 Å) were generated at 40.0 kV and 40 mA, and reflections were recorded in the 2θ range of 5°–80° at a scan speed of 6° min−1. N2 adsorption–deposition isotherms were collected with a Micromeritics TriStar II 3020 system mode at 77 K. All the samples were degassed at 100 °C for 15 h under flowing N2 before measurement. Field-emission scanning electron microscopy (FE-SEM) images and transmission electron microscopy (TEM) images were obtained using a Hitachi S4800 electron microscope (Japan) and a JEOL-2100F electron microscope (Japan), respectively. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Thermo Escalab 250 system. Raman spectra were measured with an excitation laser wavelength of 514.5 nm at room temperature using a LabRAM HR. Thermogravimetric (TG) analyses of the Edg-MoS2/C HMs and the CNT/S composite were performed in a PerkinElmer (TA Instruments) up to 650 °C at a heating rate of 10 °C min−1 in air and N2, respectively.
3 Results and Discussion
As shown in Fig. 1c, the Edg-MoS2/C HMs were synthesized by the facile hydrothermal treatment of MoO3–AN nanowires with thiourea and sucrose and a subsequent carbonization process. During the hydrothermal treatment process, MoO3–AN nanowires were first transformed into solid microspheres (Fig. S1b) and then gradually transformed into hollow microspheres (Fig. S1b–g) by the Kirkendall effect, yielding Edg-MoS2/PC HMs (Fig. S1 g) [46,47,48]. Subsequently, carbonization was conducted to enhance the degree of graphitization of the carbon network in the Edg-MoS2/C HMs. In addition, MoS2 in the Edg-MoS2/C HMs was removed using aqua regia, and the CN alone was, thus, obtained.
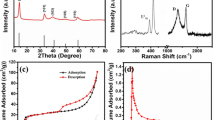
The TEM image in Fig. 2a and the SEM images in Fig. 2c, d show the hollow structure of the Edg-MoS2/C HMs where a number of nanosheets are attached to the shells. High-magnification TEM images (Figs. 2b and S2) further reveal that the shells of the Edg-MoS2/C HMs are composed of edge-rich MoS2/C nanosheets and a carbon network. The interlayer distance of the MoS2 (002) crystalline planes was measured to be around 9.7 Å. As illustrated in the low-magnification TEM image (Fig. 2a), the average diameter of the Edg-MoS2/C HMs particles is a few hundreds of nanometers, and the average thickness of the edge-rich MoS2/C nanosheets is about 10 nm. The observed uniformly distributed edge sites in the MoS2/C nanosheets provide abundant Li2S deposition sites. The hollow structure and ultrathin edge-rich MoS2/C nanosheets increase the utilization of active edge sites, thus reducing the weight of the interlayers while ensuring excellent performance of Li–S batteries. The MoS2 content in the Edg-MoS2/C HMs was calculated to be about 73% (Fig. S6a). The powder XRD pattern (Fig. 2k) illustrates the well-defined characteristic peaks of hexagonal MoS2, except for the (002) peak, which can be attributed to the expansion of MoS2 (002) crystal planes by the insertion of a carbon layer [49]. The presence of C–OH/C–O–Mo (285.3 eV) peaks in the C 1s XPS spectrum (Fig. S4c) and a C–O–Mo (532.7 eV) peak in the O 1s XPS spectrum (Fig. S4d) also confirm the chemical combination of hexagonal MoS2 and a carbon layer in the Edg-MoS2/C HMs [20, 50]. The Raman spectrum of the as-obtained Edg-MoS2/C HMs shows the existence of the carbon network and molybdenum disulfide (Fig. 2l) [49, 50]. In particular, the intensity of the D-band (corresponding to the A1g vibration mode caused by defective sp2 carbon rings) is larger than that of the G-band (corresponding to the E12g vibration mode of sp2 carbon rings without defects), having a ID/IG value of 0.99, which demonstrates the degree of graphitization of the carbon network [49, 50] and this means the enhancement of the overall conductivity of the Edg-MoS2/C HMs. The high specific surface area (28.8 m2 g−1) and uniform mesoporous distribution (approximately 4 nm) of the Edg-MoS2/C HMs (Fig. S5 and Table S1) are expected to provide abundant reaction sites of sulfur species in the charge–discharge processes [7].

Characterization of the Edg-MoS2/C HMs and Edg-MoS2/C@PP separators. TEM images a at low and b high magnifications, and SEM images c at low and d high magnifications of the Edg-MoS2/C HMs. Top-down SEM images of e the pristine PP separator and f Edg-MoS2/C@PP separator. g Low- and h high-magnification cross-sectional SEM images of the Edg-MoS2/C@PP separator. i Optical photographs of Edg-MoS2/C@PP separator. j Contact angle between electrolyte and PP separator (top), electrolyte and Edg-MoS2/C@PP separator (bottom). k XRD pattern, l Raman spectrum of the Edg-MoS2/C HMs
On the basis of these results, Edg-MoS2/C HMs were employed to modify commercial PP separators for Li–S batteries. Unlike the macroporous structure of the PP separator (Fig. 2e), the Edg-MoS2/C interlayer was constructed of homogeneously distributed Edg-MoS2/C HMs with Super P (Fig. 2f–h), as shown in both the top-down and cross-sectional SEM images. In the Janus-structured Edg-MoS2/C-functionalized separator (Edg-MoS2/C@PP) (Fig. 2g), the Edg-MoS2/C interlayer side toward the cathode can be used to block the diffusion of polysulfides and improve the homogeneous deposition of Li2S, whereas the non-conductive PP side toward the anode prevents direct contact between the cathode and anode [7, 10, 11, 25]. The thickness of Edg-MoS2/C interlayer is as low as about 15 μm (Fig. 2g), and its areal weight density was calculated to be about 0.34 mg cm−2. Moreover, the Edg-MoS2/C@PP separator can be bent significantly (Fig. 2i), indicating its good mechanical stability. Compared to that between the electrolyte and the pristine PP, the much smaller 5.1° contact angle (Fig. 2j) between the electrolyte and Edg-MoS2/C@PP guarantees the enhanced electrolyte wettability of the Edg-MoS2/C interlayer.
