
Abstract
Fully lithiated lithium sulphide (Li2S) is currently being explored as a promising cathode material for emerging energy storage applications. Like their sulphur counterparts, Li2S cathodes require effective encapsulation to reduce the dissolution of intermediate lithium polysulphide (Li2Sn, n=4–8) species into the electrolyte. Here we report, the encapsulation of Li2S cathodes using two-dimensional layered transition metal disulphides that possess a combination of high conductivity and strong binding with Li2S/Li2Sn species. In particular, using titanium disulphide as an encapsulation material, we demonstrate a high specific capacity of under high C-rate conditions (4C) as well as high areal capacity of 3.0 mAh cm−2 under high mass-loading conditions (). This work opens up the new prospect of using transition metal disulphides instead of conventional carbon-based materials for effective encapsulation of high-capacity electrode materials.
Similar content being viewed by others

Introduction
Rechargeable lithium-ion batteries have revolutionized the world of consumer electronics by providing a lightweight means of powering small, portable electronic devices1,2,3,4,5,6,7. However, the ever-increasing demand for large-scale energy storage and sustainable transport today has triggered the quest for alternative battery technologies with much higher specific energy. The main limiting factor in lithium-ion batteries today is the low theoretical specific capacity (~300 mAh g−1) of conventional intercalation cathodes based on transition metal oxides and phosphates1,2,3,4,5,6,7. Sulphur is a well-known, high-capacity cathode material that has been under active study (S8+16Li↔8Li2S)8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34, but further advancement is hindered by the need for pairing with a metallic lithium anode that is prone to dendrite formation and safety challenges. In this respect, fully lithiated lithium sulphide (Li2S), with its high theoretical specific capacity of 1,166 mAh g−1, represents a more attractive cathode material because of its compatibility with safer lithium metal-free anodes35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55. Moreover, since Li2S is already in its fully lithiated and fully expanded state, it circumvents the volumetric expansion problem in sulphur cathodes, thus minimizing structural damage at the electrode level15,16.
Similar to their sulphur counterparts, Li2S cathodes are plagued with two main challenges: (1) the insulating nature of Li2S, and (2) uncontrolled dissolution and loss of intermediate lithium polysulphide (Li2Sn, n=4–8) species into the electrolyte, leading to fast capacity decay and low Coulombic efficiency35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55. To address these challenges, the typical strategy is to encapsulate Li2S cathodes with conductive coating materials in an attempt to improve their conductivity as well as physically entrap intermediate Li2Sn species within the shell during cycling35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50. Besides acting as a physical barrier, an ideal encapsulation material should also possess strong chemical interaction with polar Li2S and Li2Sn species in order to further bind and confine these species within the shell during cycling. Out of the ~16 papers in the literature on encapsulating Li2S cathodes thus far, the vast majority (15 of them) use electronically conductive carbon-based materials such as graphene and porous carbon for encapsulation35,36,37,38,39,40,41,42,43,44,45,46,47,48,49 (the last paper uses Li3PS4, a lithium superionic conductor/solid electrolyte, which is a poor electronic conductor50). Although carbon-based materials are conductive, their nonpolar nature leads to weak interaction with polar Li2S and Li2Sn species17, which greatly reduces their ability to bind and confine these species within the shell during cycling. While it is possible to introduce polar oxygen-containing functional groups into carbon to improve its interaction with Li2S/Li2Sn species (for example, by using graphene oxide42), such oxidation treatment decreases the conductivity of carbon drastically. In using carbon-based materials as encapsulation materials for Li2S cathodes, there is a major constraint involved: it is difficult to achieve both high conductivity and strong binding with Li2S/Li2Sn species simultaneously. This greatly limits the cycling performance of Li2S cathodes so far, especially under high C-rate and high mass-loading conditions. Thus, there is an urgent need for a new class of encapsulation materials for Li2S cathodes that can overcome this major constraint associated with conventional carbon-based materials.
Herein we demonstrate, for the first time, the use of titanium disulphide (TiS2), a two-dimensional (2D) layered transition metal disulphide, as an effective encapsulation material for Li2S cathodes to overcome this significant constraint. TiS2 possesses a combination of high conductivity2,7 and polar Ti–S groups that can potentially interact strongly with Li2S/Li2Sn species. In this work, we synthesized Li2S@TiS2 core–shell nanostructures that exhibited 10 orders of magnitude higher electronic conductivity compared with pure Li2S. The results of ab initio simulations also show strong binding between Li2S and TiS2, with a calculated binding energy 10 times higher than that between Li2S and carbon-based graphene, a very common encapsulation material used in the literature29. Using the Li2S@TiS2 nanostructures as a cathode material, we achieve an unprecedented specific capacity of under high C-rate conditions (4C), as well as unprecedented areal capacity of 3.0 mAh cm−2 under high mass-loading conditions (), both of which are the highest values reported to date for Li2S cathodes on metal foil. Finally, using zirconium disulphide (ZrS2) and vanadium disulphide (VS2) as further examples, we open up the new concept of using 2D layered transition metal disulphides as a general class of effective encapsulation materials for achieving high performance in Li2S cathodes.
