
Abstract
Natural gas extraction from unconventional shale gas reservoirs is the subject of considerable public debate, with a key concern being the impact of leaking fugitive natural gases on shallow potable groundwater resources. Baseline data regarding the distribution, fate, and transport of these gases and their isotopes through natural formations prior to development are lacking. Here, we define the migration and fate of CH4 and δ13C-CH4 from an early-generation bacterial gas play in the Cretaceous of the Williston Basin, Canada to the water table. Our results show the CH4 is generated at depth and diffuses as a conservative species through the overlying shale. We also show that the diffusive fractionation of δ13C-CH4 (following glaciation) can complicate fugitive gas interpretations. The sensitivity of the δ13C-CH4 profile to glacial timing suggests it may be a valuable tracer for characterizing the timing of geologic changes that control transport of CH4 (and other solutes) and distinguishing between CH4 that rapidly migrates upward through a well annulus or other conduit and CH4 that diffuses upwards naturally. Results of this study were used to provide recommendations for designing baseline investigations.
Similar content being viewed by others
Introduction
Global natural gas reserves in organic-rich, low-permeability shales are estimated at 716 trillion m3 1. Natural gas extracted from these unconventional shale gas reservoirs is an important energy resource being developed in several countries and is under consideration in several others2. However, animated public debate continues over possible environmental and human impacts3, in particular the impact of leaking fugitive natural gases (i.e., methane, C1; ethane, C2; propane, C3; butane, C4; and pentane, C5) and fluids from the development of unconventional gas plays on shallow potable groundwater resources4,5,6. Evaluation of these impacts relies on baseline data regarding the distribution, fate, and transport of these gases and their isotopes through natural formations prior to development. Such data are lacking, both in regard to detailed site-specific cross-formational distributions of organic gases as well as distributions within different geographic areas4, 7. Here, we define the migration and fate of CH4, the dominant unconventional gas, and δ13C-CH4 from an early-generation bacterial gas play in the Cretaceous of the Williston Basin (WB), Canada8 to the water table and provide guidance on designing baseline investigations.
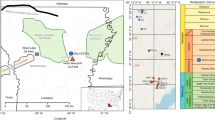
The WB is an important reservoir of unconventional energy in North America underlying 250,000 km2 of Montana and North Dakota in the USA and Saskatchewan and Manitoba in Canada (Fig. 1a). The dominant unconventional energy play in the WB is in the Bakken Formation9, 10 (Fig. 1a) and is characterized as thermogenic in origin11. Unconventional gas-producing zones also exist near the base of thick, laterally extensive, younger Cretaceous-age shales in the WB and Western Canadian Sedimentary Basin (Fig. 1a). We collected detailed gas molecular and isotopic composition profiles from the overlying glacial deposits, that are widespread throughout the WCSB, with thicknesses up to 300 m12, through the Cretaceous shale at two sites, Sites 2 and 5, to depths of 150 and 200 m below ground surface (BG), respectively. We also collected the same data from the water table through the gas source zone at two additional sites, Sites 6 and 7, to 971 m BG. All sites were located near Weyburn, Saskatchewan (lat: 49.6633; long: 103.8533) (Fig. 1b).

Location map. (a) Extent of the Western Canadian Sedimentary Basin (yellow), Williston Basin (blue)43, and Bakken shale play (orange)3 in northwestern USA and western Canada, and location of study area. (b) Location of Sites 1–415, Site 532, and Sites 6 and 7. Highways are shown as solid black lines in (b). Adapted with permission from ref. 3 and ref. 54. Copyright 2014 American Chemical Society and 2012 Springer International, respectively. Map was created with ArcMap version 10.5 (http://esri.ca/en/products/whats-new-in-arcgis-105).
Overall, the geology across all sites is consistent (Figs 2–4). The upper 35 to 83 m consists of clay-rich, Quaternary-aged glacial sediments of the Saskatoon Group (0.01–0.6 Ma BP) and Sutherland Group (0.6–1.6 Ma BP)13. The surficial 8–10 m of till is brown (oxidized) and visibly fractured and the underlying till and bedrock is dark grey (anoxic) and nonfractured. The water table is located within the oxidized till zone (~6.4 and ~7.3 m BG at Sites 2 and 5, respectively). Cretaceous shale formations disconformably underlie the tills. The thick Pierre Shale Fm (~650 m thick; 72.1–83.6 Ma BP) directly underlies the tills. It consists of non-calcareous, grey to dark grey, hard, high plasticity silt and clay14. The Mannville Gr (~115 m thick; 100.5–145 Ma BP), located at the base of the Cretaceous, is a regionally extensive, sand-dominated aquifer and defines the base of our investigation.