To validate the effects of the Edg-MoS2/C@PP separators on the electrochemical performances of Li–S cells, Li–S coin cells with the same CNT/S cathodes (sulfur areal loading of 1.7 mg cm−2) and Li foil anodes were assembled. Depending on the separator used, the assembled coin cells are designated Edg-MoS2/C@PP, CN@PP, and PP, respectively. As shown in Fig. 3a, CV measurements were used to explore the electrochemical processes occurring in the Li–S cells. Compared to those in the CV curves of CN@PP (1.98 and 2.31 V) and PP (1.94 and 2.24 V), the significantly enhanced current intensities and higher peak potentials (2.01 and 2.31 V) of the two cathodic peaks in the CV curve of Edg-MoS2/C@PP indicate the effective restriction and significantly improved polysulfide conversion kinetics [7, 9]. In the subsequent anodic scan, a lower potential (2.36 V) and larger peak area of the anodic peak in the CV curve of Edg-MoS2/C@PP were observed, and these originate from the accelerated dissolution and oxidation reaction kinetics of Li2S [9]. The much lower polarization potential (50 mV) of Edg-MoS2/C@PP (CN@PP: 90 mV and PP: 260 mV) reveals the impressive enhancement in the redox kinetics of the sulfur species [34].

Electrochemical performances of the CNT/S cathodes with the Edg-MoS2/C@PP and PP separators. a Cyclic voltammogram profiles of the Edg-MoS2/C@PP, CN@PP, and PP cells at a scan rate of 0.1 mV s−1. b Rate capabilities and c corresponding galvanostatic discharge–charge profiles at 0.2 C and 2.0 C (1 C = 1675 mA g−1) of the Edg-MoS2/C@PP, CN@PP, and PP cells. Cycling performances of d the Edg-MoS2/C@PP cells at 0.2 C, e the Edg-MoS2/C@PP, CN@PP, and PP cells at 1.0 C, f the Edg-MoS2/C@PP cells at 5.0 C. The sulfur loadings of the CNT/S cathodes in a–f are 1.7 mg cm−2. Cycling performance of the Edg-MoS2/C@PP cells g at 0.2 C with a high sulfur loading of 3.5 mg cm−2, h at 0.5 C with sulfur loadings of 1.7, 3.5, and 6.1 mg cm−2, respectively. The weight of the interlayers in all cells is 0.34 mg cm−2
To evaluate the role of the Edg-MoS2/C@PP separators on the redox reactions of sulfur species in Li–S cells further, the rate capabilities of Edg-MoS2/C@PP, CN@PP, and PP were measured. As shown in Fig. 3b, an initial discharge capacity of 1327 mAh g−1 for Edg-MoS2/C@PP was delivered at 0.05 C (1.0 C = 1675 mAh g−1). On increasing the current density to 0.2 C, 0.5 C, and 1.0 C, discharge capacities of 1014, 896, and 780 mAh g−1 were obtained, respectively. Even at a high current density of 2.0 C, the Edg-MoS2/C@PP still displayed a discharge capacity of 605 mAh g−1. On reducing the current densities to 1.0 C, 0.5 C, and 0.2 C, high reversible discharge capacities of 734, 823, and 934 mAh g−1 were maintained after the high rate test, corresponding to nearly 92% capacity recovery. In contrast, CN@PP displayed lower discharge capacities of 1020.5, 864.2, 705.4, 607.9, and 495.9 mAh g−1 at 0.05 C, 0.2 C, 0.5 C, 1.0 C, and 2.0 C, which was ascribed to the physical adsorption of polysulfides by the CN. PP exhibited obviously reduced capacities and rapid capacity degradation with increasing current density, exhibiting a much lower discharge capacity of 153 mAh g−1 at 2.0 C. The outstanding rate capability and capacity restoration of Edg-MoS2/C@PP can be attributed to the chemical affinity for polysulfides of Edg-MoS2/C HMs and the accelerated redox kinetics of the sulfur species in Edg-MoS2/C@PP. The galvanostatic charge–discharge plateaus of Edg-MoS2/C@PP, CN@PP, and PP show remarkable differences at current densities of 0.2 C and 2.0 C (Fig. 3c). Unlike those of CN@PP and PP, higher discharge plateaus and lower charge plateaus were exhibited in Edg-MoS2/C@PP. Even at 2.0 C, Edg-MoS2/C@PP still showed an apparent second discharge plateau, whereas CN@PP displayed a much lower second discharge plateau and PP had no second discharge plateau. These results demonstrate the rapid conversion kinetics of the polysulfides in the discharge process of Edg-MoS2/C@PP and that of Li2S in the charge process of Edg-MoS2/C@PP.