Results
Synthesis and characterization
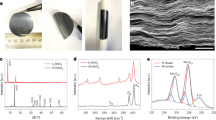
The Li2S@TiS2 core–shell nanostructures were synthesized as shown schematically in Fig. 1a (see Methods section for details). In brief, ball-milled commercial Li2S particles were dispersed in anhydrous ethyl acetate, followed by the addition of a controlled amount of TiCl4 precursor to react directly with some of the Li2S on the surface to form a TiS2 coating (TiCl4+2Li2S→ TiS2+4LiCl; Fig. 1a)56. The in situ and direct reaction of TiCl4 with Li2S ensures a uniform coating of TiS2 on the surface of the Li2S particles. The as-synthesized Li2S@TiS2 nanostructures were then washed by centrifugation and heated at 400 °C in an argon atmosphere to allow crystallization of the TiS2 shell (Fig. 1a). Figure 1b shows a scanning electron microscopy (SEM) image of the Li2S@TiS2 nanostructures. The transmission electron microscopy (TEM) image in Fig. 1c shows the Li2S particles (mostly <1 μm in size) encapsulated within TiS2 shells (~20 nm thick; Supplementary Fig. 1a). From the high-resolution TEM image, we see that the TiS2 shell is layered and crystalline in nature with interlayer spacing of 0.57 nm (Fig. 1d), which is consistent with that of TiS2 (ref. 57). The crystalline nature and identity of the TiS2 shell was also ascertained using X-ray diffraction (XRD; Fig. 1e). The uniform surface coating of TiS2 on the Li2S particles was further confirmed using Raman spectroscopy. We see that the Raman spectrum of bare Li2S shows the characteristic T2g peak of Li2S at 372 cm−1 (Fig. 1f)42,47. On the other hand, for the Li2S@TiS2 structures, the A1g peak of TiS2 at 335 cm−1 can be clearly seen58 while the peak of Li2S becomes less prominent (Fig. 1f). Since Raman spectroscopy is a surface-sensitive technique, this observation indicates that the Li2S particles have been uniformly encapsulated with TiS2 shell. Using energy-dispersive X-ray spectroscopy (EDX), the ratio of Li2S:TiS2 in the composite was determined to be 78:22 by weight, which is consistent with the amount of TiCl4 precursor added to react with Li2S based on the reaction stoichiometry (see Supplementary Fig. 2 for details).

(a) Schematic of the synthesis process, (b) SEM image and (c) TEM image of Li2S@TiS2 structures. Scale bars, 5 μm (b) and 500 nm (c). (d) High-resolution TEM image of the TiS2 shell, showing the interlayer spacing of 0.57 nm. Scale bar, 5 nm. (e) XRD spectrum of Li2S@TiS2 structures (JCPDS files 00-026-1188 and 04-004-6690 were used as references for Li2S and TiS2, respectively). (f) Raman spectra of bare Li2S and Li2S@TiS2 structures. (g) Ab initio simulations showing the most stable binding configuration of Li2S with a single layer of TiS2, with a calculated binding energy of 2.99 eV. RT, room temperature.
D.C. conductivity measurements of the Li2S@TiS2 structures were performed at 25 °C after the samples were dry-pressed into a pellet and sandwiched between two blocking electrodes (Supplementary Fig. 3). The electronic conductivity of the Li2S@TiS2 structures was measured to be 5.1 × 10−3 S cm−1, which is ~10 orders of magnitude higher than that of bare Li2S (10−13 S cm−1)50,52. In terms of confining Li2S and Li2Sn species, the unique 2D layered structure of TiS2 would enable it to act as a good physical barrier to entrap these species within the shell during cycling. To probe the further possibility of chemical interaction between Li2S and TiS2, ab initio simulations were carried out in the framework of density functional theory (see Methods section for details)59,60,61,62. The results of the simulations in Fig. 1g indicate strong Li–S interaction (between the Li atoms in Li2S and S atoms in TiS2), as well as strong S–S interaction (between the S atoms in Li2S and S atoms in TiS2). The binding energy between Li2S and a single layer of TiS2 was calculated to be 2.99 eV. This value is 10 times higher than that between Li2S and a single layer of carbon-based graphene (0.29 eV; ref. 17), which is a very common encapsulation material used in the literature29. The much stronger interaction between Li2S and TiS2 can be explained by their similar ionic bonding and polar nature, unlike graphene which is covalently bonded and nonpolar in nature. Previous results have shown that entities that bind strongly to Li2S exhibit strong binding with Li2Sn species as well owing to their similar chemical bonding nature27,53. Overall, the dual effect of physical entrapment and chemical binding would enable TiS2 to be effective in confining Li2S and Li2Sn species within the shell during cycling.
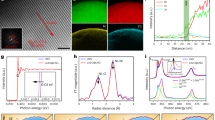
To verify the above-mentioned point, the Li2S@TiS2 structures and bare Li2S particles were dispersed directly onto conductive carbon fibre substrates to prepare working electrodes, and pouch cells were assembled using lithium foil as the counter electrodes. Some cells were charged to a high cutoff voltage of 3.8 V versus Li+/Li to delithiate the Li2S (ref. 52) and then disassembled in the charged state. Others were charged in the same way and then discharged to 1.8 V versus Li+/Li for lithiation to re-form Li2S, before being disassembled in the discharged state (see Supplementary Fig. 4 for the voltage profiles). After disassembly of the cells, the working electrodes were examined using SEM. The SEM image in Fig. 2a shows the bare Li2S particles on carbon fibre substrate before cycling. At the end of charge, only the underlying carbon fibre substrate could be observed, but not the particles that were originally on the surface (Fig. 2b). This is attributed to the delithiation of Li2S upon charging to form Li2Sn species (n=4–8) that dissolve readily into the electrolyte owing to the complete lack of encapsulation (it is known that solid S8 is not re-formed at the end of initial charge52). At the end of discharge, the original Li2S particle morphology was not recovered as well; irregularly shaped and -sized Li2S particles were formed on the electrode surface instead (Fig. 2c). This is due to uncontrolled dissolution of intermediate Li2Sn species leading to random precipitation of Li2S all over the electrode upon discharge. On the other hand, in the case of the Li2S@TiS2 structures, little change in particle morphology was observed before cycling versus at the end of charge as seen under SEM (Fig. 2d,e). TEM imaging also shows that core–shell structure was preserved and the protective TiS2 shell remained intact (Fig. 2g,h and Supplementary Fig. 1b,c). At the end of discharge (lithiation to re-form Li2S), the Li2S@TiS2 particles were seen to maintain their original morphology as well, as evidenced by both SEM and TEM (Fig. 2f,i and Supplementary Fig. 1d). This indicates that TiS2 is effective in confining the charge and discharge species (Li2Sn and Li2S, respectively) within the shell during the charge–discharge cycle, hence resulting in the observed preservation of morphology.

