
Abstract
New antibiotics are necessary to treat microbial pathogens that are becoming increasingly resistant to available treatment. Despite the medical need, the number of newly approved drugs continues to decline. We offer an overview of the pipeline for new antibiotics at different stages, from compounds in clinical development to newly discovered chemical classes. Consistent with historical data, the majority of antibiotics under clinical development are natural products or derivatives thereof. However, many of them also represent improved variants of marketed compounds, with the consequent risk of being only partially effective against the prevailing resistance mechanisms. In the discovery arena, instead, compounds with promising activities have been obtained from microbial sources and from chemical modification of antibiotic classes other than those in clinical use. Furthermore, new natural product scaffolds have also been discovered by ingenious screening programs. After providing selected examples, we offer our view on the future of antibiotic discovery.
Similar content being viewed by others
Main
Medical progress in the prevention and treatment of many diseases, which have resulted in significantly increasing life expectancy, may be put at risk without the introduction into clinical practice of new antibiotics effective against multidrug-resistant (MDR) pathogens. Although most stakeholders agree that new antibiotics could tackle this unmet medical need, opinions vary on how new antibiotics could be discovered and brought into the market in a cost-effective manner.1, 2, 3 Two considerations would probably meet with unanimous consensus: the golden era of antibiotic discovery is gone and it will not be repeated; and genomics, combinatorial chemistry and high-throughput screening do not represent the magic bullet to fill the pipeline with new developmental drug candidates. In this respect, it is important to underline the contribution that natural products, especially those of microbial origin, can provide to antibiotic discovery, as advocated by Demain4, 5 on several occasions. The decreasing number of drugs approved for clinical use, year after year, suggests that the ‘ailing pharmaceutical industry’ is not yet following the ‘prescription’ of Demain,6 as spelled out in 2002.
The purpose of this review is to highlight some of today's features of antibiotic discovery in the context of the current medical needs and the existing pipeline of antibacterial agents in clinical development. Our main focus will be on chemical classes that, if developed into drugs, would be new to the clinic. However, these classes would not necessarily be new to science. For example, a ‘look-back’ strategy was applied to antibiotics discovered during the golden era, which were then reexamined using contemporary tools in the light of current medical needs.7 Although some important breakthroughs have also been made in identifying new promising drug candidates from synthetic origin, for reason of space, and in the spirit of the important contributions to the field by Demain, we would limit ourselves to antibiotics of microbial origin and their derivatives reported since 2005.
Current antibiotic pipeline
Infections due to methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecium (VRE) and fluoroquinolone-resistant Pseudomonas aeruginosa are rapidly increasing in US hospitals, and even more frightening is the recent occurrence of panantibiotic-resistant infections, involving Acinetobacter species, MDR P. aeruginosa and carbapenem-resistant Klebsiella species.8, 9 Although antibiotic resistance continues to grow in hospitals and in the community, involving both Gram-positive and Gram-negative pathogens, the number of newly approved agents has been decreasing, with only six new antibiotics approved since 2003.
In the late 90s, following the global concern regarding the rapid increase in MRSA, many companies redirected their attention to target Gram-positive pathogens, particularly MRSA, VRE and penicillin-resistant Streptococcus pneumoniae, as evidenced by the commercial and clinical success of linezolid and daptomycin, the only antibiotics belonging to new classes introduced in clinical practice since the early 1960s. However, most antibiotics currently under development for Gram-positive infections are improved derivatives of existing drugs (see Table 1). As vancomycin has been increasingly used for the treatment of a wide range of infections, second-generation glycopeptides with improved profile over vancomycin were developed. Among them, telavancin, a once-a-day derivative of vancomycin, was approved by the US Food and Drug Administration (FDA) in 2009. Oritavancin, derived from the vancomycin-related glycopeptide chloroeremomycin, is highly active against VRE strains and shows a long plasma half-life. However, in 2008, the FDA did not authorize its commercialization. The long-acting glycopeptide dalbavancin, a derivative of the teicoplanin-related glycopeptide A40926, was also not approved by FDA, because of insufficient clinical evidence of efficacy. If approved, dalbavancin would be the first antibiotic to be administered once weekly.10
Resistance to methicillin in S. aureus is mediated by the production of a penicillin-binding protein with reduced affinity for β-lactams. The most recent cephalosporins, ceftobiprole and ceftaroline (Table 1), have been specifically designed to enhance activity against MRSA and, thanks to their oral availability, are particularly attractive for the community setting. Ceftobiprole is quickly bactericidal against a wide range of Gram-positive pathogens, including MRSA and VRE and has been approved in Canada and Switzerland.11 However, early in 2010, the FDA did not grant market authorization to ceftobiprole, and later the European authority issued a negative opinion on this compound. Ceftaroline, which is active against most Gram-positive pathogens with the exclusion of enterococci, has completed phase III studies and may be submitted for FDA approval.12 Both cephalosporins, however, lose potency against MRSA compared with methicillin-susceptible S. aureus strains. The injectable carbapenem PZ-601 has shown potent activity against drug-resistant Gram-positive pathogens, including MRSA, and is currently undergoing phase II studies.13
After the success of linezolid, many new oxazolidinones are being developed for Gram-positive infections. Radezolid14 and torezolid15 are currently in phase II trials, whereas RWJ-416457 has completed the phase I trial. Despite the fact that the use of fluoroquinolones has been associated with increased incidence of MRSA,16 several new members of this class are under development: delafloxacin, nemonoxacin, zabofloxacin and WCK-771 (Table 1) are the most advanced.
