
Abstract
The prototypical photocatalyst TiO2 exists in different polymorphs, the most common forms are the anatase- and rutile-crystal structures. Generally, anatase is more active than rutile, but no consensus exists to explain this difference. Here we demonstrate that it is the bulk transport of excitons to the surface that contributes to the difference. Utilizing high –quality epitaxial TiO2 films of the two polymorphs we evaluate the photocatalytic activity as a function of TiO2-film thickness. For anatase the activity increases for films up to ~5 nm thick, while rutile films reach their maximum activity for ~2.5 nm films already. This shows that charge carriers excited deeper in the bulk contribute to surface reactions in anatase than in rutile. Furthermore, we measure surface orientation dependent activity on rutile single crystals. The pronounced orientation-dependent activity can also be correlated to anisotropic bulk charge carrier mobility, suggesting general importance of bulk charge diffusion for explaining photocatalytic anisotropies.
Similar content being viewed by others

Introduction
Titania (TiO2) is the most widely used photocatalyst1,2,3 for decomposition of organic pollutants because it is chemically stable and biologically benign. The band gap of TiO2 is larger than 3 eV (~3.0 for rutile and ~3.2 for anatase), thus making pure TiO2 primarily active for UV light. The most common commercial photocatalyst is the Degussa P-25, a powder consisting of both rutile and anatase crystallites4. The phase mixture of different polymorphs is known to have synergistic effects and an increased photocatalytic activity is observed compared to pure phases5. However, for pure phases it is generally accepted that anatase exhibits a higher photocatalytic activity compared to rutile TiO26. Furthermore, not only do the two polymorphs show varying photoactivity, but the different crystallographic orientations of the same material may exhibit different activities7,8,9,10,11,12. Despite the intensive study of TiO2 there is no generally accepted explanation for the differences of photocatalytic activity of different polymorphs or surface orientations. Possible explanations may be categorized as follows:
-
Anatase has a larger band gap than rutile TiO2. While this reduces the light that can be absorbed, it may raise the valence band maximum to higher energy levels relative to redox potentials of adsorbed molecules. This increases the oxidation ‘power’ of electrons and facilitates electron transfer from the TiO2 to adsorbed molecules13. This explanation has also been expanded to explain surface orientation dependent activities by suggesting that different surfaces exhibit different band gaps14.
-
Surface properties may play a role in the adsorption of molecules and subsequent charge transfer to the molecule. The surface properties may not just be polymorph dependent but may differ largely for the same material for different surface orientations or reconstructions15,16 and consequently may contribute to the observation of pronounced surface effects in photocatalytic activities. Surface properties may again be sub-divided into (i) chemical effects, e.g. coordination structure of surfaces that controls adsorption of molecules17, (ii) electronic structure of the clean surface18 or defects and adsorbate (e.g. hydroxyl)-induced states that may be crucial for charge trapping and separation at the surface19, (iii) interaction of molecules with surface defects6,20 and (iv) surface potential differences (such as work function differences measured in vacuum or flat band potentials in aqueous solution)21,22 may affect charge transfer from the photocatalyst to molecules. It should be mentioned that the relative position of the conduction band minimum (CBM) in rutile and anatase is still controversial, while the large band gap of anatase might suggest the CBM in anatase to be higher than for rutile and this has been so far the general perception11, recent results are suggesting that conduction band of anatase is actually lower than that of rutile5.
-
Anatase exhibits an indirect band gap that is smaller than its direct band gap. For rutile, on the other hand, its fundamental band gap is either direct or its indirect band gap is very similar to its direct band gap. Semiconductors with indirect band gap generally exhibit longer charge carrier life times compared to direct gap materials. A longer electron-hole pair life in anatase than in rutile would make it more likely for charge carriers to participate in surface reactions. One evidence for longer charge carrier lifetimes in anatase than in rutile comes from transient photoconductivity measurements on single crystalline samples23.
-
Charge transport may differ for different polymorphs. In addition to the exciton lifetime the exciton mobility needs to be taken into account. Only excitons that efficiently diffuse can reach the surface within their life time. Preferential diffusion of excitons along certain crystallographic directions has been proposed for other photocatalysts to be important to explain surface orientation dependencies in their oxidation/reduction behavior11. One measure for exciton mobility is the polaron effective mass. Although contradicting values for effective masses are reported, generally a higher effective mass is reported for rutile than for anatase. The polaron effective mass for rutile is ~7–8 m0 (where m0 is the electron mass) while anatase exhibits a polaron effective mass of ~m024,25,26. In addition, in rutile a strong anisotropy for the effective electron masses exists and consequently, its charge mobility is reported, with values ~2–4 m0 along the <001> direction and ~10–15 m0 along the <100> direction27,28. No values are reported for other crystallographic directions. Here we demonstrate that bulk charge carrier transport indeed explains the difference between rutile and anatase and furthermore is consistent with orientation dependent activity variations in rutile.