The improvement in the polysulfide shuttling through the Edg-MoS2/C HMs and the polysulfide and Li2S redox reaction kinetics were also investigated using long-term cycling tests of the Li–S cells. After three activation cycles at 0.05 C, Edg-MoS2/C@PP delivered an initial discharge capacity of 1106 mAh g−1 at 0.2 C (Fig. 3d). The discharge capacity of 957 mAh g−1 was retained in Edg-MoS2/C@PP after 100 cycles, accounting for 86.5% of the initial capacity, and the corresponding capacity decay was as low as 0.13% per cycle. This excellent behavior can probably be ascribed to the strong chemical affinity and excellent electrocatalytic performance toward polysulfides, as well as the enhanced Li2S conversion kinetics, of the Edg-MoS2/C HMs. The long-term cycling stability of Edg-MoS2/C@PP was further tested at 1.0 C for 1000 cycles. As shown in Fig. 3e, Edg-MoS2/C@PP displayed an initial discharge capacity of 935 mAh g−1 at 1.0 C after three cycles at 0.05 C. A reversible discharge capacity of 494 mAh g−1 with a coulombic efficiency above 97% was retained after 1000 cycles, demonstrating the long-term effectiveness of the Edg-MoS2/C HMs in promoting the redox reactions in Li–S cells. In contrast, the discharge capacities of CN@PP and PP were only 365.6 and 235 mAh g−1 at 1.0 C after 300 cycles, respectively, which could be ascribed to the sluggish conversion reaction and severe polysulfide shuttling, which would result in the low utilization of active sulfur, the corrosion of the Li anode, and the formation of lithium dendrites. This is confirmed by the SEM images of the Li anodes (Fig. S9), which were obtained by dissembling Li–S cells after 10 cycles at 1.0 C. The EIS measurements reveal the accelerated redox kinetics of the sulfur intermediates when using the Edg-MoS2/C HMs (Fig. S10). On increasing to 5.0 C, a discharge capacity of 602 mAh g−1 was obtained for Edg-MoS2/C@PP after three cycles at 0.05 C and 12 cycles at 5.0 C (Fig. 3f). After 500 continuous cycles, an excellent reversible capacity of 393 mAh g−1 remained, demonstrating the outstanding redox reaction kinetics of the Edg-MoS2/C HMs, even at high rates.
The mass ratio of MoS2 to C in the composite microspheres was adjusted to achieve the best performing Li–S battery. As illustrated in Fig. S15 and Table S2, Edg-MoS2/C400 HMs (44.4% MoS2) showed poor electrochemical performance because of the weak limiting effect on lithium polysulfides resulting from its low MoS2 content. Because the MoS2 MFs (98.1% MoS2) are solid structures with fewer active sites and poor conductivity arising from the ultra-low carbon content, this material also demonstrated poorer electrochemical performance than Edg-MoS2/C HMs (72.9% MoS2). Consequently, the Edg-MoS2/C HMs (72.9% MoS2) presented the best performance among these samples.
To explore the potential practical applications of Edg-MoS2/C@PP separators, the electrochemical performance of Li–S cells with higher sulfur areal loadings were tested. CNT/S cathodes with sulfur loadings of 1.7, 3.5, and 6.1 mg cm−2 were fabricated, and Li–S coin cells were assembled using the corresponding Edg-MoS2/C@PP separators, denoted CNT/S-1.7, CNT/S-3.5, and CNT/S-6.1, respectively. After three-cycle activation at 0.05 C, CNT/S-3.5 delivered an initial discharge capacity of 839 mAh g−1 (Fig. 3g) and a high capacity of 677 mAh g−1 with a coulombic efficiency of 98.6% after 300 cycles at 0.2 C, thus displaying a high capacity retention of 80.7% and a low capacity decay of 0.064% per cycle. Even at a high current density of 0.5 C, high discharge capacities of 594, 539, and 478 mAh g−1 (corresponding to areal capacities of 1.01, 1.89, and 2.92 mAh cm−2, respectively) were obtained for CNT/S-1.7, CNT/S-3.5, and CNT/S-6.1 after 300 cycles, respectively (Figs. 3h and S11), confirming the effective restriction of polysulfides and the enhancement in the reaction kinetics induced by the Edg-MoS2/C HMs, even in Li–S cells with high sulfur mass loadings and operated at a high rate. These results demonstrate that the Edg-MoS2/C@PP separators have potential practical applications in Li–S batteries. The rate performances and cycle stabilities of Li–S cells with Edg-MoS2/C@PP separators are comparable to those of recently reported Li–S cells with functionalized separators, as summarized in Tables 1 and S3 [19, 20, 24, 27, 32, 33, 51,52,53,54,55,56]. As illustrated in Table 1, although some of the interlayers of the listed MoS2-based materials are relatively thin and light, the performances of cells with these interlayer-modified separators at high areal sulfur cathode loadings were not provided, and this information is very important for assessing possible practical applications of Li–S batteries. However, the cells containing Edg-MoS2/C HM-functionalized separators exhibited excellent performance compared to those of other Li–S batteries at high sulfur contents and high areal sulfur loadings of the cathodes.
To investigate the mechanism of interaction between the Edg-MoS2/C HMs and the sulfur species during the redox reactions of the Li–S cells, visual adsorption tests on Edg-MoS2/C HMs and CN of the same weight and lithium polysulfide content were carried out. After static adsorption for 6 h, the Li2S6 solution (Fig. 4a) with Edg-MoS2/C HMs was discolored, whereas the color of the Li2S6 solution with CN had not changed, indicating the strong chemical absorption of the soluble polysulfides by the Edg-MoS2/C HMs [7, 57]. Thus, the Edg-MoS2/C HMs can effectively prevent polysulfides from dissolving into the electrolyte, increasing the utilization of active sulfur and providing the preconditions for further conversion reactions of polysulfides.