Geology (Site 2) and dissolved CH4 concentrations (a), Cl− concentrations (b), and δ13C-CH4 values (c) vs. depth below the top of the Pierre Shale at Sites 2 and 6. The Isojar® data from Site 2 are shown as solid red dots, and mud gas data from Sites 2 and 6 as solid green squares and solid blue triangles, respectively.

Geology (Site 5) and dissolved CH4 concentrations (a), Cl− concentrations (b), and δ13C-CH4 values (c) vs. depth below the top of the Pierre Shale at Site 5. Site 6 data from Fig. 2 are added in this figure to show the different CH4 and Cl− profiles at Site 5 versus Sites 2 and 6. The Isojar® data from Site 5 are shown as solid red dots, and mud gas data from Sites 5 and 6 as solid green squares and solid blue triangles, respectively.

Geology and dissolved CH4 concentrations (a), Cl− concentrations (b), and δ13C-CH4 values (c) through the Quaternary deposits and Cretaceous shales at Sites 2, 5, 6, and 7. In (a) and (b), Isojar® data from Sites 2 and 5 are shown as solid blue diamonds and grey stars, respectively, and calibrated mud gas data from Sites 2, 5, and 6 as dashed black line, solid black line, and solid red circles, respectively. Measured Cl− concentrations from the base of the Pierre Fm to the Joli Fou Fm from a drill site located 250 km SE of the study area22 are presented as solid black squares in (b). In (c), IsoTube® data from Sites 2 and 7 are presented as solid blue diamonds and green crosses, respectively, and mud gas data from Sites 5 and 6 as solid pink inverted triangles and solid red circles, respectively. The best-fit 1-D diffusive modeling results (1.6 Ma and 0.3 Ma for Sutherland and Saskatoon Group tills, respectively) are presented as solid lines (Sites 2 and 6) and dashed lines (Site 5) in (a), (b), and (c).
We measured similar CH4 concentration-depth profiles below the water table at Sites 2, 5, and 6 (Figs 2–4). The upper portion of the CH4 profiles are concave downwards and increasing with depth from low (typically < 1.0 mg L−1) above the till-shale contact at Sites 2 and 6 or slightly below the anoxic-oxic till contact (12 m BG) at Site 5 to about 225 mg L−1 at 200 m below the top of the Pierre Shale Fm (BTS). The concentrations increase in a linear manner (y = −2.6x–220; n = 106; R2 = 0.66) from 250 m BTS to approximately 300 mg L−1 near the base of the Pierre Shale Fm (600 m BTS). The concentrations increase rapidly to > 1000 mg L−1 across the Colorado to the Base Fish Scales Fm (600–800 m BTS) with maximum concentrations in the Second White Specks and Lower Colorado Sand Fms (707–763 m BTS) where the hydrocarbons are generated (Fig. 4). Below the Base Fish Scales Fm, the concentrations decrease through the Viking and Joli Fou Fms to 236 mg L−1 at the top of the Mannville Gr (881 m BTS). Although the datasets are limited in areal extent, the consistency between the CH4 depth profiles from all sites and the fact we observe elevated CH4 concentrations in the Second White Specks Fm at a site located 250 km NE of the study area15 suggest our data represent general CH4 conditions in the Cretaceous shales across the WB.
Concentrations of CH4 through the Quaternary and shale deposits are below the solubility limit (Fig. S1) as estimated from in vitro gas concentrations corrected for in situ temperatures and pressures. From the Second White Specks Fm to the Base Fish Scales Fm, however, CH4 concentrations are equal to or exceed the estimated solubility limit suggesting the presence of free CH4 gas in these formations. This determination is supported by the presence of seismic reflectors believed to be associated with free gas in the same formations at other sites in the WB (pers. comm. D Gendzwill; pers. comm. J. Szmigielski). The Second White Specks Fm is a known early-generation biogenic gas reservoir in Cretaceous sediments of Western Canada8.