One main obstacle that has prevented a better fundamental description of titania photocatalysis is the masking of bulk properties by complex surface effects. Here we describe a new approach that enables separating surface from bulk effects in describing the photocatalytic activity and to compare photoactivity on rutile and anatase TiO2. Utilizing thin epitaxial films of anatase and rutile we evaluate the photocatalytic activity as a function of film thickness. Since the surface properties are the same for any film thickness of the same material any change in the photoactivity can be solely ascribed to the increased bulk volume. The increase in the photocatalytic activity with film thickness is thus a consequence of more excitons, generated by photo-absorption in the bulk, reaching the surface. In this case the photoactivity-increase saturates for TiO2-films that are thicker than the layer that contributes charges for surface reactions, i.e. when the film becomes thicker than the maximum exciton diffusion length. Consequently, this approach enables us to measure for the first time quantitatively the surface region that contributes to photocatalytic reactions. We demonstrate that this surface region is larger for anatase than for rutile and this difference contributes to the different photocatalytic performances of these two TiO2 polymorphs. Furthermore, we investigate different crystallographic orientations for rutile and find that the orientation dependency may also be correlated to bulk anisotropies in exciton diffusion.
Results
We first describe the structure and morphology of the thin rutile and anatase TiO2 films. This is followed by measurements of the thin films' photocatalytic activity and the dependence of it on the film thickness. The relationship between photocatalytic activity and film thickness contains information on the bulk exciton diffusion length in the two different TiO2 polymorphs. In order to connect the findings on the polymorph-dependency of photocatalytic activity of TiO2 with crystallographic anisotropies we subsequently performed measurements on rutile single crystal samples with different crystallographic orientations.
Structure and properties of TiO2 films
Epitaxial rutile and anatase TiO2 films have been grown by a variety of physical vapor deposition methods including (oxygen plasma assisted) molecular beam epitaxy ((OPA)MBE)29 and sputter deposition. Rutile TiO2(101) has been grown on r-cut sapphire (α-Al2O3) (1-102)30,31,32 while anatase TiO2(001) has been previously synthesized on SrTiO3 or LaAlO3 (100) substrates33,34,35,36,37,38. For anatase TiO2 on LaAlO3 the crystallographic relationship is (001)[-110]anatase//(001)[110]LAO and for rutile TiO2 on sapphire the crystallographic relationship is (101)[-111]rutile//(-1102)[20-2-1]sapphire. For rutile TiO2(101) grown on r-cut sapphire it is known that it forms twin domain structures with coherent boundaries in {101} planes30. In the studies reported here mainly LaAlO3 and Al2O3 are used, because these substrates (contrary to SrTiO3) are wide band gap materials and do not exhibit any photocatalytic properties by themselves. Furthermore, the large band-gap of the substrate prevents charges to be transferred to the substrate.
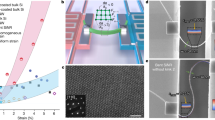
Central to the success of measuring photocatalytic activity as a function of film thickness is the growth of well-defined TiO2-films and thus we briefly present key characterizations of the prepared films. Fig. 1 shows characterization of anatase films and Fig. 2 for rutile films. Reflection high energy electron diffraction (RHEED) patterns of the as prepared films are shown in Fig. 1 (a) and 2(a) for the anatase (001) and rutile (101) samples, respectively. For the anatase sample a 4 × 1 superstructure is observed in the RHEED pattern. This is the typical surface reconstruction for the anatase (001) surface in vacuum15,39,40 and the fairly sharp diffraction pattern confirms a good surface quality of the film. For the rutile (101) surface no superstructure spots are observed despite the fact that the rutile (101) surface is known to reconstruct into a 2 × 1 superstructure41,42,43. Absence of surface superstructure spots is consistent with the diffraction pattern exhibiting bulk-like diffraction and thus may indicate a somewhat larger surface roughness.

Characterization of anatase (001) films.
(a) and (b) RHEED pattern along the <101> and <110> azimuths, respectively. Note the superstructure streaks in (a) indicating the 4 × 1 surface reconstruction. (b) and (c) show ambient AFM images, indicating atomically flat terraces. (e) cross-sectional TEM of the LAO/anatase interface, with (f) showing the diffraction pattern of the interface indicating the epitaxial alignment of the anatase film.

Characterization of rutile (101) films.
(a) and (b) RHEED pattern along the <010> and <−101> azimuths, respectively. (b) and (c) show ambient AFM images, indicating two crystal orientations due to twinning in the film. (e) cross-sectional TEM of the Al2O3/rutile interface, with (f) showing the diffraction pattern of the interface indicating the epitaxial alignment of the rutile film.
The surface roughness has been characterized by atomic force microscopy (AFM) for every sample. Fig. 1(b) and 2(b) show typical images for anatase and rutile films, respectively. On the anatase films, flat terraces with mono-atomic steps are observed indicating a well-defined crystalline surface quality in agreement with the RHEED pattern. The rutile samples exhibits slightly higher surface roughness, with some ~40 nm wide ‘grains’ with roughness ~2 nm for a 12 nm thick film. The ‘grains’ have a slightly rectangular shape and two kinds of rectangular grains oriented 90° to each other are observed. These are due to the before mentioned twinning of the film30. As the zoomed-in image in Fig. 2(b) shows the individual ‘grains’ are flat and atomic-height step edges can be imaged.