Investigation of the mechanism of interaction between the Edg-MoS2/C HMs and sulfur species during the redox reactions of the Li–S cells. a Visual adsorption tests of Li2S6 by pristine Edg-MoS2/C HMs and CN. b CV curves of symmetric dummy cells employing Edg-MoS2/C HMs and CN electrodes at a high scan rate of 2000 mV s−1. c CV curves of the Edg-MoS2/C HMs symmetric cells at different scan rates. d The Mo 3d, S 2s, and e S 2p spectrum of the Edg-MoS2/C-functionalized separator before and after the first 100% discharge at 0.05 C
XPS measurements were employed to determine the chemical interaction between MoS2 and the polysulfides. The high-resolution Mo 3d XPS spectrum (Fig. S13a) contains three typical peaks at 232.6, 229.4, and 235.4 eV, corresponding to Mo 3d3/2, Mo 3d5/2, and the + 6 oxidation state of Mo, respectively [58]. The Mo 3d3/2 and Mo 3d5/2 peaks could be assigned to Mo4+ [58]. After the polysulfide adsorption experiments, all three peaks (especially the Mo6+ peak) were downshifted to lower energies, indicating the intense chemical interaction of exposed Mo sites with the sulfur of the polysulfides [58]. The S 2s peak at 226.6 eV (Fig. S13a), S 2p1/2 peak at 163.4 eV, and S 2p3/2 peak at 162.2 eV (Fig. S13b) correspond to S2− in MoS2. The three peaks upshifted to 226.9, 163.6, and 162.5 eV, respectively, after adsorption, demonstrating the electron transfer from surface-exposed S atoms in MoS2 to Li atoms in polysulfides [58]. Notably, two new peaks at 164.0 and 164.8 eV can be attributed to the polysulfides [58], and a further two new peaks at 169.3 and 170.5 eV were assigned to the S–O bond in the oxidized sulfur reaction intermediates of polysulfides and MoS2, along with the reduction in the Mo oxidation state [58]. The chemical interaction between Edg-MoS2/C HMs and polysulfides significantly restricted the polysulfide shuttle effect.
The polysulfide conversion rate in the discharge process has a tremendous effect on the rate capabilities of the Li–S cells. To probe the effect of the Edg-MoS2/C HMs on the polysulfide conversion reaction, CV measurements of symmetric dummy cells containing the Li2S6 electrolyte were carried out [7, 25, 59]. In comparison with that of the CN electrode, the Edg-MoS2/C HMs electrode exhibits a much higher current density at a high scan rate of 2000 mV s−1 (Fig. 4b), suggesting enhanced conversion kinetics between the soluble polysulfides [7]. To explore the electrocatalytic performance of the Edg-MoS2/C HMs on the conversion of soluble polysulfides further, CV profiles at different scan rates were obtained (Fig. 4c). The current densities of the reduction and oxidation peaks were found to increase remarkably at scan rates from 50 to 1000 mV s−1, implying the significantly rapid redox reaction kinetics of the sulfur intermediates, even at high scan rates. Generally, at a fast scan rate, the redox reactions are severely constrained by the diffusion rate of the reacting substances. However, distinguishable redox peaks (Fig. 4c) could be still observed in the CV curve of the Edg-MoS2/C HM electrode at a high scan rate of 1000 mV s−1, indicating the excellent electrocatalytic effectiveness of the Edg-MoS2/C HMs toward soluble lithium polysulfides [7]. These effects mainly originate from the uniformly distributed edge sites resulting from the combination of MoS2 and a carbon network, which provides more active sites for electrocatalysis and accelerates the diffusion rate of electrons and Li+, thus boosting polysulfide conversion.
The chemical interaction between the edge sites in the Edg-MoS2/C HMs and Li2S was verified by the XPS results (Fig. 4d). Before discharge, the Mo 3d3/2, Mo 3d5/2, and Mo6+ peaks were located at 232.5, 229.4, and 235.6 eV in the Mo 3d spectrum, respectively. After the first 100% discharge at 0.05 C, the corresponding peaks downshifted to 232.2, 228.9, and 234.9 eV, respectively, which is attributed to the chemical affinity of the Mo atoms in the Edg-MoS2/C HMs and S atoms in the Li2S. In addition, the S 2s peak upshifted from 226.6 eV before discharge to 226.8 eV after the first 100% discharge at 0.05 C, which is due to the chemical interactions between the S atoms in the Edg-MoS2/C HMs and the Li atoms in the Li2S via chemical absorption. As shown in Fig. 4e, the S 2p1/2 peak at 163.4 eV and the S 2p3/2 peak at 162.2 eV in the S 2p spectrum correspond to S2− in MoS2, and the corresponding peaks shifted to higher energies of 163.6 and 162.5 eV, respectively, after adsorption, demonstrating electron transfer from the surface-exposed S atoms in MoS2 to Li atoms in Li2S [58]. In addition, some new peaks emerged. The two peaks at 167.3 and 165.3 eV were attributed to Li2S [60], and a further two peaks at 169.1 and 170.3 eV were assigned to the S–O bonds in the oxidized sulfur species [58]. Therefore, a strong chemical interaction exists between Edg-MoS2/C HMs and Li2S, which facilitates the nucleation of Li2S on the Edg-MoS2/C HMs.
The rapid conversion kinetics of the lithium polysulfides and the strong chemical interactions between the Edg-MoS2/C HMs and Li2S create favorable conditions for the well-distributed nucleation and growth of Li2S in the second stage of the discharge process. To examine the deposition of Li2S on the matrix, the cell was disassembled after the first 100% discharge at 0.05 C. Impressively, the deep discharge product Li2S in Edg-MoS2/C@PP was deposited evenly along the surface of Edg-MoS2/C HMs in the Edg-MoS2/C interlayer and CNTs in the cathode (Fig. 5e, f), which is because the uniform edge sites of the Edg-MoS2/C HMs provided a large number of nucleation sites [40], and the strong chemical absorption and fast reduction capability for soluble polysulfides by Edg-MoS2/C HMs guided the uniform growth of Li2S. Sulfur particles were uniformly deposited on the cathode and the Edg-MoS2/C-modified separator (Fig. 5g, h). This is because the well-distributed Li2S precipitates in the discharge process are beneficial for the accelerated dissolution of Li2S, and the conversion of short-chain polysulfides to long-chain polysulfides is enhanced by the excellent electrocatalytic performance of the Edg-MoS2/C HMs. In contrast, it was found that the very large Li2S precipitates (Fig. 5b) (dozens of micrometers) were deposited on the surface of the cathode in PP, whereas the large Li2S precipitates measuring several micrometers (Fig. 5c, d) were deposited on the surfaces of the cathode and separator for the CN@PP cell. This was caused by the low density of nucleation sites in the cathodes with CN@PP and PP, as well as the sluggish polysulfide reduction kinetics. In addition, the physical adsorption of polysulfides by the CN interlayer occurred in CN@PP.