The extensive use of fluoroquinolones and other wide-spectrum antibiotics such as cephalosporins, by affecting the normal gut flora, has led to the rapid diffusion of Clostridium difficile-associated diarrhea, particularly in elderly and immunocompromised patients. Difimicin, currently in phase III, and ramoplanin, with phase II completed, are microbial products under development for prevention and treatment of C. difficile-associated diarrhea, acting locally by decolonizing the gut (Table 1).
Other compounds which have completed phase I clinical trials include the oral and injectable pleuromutilin BC-3205,17 the FabI inhibitor AFN-1252 targeting staphylococcal infections18 and the lipopeptide friulimicin (Table 1).19
The scenario is even more disappointing for compounds targeting Gram-negative pathogens, in which old drugs have been revamped for new uses, and none of them has reached phase III yet (Table 1). Ceftazidime is a marketed cephalosporin being developed in combination with NXL104, a representative of a new class of β-lactamase inhibitors,20 which renders cephalosporin effective against most β-lactamase-producing enterobacteria. If approved, this combination would be the first alternative to piperacillin/tazobactam. NXL104 is also under investigation in combination with ceftaroline.21 CXA-101 is a ceftazidime-like compound with improved stability against the AmpC β-lactamase, but it shows no improvements against MDR P. aeruginosa,22 unless administered in combination with tazobactam. The new aminoglycoside ACHN-490, effective against pathogens resistant to this class, has recently completed phase I.23 The new monobactam BAL-30072, stable against metalloenzymes, is ready to start clinical development against difficult-to-treat Gram negatives, including Pseudomonas and Acinetobacter.24
The increasing spread of MDR Gram-negative pathogens, particularly P. aeruginosa, Acinetobacter spp. and some Enterobacteriaceae has renewed the interest toward narrow-spectrum compounds, to avoid other clinical conditions associated with the use of broad-spectrum antibiotics. However, because of a long history of success in the empirical treatment of infections, many hospitals lack rapid and effective tools for identifying etiological agents. This limitation poses significant hurdles for the clinical development of narrow-spectrum compounds.
Approaches leading to new antibiotic classes
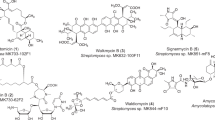
It is generally agreed that the best way to overcome the decreasing efficacy of existing antibacterial agents is to introduce into practice compounds belonging to classes that are new to the clinic. Microbial sources can provide a rich reservoir of such compounds, and the different approaches used usually aim at discovering either a novel class or an improved variant of a poorly explored class. However, this must be carried out in a high background of many known compounds, some of which are encountered in random screening programs at a relatively high frequency. Thus, the discovery of an antibacterial agent belonging to a new chemical class or an improved variant of an existing class is a rare event, and the approaches described below reflect strategies designed and implemented to capture this rare event. Appropriate strategies include retrieving microbial strains from underexplored environments, screening new microbial taxa, mining microbial genomes and using innovative assays. These strategies have led to some novel chemical classes, as illustrated in Figure 1.

Examples of new chemical classes discovered from microorganisms. Only one congener is reported for each class.
As an example of the first strategy, investigation of deep-sea sediment samples led to the discovery of abyssomicins (Figure 1), which are polycyclic antibiotics from the new marine actinomycete taxon Verrucosispora.25 The compounds were discovered using a simple agar diffusion assay, which involved pursuing antibiotics the action of which could be reverted upon addition of p-aminobenzoic acid. Abyssomicins represent a new chemical class, and preliminary studies indicate that they act as substrate mimics of chorismate. Interestingly, only abyssomicin C and its atrop stereoisomer show antibiotic activity against Gram-positive bacteria, including MDR S. aureus.26
An additional example of a new chemical class discovered by screening new taxa is represented by thuggacins (Figure 1), which are thiazole-containing macrolides produced by the myxobacteria Sorangium cellulosum and Chondromyces crocatus.27 These compounds show activity against Mycobacterium tuberculosis and their target appears to be the electron transport chain.