Transmission electron microscopy (TEM) imaging and selected area electron diffraction (SAED) of 25 nm thick films further corroborate the high crystalline order and epitaxial relationship between substrate and film. Fig. 1(e), (f) and 2(e), (f) show TEM images and corresponding diffraction patterns (DPs) for electron beam along the <0–10>LAO/<100>TiO2 and <110>Al2O3/<010>TiO2 for the anatase and rutile films, respectively. The SAED were taken with a ~500 nm aperture at several points along the film. In addition a 20 nm diameter electron beam was scanned along the film and the diffraction pattern monitored. No other phases were detected in the diffraction patterns, in particular the anatase films were phase-pure and formation of any rutile inclusions can be excluded. X-ray photoemission spectra (XPS) of the films are compared to those of rutile single crystal samples and no discernible difference is observed indicating the formation of stoichiometric TiO2 within the sensitivity of XPS.
Photocatalytic activity of rutile(101) and anatase(001) films
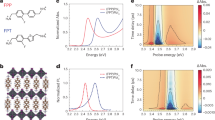
The photocatalytic activity of the films is measured by photocatalytic decomposition of an organic dye (methyl orange). Fig. 3 (a) and (b) illustrate a typical measurement of the methyl orange concentration versus reaction time for a TiO2 film. From the rate of methyl orange decomposition the photocatalytic activity for different TiO2 films is determined and this information is plotted in Fig. 3 (c) as a function of the film thickness. For thick films, it is apparent that the photoactivity for the anatase films is about twice the activity of rutile in agreement with the general notion that anatase is the photocatalytically more active material. Important for this study is, however, the dependence of the photoactivity with film thickness. For the rutile films, the photocatalytic activity does not change significantly for films thicker than ~2.5 nm. Only for the very thinnest films the activity drops. This suggests that for thick films excitons generated deeper than ~2.5 nm from the surface do not reach the surface, i.e. they recombine before reaching the surface and thus do not contribute to photoreactions. For anatase films, on the other hand, the photocatalytic activity increases to film thickness larger than 5 nm. This indicates that in anatase charge carriers from deeper in the bulk reach the surface compared to rutile.

Evaluation of photocatalytic activity of samples by decomposition of an organic dye (methyl orange).
(a) shows the absorption spectra for the methyl orange solution for different irradiation times. The peak area of the absorption spectra is a direct measurement of the molecule concentration and thus its decrease with UV-irradiation time is a measure of the photocatalytic decomposition of the molecule. In (b) the absorption peak area is plotted versus irradiation time for anatase films with different film thicknesses. Fitting an exponential decay function gives the photocatalytic decomposition rate for the different samples. This measured rate is plotted in (c) as a function of film thickness for the rutile and anatase films. The anatase films reach a higher photocatalytic activity for thick films. However, the maximum activity is reached already for ~2.5 nm thick films for rutile, while the maximum activity is only reached for ~5 nm thick films for anatase.
Dependence of photocatalytic activity on surface orientation and sample preparation conditions of rutile single crystals
High quality epitaxial thin films may only be grown with a few surface orientations. Thus, in order to investigate the variation of the photocatalytic activity as a function of surface orientation, we resort to studies of single crystal rutile TiO2. No anatase single crystals of large-enough size are available to investigate surface dependence of the photocatalytic activity on anatase with our approach. However, it should be mentioned that there does exist some interesting investigations on surface engineered powder samples that exhibit preferential surface orientations44,45,46,47,48,49,50,51,52.
Figure 4 (a), shows the measured photocatalytic activity for rutile samples with different surface orientations for different surface preparation methods. For all sample preparation procedures, with exception of HF-etched and tube furnace annealed samples, the activity follows the order (101) > (110) > (001) > (100) for photocatalytic degradation of organics. For HF etched and tube furnace annealed samples the (001) orientation exhibited a slightly higher activity than the (110) sample. Remarkably, the photoactivity of the ‘as-received’ samples are as much as 30% higher than the samples after HF-etching and tube furnace annealing, which results in a much better defined surface as indicated in the AFM images shown in Fig. 4(b) and (c). Formation of surface defects has been discussed in several publications to affect surface charge trapping and charge transfer to adsorbates and/or water53,54,55,56,57. Our observation of a variation of the overall activity of the single crystals on the surface preparation is in agreement with such an influence of the surface morphology. This further underlines the challenge in separating bulk from surface effects for photoactivity measurements and illustrates the need of identical sample preparation to enable quantitative comparisons.

Photocatalytic activity measurements on rutile single crystals with four different surface orientations and three different sample preparation conditions.
For all preparation conditions the (101) orientation is the most active surface. (b) and (c) shows AFM images for as-received and after HF and annealing treatment for all four sample orientations.
Importantly, the single crystal studies on rutile (101) show very similar photocatalytic activity as those of the rutile (101) films. The same photocatalytic activity of the films and the single crystal demonstrates that the films are of single-crystal quality in terms of photocatalytic activity. In particular, this implies that the twin-boundary structure and the slightly increased surface roughness of the rutile films compared to the single crystal surfaces does not adversely affect the photocatalytic activity of the films. We also point out that the (101) surface is the most photocatalytically active surface of all the rutile surface orientations studied, in agreement with previous reports7,8,10. This is important for comparing the overall photocatalytic activity of the rutile and anatase films. The fact that the most active rutile surface is significantly less active than the anatase (001) surface (which according to some reports12 is only the second most active anatase surface) further validates the fact that anatase is photocatalytically more active than rutile.