SEM images of Li2S and S deposition in the redox reactions of the Li–S cells. a SEM images of the pristine CNT/S-1.7 cathode. b SEM images of Li2S deposited on the CNT/S-1.7 cathode in the PP coin cell after the first 100% discharge at 0.05 C. SEM images of Li2S deposition on the CNT/S-1.7 cathode c and the separator d in the CN@PP coin cell after the first 100% discharge at 0.05 C. SEM images of Li2S deposition onto the CNT/S-1.7 cathode e and the separator f in the Edg-MoS2/C@PP coin cell after the first 100% discharge at 0.05 C. SEM images of the CNT/S-1.7 cathode g and the separator h in the Edg-MoS2/C@PP coin cell after the first 100% charge at 0.05 C
On the basis of our results, the well-distributed deposition of Li2S plays a significant role in the discharge and charge processes. Therefore, it is necessary to guide the uniform deposition of Li2S to achieve high-performance Li–S batteries. The binding affinity for polysulfides, electric conductivity, electrocatalytic performance of the matrix, and the density of Li2S binding sites, as well as their distribution on the matrix, are the key factors for controlling the deposition process of Li2S. In detail, (1) the hollow, edge-rich MoS2/C microspheres afford abundant chemical absorption sites for polysulfides, thus effectively restricting the polysulfide shuttle effect and providing the preconditions for the well-distributed deposition of Li2S. (2) The abundant edge sites exposed on the surface of Edg-MoS2/C HMs facilitate the nucleation and dissolution of Li2S, thus improving the reversibility or the phase conversion of the active species. (3) The intrinsic electrocatalytic performance of MoS2 and the enhanced electrical conductivity achieved because of the combination of MoS2 and the carbon network in the hollow, edge-rich MoS2/C microspheres, accelerated the conversion rate of sulfur intermediates, and guided the uniform growth of Li2S, which increased the utilization of active sulfur and improved the specific capacity and rate performance of the Li–S batteries. As a result, hollow, edge-rich MoS2/C microspheres can effectively regulate the uniform deposition of Li2S, resulting in high-performance Li–S batteries.
4 Conclusion
In summary, hollow, edge-rich MoS2/C microspheres were successfully used to functionalize separators to regulate the uniform deposition of Li2S in lithium–sulfur batteries. We confirmed that Edg-MoS2/C HMs could restrict the polysulfide shuttle effect effectively and enhance the conversion kinetics of sulfur intermediates. More importantly, the uniform edge sites on the MoS2/C HMs provide abundant Li2S nucleation sites that guide the growth of Li2S, leading to well-distributed Li2S precipitates on the matrix. The cell containing a separator functionalized with hollow, edge-rich MoS2/C microspheres displayed an initial discharge capacity of 935 mAh g−1 at 1.0 C and maintained a capacity of 494 mAh g−1 after 1000 cycles at a sulfur loading of 1.7 mg cm−2. Impressively, at a high sulfur loading of 6.1 mg cm−2 and high rate of 0.5 C, the cell still delivered a high reversible discharge capacity of 478 mAh g−1 after 300 cycles. This work provides fresh views and solutions to the problems resulting from the complex phase conversion processes in energy storage systems.
References
J. Lu, Z. Chen, Z. Ma, F. Pan, L.A. Curtiss, K. Amine, The role of nanotechnology in the development of battery materials for electric vehicles. Nat. Nanotechnol. 11, 1031–1038 (2016). https://doi.org/10.1038/nnano.2016.207
Z.W. Seh, Y. Sun, Q. Zhang, Y. Cui, Designing high-energy lithium–sulfur batteries. Chem. Soc. Rev. 45(20), 5605–5634 (2016). https://doi.org/10.1039/C5CS00410A
X. Liu, J.Q. Huang, Q. Zhang, L. Mai, Nanostructured metal oxides and sulfides for lithium-sulfur batteries. Adv. Mater. 29(20), 1601759 (2017). https://doi.org/10.1002/adma.201601759
H. Wang, W. Zhang, J. Xu, Z. Guo, Advances in polar materials for lithium–sulfur batteries. Adv. Funct. Mater. 28(38), 1707520 (2018). https://doi.org/10.1002/adfm.201707520
B. Li, H. Xu, Y. Ma, S. Yang, Harnessing the unique properties of 2D materials for advanced lithium–sulfur batteries. Nanoscale Horiz. 4(1), 77–98 (2019). https://doi.org/10.1039/C8NH00170G
H.-J. Peng, J.-Q. Huang, X.-B. Cheng, Q. Zhang, Review on high-loading and high-energy lithium-sulfur batteries. Adv. Energy Mater. 7(24), 1700260 (2017). https://doi.org/10.1002/aenm.201700260