Another successful approach has been exploring microbial genomes for the presence of secondary metabolite pathways. As the corresponding genes are organized in clusters and bioinformatic tools allow a reasonable prediction of the pathway product, this bioactivity-independent approach can directly target structural novelty. On a pioneering work of this type, scientists at Ecopia Biosciences (now Thallion Pharmaceuticals, Montreal, QC, Canada) identified ECO-02301, a linear polyene from Streptomyces aizunensis with antifungal activity28 and ECO-0501, a glycosidic polyketide from Amycolatopsis orientalis with activity against Gram-positive pathogens, including MDR isolates (Figure 1).29 In a similar approach, a novel cyclic lipopeptide, designated orfamide (Figure 1), was identified from the Pseudomonas fluorescens genome.30 In this case, the bioinformatic prediction that the peptide contained four leucine residues suggested feeding with 15N-Leu, which facilitated compound purification and characterization. Orfamide shows a moderate antifungal activity against amphotericin-resistant strains of Candida albicans and may prove beneficial in agriculture and crop protection.
Another important strategy for discovering new classes of antibiotics has been the implementation of increased-sensitivity assays in screening programs. One such approach relied on the antisense technology. When the level of a desired bacterial target is depleted by overexpression of the cognate antisense mRNA, the strain becomes hypersensitive to compounds acting on that target. By using a target against which few compounds are known to act, the increased sensitivity of the assay should allow the identification of compounds routinely missed with growth inhibition assays on the wild-type strain.31 One assay involved the FabH/FabF enzyme, required for fatty acid biosynthesis in bacteria. Antimicrobial activities were detected by agar diffusion in a two-plate assay, in which one plate was inoculated with S. aureus carrying the antisense construct and the other plate with an S. aureus control. Different inhibition halos in the two plates indicated an increased sensitivity of the ‘antisense strain.’ After screening >250 000 microbial product extracts, the assay led to the identification of a new chemical class that includes platensimycin (Figure 1), produced by Streptomyces platensis, and related compounds. Platensimycin shows antibacterial activity against Gram-positive pathogens, including MDR strains, and was also effective in an experimental model of infection.32
In another increased-sensitivity assay, a high-throughput screening program was implemented to identify inhibitors of a cell-free translational system affecting steps other than elongation. The assay made use of a model ‘universal’ mRNA that could be translated with similar efficiency by cell-free extracts from bacterial, yeast or mammalian cells. The rationale behind the approach was to use a sensitive assay and to discard frequently encountered compounds using a polyU-based assay. This program led to the identification of GE81112 (Figure 1), a novel tetrapeptide produced by a Streptomyces sp., which targets specifically the 30S ribosomal subunit by interfering with fMet-tRNA binding to the P-site.33 The compound was highly effective against a few Gram-positive and Gram-negative strains, if grown in minimal or chemically defined medium, suggesting active uptake by the cells.34
The above examples illustrate how different approaches can lead to novel antibiotic classes. Usually, when unexploited microbial diversity is accessed, there is no need for specific, high-sensitivity assays. Whichever the approach chosen, there is no guarantee of success. The reader is referred to a recent review for suggestions on how to increase the probabilities of success.35
Improved variants from microbial sources
New variants of known classes can be found by screening microbial strains, by varying cultivation procedures or by manipulating the biosynthetic pathway. There is an increasing amount of literature related to pathway manipulation and this trend is likely to continue as methodological advancements result in increased success rates. In some cases, the desired variant might not be a more active compound, but a molecule carrying functional groups suitable for further chemical modifications. As the antibiotics in clinical use belong to a few classes, which have been extensively explored by screening and chemical modification, there is probably little space for finding improved variants within those classes. We provide selected examples of microbial strains producing improved variants of chemical classes not yet in clinical use.
Lantibiotics, which are ribosomally synthesized peptides that undergo posttranslational modifications to yield the active structures containing the typical thioether-linked (methyl)lanthionines, are produced mostly from strains belonging to the Firmicutes and, to a lesser extent, to the Actinobacteria. Their antimicrobial activity is limited to Gram-positive bacteria. The prototype molecule is nisin, discovered in the 1920s and used as a food preservative for >40 years.36 Lantibiotics with antibacterial activity are divided into two classes according to their biogenesis: lanthionine formation in class I compounds requires two separate enzymes, a dehydratase and a cyclase, whereas a single enzyme carries both activities for class II lantibiotics. Until recently, the occurrence of class I compounds was limited to the Firmicutes (see below). Although compounds from both classes exert their antimicrobial activity by binding to Lipid II, they do so by binding to different portions of this key peptidoglycan intermediate.