(a–c) SEM images of bare Li2S particles (a) before cycling, (b) at the end of charge and (c) at the end of discharge. (d–i) SEM images of Li2S@TiS2 structures (d) before cycling, (e) at the end of charge and (f) at the end of discharge, as well as (g–i) their corresponding TEM images. Scale bars, 5 μm (a–f) and 500 nm (g–i).
Electrochemical performance
To evaluate their electrochemical performance, working electrodes were prepared by mixing the Li2S@TiS2 nanostructures with conductive carbon black and polyvinylidene fluoride (PVDF) binder (65:30:5 by weight) in N-methyl-2-pyrrolidinone to form a slurry, which was then coated onto aluminium foil and dried in a glove box. Bare Li2S cathodes were also prepared in the same way for comparison. 2032-Type coin cells were then assembled using lithium foil as the counter electrode. The electrolyte used was lithium bis(trifluoromethanesulphonyl)imide (1 M) in 1:1 (v/v) 1,2-dimethoxyethane and 1,3-dioxolane, with LiNO3 additive (1 wt%) to help passivate the lithium anode surface21. The typical mass loading was unless otherwise stated.
The impedance spectrum of the as-assembled Li2S@TiS2 cells at open-circuit voltage showed a smaller semicircle in the high-frequency region compared with bare Li2S cells, which indicates lower charge-transfer resistance in the former case (Fig. 3a and Supplementary Fig. 5)13. This is consistent with the much higher conductivity of the Li2S particles upon TiS2 coating. The as-assembled cells were first charged to a high cutoff voltage of 3.8 V versus Li+/Li to delithiate the Li2S (ref. 52). In the case of bare Li2S cathodes, a high potential barrier at ~3.5 V was observed during initial charging (Fig. 3b) owing to the high charge-transfer resistance involved in delithiating Li2S, which is insulating in nature (Li2S→ Li2−xS+xLi++xe−)52. On the other hand, the height of the potential barrier was found to be reduced to 3.0 V in the case of the Li2S@TiS2 cathodes (Fig. 3b), indicating improved conductivity and lowered charge-transfer resistance upon TiS2 coating, which is consistent with results of the impedance measurements. After initial charging, galvanostatic cycling of the Li2S@TiS2 cells was then carried out from 1.8 to 2.6 V versus Li+/Li at 0.2C, where 1C=. The discharge cutoff voltage was set at 1.8 V to avoid the irreversible reduction of LiNO3 additive in the electrolyte21. The capacity contribution of TiS2 was found to be negligible in this voltage range (Supplementary Fig. 6). As shown in Fig. 3c, the Li2S@TiS2 cathodes exhibited high initial specific capacity of () and stable cycling performance at 0.2C. Relative to the initial cycle, the capacity retentions achieved at the end of 50, 100 and 150 cycles were 90%, 89% and 86%, respectively. In comparison, the bare Li2S cathodes showed lower initial capacity and greater capacity decay under identical testing conditions (Fig. 3c). Relative to the initial specific capacity of , the capacity retention of the bare Li2S cathodes was only 66% after 100 cycles (compared with 89% for Li2S@TiS2 structures). This indicates a greater degree of dissolution of Li2Sn species into the electrolyte in the case of bare Li2S cathodes.

(a) Impedance spectra at open-circuit voltage, (b) first cycle charge voltage profiles and (c) specific capacities at 0.2 C (1 C=) for Li2S@TiS2 and bare Li2S cathodes. (d) Typical discharge–charge voltage profile showing various DoD and SoC (points 1–10) and the corresponding percentage of sulphur measured in the electrolyte relative to the total sulphur mass on the electrode at these various points for Li2S@TiS2 and bare Li2S cathodes. (e,f) Low- and high-magnification (inset) SEM images of the surface of (e) bare Li2S and (f) Li2S@TiS2 cathodes after 100 cycles in the discharged state. Scale bars, 100 μm in (e,f) and 1 μm (e,f; inset).