Discussion
The general perception that anatase has a higher photocatalytic activity compared to rutile TiO2 is confirmed by our measurements on extended planar epitaxial thin films. The anatase (001) films (in the thick-limit: ~20 nm) exhibit around twice the activity for photocatalytic decomposition of organic molecules than the rutile (101) films grown under identical conditions. Importantly, the film thickness-dependence of the photocatalytic activity demonstrates that this difference in the photocatalytic activity is at least partially a bulk property of the two forms of TiO2. In particular the measurements show similar activity (or slightly higher activity for rutile) for very thin films (less than 2 nm) but while the activity for rutile films remains almost unchanged for films thicker than 2 nm the activity for anatase films keeps increasing and only saturates for films thicker than ~5 nm. This behavior indicates that charge carriers for photocatalytic reactions can originate from much deeper in the bulk for anatase than for rutile. The film thicknesses are much smaller than the absorption depth of light and thus light absorption cannot be responsible for the saturation of the photocatalytic activity. Also, the film thickness is smaller than typical depletion regions in oxides, which excludes band-bending effects for charge separation. The studies reported here also compare favorably with previously reported work on photocatalytic activity as a function of film thickness for rutile films using photoreduction of Ag ions as a measure of the photoactivity6. In these studies a sharp increase in the photoactivity for thin films up to less than 10 nm thickness was reported which then plateaued. This is very similar to the results presented here, however, different to the studies shown here a further increase in the activity at a lower rate has also been observed. The two different rates of increase in photoactivity suggest two different mechanisms at work. In studies reported here the increase for much thicker TiO2 samples could not be observed and the photocatalytic activity truly saturates at less than 10 nm. This difference in the two investigations is likely a consequence of the different photoreactions studied. In particular Ag-clusters that formed during photoreaction in previous work will modify the photocatalyst and this can give rise to additional phenomena.
In order to quantify the charge diffusion length normal to the surface of our macroscopically planar samples, we fit the increase in photocatalytic activity with increasing film thickness by an exponential dependence of the form: k = C [1 − exp(−d/λ)], where k is the photocatalytic activity of the films (equivalent to the measured decomposition rate constant) and d is the film thickness. C and λ are fitting parameters, where C corresponds to the activity for very thick films (or bulk samples). The best fit parameters give a value of Canatase = 0.0033 ± 0.0003, λanatase = 3.2 ± 0.6 nm and Crutile = 0.0018 ± 0.0001, λrutile = 1.6 ± 0.4 nm, for anatase and rutile respectively. The parameter λ may be interpreted as the (surface-normal) charge diffusion length and its value indicates the distance from the surface at which a generated charge carrier has a probability of 1/e to reach the surface. The films studied here differ from pure TiO2 by the presence of an interface with a substrate. Consequently it may be important to consider how this interface may affect our observations. There are three main potential contributions by which the interface could distort the measured photocatalytic properties compared to a hypothetical ideal case of a ‘free’ TiO2 sheet. Firstly, charge carriers may be trapped and recombine at the interface and the rate of this process may be different for the LaAlO3 or Al2O3 substrates. Secondly, the lattice matching at the interface will induce some strain in the film that could affect the exciton diffusion to the surface. Thirdly, the lattice mismatch will facilitate point-defect formation in the film that varies with film thickness. All three of these effects are likely present to some extent but we argue that they do not obscure the main conclusion of a twice longer exciton diffusion length in anatase compared to rutile. The observation that rutile films, only 5 nm thick (a thickness where substrate induced strain is expected to be still present), exhibit a photocatalytic activity that is identical as that for rutile single crystal samples, indicates that strain in the films does not significantly alter the photocatalytic properties. In terms of charge trapping and recombination at the interface, it is important to realize that this effect can only modify the photocatalytic activity for films appreciably thinner than the charge diffusion length (d < λ) and any influence from charge recombination at the interface diminishes as the film thickness approaches λ. Thus exciton recombination at the interface may only contribute to a deviation from the ideal exponential-functional dependence used to describe the behavior of the photocatalytic activity versus film thickness, but it will not change the film thickness at which the photocatalytic activity saturates. In order to assess the general possibility of lattice mismatch induced variation in the film properties, including formation of point defects, we compared the activity of anatase films grown on LaAlO3(001) with those grown on SrTiO3(001). These substrates exhibit largely varying lattice mismatch with respect to anatase films of 0.1% and 3.1%, respectively36. Despite this large misfit for SrTiO3, we measure the same (within 5%) photocatalytic activity as for anatase films on LaAlO3 if the films thicker than 5 nm. For thinner films charge carriers generated in the photocatalytically active SrTiO3 substrate can contribute to the photocatalytic reactions and thus a slightly larger activity is measured compared to LaAlO3 substrates for very thin films. Thus from these arguments and test-studies we conclude that interface effects will not affect the key result of a larger λ for anatase than rutile and consequently our measurements show, for the first time conclusively, that the material-volume that contributes to the photocatalytic activity is significantly larger for anatase than for rutile.