H. Yuan, H.-J. Peng, B.-Q. Li, J. Xie, L. Kong et al., Conductive and catalytic triple-phase interfaces enabling uniform nucleation in high-rate lithium-sulfur batteries. Adv. Energy Mater. 9(1), 1802768 (2019). https://doi.org/10.1002/aenm.201802768
G. Li, S. Wang, Y. Zhang, M. Li, Z. Chen, J. Lu, Revisiting the role of polysulfides in lithium–sulfur batteries. Adv. Mater. 30(22), 1705590 (2018). https://doi.org/10.1002/adma.201705590
H.-J. Peng, G. Zhang, X. Chen, Z.-W. Zhang, W.-T. Xu, J.-Q. Huang, Q. Zhang, Enhanced electrochemical kinetics on conductive polar mediators for lithium–sulfur batteries. Angew. Chem. Int. Ed. 55(42), 12990–12995 (2016). https://doi.org/10.1002/anie.201605676
L.C.H. Gerber, P.D. Frischmann, F.Y. Fan, S.E. Doris, X. Qu et al., Three-dimensional growth of Li2S in lithium–sulfur batteries promoted by a redox mediator. Nano Lett. 16(1), 549–554 (2016). https://doi.org/10.1021/acs.nanolett.5b04189
F.Y. Fan, W.C. Carter, Y.M. Chiang, Mechanism and kinetics of Li2S precipitation in lithium-sulfur batteries. Adv. Mater. 27(35), 5203–5209 (2015). https://doi.org/10.1002/adma.201501559
K. Mi, S. Chen, B. Xi, S. Kai, Y. Jiang, J. Feng, Y. Qian, S. Xiong, Sole chemical confinement of polysulfides on nonporous nitrogen/oxygen dual-doped carbon at the kilogram scale for lithium–sulfur batteries. Adv. Funct. Mater. 27(1), 1604265 (2017). https://doi.org/10.1002/adfm.201604265
Y. Liu, G. Li, J. Fu, Z. Chen, X. Peng, Strings of porous carbon polyhedrons as self-standing cathode host for high-energy-density lithium–sulfur batteries. Angew. Chem. Int. Ed. 56(22), 6176–6180 (2017). https://doi.org/10.1002/anie.201700686
Z. Li, B.Y. Guan, J. Zhang, X.W. Lou, A compact nanoconfined sulfur cathode for high-performance lithium-sulfur batteries. Joule 1(3), 576–587 (2017). https://doi.org/10.1016/j.joule.2017.06.003
J.S. Lee, A. Manthiram, Hydroxylated N-doped carbon nanotube-sulfur composites as cathodes for high-performance lithium-sulfur batteries. J. Power Sources 343, 54–59 (2017). https://doi.org/10.1016/j.jpowsour.2017.01.049
C. Jin, W. Zhang, Z. Zhuang, J. Wang, H. Huang et al., Enhanced sulfide chemisorption using boron and oxygen dually doped multi-walled carbon nanotubes for advanced lithium–sulfur batteries. J. Mater. Chem. A 5(2), 632–640 (2017). https://doi.org/10.1039/C6TA07620C
H. Al Salem, G. Babu, C.V. Rao, L.M.R. Arava, Electrocatalytic polysulfide traps for controlling redox shuttle process of Li–S batteries. J. Am. Chem. Soc. 137(36), 11542–11545 (2015). https://doi.org/10.1021/jacs.5b04472
Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H.-M. Cheng, F. Li, Conductive porous vanadium nitride/graphene composite as chemical anchor of polysulfides for lithium-sulfur batteries. Nat. Commun. 8, 14627 (2017). https://doi.org/10.1038/ncomms14627
G. Zhou, L. Li, D.-W. Wang, X.-Y. Shan, S. Pei, F. Li, H.-M. Cheng, A flexible sulfur-graphene-polypropylene separator integrated electrode for advanced Li–S batteries. Adv. Mater. 27(4), 641–647 (2014). https://doi.org/10.1002/adma.201404210
L. Tan, X. Li, Z. Wang, H. Guo, J. Wang, Lightweight reduced graphene oxide@MoS2 interlayer as polysulfide barrier for high-performance lithium–sulfur batteries. ACS Appl. Mater. Interfaces 10(4), 3707–3713 (2018). https://doi.org/10.1021/acsami.7b18645
Z.Z. Pan, W. Lv, Y.B. He, Y. Zhao, G. Zhou et al., A nacre-like carbon nanotube sheet for high performance Li-polysulfide batteries with high sulfur loading. Adv. Sci. 5(6), 1800384 (2018). https://doi.org/10.1002/advs.201800384
Q. Li, Y. Song, R. Xu, L. Zhang, J. Gao et al., Biotemplating growth of nepenthes-like N-doped graphene as a bifunctional polysulfide scavenger for Li-S batteries. ACS Nano 12(10), 10240–10250 (2018). https://doi.org/10.1021/acsnano.8b05246
J. He, Y. Chen, A. Manthiram, Vertical Co9S8 hollow nanowall arrays grown on a celgard separator as a multifunctional polysulfide barrier for high-performance Li–S batteries. Energy Environ. Sci. 11(9), 2560–2568 (2018). https://doi.org/10.1039/c8ee00893k
H. Wang, W. Zhang, H. Liu, Z. Guo, A strategy for configuration of an integrated flexible sulfur cathode for high-performance lithium-sulfur batteries. Angew. Chem. Int. Ed. 55(12), 3992–3996 (2016). https://doi.org/10.1002/anie.201511673
H.J. Peng, Z.W. Zhang, J.Q. Huang, G. Zhang et al., A cooperative interface for highly efficient lithium-sulfur batteries. Adv. Mater. 28(43), 9551–9558 (2016). https://doi.org/10.1002/adma.201603401
H.J. Peng, D.W. Wang, J.Q. Huang, X.B. Cheng, Z. Yuan, F. Wei, Q. Zhang, Janus separator of polypropylene-supported cellular graphene framework for sulfur cathodes with high utilization in lithium-sulfur batteries. Adv. Sci. 3(1), 1500268 (2016). https://doi.org/10.1002/advs.201500268