The above conclusion is supported by disassembly of the cells at various intermediate stages of cycling and testing the electrolyte for sulphur content using inductively coupled plasma-optical emission spectroscopy (ICP-OES; see Methods section for details)42,47. Points 1–10 in Fig. 3d correspond to various depths of discharge (DoD) and states of charge (SoC) during cycling, where 100% DoD (point 6) and 100% SoC (point 10) refer to the maximum discharge and charge capacities attained, respectively. The ICP-OES results for the bare Li2S cathodes showed a huge variation in sulphur content detected in the electrolyte at various stages of cycling (Fig. 3d) owing to lack of control over the Li2Sn dissolution process. On the other hand, in the case of Li2S@TiS2 cathodes, we see significantly less variation and lower percentage loss of sulphur into the electrolyte during cycling (Fig. 3d). For example, at points 3, 6 and 9 during the cycling process (25% DoD, 100% DoD and 75% SoC), we measured 17%, 9% and 18% of the total sulphur mass on the electrode dissolved into the electrolyte, respectively, for the Li2S@TiS2 cathodes (Fig. 3d). In comparison, the percentage loss of total sulphur mass into the electrolyte was found to be much higher in the case of bare Li2S cathodes: 91%, 35% and 93% at points 3, 6 and 9, respectively, (Fig. 3d). The cells were also disassembled in the discharged state at the end of 100 cycles to examine the morphology of the cathode surface after cycling. In the case of bare Li2S cathodes, an insoluble dense film of Li2S was formed on the surface owing to uncontrolled dissolution of intermediate Li2Sn species into the electrolyte (Fig. 3e; see inset for a high-magnification image). The presence of this insulating Li2S film passivates the electrode surface, thereby leading to low active material utilization and rapid capacity decay upon cycling8,9. On the other hand, there was no such passivating Li2S film formed on the surface of the Li2S@TiS2 cathodes after 100 cycles (Fig. 3f and Supplementary Fig. 7), thus ensuring continued good access of electrolyte and Li+ to the active materials throughout the cycling process.
To further evaluate their electrochemical cycling performance, the Li2S@TiS2 cathodes were subject to prolonged cycling. From Fig. 4a, we see that the Li2S@TiS2 cathodes showed stable cycling performance at 0.5C over 400 charge–discharge cycles. Relative to the initial specific capacity of 666 (~956 mAh g−1S) at 0.5C, the cells retained 77% of their capacity at the end of 400 cycles, which corresponds to a small average capacity decay of 0.058% per cycle. The average Coulombic efficiency was calculated to be 98% (Fig. 4a). Next, the Li2S@TiS2 cathodes were cycled at different C-rates to evaluate their rate capability and electrode kinetics (Fig. 4b,c). When the C-rate was increased successively from 0.2C to 0.5C, 1C, 2C and 3C, the cells delivered high stabilized specific capacities of 700, 650, 608, 595 and 560 , respectively (Fig. 4b,c). The capacities achieved at 0.5C, 1C, 2C and 3C correspond to 93%, 87%, 85% and 80%, respectively, of the capacity that was reached at 0.2C. Even at a high C-rate of 4C (4,664 ), a reversible specific capacity of 503 mAh (~722 ) could be attained (Fig. 4b,c). To the best of our knowledge, this is the first time such a high specific capacity has been achieved at 4C for Li2S cathodes on metal foil, which indicates good conductivity and charge-transfer kinetics in our Li2S@TiS2 cathodes. When the C-rate was switched abruptly from 4C back to 0.2C again, the original capacity was mostly recovered (Fig. 4b), indicating robustness and stability of the cathode material.

(a) Specific capacity and Coulombic efficiency of Li2S@TiS2 cathodes upon prolonged 400 charge–discharge cycles at 0.5 C (1 C=). (b) Specific capacity and (c) charge–discharge voltage profiles of Li2S@TiS2 cathodes cycled from 0.2 to 4 C. (d) First cycle discharge voltage profile and (e) areal capacity of Li2S@TiS2 cathodes with high mass-loading cycled from 0.1 to 0.6 mA cm−2.
We note that publications on Li2S cathodes in the literature typically only report specific capacity values that are normalized by the mass of Li2S (refs 41, 42, 43, 44, 45, 46, 47). The mass loading of Li2S on metal foil used in the literature is usually <1 cm−2, which limits the typical areal capacity values to <1 mAh cm−2 (refs 41, 42, 43, 44, 45, 46, 47). However, high mass loadings and high areal capacities are essential for practical applications in commercial batteries. Therefore, we tested thick Li2S@TiS2 cathodes on aluminium foil with much higher mass loading of 5.3 cm−2. Upon cycling at 0.1 mA cm−2, an initial specific capacity of 566 was achieved that corresponds to an unprecedented areal capacity of 3.0 mAh cm−2 (Fig. 4d). This is the highest areal capacity value reported for Li2S cathodes on metal foil so far. Upon cycling at higher current densities of 0.3 and 0.6 mA cm−2, areal capacities of 2.2 and 1.6 mAh cm−2 were achieved, respectively, (Fig. 4e). Cycling of a high mass-loading Li2S cathode places high demands on the conductivity of the active materials, as well as the ability to confine Li2S and Li2Sn species within the shell. The performance demonstrated in this work shows the importance of selecting an effective encapsulation material such as TiS2 in order to achieve stable cycling and high areal capacity in Li2S cathodes.
Generalization to other transition metal disulphides
Finally, to show the generality of using 2D transition metal disulphides as encapsulation materials for Li2S cathodes, we also synthesized Li2S@ZrS2 and Li2S@VS2 core–shell structures (Fig. 5a,b) using the same reaction mechanism: MCl4+2Li2S → MS2+4LiCl (M=Ti, Zr and V)56. For further characterization of these materials using TEM, XRD and EDX, please refer to Supplementary Fig. 8. The conductivities of the Li2S@ZrS2 and Li2S@VS2 structures were measured to be 4.0 × 10−9 and 3.8 × 10−9 S cm−1, respectively, which are ~4 orders of magnitude higher than that of bare Li2S (10−13 S cm−1; refs 50, 52). The results of ab initio simulations also show strong binding of Li2S to ZrS2 and VS2, with calculated binding energies of 2.70 and 2.94 eV, respectively, (Fig. 5c,d), which are ~9–10 times higher than that between Li2S and carbon-based graphene (0.29 eV; ref. 17). Working electrodes on aluminium foil were prepared for the Li2S@ZrS2 and Li2S@VS2 structures, and the cells were subject to galvanostatic cycling as well. Using , the Li2S@ZrS2 and Li2S@VS2 cathodes exhibited high initial specific capacities of 777 and , respectively, at 0.2C, with capacity retentions of 85% and 86%, respectively, after 100 cycles (Fig. 5e), both of which are much higher than in the case of bare Li2S cathodes (66% after 100 cycles). The morphologies of the electrode surfaces were also examined after 100 cycles in the discharged state, both of which show the absence of a passivating Li2S film (Supplementary Fig. 9), similar to the case of Li2S@TiS2 cathodes after cycling. Both the Li2S@ZrS2 and Li2S@VS2 cathodes were also subject to cycling under high mass-loading conditions. Using for the Li2S@ZrS2 cathodes and current densities of 0.1, 0.3 and 0.6 mA cm−2, we achieve areal capacities of 2.7, 2.0 and 1.7 mAh cm−2, respectively, (Fig. 5f). The areal capacities attained are similarly high in the case of Li2S@VS2 cathodes containing : 2.7, 1.9 and 1.6 mAh cm−2, respectively, at the above-mentioned current densities (Fig. 5f).