As lantibiotics bind Lipid II at a site different from that affected by vancomycin and related glycopeptides, they are active against MDR Gram-positive pathogens and have attracted attention as potential drug candidates. The compound NVB302, a derivative of deoxyactagardine B (Figure 2a) produced by a strain of Actinoplanes liguriae, is currently a developmental candidate for the treatment of C. difficile-associated diarrhea.37 Independently, a screening program, designed to detect cell-wall-inhibiting compounds turned out to be very effective in identifying lantibiotics from actinomycetes.38 It consisted of identifying extracts active against S. aureus but inactive against isogenic L-forms, discarding extracts the activity of which was abolished by β-lactamases or by excess N-caproyl-D-alanyl-D-alanine. Among the new lantibiotics identified, the most active compound was NAI-107 (Figure 2a), produced by Microbispora sp.39 This compound represents the first example of a class I lantibiotic produced by actinomycetes. It is currently a developmental candidate for the treatment of nosocomial infections by Gram-positive pathogens.40 The same screening program led to the identification of additional class I lantibiotics from actinomycetes. Among them, the compound 97518 (Figure 2a), structurally related to NAI-107,41 afforded improved derivatives by chemical modification.42 Another interesting advancement in the lantibiotic field has been the discovery of two-component lantibiotics produced by members of the class Bacilli. The best characterized compound is haloduracin43, 44 (Figure 2a), whereas lichenicidin has been proposed from genomic studies but has not yet confirmed by structural elucidation.45 Although their antimicrobial activities have not been described in detail, recent work suggests similar activities for haloduracin and nisin.44

(a) Examples of lantibiotics. (b) Recently discovered thiopeptides.
Thiazolylpeptides are highly modified, ribosomally synthesized peptides that inhibit bacterial protein synthesis by affecting either one of two targets: elongation factor Tu, as for GE2270 and related compounds; or the loops defined by 23S rRNA and the L11 protein, exemplified by thiostrepton. Most thiazolylpeptides show potent activity against Gram-positive pathogens, yet their poor solubility has limited clinical progress, and only a derivative of GE2270 has entered clinical trials for the topical treatment of acne.46 Novel members of this class have been described (Figure 2b): thiomuracins47 belong to the subgroup targeting EF-Tu, with an antibacterial profile similar to GE2270; thiazomycin48 and philipimycin,49 which target the 50S subunit, show high activity against Gram-positive strains, and a similar profile to thiostrepton.
For ribosomally synthesized peptides, such as lantibiotics and thiopeptides, new representatives can be generated by site-directed mutagenesis of the corresponding structural genes. Libraries of new molecules have been obtained, many of which, as in the examples of actagardine50 and thiocillin,51 retained antibiotic activities comparable with those of the parent molecule.
Chemical derivatives
Many papers have been published in the past 5 years reporting chemical programs aimed at overcoming the prevailing resistance mechanisms and/or to improve the drug profile of known microbial products. Novel approaches included the use of new tools, such as click chemistry and total synthesis. For the classical approach of semi-synthesis, we will limit the examples to selected compounds not yet in clinical use.
Click chemistry is a new synthetic approach that can accelerate drug discovery by using a few practical and reliable reactions. A ‘click’ reaction must be of wide scope, giving consistently high yields with various starting materials; it must be easy to perform, insensitive to oxygen or water and use only readily available reagents; finally, reaction work-up and product isolation must be simple, without chromatographic purification.52 As an example, this approach was used to produce new lipophilic teicoplanin and ristocetin aglycons with improved activity against Gram-positive bacteria, including VRE.53 For aminoglycosides, which usually require multiple protection–deprotection steps to selectively manipulate the desired amino and hydroxyl groups, click chemistry allowed the transformation of neomycin B into several novel building blocks that were used for the specific modification of the ring systems, thus generating new neomycin analogs the biological activity of which is currently under investigation.54
For some low-molecular-weight compounds, total synthesis has become available and will be useful to design preliminary SAR for new classes of antibiotics (such as platensimycin) or to access new derivatives for already known classes (such as tetracyclines). Indeed, the novel scaffold and intriguing biological property of platensimycin captured the interest of several research groups, which reported different elegant total syntheses.55 In addition, medicinal chemistry studies have been conducted, and the design, synthesis and biological evaluation of several platensimycin analogs incorporating varying degrees of molecular complexity have been reported.56, 57, 58 Preliminary data indicate that certain modifications of the intricate cage region can be made without detrimental effects on potency, whereas even small modifications of the benzoic acid region result in a drastic loss of activity (Figure 1). Another remarkable chemical improvement in the synthesis of natural product analogs was a short and enantioselective synthetic route to a diverse range of 6-deoxytetracycline antibiotics (Figure 3a). This new approach targeted not a single compound but a group of structures with the D ring as a site of structural variability. A late-stage, diastereoselective C-ring construction was used to couple structurally varied D-ring precursors with an AB precursor containing much of the essential functionality for binding to the bacterial ribosome. Results of antibacterial assays and preliminary data obtained from a murine septicemia model show that many of the novel tetracyclines synthesized have potent antibiotic activities. This synthetic platform gives access to a broad range of tetracyclines that would be inaccessible by semi-synthesis and provides a powerful engine for the discovery of new tetracyclines.59, 60

(a) Total synthesis of new tetracyclines. (b) Semi-synthetic derivatives of known antibiotics. Modified portions are in bold type.