The charge diffusion length λ is a convolution of charge carrier life time and charge carrier diffusivity. Arguably, it is the diffusion length λ that is the important property for characterizing the efficiency of a photocatalyst. The difference in the charge diffusion length between rutile and anatase may have its origin in the longer life-time of charge excitations and/or higher charge carrier mobilities in anatase than in rutile. Both properties have been previously reported3 but could not be unambiguously linked to photocatalytic activity differences between polymorphs.
Our determination that bulk properties are important to explain differences in the photoactivity of different polymorphs of TiO2 can also be applied to the measured surface orientation dependence for the rutile samples. The known bulk anisotropy in the effective masses and charge mobility in rutile along and perpendicular to the c-axis, i.e. (001) and (100) surface orientations, respectively, correlates well with the observed photocatalytic activity measurements. Independently from the surface preparation, we consistently measure higher photocatalytic activity for the (001) direction than for the (100) direction for which room temperature mobilities of μ(001) = 8 cm2/V s and μ(100) = 1.4 cm2/V s are reported respectively26. Unfortunately, to the best of our knowledge, no charge mobility data for the (101) or (110) direction are known.
In conclusion, this investigation demonstrates the importance of bulk properties for the production of more efficient photocatalysts. For TiO2, it appears that a surface region of only a few nanometer depths contributes charge carriers to photoreactions. Higher activity in e.g. ZnO5 may be attributed to higher charge mobility in ZnO and thus the search for better photocatalysts should take charge mobilities and exciton life times into account. Finally, the approach described here for determining the active surface regions may not only be applied to pure materials but also to bulk dopant modified photocatalysts. This may enable future studies to extract information on the influence of dopants on the overall photocatalytic performance.
Methods
Epitaxial TiO2 film growth and characterization
LaAlO3(100), SrTiO3(100) and  (r-cut) substrates (MTI Corp.) were used for TiO2 growth. The substrates were ultrasonically cleaned in acetone and ethanol to remove any residual surface contaminants. To ensure identical growth conditions and identical film thicknesses a LaAlO3(100) and an
(r-cut) substrates (MTI Corp.) were used for TiO2 growth. The substrates were ultrasonically cleaned in acetone and ethanol to remove any residual surface contaminants. To ensure identical growth conditions and identical film thicknesses a LaAlO3(100) and an  substrates were mounted together and the TiO2 film was grown on both substrates at the same time. Before growth the samples were heated in the growth chamber at 600°C in a 2 × 10−6 Torr O2 atmosphere for 3 hours.
substrates were mounted together and the TiO2 film was grown on both substrates at the same time. Before growth the samples were heated in the growth chamber at 600°C in a 2 × 10−6 Torr O2 atmosphere for 3 hours.
TiO2 films were grown by pulsed laser deposition (PLD). The PLD ultra high vacuum (UHV) chamber was equipped with quartz-micro balance for calibrating and monitoring the deposition rate and a reflection high energy electron diffraction (RHEED) optics. A long target-to-substrate distance (8 cm) reduces the growth rate and eliminated the deposition of particulates from the ablation process. The target was ablated with a 355 nm Nd:YAG laser (Symphotic Tii). The TiO2 films were grown with the substrates at 600°C and an oxygen background pressure of 2 × 10−6 Torr. The deposition rate was in a range of 0.07 to 0.09 nm/min. The film thickness derived from the microbalance readings was checked on selected samples by ellipsometry and compared to cross-sectional TEM images for one sample.
Ambient atomic force microscopy (AFM) (Park Scientific XE 70) characterizations were performed on all samples to ensure comparable surface morphologies and the measured rms-roughness was used to estimate the uncertainty in film thickness.
TEM characterization was conducted at the Center for Functional Nanomaterials (CFN) at Brookhaven National Laboratory, Upton, NY. Thin sections for TEM were prepared by focused ion beam milling. High-resolution TEM (HRTEM) imaging and selected area electron diffraction were performed in a JEOL JEM-2100F at 200 kV.
Rutile single crystal preparation
In addition to thin epitaxial films we also conducted studies on rutile single crystals with (011), (110), (100) and (001) orientation. Epi-polished crystals were obtained from MTI-corp. and their orientations were checked with x-ray diffraction. The as received crystals were flat but did not exhibit a clearly defined step structure as the AFM images in Fig. 4 show. To improve and obtain better defined surface morphology a slightly modified method of a previously reported procedure58,59 was used. Briefly, the substrates were etched in 10% HF for 30 min, cleaned in ethanol and rinsed with DI-water and subsequently annealed in 200 mTorr O2 at 800°C for 1 h. This procedure resulted in well-defined stepped surfaces.
We also measured the activity of the samples after annealing in ultra-high vacuum at 600°C for 30 min. This causes a slight reduction of the samples as was evident from a change in color from transparent (slight yellowish) to a blue hue.