(a,b) SEM images of (a) Li2S@ZrS2 and (b) Li2S@VS2 structures. Scale bars, 5 μm. (c,d) Ab initio simulations showing the most stable binding configuration of Li2S with a single layer of (c) ZrS2 and (d) VS2 with calculated binding energies of 2.70 and 2.94 eV, respectively. (e) Specific capacity of Li2S@ZrS2 and Li2S@VS2 cathodes cycled at 0.2 C (1 C=). (f) Areal capacity of Li2S@ZrS2 and Li2S@VS2 cathodes with high mass loading cycled from 0.1 to 0.6 mA cm−2.
Discussion
In this work, we have presented the electrochemical performance of Li2S@TiS2 structures with Li2S core size of <1 μm and TiS2 shell thickness of ~20 nm (Li2S:TiS2=78: 22 by weight). For practical applications in which high percentage of active material (Li2S) is desired, the ratio of Li2S:TiS2 should be further optimized. To this end, we investigated the cycling performance of Li2S@TiS2 composites with three different TiS2 shell thicknesses: ~11, 20 and 55 nm, which were synthesized by varying the amount of TiCl4 precursor added (Supplementary Fig. 10a–c). Upon galvanostatic cycling at 0.2C, the specific capacity of the Li2S@TiS2 cathode with a 11-nm thin shell was found to fade faster than that of a 20-nm shell (Supplementary Fig. 10d), because the former shell is not thick enough to act as a physical barrier to the dissolution of Li2Sn species. On the other hand, the cycling performance of the cathode with a 55-nm thick shell was comparable to that of a 20-nm shell (Supplementary Fig. 10d), which explains our choice of 20 nm as the TiS2 shell thickness in this work.
Instead of decreasing the TiS2 shell thickness, another way to increase the percentage of Li2S is to increase the size of the Li2S core. To this end, we coated TiS2 onto larger, non-ball-milled Li2S particles that are >1 μm in size (Supplementary Fig. 11a). The percentage of Li2S in the resulting composite was determined to be higher using EDX (Li2S:TiS2=87: 13 by weight; Supplementary Fig. 11b). Upon galvanostatic cycling at 0.2C, 0.5C and 1C, similarly good cycling performance can be achieved for the Li2S@TiS2 cathodes with larger versus smaller Li2S core (Supplementary Fig. 11c). However, at a higher C-rate of 2C, the cathode with smaller Li2S core exhibited higher specific capacity and better cycling stability (Supplementary Fig. 11c) because of the shorter Li+ diffusion lengths in smaller Li2S particles. There is a compromise to be reached in terms of the size of the Li2S particles and their rate capability.
In conclusion, we have demonstrated the use of 2D layered transition metal disulphides for effective encapsulation of Li2S cathodes owing to their combination of high conductivity and strong binding with Li2S/Li2Sn species. Further work is currently ongoing to pair these stable-cycling Li2S@MS2 cathodes with lithium metal-free anodes (such as silicon) to achieve a full-cell configuration. This work overcomes the major constraint associated with conventional carbon-based materials and opens up the new prospect of using transition metal disulphides for effective encapsulation of other high-capacity electrode materials.
Methods
Materials synthesis
Owing to the sensitivity of Li2S to moisture, all the material synthesis procedures were carried out in an argon-filled glove box with moisture and oxygen levels below 0.5 p.p.m. Commercial Li2S particles (99.9%, Alfa Aesar) were first ball-milled for 1 h using a SPEX 5100 Mixer/Mill. The ball-milled Li2S particles (10 mmol) were then dispersed in anhydrous ethyl acetate, followed by addition of MCl4 precursor (M=Ti, Zr and V; 1 mmol) using a glass syringe to convert 20% of the Li2S on the surface into MS2 (MCl4+2Li2S → MS2+4LiCl; ref. 56). After overnight reaction at room temperature, the as-synthesized Li2S@MS2 core–shell structures were washed by centrifugation three times using anhydrous tetrahydrofuran to remove the LiCl side product. These were then heated at 400 °C under argon gas atmosphere to remove the ethyl acetate solvent and allow crystallization of MS2. The ratio of Li2S: MS2 in the Li2S@MS2 structures was determined using EDX to be 78:22, 72:28 and 75:25 by weight for M=Ti, Zr and V, respectively, all of which are consistent with the amount of MCl4 precursor added to react with Li2S based on the reaction stoichiometry (Supplementary Figs 2 and 8c,f).
