
Abstract
Thread-like chains of flexible polymers that adsorb to a solid surface assume a flat ‘pancake’ conformation1 when the surface coverage is low and are only able to diffuse in two dimensions because so many segments are adsorbed. Here we show that the centre-of-mass diffusion coefficient of the polymer chain, measured at dilute coverage to ensure minimal chain–chain interaction, has a strong power-law dependence on the degree of polymerization. This nonlinear dependence of polymer diffusion on a solid surface contrasts with the linear dependence observed on a fluid membrane2.
Similar content being viewed by others

Main
Our system consisted of polyethylene glycol (PEG) adsorbed from aqueous solution onto a monolayer surface of self-assembled octadecyltriethoxysilane3 coated onto a fused silica coverslip to render it hydrophobic (because PEG does not adsorb from aqueous solution to hydrophilic fused silica at high pH).
Diffusion was investigated by fluctuation correlation spectroscopy4,5,6 of a mono-end-labelled fluorescent probe after two-photon excitation. The focused two-photon beam creates an illuminated spot with a beam waist of about 0.3 μm. The small number of fluorophores contained within a given volume (typically 3–10) fluctuates as polymers diffuse in and out. The fit to the autocorrelation function determines the mutual diffusion coefficient (DM) of the fluorescing species, and DM≈D, the centre-of-mass diffusion coefficient, because the system is dilute. Experiments were followed using a Zeiss microscope with a 63× Plan Apochromat objective (numerical aperture 1.4).
We varied the PEG polymer chain length (Fig. 1 legend) by a factor of 15. Polymers were allowed to adsorb for less than 5 min from dilute (1–10 nM) solution in 0.01 M aqueous phosphate buffer, pH 8.4. To study bulk solutions, PEG samples were labelled with fluorescein 5-isothiocyanate, and for monitoring surface diffusion, we used an Alexa-488 label (from Molecular Probes), a derivatized rhodamine-green molecule. Control experiments demonstrated that unattached fluorescent labels did not adsorb, confirming that adsorption was controlled by polymer–surface attraction. At this dilute surface coverage, 0.5–2% of the saturating amount of polymer adsorbed.

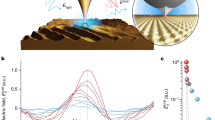
a, Flexible polymer chains that adsorb are nearly flat at dilute surface coverage (‘de Gennes pancake’). The sticking energy for each segment is small, so no single segment is bound tightly, but the molecular sticking energy is large. b, Diffusion coefficients (D) in dilute solution (circles) and at dilute coverage on a solid surface (squares) are plotted on log–log scales against degree of polymerization (N) at 22 °C, for chains of weight-average molecular weights, Mw, of 2,200, 5,000, 10,800, 20,100 and 30,500 g-mol−1 and Mw/Mn values of 1.01–1.03 (where Mn is the number-average molecular weight).
We estimated the ‘sticking energy’ of the polymer as 0.5–1 kBT per segment, where kB is Boltzmann's constant at T the absolute temperature. This emerged consistently from analysis of the Langmuir isotherm at low surface coverage, and from Arrhenius analyses of the very slow desorption rate and of the data presented below after extrapolation to the molecular weight of a single segment. Polymer chains thus adsorb, at low coverage, in a flat ‘pancake’ conformation1 (Fig. 1a).
Measurements of D (Fig. 1b) reveal a power-law scaling with the number of chain segments (N), so D≈N−3/2. This is strikingly stronger than the D≈N−1 relation observed for charged, semi-flexible DNA obeying excluded volume statistics but adsorbed by Coulombic attraction on a fluid lipid membrane2. The key difference is that adsorption sites on a solid surface are static, so the rate-limiting events concern the polymer rather than surface. For diffusion in solution, our results are consistent with standard hydrodynamic results for chains of moderate length, when D≈N−1/2 (refs 7,8).
Reptation (the diffusion of a chain, snakelike, along its own length) may explain this stronger dependence of D on N for polymers under our conditions. In this model1, the terminal relaxation time scales as τrept≈N3. Knowing that the radius of gyration (Rg) scales as Rg≈N3/4 in a good solvent in two dimensions2,9, and arguing that D≈Rg2/τrept, it follows that D≈N−3/2 for chains with excluded-volume statistics, as we find here. A simulation for a single self-avoiding chain diffusing among regularly spaced obstacles in two dimensions also gives D≈N−3/2 (ref. 10). But interpreting the value of scaling exponents is a problem, particularly as they seem to depend on obstacle density11.
Reptation may be considered surprising in a dilute system where the physical origin of the static constraints that suppress lateral motion is unclear. But if there were some slack between sticking points — for example, loops of an isolated flexible chain might propagate with high probability along its length in a caterpillar-like fashion — the mathematics of the reptation model would then apply, despite the unconventional physical situation.
References
de Gennes, P.-G Scaling Concepts in Polymer Physics (Cornell Univ. Press, Ithaca, NY, 1979).
Maier, B. & Rädler, O. Phys. Rev. Lett. 82, 1911–1914 (1999).
Kessel, C. R. & Granick, S. Langmuir 7, 532–538 (1991).
Magde, D., Elson, E. L. & Webb, W. W. Biopolymers 13, 29–61 (1974).
Berland, K. M., So, P. T. C. & Gratton, E. Biophys. J. 68, 694–701 (1995).
So, P. T. C. et al. Bioimaging 3, 1–15 (1995).
Yuko, K. & Chikako, H. Polym. Commun. 25, 154–157 (1984).
Huber, K., Bantle, S., Lutz, P. & Burchard, W. Macromolecules 18, 1461–1467 (1985).
des Cloizeaux, J. & Jannink, G. Polymers in Solution (Clarendon, Oxford, 1990).
Azuma, R. & Takayama, H. J. Chem. Phys. 111, 8666–8671 (1999).
Slater, G. W. & Wu, S. Y. Phys. Rev. Lett. 75, 164–168 (1995).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Sukhishvili, S., Chen, Y., Müller, J. et al. Diffusion of a polymer ‘pancake’. Nature 406, 146 (2000). https://doi.org/10.1038/35018166
Issue Date:
DOI: https://doi.org/10.1038/35018166
This article is cited by
-
Probing the interplay between chain diffusion and polymer crystal growth under nanoscale confinement: a study by single molecule fluorescence microscopy
Science China Chemistry (2018)
-
Polymers in focus: fluorescence correlation spectroscopy
Colloid and Polymer Science (2014)
-
Molecular weight dependence of crystal pattern transitions of poly(ethylene oxide)
Chinese Journal of Polymer Science (2013)
-
Aggregation behavior of thermo-responsive poly(2-oxazoline)s at the cloud point investigated by FCS and SANS
Colloid and Polymer Science (2012)
-
A molecular-dynamics simulation study of diffusion of a single model carbonic chain on a graphite (001) surface
Journal of Molecular Modeling (2006)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