The dissolved chloride (Cl−) concentration-depth profiles at Sites 2 and 5 (Figs 2 and 3) positively correlate with the CH4 data; cross-plotted CH4 vs. Cl− concentrations yield strong linear relationships (y = 0.04x + 3.4, R2 = 0.90, n = 15 and y = 0.06x + −28.5, R2 = 0.89, n = 35, respectively; Fig. S2. Given that Cl− is a conservative species, these strong linear relationships suggest, on the whole, the same transport mechanism is operative for both Cl− and CH4 with no indication of losses of CH4 from biogeochemical activity (i.e., no production or removal) in the CH4 concentration-depth profiles in the till and shale. The lack of sorption-desorption reactions for CH4 is supported by laboratory experiments and consistent with low concentrations of total organic carbon in the Pierre Shale (~0.8% wt)16.
The strong linear correlations over the entire data sets, however, do not apply at the upper boundaries for Cl− and CH4 profiles at all sites (Fig. S2). We compared the upper boundaries for the Cl− and CH4 profiles at Sites 2 and 5 to identify the controls on CH4 depletion at these locations (by either a biological or physical mass loss process). The upper boundaries for both the increasing Cl− and CH4 depth profiles at Site 2 are at the till-shale interface (about 31 m BG). That both the Cl− (240 mg/L) and CH4 (1.3 mg/L) concentrations were low and constant at and above this depth suggests the dominant control on both solutes at Site 2 is flushing/dilution by lateral water migration, likely through more permeable sediments in the glacial till16. In contrast to Site 2, the upper boundaries for Cl− (450 mg/L) and CH4 (3.4 mg/L) at Site 5 are located at different depths in the till. The upper boundary for the Cl− profile is located at the base of the oxidized, fractured till zone (about 4.5 m BG) whereas the upper boundary for the CH4 profile is in the underlying anoxic till (about 12 m BG). Unlike at Site 2, the extension of both Cl− and CH4 profiles well into the till at Site 5 is attributed to the lack of permeable zones in the till above the shale (i.e., no permeable layers were observed in continuous core samples from the till). The upward migration of solutes from the shale into the overlying till at Site 5 is observed in other tills and also attributed to a lack of permeable zones17, 18. The presence of the upper boundary for the Cl− profile in the oxidized till at Site 5 is attributed to the presence of fractures (higher K) in this zone that allows solutes to be transported laterally with flowing groundwater19. In contrast, the presence of the upper CH4 boundary in the underlying anoxic till is attributed to CH4 oxidation associated with the reduction of SO4 2− generated in the oxidized till by the oxidation of reduced sulphur during the Altithermal Period, a mid-Holocene warming period between 9000 and 5000 years before present20,21,22.
Although the shallow data at Site 6 are limited, the trend in CH4 at the upper boundary is consistent with Site 2. The geologic log for Site 6 also shows the till has a high sand content, indicative of the presence of permeable sand layers. The fact that the upper boundaries for Cl− and CH4 at Sites 3 and 416, also located in our study area (Fig. 1b), coincide with the presence of a permeable zone at the top of the Pierre Shale Fm suggests that physical control (i.e., lateral flushing) may be more common at the upper boundary for CH4 than the biogeochemical reactions (i.e., methane oxidation) observed at Site 5.
The overall strong correlation between CH4 and Cl− profiles at Sites 2 and 5 was used to estimate Cl− concentrations through the Pierre Shale at Site 6. The good visual fits between the estimated Cl− profile and those measured on squeezed samples from Sites 2 and 5 (Figs 2b and 3b) and the estimated Cl− concentrations being consistent with those measured through the lower 275 m of Cretaceous shales at a site ~250 km SE of the study area23 (Fig. 4b) provide confidence in the calculated Cl− profile for Site 6. As was the case for the CH4 profiles, the Cl− profiles exhibit well-defined, slight curvilinear (concave down) depth trends to about 200 m BTS after which they increase in a linear manner from 250 m BTS to near the base of the Pierre Shale Fm (600 m BTS) (Fig. 4b). Based on Cl− concentration profiles with depth through the WB, the source of Cl− in the Cretaceous shales of the WB was determined to be the Prairie Evaporite Fm, located about 1800 m BG in our study area23.