Even on larger molecules, semi-synthetic and synthetic chemistry has been successfully applied to study and optimize lead compounds. The lipoglycodepsipeptide ramoplanin (Figure 3b) is 2–10 times more active than vancomycin against Gram-positive bacteria and maintains full activity against VRE and all MRSA strains. However, its systemic use has been prevented by its low tolerability at the injection site, apparently related to the length of the fatty acid side chain. To overcome this problem, the fatty acid side chain was selectively removed and replaced with different carboxylic acids. Many derivatives showed an antimicrobial activity similar to that of the precursor, and a significantly improved local tolerability.61 The recently described, fully synthetic lactam analog of ramoplanin showed the same biological activity as the natural product. Moreover, a set of alanine analogs, obtained by total synthesis, has provided insights into the importance of individual amino-acid residues on ramoplanin activity. The MICs of each alanine-containing analog parallels its ability to bind Lipid II. Apart from positions 5, 6 and 9, which can tolerate alanine substitutions, MICs increased >15-fold upon alanine replacement, with dramatic effects observed for positions 4, 8, 10 and 12. The new data thus confirm the importance of the ornithine residues at positions 4 and 10, with the latter directly involved in target binding, most likely by ion pairing with the diphosphate of Lipid II.62, 63
The mannopeptimycins, which were originally isolated in the late 1950s from Streptomyces hygroscopicus, have been recently revived because of their promising activity against clinically important Gram-positive pathogens, including S. pneumoniae, MRSA and VRE. They also bind to Lipid II, but in a manner different from ramoplanin, mersacidin and vancomycin. Multiple approaches have been used to optimize the mannopeptimycin activity profile, including selective chemical derivatization, precursor-directed biosynthesis and pathway engineering. The SAR data have shown that substitution of a hydrophobic ester group on the N-linked mannose or serine moieties suppressed antibacterial activity, whereas hydrophobic acylation on either of the two O-mannoses, particularly the terminal mannose, significantly enhanced activity. AC98-6446 (Figure 3b) represents an optimized lead obtained by adamantyl ketalization of a cyclohexyl analog prepared by cyclohexylalanine-directed biosynthesis. AC98-6446 showed superior antimicrobial potency and properties, both in vitro and in vivo.7, 64
Laspartomycin is active against VRE and MRSA strains. Recently, enzymatic cleavage of its lipophilic moiety allowed the synthesis of various acylated derivatives (Figure 3b), even if none was more potent than the parent antibiotic.65 The cyclic heptapeptide GE23077 is a potent and selective inhibitor of bacterial RNA polymerase that, probably because of its hydrophilicity, is unable to cross bacterial membranes. New derivatives obtained by modifying different moieties were reported. Although many of them retained activity on the enzyme, none showed a significant antibacterial activity apart from marginal inhibition of Moraxella catarrhalis growth (Figure 3b).66
Future perspectives
This brief and nonexhaustive excursus on the present and future pipeline of antibacterial agents for treating human diseases provides opportunities for additional considerations. The first is that, of the antibiotics under clinical development (Table 1), 67% are natural products themselves, or natural product-derived compounds, a percentage perfectly in line with that found with exisiting drugs.67 The second consideration is that the major players in antibacterial development are small companies, which are not deterred by the small market size for these drugs. However, it should be noted that a significant number of the compounds listed in Table 1 were not discovered by small companies, but actually represent projects abandoned by large pharmaceuticals companies. Thus, it remains to be seen whether small biotechs will dedicate sufficient resources and be successful in discovering and developing novel antibacterial agents.
In this relatively grim scenario, microbial products continue to provide new chemical classes or unexpectedly active variants of chemical classes already known to science. New technologies can now provide access to unexplored microbial diversity or to hypersensitive assays to detect bioactive compounds. Furthermore, the information derived from rapidly accessing the genome of many microbial strains can provide new routes to natural product discovery, as well as making more effective traditional, bioassay-based screening efforts. In our opinion, no single technology will represent the magic bullet for antibiotic discovery, but only the painstaking integration of a multidiscplinary team with profound knowledge of microbiology, chemistry and bioinformatics will ultimately lead to new antibacterial agents of medical relevance and commercial success.