Characterization
To prevent moisture contamination of Li2S, special precautions were taken during characterization. SEM, EDX and TEM were performed using a FEI Quanta 250 FEG, FEI XL30 Sirion and Tecnai G2 F20 X-TWIN, respectively. The samples were first tightly sealed in foil/polybags before being transferred into the microscope chamber via an argon-filled glove bag. XRD patterns were obtained on a PANalytical X’Pert diffractometer (Cu Kα radiation) after the samples were tightly sealed using Kapton tape. Raman spectra were obtained using a WITEC Raman spectrometer (531 nm excitation laser) after the samples were tightly sealed in a glass holder. D.C. conductivity measurements were performed at 25 °C in an argon-filled glove box using a Bio-Logic VMP3 potentiostat after the samples were dry-pressed into a pellet and sandwiched between two pieces of carbon-coated aluminium foil as the blocking electrodes.
Ab initio simulations
Ab initio simulations were performed using the Vienna Ab Initio Simulation Package in the framework of density functional theory59,60. The projector augmented-wave pseudopotential61 and the generalized gradient approximation exchange-correlation function described by Perdew–Burke–Ernzerhof62 were adopted. To ensure convergence, 500 eV was chosen as the cutoff energy of the plane-wave basis. The vacuum between MS2 (M=Ti, Zr and V) and its image exceeds 30 Å, while the distance between Li2S and its image is no <15 Å along the periodic direction. These systems were large enough to avoid any artificial interaction caused by periodicity. The binding energy, Eb, was defined as the energy difference between the Li2S–MS2 adsorbed system (Etot) and the summation of pure Li2S () and pure MS2 (EMS2): Eb=(+EMS2)−Etot. The 3D visualization models were constructed using VESTA 3 software63.
Electrochemical measurements
Owing to the sensitivity of Li2S to moisture, all the electrode preparation and cell assembly procedures were carried out in an argon-filled glove box with moisture and oxygen levels below 0.5 p.p.m. The Li2S@MS2 structures (M=Ti, Zr and V) were mixed with conductive carbon black (Super P) and PVDF binder in a weight ratio of 65:30:5, followed by dispersion in N-methyl-2-pyrrolidinone to form a slurry. After overnight stirring, the slurry was then coated onto aluminium foil (carbon-coated) using doctor blade and dried at 60 °C to form the working electrodes. Based on the Li2S content in the Li2S@MS2 structures (Supplementary Figs 2 and 8c,f), we can determine the weight percentage of Li2S in the electrodes to be ~51%, 47% and 49% for M=Ti, Zr and V respectively. The typical mass loading was unless otherwise stated. The high mass-loading electrodes were prepared on aluminium foil in the same way. For comparison, bare Li2S cathodes were also prepared by mixing ball-milled Li2S with Super P and PVDF binder in the same weight ratio of 65:30:5 and coating the slurry onto aluminium foil. 2032-Type coin cells were then assembled using lithium foil as the counter electrode. The electrolyte used was a solution of lithium bis(trifluoromethanesulphonyl)imide (1 M) in 1:1 (v/v) 1,2-dimethoxyethane and 1,3-dioxolane containing LiNO3 additive (1 wt%). Using eight-channel MTI battery testers, the cathodes were first activated at C/20 (1C=) by charging to a high cutoff voltage of 3.8 V versus Li+/Li52. Galvanostatic cycling was then carried out at the specified C-rate from 1.8 to 2.6 V versus Li+/Li. Impedance measurements were carried out using a Bio-Logic VMP3 potentiostat in the frequency range from 200 kHz to 100 mHz. For analysis of sulphur content in the electrolyte, all the cell assembly procedures are the same as described above except that a sulphur-free lithium salt of LiClO4 was used as the electrolyte salt instead of lithium bis(trifluoromethanesulphonyl)imide42,47. The cells were disassembled at various points 1–10 during a discharge–charge cycle and the contents (cathode, anode and electrolyte-soaked separator) were washed with 1,3-dioxolane. This polysulphide-containing solution was then oxidized with concentrated HNO3 and diluted with deionized water for analysis of sulphur content using ICP-OES (Thermo Scientific ICAP 6,300 Duo View spectrometer).
Additional information
How to cite this article: Seh, Z. W. et al. Two-dimensional layered transition metal disulphides for effective encapsulation of high-capacity lithium sulphide cathodes. Nat. Commun. 5:5017 doi: 10.1038/ncomms6017 (2014).
References
Bruce, P. G., Freunberger, S. A., Hardwick, L. J. & Tarascon, J.-M. Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 11, 19–29 (2012).
Whittingham, M. S. Electrical energy-storage and intercalation chemistry. Science 192, 1126–1127 (1976).
Armand, M. & Tarascon, J. M. Building better batteries. Nature 451, 652–657 (2008).
Chiang, Y.-M. Building a better battery. Science 330, 1485–1486 (2010).
Lee, J. et al. Unlocking the potential of cation-disordered oxides for rechargeable lithium batteries. Science 343, 519–522 (2014).
Goodenough, J. B. & Park, K.-S. The Li-ion rechargeable battery: a perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013).
Whittingham, M. S. Lithium batteries and cathode materials. Chem. Rev. 104, 4271–4301 (2004).
Ji, X., Lee, K. T. & Nazar, L. F. A highly ordered nanostructured carbon-sulphur cathode for lithium-sulphur batteries. Nat. Mater. 8, 500–506 (2009).
Ji, X., Evers, S., Black, R. & Nazar, L. F. Stabilizing lithium-sulphur cathodes using polysulphide reservoirs. Nat. Commun. 2, 325 (2011).
Schuster, J. et al. Spherical ordered mesoporous carbon nanoparticles with high porosity for lithium-sulfur batteries. Angew. Chem. Int. Ed. 51, 3591–3595 (2012).