Photocatalytic activity measurement and analysis
The photocatalytic activity of different samples has been evaluated by measuring the photocatalytic decomposition of methyl orange under UV illumination60. A 100 W Hg arc lamp (Oriel) equipped with a water-cooled IR filter was used as the light source. The 5 × 5 mm square samples were suspended within a closed glass cuvette in a methylene orange (Fisher Scientific) solution. The glass cuvette has a transmission cut-off at ~350 nm (3.54 eV) so that only the near UV portion of the spectrum of our UV-lamp was transmitted and reached the sample. At regular time intervals the sample was taken out of the cuvette and the transmission of the methyl orange solution was measured with UV-Vis spectrometer. The intensity of the orange absorption of the solution at a wavelength of ~489 nm is a direct measure of the decomposition of the dye and thus of the photocatalytic activity. A base line of the decomposition of the methyl orange without a photocatalytically active sample, e.g. a bare LaAlO3 substrate, shows a very small decrease in the orange absorption with irradiation time (see Fig. 3(b)). This base-line has been subtracted from all other measurements in order to only monitor the methyl orange decomposition due to photocatalytic action only. The intensity of the absorption peak is plotted as a function of irradiation time and the decrease is fitted with an exponential decay function (see Fig. 3(b)) in order to measure the rate constant. The rate constant of the decomposition has been used as a measure for the photoactivity of the films and single crystal samples. Since all the samples have identical exposed surface area, the decay time is directly used for comparing the photocatalytic activity of different samples. For TiO2-films this photocatalytic activity value was plotted as a function of film thickness to derive information on the volume of the TiO2 that contributes to the photoactivity.
Uncertainties in the measured photocatalytic activity are determined from the standard deviation in the photoactivity of the samples thicker than ten nanometers, i.e. samples in the thick limit where the activity does not increase anymore. The error bars for the film thickness shown in Fig. 3(c) are the rms-roughness of the films measured by AFM.
References
Fujishima, A., Zhang, X. & Tryk, D. A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 63, 515–582 (2008).
Linsebigler, A. L., Lu, G. Q. & Yates, J. T. Photocatalysis on TiO2 surfaces-principles, mechanisms and selected results. Chem. Rev. 95, 735–758 (1995).
Henderson, M. A. A surface science perspective onTiO2 photocatalysis Surf. Sci. Rep. 66, 185–297 (2011).
Ohno, T., Sarukawa, K., Tokieda, K. & Matsumura, M. Morphology of a TiO2 photocatalyst (Degussa, P-25) consisting of anatase and rutile crystalline phases. J. Catal. 203, 82–86 (2001).
Scanlon, D. O. et al. Band alignement of rutile and anatase TiO2 . Nature Mater. 12, 798–801 (2013).
Liu, L., Zhao, H., Andino, J. M. & Li, Y. Photocatalytic CO2 reduction with H2O on TiO2 nanocrystals: Comparison of anatase, rutile and brookite polymorphs and exploration of surface chemistry. ACS Catal. 2, 1817–1828 (2012).
Kislov, N. et al. Photocatalytic Degradation of Methyl Orange over Single Crystalline ZnO: Orientation Dependence of Photoactivity and Photostability of ZnO. Langmuir 25, 3310–3315 (2009).
Morris Hotsenpiller, P. A., Bolt, J. D., Farneth, W. E., Lowekamp, J. B. & Rohrer, G. S. Orientation dependence of photochemical reactions on TiO2 surfaces. J. Phys. Chem. B 102, 3216–3226 (1998).
Lowekamp, J. B., Rohrer, G. S., Morris Hotsenpiller, P. A., Bolt, J. D. & Farneth, W. E. Anisotropic photochemical reactivity of bulk TiO2 crystals. J. Phys. Chem. B 102, 7323–7327 (1998).
Ohno, T., Sarukawa, K. & Matsumura, M. Crystal faces of rutile and anatase TiO2 particles and their roles in photocatalytic reactions. New J. Chem. 26, 1167–1170 (2002).
Giocondi, J. L., Salvador, P. A. & Rohrer, G. S. The origin of photochemical anisotropy in SrTiO3 . Top. Catal. 44, 529–533 (2007).
Yamamoto, Y., Nakajima, K., Ohsawa, T., Matsumoto, Y. & Koinuma, H. Preparation of atomically smooth TiO2 single crystal surfaces and their photochemical property. Jap. J. Appl. Phys. 44, L511–L514 (2005).
Batzill, M. Fundamental aspects of surface engineering of transition metal oxide photocatalysts. Energy Environm. Sci. 4, 3275–3286 (2011).
Pan, J., Liu, G., Lu, G. Q. & Cheng, H.-M. On the true photoreactivity order of {001},{010} and {101} facets of anatase TiO2 crystals. Angew. Chem., Int. Ed. 50, 2133–2137 (2011).
Wilson, J. N. & Idriss, H. Effect of surface reconstruction of TiO2(001) single crystal on the photoreaction of acetic acid. J. Catal. 214, 46–52 (2003).
Wilson, J. N. & Idriss, H. Structure sensitivity and photocatalytic reactions of semiconductors. Effect of the last layer atomic arrangement. J. Amer. Chem. Soc. 124, 11284–11285 (2002).
Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 48, 53–230 (2003).
Tao, J. G. & Batzill, M. Role of surface structure on the charge trapping in TiO2 photocatalysts. J. Phys. Chem. Lett. 1, 3200–3206 (2010).