References
Payne, D. J., Gwynn, M. N., Holmes, D. J. & Pompliano, D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40 (2007).
Projan, S. J. & Bradford, P. A. Late stage antibacterial drugs in the clinical pipeline. Curr. Opin. Microbiol. 10, 441–446 (2007).
Theuretzbacher, U. Future antibiotics scenarios: is the tide starting to turn? Int. J. Antimicrob. 34, 15–20 (2009).
Demain, A. L. Microbial natural products: alive and well in 1998. Nat. Biotechnol. 16, 3–4 (1998).
Demain, A. L. & Adrio, J. L. Contributions of microorganisms to industrial biology. Mol. Biotechnol. 38, 41–55 (2008).
Demain, A. L. Prescription for an ailing pharmaceutical industry. Nat. Biotechnol. 20, 331 (2002).
Koehn, F. E. New strategies and methods in the discovery of natural product anti-infective agents: the mannopeptimycins. J. Med. Chem. 51, 2613–2617 (2008).
Souli, M., Galani, I. & Giamarellou, H. Emergence of extensively drug-resistant and pandrug resistant Gram-negative bacilli in Europe. Euro Surveill. 13, 19045 (2008).
Arias, C. A. & Murray, B. E. Antibiotic-resistant bugs in the 21st century-a clinical super-challenge. N. Engl. J. Med. 360, 439–443 (2009).
Dorr, M. B. et al. Human pharmacokinetics and rationale for once-weekly dosing of dalbavancin, a semisynthetic glycopeptide. J. Antimicrob. Chemother. 55 (Suppl. 2), 25–30 (2005).
Noel, G. J. et al. A randomized, double-blind trial comparing ceftobiprole medocaril with vancomycin plus ceftazidime for the treatment of patients with complicated skin and skin-structure infections. Clin. Infect. Dis. 46, 647–655 (2008).
Eckburg, P. et al. Focus 1 and 2: randomized double-blinded, multicenter phase III trials of the efficacy and safety of ceftaroline (CPT) vs ceftriaxone (CRO) in community-acquired pneumonia (CAP). Abstracts of papers of 49th Intersci Conf on Antimicrob Agents Chemother, No. L1-345a, San Francisco (2009).
Bhavnani, S. M. et al. Population pharmacokinetic and Monte Carlo simulation analyses to support phase 2/3 PZ-601 (SMP-601) dosing strategies for complicated skin and skin-structure infections. Abstracts of papers of 47th Intersci Conf on Antimicrob Agents Chemother, No. 40, Chicago (2007).
File, T. et al. A phase study comparing two doses of radezolid to linezolid in adults with uncomplicated skin and skin structure infections (uSSSI). Abstracts of papers of 48th Intersci Conf on Antimicrob Agents Chemother, No. L-1515c, Washington, DC (2008).
Surber, J. et al. Efficacy and safety of torezolid phosphate (torezolid) in a dose-ranging phase 2 randomized, double-blind study in patients with severe complicated skin and skin structure infections (cSSSI). Abstracts of papers of 49th Intersci Conf on Antimicrob Agents Chemother, No. L1-335, San Francisco (2009).
Saxton, K., Baines, S. D., Freeman, J., O'Connor, R. & Wilcox, M. H. Effects of exposure of Clostridium difficile PCR ribotypes 027 and 001 to fluoroquinolones in a human gut model. Antimicrob. Agents Chemother. 53, 412–420 (2009).
Biedenbach, D. J., Jones, R. N., Ivezic-Schoenfeld, Z., Paukner, S. & Novak, R. In vitro antibacterial spectrum of BC-3205, a novel pleuromutilin derivative for oral use in humans. Abstracts of papers of 49th 49th Intersci Conf on Antimicrob Agents Chemother, No. FI-1513, San Francisco (2009).
Karlowsky, J. A. In vitro activity of API-1252, a novel FabI inhibitor, against clinical isolates of Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 51, 1580–1581 (2007).
Schneider, T. et al. The lipopeptide antibiotic Friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob. Agents Chemother. 53, 1610–1618 (2009).
Stachyra, T. et al. In vitro activity of the β-lactamase inhibitor NXL104 against KPC-2 carbapenemase and Enterobacteriaceae expressing KPC carbapenemases. J. Antimicrob. Chemother. 64, 326–329 (2009).
Livermore, D. M., Mushtaq, S., Warner, M., Miossec, C. & Woodford, N. NXL-104 combinations versus Enterobacteriaceae with CTX-M extended-spectrum- β-lactamases and carbapenemases. J. Antimicrob. Chemother. 62, 1053–1066 (2008).
Takeda, S., Nakai, T., Wakai, Y., Ikeda, F. & Hatano, K. In vitro and in vivo activities of a new cephalosporin, FR264205, against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 51, 826–830 (2007).