Yamin, H., Gorenshtein, A., Penciner, J., Sternberg, Y. & Peled, E. Lithium sulfur battery—oxidation reduction-mechanisms of polysulfides in THF solutions. J. Electrochem. Soc. 135, 1045–1048 (1988).
Elazari, R., Salitra, G., Garsuch, A., Panchenko, A. & Aurbach, D. Sulfur-impregnated activated carbon fiber cloth as a binder-free cathode for rechargeable Li-S batteries. Adv. Mater. 23, 5641–5644 (2011).
Su, Y.-S. & Manthiram, A. Lithium-sulphur batteries with a microporous carbon paper as a bifunctional interlayer. Nat. Commun. 3, 1166 (2012).
Su, Y.-S., Fu, Y., Cochell, T. & Manthiram, A. A strategic approach to recharging lithium-sulphur batteries for long cycle life. Nat. Commun. 4, 2985 (2013).
Seh, Z. W. et al. Sulphur-TiO2 yolk-shell nanoarchitecture with internal void space for long-cycle lithium-sulphur batteries. Nat. Commun. 4, 1331 (2013).
Li, W. et al. High-performance hollow sulfur nanostructured battery cathode through a scalable, room temperature, one-step, bottom-up approach. Proc. Natl Acad. Sci. USA 110, 7148–7153 (2013).
Zheng, G. et al. Amphiphilic surface modification of hollow carbon nanofibers for improved cycle life of lithium sulfur batteries. Nano Lett. 13, 1265–1270 (2013).
Li, W. et al. Understanding the role of different conductive polymers in improving the nanostructured sulfur cathode performance. Nano Lett. 13, 5534–5540 (2013).
Liang, Z. et al. Sulfur cathodes with hydrogen reduced titanium dioxide inverse opal structure. ACS Nano 8, 5249–5256 (2014).
Yao, H. et al. Improving lithium-sulphur batteries through spatial control of sulphur species deposition on a hybrid electrode surface. Nat. Commun. 5, 3943 (2014).
Zhang, S. S. Effect of discharge cutoff voltage on reversibility of lithium/sulfur batteries with LiNO3-contained electrolyte. J. Electrochem. Soc. 159, A920–A923 (2012).
Zhang, B., Qin, X., Li, G. R. & Gao, X. P. Enhancement of long stability of sulfur cathode by encapsulating sulfur into micropores of carbon spheres. Energy Environ. Sci. 3, 1531–1537 (2010).
Ji, L. et al. Graphene oxide as a sulfur immobilizer in high performance lithium/sulfur cells. J. Am. Chem. Soc. 133, 18522–18525 (2011).
Song, M.-K., Zhang, Y. & Cairns, E. J. A long-life, high-rate lithium/sulfur cell: a multifaceted approach to enhancing cell performance. Nano Lett. 13, 5891–5899 (2013).
Huang, C. et al. Manipulating surface reactions in lithium-sulphur batteries using hybrid anode structures. Nat. Commun. 5, 3015 (2014).
Xiao, L. et al. A soft approach to encapsulate sulfur: polyaniline nanotubes for lithium-sulfur batteries with long cycle life. Adv. Mater. 24, 1176–1181 (2012).
Zheng, J. et al. Lewis acid-base interactions between polysulfides and metal organic framework in lithium sulfur batteries. Nano Lett. 14, 2345–2352 (2014).
Xin, S. et al. Smaller sulfur molecules promise better lithium-sulfur batteries. J. Am. Chem. Soc. 134, 18510–18513 (2012).
Yin, Y.-X., Xin, S., Guo, Y.-G. & Wan, L.-J. Lithium-sulfur batteries: electrochemistry, materials, and prospects. Angew. Chem. Int. Ed. 52, 13186–13200 (2013).
Suo, L., Hu, Y.-S., Li, H., Armand, M. & Chen, L. A new class of solvent-in-salt electrolyte for high-energy rechargeable metallic lithium batteries. Nat. Commun. 4, 1481 (2013).
Moon, S. et al. Encapsulated monoclinic sulfur for stable cycling of Li-S rechargeable batteries. Adv. Mater. 25, 6547–6553 (2013).
Hwang, T. H., Jung, D. S., Kim, J.-S., Kim, B. G. & Choi, J. W. One-dimensional carbon-sulfur composite fibers for Na-S rechargeable batteries operating at room temperature. Nano Lett. 13, 4532–4538 (2013).
Chen, R. J. et al. Graphene-based three-dimensional hierarchical sandwich-type architecture for high-performance Li/S batteries. Nano Lett. 13, 4642–4649 (2013).
Zhao, M.-Q. et al. Unstacked double-layer templated graphene for high-rate lithium-sulphur batteries. Nat. Commun. 5, 3410 (2014).
Hassoun, J. & Scrosati, B. A high-performance polymer tin sulfur lithium ion battery. Angew. Chem. Int. Ed. 49, 2371–2374 (2010).
Agostini, M. et al. A lithium-ion sulfur battery based on a carbon-coated lithium-sulfide cathode and an electrodeposited silicon-based anode. ACS Appl. Mater. Interfaces 6, 10924–10928 (2014).
Takeuchi, T. et al. Preparation of electrochemically active lithium sulfide-carbon composites using spark-plasma-sintering process. J. Power Sources 195, 2928–2934 (2010).
Nagao, M., Hayashi, A. & Tatsumisago, M. High-capacity Li2S-nanocarbon composite electrode for all-solid-state rechargeable lithium batteries. J. Mater. Chem. 22, 10015–10020 (2012).