Tao, J. G., Luttrell, T. & Batzill, M. A two-dimensional phase of TiO2 with a reduced bandgap. Nature Chem. 3, 296–300 (2011).
Setvín, M. et al. Reaction of O2 with Subsurface Oxygen Vacancies on TiO2 Anatase (101). Science 341, 988–991 (2013).
Bullard, J. W. & Cima, M. J. Orientation dependence of isoelectric point of TiO2 (rutile) surfaces. Langmuir 22, 10264–10271 (2006).
Hengerer, R., Kavan, L., Krtil, P. & Grätzel, M. Orientation dependence of charge-transfer processes on TiO2 (anatase) single crystal. J. Electrochem. Soc. 147, 1467–1472 (2000).
Xu, M. et al. Photocatalytic activity of bulk TiO2 anatase and rutile single crystals using infrared absorption spectroscopy. Phys. Rev. Lett. 106, 138302 (2011).
Tang, H., Prasad, K., Sanjines, R., Schmid, P. E. & Levy, F. Electrical and optical properties of TiO2 anatase thin films. J. Appl. Phys. 75, 2042 (1994).
Furubayashi, Y. et al. Transport properties of d-electron-based transparent conducting oxide: Anatase Ti1−xNbxO2 . J. Appl. Phys. 101, 093705 (2007).
Thulin, L. & Guerra, J. Calculations of strain-modified anatase TiO2 band structures. Phys. Rev. B 77, 195112 (2008).
Yagi, E., Hasiguti, R. R. & Aono, M. Electronic conduction above 4 K of slightly reduced oxygen-deficient rutile TiO2−x . Phys. Rev. B 54, 7945 (1996).
Hendry, E., Wang, F., Shan, J., Heinz, T. F. & Bonn, M. Electron transport in TiO2 probed by THz time-domain spectroscopy. Phys. Rev. B 69, 081101(R) (2004).
Chambers, S. A. Epitaxial growth and properties of thin film oxides. Surf. Sci. Rep. 39, 105–180 (2000).
Jalan, B., Engel-Herbert, R., Cagnon, J. & Stemmer, S. Growth modes in metal-organic molecular beam epitaxy of TiO2 on r-plane sapphire. J. Vac. Sci. Technol. A 27, 230–233 (2009).
Huang, J. Y. et al. High-resolution transmission electron microscopy study of defects and interfaces in epitaxial TiO2 films on sapphire and LaAlO3 . Philosph. Mag. A 82, 735–749 (2002).
Engel-Herbert, R., Jalan, B., Cagnon, J. & Stemmer, S. Microstructure of epitaxial rutile TiO2 films grown by molecular beam epitaxy on r-plane Al2O3 . J. Crystal Growth 312, 149–153 (2009).
Sasahara, A., Droubay, T. C., Chambers, S. A., Uetsuka, H. & Onishi, H. Topography of anatase TiO2 film synthesized on LaAlO3(001). Nanotechnol. 16, S18–S21 (2005).
Liang, Y., Gan, S. P., Chambers, S. A. & Altman, E. I. Surface structure of anatase TiO2(001): Reconstruction, atomic steps and domains. Phys. Rev. B 63, 235402 (2001).
Chambers, S. A. et al. Epitaxial growth and properties of ferromagnetic co-doped TiO2 anatase. Appl. Phys. Lett. 79, 3467–3469 (2001).
Kennedy, R. J. & Stampe, P. A. The influence of lattice mismatch and film thickness on the growth of TiO2 on LaAlO3 and SrTiO3 substrates. J. Cryst. Growth 252, 333–342 (2003).
Jeong, B. S., Budai, J. D. & Norton, D. P. Epitaxial stabilization of single crystal anatase films via reactive sputter deposition. Thin Solid Films 422, 166–169 (2002).
Yamamoto, S., Sumita, T., Sugiharuto, Miyashita, A. & Naramoto, H. Preparation of epitaxial TiO2 films by pulsed laser deposition technique. Thin Solid Films 401, 88–93 (2001).
Herman, G. S., Sievers, M. R. & Gao, Y. Structure determination of the two-domain (1 × 4) anatase TiO2(001) surface. Phys. Rev. Lett. 84, 3354–3357 (2000).
Lazzeri, M. & Selloni, A. Stress-driven reconstruction of an oxide surface: The anatase TiO2(001)-(1 × 4) surface. Phys. Rev. Lett. 87, 266105 (2001).
Beck, T. J. et al. Surface structure of TiO2(011)-(2 × 1). Phys. Rev. Lett. 93, 036104 (2004).
Torrelles, X. et al. Geometric structure of TiO2(011)(2 × 1). Phys. Rev. Lett. 101, 185501 (2008).
Cuan, Q., Tao, J. G., Gong, X. Q. & Batzill, M. Adsorbate induced restructuring of TiO2(011)-(2 × 1) leads to one-dimensional nanocluster formation. Phys. Rev. Lett. 108, 106105 (2012).
Yang, H. G. et al. Solvothermal synthesis and photoreactivity of anatase TiO2 nanosheets with dominant {001} facets. J. Amer. Chem. Soc. 131, 4078–4083 (2009).