The shapes of the CH4 and Cl− concentration profiles suggest the dominant transport mechanism for these species is diffusion. Diffusive transport in the Pierre Shale Fm and anoxic tills is supported by low measured hydraulic conductivity (K) values of Cretaceous shale (10−12–10−13 m/s)24 and anoxic tills (10−10–10−11 m/s)25, 26 in the WB. The dominance of diffusive transport in low K argillaceous sediments is consistent with findings from other argillaceous systems in the WB or proximal to it15, 23, 27, 28 and in other areas of the world29,30,31,32,33.
We modeled the CH4 and Cl− concentration profiles in the Pierre Shale Fm and Quaternary tills at Sites 2 and 5 assuming diffusion to be the dominant solute transport mechanism. Results of modelled scenarios are summarized in Figs 4 and S6 to S11 and the model parameters used are summarized in Table S1. The curvilinear profiles in the upper 200 m of the profiles developed in response to assumed abrupt changes in the upper boundary condition. These abrupt changes are attributed to the effects of glaciation17, 27. The good fits between the modeling results and measured data for time frames consistent with known ages of shale and glacial deposition confirm that long-term upward diffusive transport over many millions of years can explain the current measured Cl− and CH4 profiles. Although the times (i.e., onset of Sutherland Gr. glaciation at 1.6 Ma and onset of Saskatoon Gr. glaciation between 0.1 and 0.6 Ma) used to simulate the measured profiles (especially the upper 200 m of Pierre Shale Fm) are consistent with the timing of the glaciations, the results cannot be considered unique because they are based on a limited understanding of the geologic history of the shale and tills. However, as we show below, the estimates of timing can also be constrained by simulating the δ13C-CH4 profiles.
The δ13C-CH4 depth profiles at all sites exhibit a well-defined curvilinear enrichment in 13C with increasing depth to about 250 m BG (Figs 2–4). Over this depth, the δ13C-CH4 values increase from about −85‰ to about −70‰. Below about 250 m BG depth, the δ13C-CH4 values are constant, ranging from −65.3 to −68.5‰ (depending on sampling method).
The δ13C-CH4 values (−72‰ to −64‰) and lower concentration ratios of CH4 to the heavier hydrocarbons (C1/C2+; >10; Fig. S12) for all samples collected in this study are consistent with early-generation bacterial gas8. Late-generation biogenic gases are often thought to be 13C-depleted relative to these values and have concentration ratios of CH4 to the heavier hydrocarbons (C1/C2+; >1000)34,35,36.
Well-defined changes in the δ13C-CH4 values with depth such as those measured across the Pierre Shale are not documented in the literature. A similar isotopic shift to that observed in the upper 250 m of shale was, however, measured on mud gas samples at a site near Brooks, Alberta (600 km NW of our study area)37. In that study, the δ13C-CH4 values increased from −80‰ at depths <50 m BG to −62‰ at about 150 m BG and remained constant at this value to 530 m BG. The similarity between the Brooks profile and our profiles, and given the 600-km distance between these sites, suggests the δ13C-CH4 profiles in our study area represent conditions across the WB. Some authors suggest changes in δ13C-CH4 values can be caused by CH4 transport in the subsurface16, 38,39,40 while others discount the effects of δ13C-CH4 migration on δ13C values41.
The constant δ13C-CH4 values measured below 250 m BG are consistent with near steady-state CH4 transport through the shale prior to glaciation (as demonstrated in the modelling). An abrupt change in the upper CH4 boundary condition, such as glaciation, would trigger transient diffusion that would cause diffusive isotopic fractionation. The isotopic shift in the δ13C-CH4 profiles to 250 m BG is consistent with experimental results that show diffusing CH4 has a significantly 13C-depleted δ13C value than its source38. Because the CH4 and Cl− profiles in our study can be explained via diffusive transport, we assessed whether diffusive transport could produce the measured δ13C-CH4 profiles by conducting diffusive transport modeling of the δ13C-CH4 depth profiles through the shale at Sites 2 and 5. The simulation results using a consistent set of transport conditions to those used in the CH4 simulations yield reasonable fits to the measured data (Fig. 4) and suggest that isotopic fractionation as a result of differences in the diffusion of 12C and 13C in the CH4 can explain the measured δ13C-CH4 profile. As was the case for the CH4 and Cl− simulations, the δ13C-CH4 simulations are non-unique.