J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert, L. Giebeler, Functional mesoporous carbon-coated separator for long-life, high-energy lithium-sulfur batteries. Adv. Funct. Mater. 25(33), 5285–5291 (2015). https://doi.org/10.1002/adfm.201502251
S.-H. Chung, A. Manthiram, Bifunctional separator with a light-weight carbon-coating for dynamically and statically stable lithium-sulfur batteries. Adv. Funct. Mater. 24(33), 5299–5306 (2014). https://doi.org/10.1002/adfm.201400845
F. Qin, X. Wang, K. Zhang, J. Fang, J. Li, Y. Lai, High areal capacity cathode and electrolyte reservoir render practical Li-S batteries. Nano Energy 38(38), 137–146 (2017). https://doi.org/10.1016/j.nanoen.2017.05.037
S.S. Zhang, Liquid electrolyte lithium/sulfur battery: fundamental chemistry, problems, and solutions. J. Power Sources 231, 153–162 (2013). https://doi.org/10.1016/j.jpowsour.2012.12.102
X. Liang, Z. Wen, Y. Liu, M. Wu, J. Jin, H. Zhang, X. Wu, Improved cycling performances of lithium sulfur batteries with LiNO3-modified electrolyte. J. Power Sources 196(22), 9839–9843 (2011). https://doi.org/10.1016/j.jpowsour.2011.08.027
Z.A. Ghazi, X. He, A.M. Khattak, N.A. Khan, B. Liang et al., MoS2/celgard separator as efficient polysulfide barrier for long-life lithium–sulfur batteries. Adv. Mater. 29(21), 1606817 (2017). https://doi.org/10.1002/adma.201606817
W. Cai, G. Li, K. Zhang, G. Xiao, C. Wang, K. Ye, Z. Chen, Y. Zhu, Y. Qian, Conductive nanocrystalline niobium carbide as high-efficiency polysulfides tamer for lithium-sulfur batteries. Adv. Funct. Mater. 28(2), 1704865 (2018). https://doi.org/10.1002/adfm.201704865
L. Zhang, X. Chen, F. Wan, Z. Niu, Y. Wang, Q. Zhang, J. Chen, Enhanced electrochemical kinetics and polysulfide traps of indium nitride for highly stable lithium-sulfur batteries. ACS Nano 12(9), 9578–9586 (2018). https://doi.org/10.1021/acsnano.8b05466
Y. Zhong, L. Yin, P. He, W. Liu, Z. Wu, H. Wang, Surface chemistry in cobalt phosphide-stabilized lithium–sulfur batteries. J. Am. Chem. Soc. 140(4), 1455–1459 (2018). https://doi.org/10.1021/jacs.7b11434
H. Yuan, X. Chen, G. Zhou, W. Zhang, J. Luo et al., Efficient activation of Li2S by transition metal phosphides nanoparticles for highly stable lithium–sulfur batteries. ACS Energy Lett. 2(7), 1711–1719 (2017). https://doi.org/10.1021/acsenergylett.7b00465
J. Zhang, Y. Shi, Y. Ding, L. Peng, W. Zhang, G. Yu, A conductive molecular framework derived Li2S/N, P-codoped carbon cathode for advanced lithium–sulfur batteries. Adv. Energy Mater. 7(14), 1602876 (2017). https://doi.org/10.1002/aenm.201602876
D. Su, M. Cortie, G. Wang, Fabrication of N-doped graphene–carbon nanotube hybrids from prussian blue for lithium–sulfur batteries. Adv. Energy Mater. 7(8), 1602014 (2017). https://doi.org/10.1002/aenm.201602014
R. Fang, S. Zhao, Z. Sun, D.-W. Wang, R. Amal, S. Wang, H.-M. Cheng, F. Li, Polysulfide immobilization and conversion on a conductive polar MoC@MoOx material for lithium-sulfur batteries. Energy Storage Mater. 10, 56–61 (2018). https://doi.org/10.1016/j.ensm.2017.08.005
H. Wang, Q. Zhang, H. Yao, Z. Liang, H.W. Lee, P.C. Hsu, G. Zheng, Y. Cui, High electrochemical selectivity of edge versus terrace sites in two-dimensional layered MoS2 materials. Nano Lett. 14(12), 7138–7144 (2014). https://doi.org/10.1021/nl503730c
M. Zhao, H.-J. Peng, Z.-W. Zhang, B.-Q. Li, X. Chen et al., Activating inert metallic compounds for high-rate lithium–sulfur batteries through in situ etching of extrinsic metal. Angew. Chem. Int. Ed. 131(12), 3819–3823 (2019). https://doi.org/10.1002/ange.201812062
Z.-W. Zhang, H.-J. Peng, M. Zhao, J.-Q. Huang, Heterogeneous/homogeneous mediators for high-energy-density lithium–sulfur batteries: progress and prospects. Adv. Funct. Mater. 28(38), 1707536 (2018). https://doi.org/10.1002/adfm.201707536
L. Kong, B.-Q. Li, H.-J. Peng, R. Zhang, J. Xie, J.-Q. Huang, Q. Zhang, Porphyrin-derived graphene-based nanosheets enabling strong polysulfide chemisorption and rapid kinetics in lithium–sulfur batteries. Adv. Energy Mater. 8(20), 1800849 (2018). https://doi.org/10.1002/aenm.201800849
H. Yuan, H.-J. Peng, J.-Q. Huang, Q. Zhang, Sulfur redox reactions at working interfaces in lithium–sulfur batteries: a perspective. Adv. Mater. Interfaces 6(4), 1802046 (2019). https://doi.org/10.1002/admi.201802046