Endimiani, A. et al. ACHN-490, a neoglycoside with potent in vitro activity against multidrug-resistant Klebsiella pneumoniae isolates. Antimicrob. Agents Chemother. 53, 4504–4507 (2009).
Mushtaq, S., Warner, M. & Livermore, D. Activity of the siderophore monobactam BAL30072 against multiresistant non-fermenters. J. Antimicrob. Chemother. 65, 266–270 (2010).
Bister, B. et al. Abyssomicin C—a polycyclic antibiotic from a marine Verrucosispora strain as an inhibitor of the p-aminobenzoic acid/tetrahydrofolate biosynthesis pathway. Angew. Chem. Int. Ed. 43, 2574–2576 (2004).
Keller, S. et al. Abyssomicins G and H and atrop-Abyssomicin C from the marine Verrucosispora strain AB-18-032. J. Antibiot. 60, 391–394 (2007).
Steinmetz, H. et al. Thuggacins, macrolide antibiotics active against Mycobacterium tuberculosis: isolation form myxobacteria, structure elucidation, conformation analysis and biosynthesis. Chem. Eur. J. 13, 5822–5832 (2007).
McAlpine, J. B. et al. Microbial genomics as a guide to drug discovery and structure elucidation: ECO-02301, a novel antifungal agent, as an example. J. Nat. Prod. 68, 493–496 (2005).
Banskota, A. H. et al. Genomic analyses lead to novel secondary metabolites. Part 3. ECO-0501, a novel antibacterial of a new class. J. Antibiot. 59, 533–542 (2006).
Gross, H. et al. The genomisotopic approach: a systematic method to isolate products of orphan biosynthetic gene clusters. Chem. Biol. 14, 53–63 (2007).
Singh, S. B., Phillips, J. W. & Wang, J. Highly sensitive target-based whole-cell antibacterial discovery strategy by antisense RNA silencing. Curr. Opin. Drug Discov. Devel. 10, 160–166 (2007).
Wang, J. et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441, 358–361 (2006).
Brandi, L. et al. Specific, efficient, and selective inhibition of prokaryotic translation inhibition of prokaryotic translation initiation by a novel peptide antibiotic. Proc. Natl Acad. Sci. USA 103, 39–44 (2006).
Brandi, L. et al. Novel tetrapeptide inhibitors of bacterial protein synthesis produced by a Streptomyces sp. Biochemistry 45, 3692–3702 (2006).
Donadio, S., Monciardini, P. & Sosio, M. Approaches to discovering novel antibacterial and antifungal agents. Methods Enzymol. 458, 3–28 (2009).
Willey, J. M. & van der Donk, W. A. Lantibiotics: peptides with diverse structure and function. Annu. Rev. Microbiol. 61, 477–501 (2007).
Appleyard, A. N. et al. NVB302: a narrow spectrum antibiotic under development for the treatment of Clostridium difficile infection. Abstracts of papers of 49th Intersci Conf on Antimicrob Agents Chemother, No. F1-1517, San Francisco (2009).
Jabes, D. & Donadio, S. Strategies for the isolation and characterization of antibacterial lantibiotics. Methods Mol. Biol. 618, 31–45 (2010).
Castiglione, F. et al. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem. Biol. 15, 22–31 (2008).
Jabes, D., Brunati, C., Guglierame, P. & Donadio, S. In vitro antibacterial profile of the new lantibiotic NAI-107. Abstracts of 49th Intersci Conf on Antimicrob Agents Chemother, No. F1-1502, San Francisco (2009).
Maffioli, S. I. et al. Structure revision of the lantibiotic 97518. J. Nat. Prod. 79, 605–607 (2009).
Maffioli, S. I., Vasile, F., Potenza, D., Brunati, C. & Donadio, S. Lantibiotic carboxyamide derivatives with enhanced antibacterial activity. WO/2010/058238 (2010).
Lawton, E. M., Cotter, P. D., Hill, C. & Ross, R. P. Identification of a novel two-peptide lantibiotic, haloduracin, produced by the alkaliphile Bacillus halodurans C-125. FEMS Microbiol. Lett. 267, 64–71 (2007).
Oman, T. J. & van der Donk, W. A. Insights into the mode of action of the two-peptide lantibiotic haloduracin. ACS Chem. Biol. 4, 865–874 (2009).
Dischinger, J., Joste, M., Szekat, C., Sahl, H. -G. & Bierbaum, G. Production of the novel two-peptide lantibiotic lichenicidin by Bacillus licheniformis DSM 13. PloS ONE 4, e6788 (2009).
Butler, M. S. Natural products to drugs: natural product-derived compounds in clinical trials. Nat. Prod. Rep. 25, 475–516 (2008).