Based on high-resolution profiling, we gained an understanding of the origin, fate, and transport of CH4 in the WB. Specifically, the study provided insights into gas migration processes, CH4 source area delineation and methods to define upper boundary controls. Findings show the source of the CH4 and δ13C-CH4 in the shallow groundwater is an early-generation bacterial gas play located in the Lower Colorado sand and Second White Specks Fms. This CH4 migrates upward from its source area through the overlying shale and into the shallow subsurface via non-reactive diffusion over millions of years. Modelling the migration of the δ13C-CH4 profiles can be used to improve the estimates of timing obtained from modelling of the CH4 and Cl− profiles. Our observations relating Cl− and CH4 concentrations in the near surface environment can be used to define the controls on CH4 (i.e., flushing vs. microbiological). The Cl− and CH4 concentrations data could also provide insight with respect to distinguishing recent CH4 production from gas migration in near surface aquifers. For example, if the concentrations of both CH4 and Cl− in aquifer waters are elevated, the CH4 may be sourced deeper in the basin. However, if Cl− concentrations are low relative to CH4 concentrations, CH4 is likely produced in the aquifer. The −15‰ diffusive shift in δ13C-CH4 over the upper 250 m of the WB can generate isotopic values comparable to those commonly associated with more recent methanogenic CH4 production in shallow potable aquifers (<−80‰)22, 36, 42; thus complicating our ability to distinguish CH4 produced in an aquifer from the impacts of CH4 that fractionated during migration from early-generation bacterial gas plays. This negative shift in carbon isotopes and co-migration of Cl− allows us to distinguish CH4 contributions from pre-development CH4, recent microbial production, and migration from shallow bacterial gas plays (13C-depleted δ13C values relative to −65‰) from fugitive gases that rapidly migrate upward through a well annulus or other conduit originating from bacterial gas plays and deeper thermogenic oil-associated production zones in the WB (13C-enriched δ13C values relative to −65‰)11, 42,43,44. Although the large δ13C-CH4 fractionation resulting from diffusion can complicate identifying bacterial gas sources, our improved understanding of the regional dynamics of gas migration provides a new means to evaluate CH4 sources in potable groundwater resources in the WB and similar basins in western North America. Data generated in our study can also inform other valuable studies including those applying the model-derived upward flux of CH4 (6.6 × 10−2 mg m−1 d−1) to estimate long-term gas production from shallow biogenic gas plays.
The challenge to define the environmental impact of anthropogenic CH4 on shallow aquifers requires differentiation between natural in situ microbial CH4 from the migration of microbial or thermogenic CH4 from depth, from thermogenic CH4 that migrates from an even greater depth due to anthropogenic activities45. Although water samples commonly collected from domestic or farm water wells are used to define baseline conditions46, 47, Jackson and Heagle48 state that such receptor monitoring does not yield baseline concentrations of the aquifer or the regional groundwater supply. We suggest that not wells, but high-resolution profiling be used to provide the much-needed information in environmentally sensitive aquifer systems. This profiling only needs to extend deep enough below the aquifer to allow its evolution to and in the aquifer to be characterized. Based on the profile development in the current study, we suggest the profiling terminate 20–50 m below the bottom of the aquifer. We also suggest, based on the profiles generated in the current study, the sample interval could be reduced from 3m in the current study to 5 m. Assuming the base of the aquifer is located about 50 m BG (a common aquifer depth reported)47, and the core hole is terminated 50 m below the base of the aquifer, 20 samples would be collected for analysis. This approach can be integrated with existing drilling programs for new assets (production wells, observation wells, groundwater wells, etc.), where the additional geochemical costs are often negligible compared to drilling costs that could be offset by the potential for major reductions in the future costs associated with fugitive gas migration source tracking and potable aquifer impact assessments.
Methods
Drilling, Sampling, and Analyses
Rotary drilling and geologic logging were performed between September 2013 and September 2014 to 150, 200, 1502, and 1432 m below ground surface (BG) at Sites 2, 5, 6, and 7, respectively (Sites 6 and 7 were located ~800 m apart) (Fig. 1). Continuous core sampling was conducted at all four sites and mud gas logging at Sites 6 and 7 only.