Q. Gao, S. Wang, H. Fang, J. Weng, Y. Zhang, J. Mao, Y. Tang, One-dimensional growth of MoOx-based organic–inorganic hybrid nanowires with tunable photochromic properties. J. Mater. Chem. 22(11), 4709–4715 (2012). https://doi.org/10.1039/C2JM15443A
J. Pu, Z. Shen, J. Zheng, W. Wu, C. Zhu, Q. Zhou, H. Zhang, F. Pan, Multifunctional Co3S4@sulfur nanotubes for enhanced lithium-sulfur battery performance. Nano Energy 37, 7–14 (2017). https://doi.org/10.1016/j.nanoen.2017.05.009
T. Chen, L. Ma, B. Cheng, R. Chen, Y. Hu et al., Metallic and polar Co9S8 inlaid carbon hollow nanopolyhedra as efficient polysulfide mediator for lithium–sulfur batteries. Nano Energy 38, 239–248 (2017). https://doi.org/10.1016/j.nanoen.2017.05.064
H. Jin Fan, M. Knez, R. Scholz, K. Nielsch, E. Pippel, D. Hesse, M. Zacharias, U. Gösele, Monocrystalline spinel nanotube fabrication based on the kirkendall effect. Nat. Mater. 5, 627–631 (2006). https://doi.org/10.1038/nmat1673
H. Jiang, D. Ren, H. Wang, Y. Hu, S. Guo, H. Yuan, P. Hu, L. Zhang, C. Li, 2D monolayer MoS2–carbon interoverlapped superstructure: engineering ideal atomic interface for lithium ion storage. Adv. Mater. 27(24), 3687–3695 (2015). https://doi.org/10.1002/adma.201501059
J. Wu, Z. Lu, K. Li, J. Cui, S. Yao et al., Hierarchical MoS2/carbon microspheres as long-life and high-rate anodes for sodium-ion batteries. J. Mater. Chem. A 6(14), 5668–5677 (2018). https://doi.org/10.1039/C7TA11119C
J. Zhu, Y. Ge, D. Kim, Y. Lu, C. Chen, M. Jiang, X. Zhang, A novel separator coated by carbon for achieving exceptional high performance lithium-sulfur batteries. Nano Energy 20, 176–184 (2016). https://doi.org/10.1016/j.nanoen.2015.12.022
Y. Zhao, M. Liu, W. Lv, Y.-B. He, C. Wang, Q. Yun, B. Li, F. Kang, Q.-H. Yang, Dense coating of Li4Ti5O12 and graphene mixture on the separator to produce long cycle life of lithium-sulfur battery. Nano Energy 30(30), 1–8 (2016). https://doi.org/10.1016/j.nanoen.2016.09.030
J.-Q. Huang, Q. Zhang, H.-J. Peng, X.-Y. Liu, W.-Z. Qian, F. Wei, Ionic shield for polysulfides towards highly-stable lithium–sulfur batteries. Energy Environ. Sci. 7(1), 347–353 (2014). https://doi.org/10.1039/C3EE42223B
L. Yan, N. Luo, W. Kong, S. Luo, H. Wu et al., Enhanced performance of lithium-sulfur batteries with an ultrathin and lightweight MoS2/carbon nanotube interlayer. J. Power Sources 389, 169–177 (2018). https://doi.org/10.1016/j.jpowsour.2018.04.015
P. Guo, D. Liu, Z. Liu, X. Shang, Q. Liu, D. He, Dual functional MoS2/graphene interlayer as an efficient polysulfide barrier for advanced lithium-sulfur batteries. Electrochim. Acta 256, 28–36 (2017). https://doi.org/10.1016/j.electacta.2017.10.003
P. Han, S.-H. Chung, A. Manthiram, Thin-layered molybdenum disulfide nanoparticles as an effective polysulfide mediator in lithium–sulfur batteries. ACS Appl. Mater. Interfaces 10(27), 23122–23130 (2018). https://doi.org/10.1021/acsami.8b05397
J. He, L. Luo, Y. Chen, A. Manthiram, Yolk–shelled C@Fe3O4 nanoboxes as efficient sulfur hosts for high-performance lithium–sulfur batteries. Adv. Mater. 29(34), 1702707 (2017). https://doi.org/10.1002/adma.201702707
J. Wu, H. Zeng, X. Li, X. Xiang, Y. Liao, Z. Xue, Y. Ye, X. Xie, Ultralight layer-by-layer self-assembled MoS2-polymer modified separator for simultaneously trapping polysulfides and suppressing lithium dendrites. Adv. Energy Mater. 8(35), 1802430 (2018). https://doi.org/10.1002/aenm.201802430
Z. Yuan, H.J. Peng, T.Z. Hou, J.Q. Huang, C.M. Chen et al., Powering lithium-sulfur battery performance by propelling polysulfide redox at sulfiphilic hosts. Nano Lett. 16(1), 519–527 (2016). https://doi.org/10.1021/acs.nanolett.5b04166
M. Fantauzzi, B. Elsener, D. Atzei, A. Rigoldi, A. Rossi, Exploiting XPS for the identification of sulfides and polysulfides. RSC Adv. 5(93), 75953–75963 (2015). https://doi.org/10.1039/C5RA14915K
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 51672083); Program of Shanghai Academic/Technology Research Leader (18XD1401400); Basic Research Program of Shanghai (17JC1404702); Leading talents in Shanghai in 2018; The 111 project (B14018); and the Fundamental Research Funds for the Central Universities (222201718002).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zheng, N., Jiang, G., Chen, X. et al. Battery Separators Functionalized with Edge-Rich MoS2/C Hollow Microspheres for the Uniform Deposition of Li2S in High-Performance Lithium–Sulfur Batteries. Nano-Micro Lett. 11, 43 (2019). https://doi.org/10.1007/s40820-019-0275-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-019-0275-z