Morris, R. P. et al. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J. Am. Chem. Soc. 131, 5946–5955 (2009).
Singh, S. B. et al. Antibacterial evaluations of thiazomycin—a potent thiazolyl peptide antibiotic from Amycolatopsis fastidiosa. J. Antibiot. 60, 565–571 (2007).
Zhang, C. et al. Isolation, structure, and antibacterial activity of philipimycin, a thiazolyl peptide discovered from Actinoplanes philippinensis MA7347. J. Am. Chem. Soc. 130, 12102–12110 (2008).
Boakes, S., Cortés, J., Appleyard, A. N., Rudd, B. A. M. & Dawson, M. J. Organization of the genes encoding the biosynthesis of actagardine and engineering of a variant generation system. Mol. Microbiol. 72, 1126–1136 (2009).
Acker, M. G., Bowers, A. A. & Walsh, C. T. Generation of thiocillin variants by prepeptide gene replacement and in vivo processing by Bacillus cereus. J. Am. Chem. Soc. 131, 17563–17565 (2009).
Kolb, H. C. & Sharpless, B. K. The growing impact of click chemistry on drug discovery. Drug Discov. Today 8, 1128–1137 (2003).
Pintér, G. et al. Diazo transfer-click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: an aggregation and receptor binding study. J. Med. Chem. 52, 6053–6061 (2009).
Quader, S., Boyd, S. E., Jenkins, I. D. & Houston, T. A. Multisite modification of neomycin B: combined Mitsunobu and click chemistry approach. J. Org. Chem. 72, 1962–1979 (2007).
Tiefenbacher, K. & Mulzer, J. Synthesis of platensimycin. J. Angew. Chem. Int. Ed. 47, 2548–2555 (2008).
Nicolaou, K. C. et al. Total synthesis and antibacterial properties of carbaplatensimycin. J. Am. Chem. Soc. 129, 14850–14851 (2007).
Nicolaou, K. C. et al. Design, synthesis, and biological evaluation of platensimycin analogues with varying degrees of molecular complexity. J. Am. Chen. Soc. 130, 13110–13119 (2008).
Shen, H. G. et al. Synthesis and biological evaluation of platensimycin analogs. Bioorg. Med. Chem. Lett. 19, 1623–1627 (2009).
Charest, M. G., Lerner, C. D., Brubaker, J. D., Siegel, D. R. & Myers, A. G. A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science 308, 395–398 (2005).
Sun, C. et al. A robust platform for the synthesis of new tetracycline antibiotics. J. Am. Chem. Soc. 130, 17913–17927 (2008).
Ciabatti, R. et al. Synthesis and preliminary biological characterization of new semisynthetic derivatives of ramoplanin. J. Med. Chem. 50, 3077–3085 (2007).
Fang, X. et al. Functional and biochemical analysis of a key series of ramoplanin analogues. Bioorg. Med. Chem. Lett. 19, 6189–6191 (2009).
Nam, J., Shin, D., Rew, Y. & Boger, D. L. Alanine scan of [L-Dap2]ramoplanin A2 aglycon: assessment of the importance of each residue. J. Am. Chem. Soc. 129, 8747–8755 (2007).
He, H. Mannopeptimycins, a novel class of glycopeptides antibiotics active against Gram-positive bacteria. Appl. Microbiol. Biotechnol. 67, 444–452 (2005).
Curran, W. V. et al. Semisynthetic approaches to laspartomycin analogues. J. Nat. Prod. 70, 447–450 (2007).
Mariani, R. et al. Antibiotics GE23077, novel inhibitors of bacterial RNA polymerase. Part 3: chemical derivatization. Bioorg. Med. Chem. Lett. 15, 3748–3752 (2005).
Newman, D. J. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J. Med. Chem. 51, 2589–2599 (2008).
Acknowledgements
SM, PM and MS were partially supported by a grant from the Italian Ministry of Research (FIRB 2007-2010).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Donadio, S., Maffioli, S., Monciardini, P. et al. Antibiotic discovery in the twenty-first century: current trends and future perspectives. J Antibiot 63, 423–430 (2010). https://doi.org/10.1038/ja.2010.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2010.62
Keywords
This article is cited by
-
Amamine, an isoquinoline alkaloid from the Kitasatospora sp. HGTA304
The Journal of Antibiotics (2023)
-
Screening of marine Actinomycetia with bioactive metabolites from nearshore and deep sea marine sediments in southwestern Taiwan
Biologia (2023)
-
Repositioning of Disulfiram in Association with Vancomycin Against Enterococcus spp. MDR and XDR
Current Microbiology (2022)
-
Microsegmented flow-assisted miniaturized culturing for isolation and characterization of heavy metal-tolerant bacteria
International Journal of Environmental Science and Technology (2020)