Yang, Y. et al. New nanostructured Li2S/silicon rechargeable battery with high specific energy. Nano Lett. 10, 1486–1491 (2010).
Jeong, S., Bresser, D., Buchholz, D., Winter, M. & Passerini, S. Carbon coated lithium sulfide particles for lithium battery cathodes. J. Power Sources 235, 220–225 (2013).
Cai, K., Song, M.-K., Cairns, E. J. & Zhang, Y. Nanostructured Li2S-C composites as cathode material for high-energy lithium/sulfur batteries. Nano Lett. 12, 6474–6479 (2012).
Seh, Z. W. et al. High-capacity Li2S-graphene oxide composite cathodes with stable cycling performance. Chem. Sci. 5, 1396–1400 (2014).
Han, K. et al. Li2S-reduced graphene oxide nanocomposites as cathode material for lithium sulfur batteries. J. Power Sources 251, 331–337 (2014).
Yang, Z. et al. In situ synthesis of lithium sulfide-carbon composites as cathode materials for rechargeable lithium batteries. J. Mater. Chem. A 1, 1433–1440 (2013).
Guo, J., Yang, Z., Yu, Y., Abruna, H. D. & Archer, L. A. Lithium-sulfur battery cathode enabled by lithium-nitrile interaction. J. Am. Chem. Soc. 135, 763–767 (2013).
Zheng, S. et al. In situ formed lithium sulfide/microporous carbon cathodes for lithium-ion batteries. ACS Nano 7, 10995–11003 (2013).
Seh, Z. W. et al. Facile synthesis of Li2S-polypyrrole composite structures for high-performance Li2S cathodes. Energy Environ. Sci. 7, 672–676 (2014).
Wu, F. et al. Harnessing steric separation of freshly nucleated Li2S nanoparticles for bottom-up assembly of high-performance cathodes for lithium-sulfur and lithium-ion batteries. Adv. Energy Mater. 4, 1400196 (2014).
Nan, C. et al. Durable carbon-coated Li2S core-shell spheres for high performance lithium/sulfur cells. J. Am. Chem. Soc. 136, 4659–4663 (2014).
Lin, Z., Liu, Z., Dudney, N. J. & Liang, C. Lithium superionic sulfide cathode for all-solid lithium-sulfur batteries. ACS Nano 7, 2829–2833 (2013).
Obrovac, M. N. & Dahn, J. R. Electrochemically active lithia/metal and lithium sulfide/metal composites. Electrochem. Solid-State Lett. 5, A70–A73 (2002).
Yang, Y. et al. High-capacity micrometer-sized Li2S particles as cathode materials for advanced rechargeable lithium-ion batteries. J. Am. Chem. Soc. 134, 15387–15394 (2012).
Seh, Z. W. et al. Stable cycling of lithium sulfide cathodes through strong affinity with a bifunctional binder. Chem. Sci. 4, 3673–3677 (2013).
Fu, Y., Zu, C. & Manthiram, A. In situ-formed Li2S in lithiated graphite electrodes for lithium-sulfur batteries. J. Am. Chem. Soc. 135, 18044–18047 (2013).
Meini, S., Elazari, R., Rosenman, A., Garsuch, A. & Aurbach, D. The use of redox mediators for enhancing utilization of Li2S cathodes for advanced Li-S battery systems. J. Phys. Chem. Lett. 5, 915–918 (2014).
Chianelli, R. R. & Dines, M. B. Low-temperature solution preparation of group 4B, 5B, and 6B transition-metal dichalcogenides. Inorg. Chem. 17, 2758–2762 (1978).
Chen, J., Li, S. L., Tao, Z. L., Shen, Y. T. & Cui, C. X. Titanium disulfide nanotubes as hydrogen-storage materials. J. Am. Chem. Soc. 125, 5284–5285 (2003).
Sandoval, S. J., Chen, X. K. & Irwin, J. C. Raman-spectra of AgxTiS2 and lattice-dynamics of TiS2 . Phys. Rev. B 45, 14347–14353 (1992).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 48, 13115–13118 (1993).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
Y.C. acknowledges the support of the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy. Z.W.S. acknowledges the support of an A*STAR National Science Scholarship. We thank Dr Yuan Yang and Dr Mauro Pasta for their helpful discussions.
Author information
Authors and Affiliations
Contributions
Z.W.S. and Y.C. conceived the idea. Z.W.S. carried out materials synthesis and electrochemical tests. Z.W.S., J.H.Y., W.L., P.-C.H., H.W., Y.S. and H.Y. performed materials characterization. Q.Z. carried out ab initio simulations. Z.W.S. and Y.C. co-wrote the paper. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-11 (PDF 7451 kb)
Rights and permissions
About this article

Cite this article
Seh, Z., Yu, J., Li, W. et al. Two-dimensional layered transition metal disulphides for effective encapsulation of high-capacity lithium sulphide cathodes. Nat Commun 5, 5017 (2014). https://doi.org/10.1038/ncomms6017
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms6017
This article is cited by
-
Engineering Strategies for Suppressing the Shuttle Effect in Lithium–Sulfur Batteries
Nano-Micro Letters (2024)
-
Optimization on transport of charge carriers in cathode of sulfide electrolyte-based solid-state lithium-sulfur batteries
Nano Research (2023)
-
Development of quasi-solid-state anode-free high-energy lithium sulfide-based batteries
Nature Communications (2022)
-
Single-dispersed polyoxometalate clusters embedded on multilayer graphene as a bifunctional electrocatalyst for efficient Li-S batteries
Nature Communications (2022)
-
V2CTx MXene and its derivatives: synthesis and recent progress in electrochemical energy storage applications
Rare Metals (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