Gordon, T. R. et al. Nonaqueous synthesis of TiO2 nanocrystals using TiF4 to engineer morphology, oxygen vacancy concentration and photocatalytic activity. J. Amer. Chem. Soc. 134, 6751–6761 (2012).
Wu, B., Guo, C., Zheng, N., Xie, Z. & Stucky, G. D. Nonaqueous production of nanostructured anatase with high-energy facets. J. Amer. Chem. Soc. 130, 17563–17567 (2008).
Han, X., Kuang, Q., Jin, M., Xie, Z. & Zheng, L. Synthesis of titania nanosheets with a high percentage of exposed (001) facets and related photocatalytic properties. J. Amer. Chem. Soc. 131, 3152–3153 (2009).
Dai, Y., Cobley, C. M., Zeng, J., Sun, Y. & Xia, Y. Synthesis of anatase TiO2 nanocrystals with exposed {001} facets. Nano Lett. 9, 2455–2459 (2009).
Jiang, H. B. et al. Anatase TiO2 crystals with exposed high-index facets. Angew. Chem. Int. Ed. 50, 3764–3768 (2011).
Tachikawa, T., Yamashita, S. & Majima, T. Evidence for Crystal-Face-Dependent TiO2 Photocatalysis from Single-Molecule Imaging and Kinetic Analysis. J. Amer. Chem. Soc. 133, 7197–7204 (2011).
D'Arienzo, M. et al. Photogenerated Defects in Shape-Controlled TiO2 Anatase Nanocrystals: A Probe To Evaluate the Role of Crystal Facets in Photocatalytic Processes. J. Amer. Chem. Soc. 133, 17652–17661 (2011).
Roy, N., Sohn, Y. & Pradhan, D. Synergy of Low-Energy {101} and High-Energy {001} TiO2 Crystal Facets for Enhanced Photocatalysis. ACS Nano 7, 2532–2540 (2013).
Pan, X. Y., Yang, M. Q., Fu, X. Z., Zhang, N. & Xu, Y. J. Defective TiO2 with oxygen vacancies: synthesis, properties and photocatalytic applications. Nanoscale 5, 3601–3614 (2013).
Zhuang, J. D., Weng, S. X., Dai, W. X., Liu, P. & Liu, Q. Effects of Interface Defects on Charge Transfer and Photoinduced Properties of TiO2 Bilayer Films. J. Phys. Chem. C 116, 25354–25361 (2012).
Chen, X. B., Liu, L., Yu, P. Y. & Mao, S. S. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals. Science 331, 746–750 (2011).
Zhuang, J. D. et al. Photocatalytic degradation of RhB over TiO2 bilayer films: Effect of defects and their location. Langmuir 26, 9686–9694 (2010).
Radoicic, M. B. et al. The role of surface defect sites of titania nanoparticles in the photocatalysis: Aging and modification. Appl. Catal. B 138, 122–127 (2013).
Nakamura, R., Okamura, T., Ohashi, N., Imanishi, A. & Nakato, Y. Molecular mechanisms of photoinduced oxygen evolution, PL emission and surface roughening at atomically smooth (110) and (100) n-TiO2 (rutile) surfaces in aqueous acidic solutions. J. Amer. Chem. Soc. 127, 12975–12983 (2005).
Yamamoto, Y., Matsumoto, Y. & Koinuma, H. Homo-epitaxial growth of rutile TiO2 film on step and terrace structured substrate. Appl. Surf. Sci. 238, 189–192 (2004).
Yu, L. et al. The degradation mechanism of methyl orange under photo-catalysis of TiO2 . Phys. Chem. Chem. Phys. 14, 3589–3595 (2012).
Acknowledgements
Financial support from DOE- BES under grant no. DE-FG02-09ER1608 is acknowledged. The TEM characterization of the TiO2 films was performed at the Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. The authors thank Kim Kisslinger for technical support.
Author information
Authors and Affiliations
Contributions
T.L. grew and characterized TiO2 films and co-wrote the manuscript, S.H. performed experiments on rutile single crystal samples, J.T. assisted with experimental set-up, E.S. did the TEM characterization, A.K. assisted with TiO2-film characterization, M.B. directed the research and wrote the manuscript. All authors discussed the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Luttrell, T., Halpegamage, S., Tao, J. et al. Why is anatase a better photocatalyst than rutile? - Model studies on epitaxial TiO2 films. Sci Rep 4, 4043 (2014). https://doi.org/10.1038/srep04043
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04043
This article is cited by
-
Detection of thin film phase transformations at high-pressure and high-temperature in a diamond anvil cell
Communications Earth & Environment (2024)
-
Understanding the light induced hydrophilicity of metal-oxide thin films
Nature Communications (2024)
-
Evaluation of antifungal activity of visible light-activated doped TiO2 nanoparticles
Photochemical & Photobiological Sciences (2024)
-
An insight into the photocatalytic degradation of the antibiotic rifampicin by titanium dioxide nanoparticles in aqueous solution under UV light irradiation
Reaction Kinetics, Mechanisms and Catalysis (2024)
-
Biosynthesis, characterization and optimization of TiO2 nanoparticles by novel marine halophilic Halomonas sp. RAM2: application of natural dye-sensitized solar cells
Microbial Cell Factories (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