Core samples (~100–150 mm long) were collected from Sites 2 and 5 at 3 m intervals and analyzed for: (1) wet and dry densities (ρ m and ρ d ) and gravimetric water contents (w) (used to calculate total porosity, n T ); (2) dissolved anion chemistry (porewater was squeezed from core samples); and (3) CH4, ethane (C2H6), and propane (C3H8) concentrations (ppmV). Gas concentrations were measured in the headspace of Isojars® flushed with inert N2 or Ar prior to sealing (~440 cm3). Core samples collected in Isojars® at Sites 2 and 5 were allowed to equilibrate with the headspace at standard temperature (25 °C) and pressure (1 atm) for 30 and 101 d, respectively, prior to analyses. For analyses, about 10 cm3 gas from the headspace was collected from each Isojar® with a 60 cm3 syringe and injected into the septa port of an Agilent 7890 gas chromatograph (GC) equipped with a flame ionization detector (FID) to measure light hydrocarbons and a thermal conductivity detector to measure O2 and N2. Based on analyses conducted on ScottyTM 17 L calibration gas standards (concentrations of 0.0010, 0.01, 0.1, and 10% CH4 and 0.0010, 0.01, 0.1, and 1.0% C2H6 and C3H8) and on replicate core samples, the accuracy of the analytical method was ± 5%. The limit of detection (LOD) and limit of quantification (LOQ) for CH4, C2H6, and C3H8 were determined to be 1.1 and 11 ppm, respectively. Concentrations measured in the headspace of the Isojars® were converted to dissolved concentrations (mg L−1) using a previously outlined method33, 49. These calculations used the core-derived nT values.
At Site 2, the mud gas collection cylinder, located in the mud tank, was connected to an Agilent 490 gas chromatograph (GC) and an IsoTube® gas-sampling manifold. A Los Gatos Research Inc. (LGR) methane carbon isotope analyzer (MCIA)-24-EP CH4 was connected in line with the outlet of the IsoTube® gas sampler at Sites 5 and 6. The C1-C5 concentrations were measured on the mud gas using the Agilent GC every 90 s with a repeatability of 0.5% at 1 mol% and an LOD of 1 ppmV. The LGR CH4 analyzer measured CH4 concentrations (0–4,000 ppmV) and δ13C-CH4 values over a CH4 concentration range of 500–10,000 ppmV every 50 s. The analytical error for the δ13C-CH4 values was ± 1.0‰, based on LGR’s model specifications; however, field conditions may have raised the analytical error to ± 1.0‰ - 2‰. Although the LGR CH4 analyzer required no internal calibration during sampling, a CH4 calibration standard (CH4 = 10,000 ppmV, δ13C-CH4 = −42.3‰) was analyzed every 6 h as a data quality assurance measure to verify instrument accuracy and precision. The δ13C-CH4 values reported from the LGR were confirmed by submitting 10 gas samples covering the range in values (−67 to −80‰) measured in the Isojars® and analyzed on the LGR at Site 5 to the Isotope Science Lab, University of Calgary. These duplicate results were in good agreement (mean difference = 0.15 ± 0.7‰). δ13C analyses were reported in permil (‰) relative to Vienna Pee Dee Belemnite (VPDB), with an accuracy and precision better than ± 0.2‰.
The CH4 concentrations from mud gas profiling at Sites 2 and 5 yielded the same depth trend as the core-jar samples but were lower; this was attributed to dilution by drilling fluid during hole advancement followed by dilution with atmospheric gases during sample collection in the mud tank32. The strong linear relationship (y = 13.6x + 65.1; R2 = 0.83, n = 76) between the dissolved CH4 concentrations (mg L−1) from core samples collected at Sites 2 and 5 vs. CH4 concentrations measured during mud gas logging (ppmV) was used to convert CH4 concentrations from mud gas to dissolved gas concentrations at Sites 2 and 5. Because the collection cylinder used to capture mud gas at Site 6 was reconfigured (to minimize atmospheric contamination into the mud gas line) from that used at Sites 2 and 5, calculated dissolved CH4 concentrations at Site 6 were determined by multiplying the equation to convert CH4 concentrations from mud gas to dissolved gas concentrations at Sites 2 and 5 (y = 13.6x + 65.1) by 0.25 to account for dilution of the mud gas concentrations during hole advancement. The strong linear relationship (y = 0.05x–17.6; R2 = 0.88, n = 56) between dissolved CH4 concentrations (mg L−1) vs. Cl− concentrations (mg L−1) measured from core samples collected at Sites 2 and 5 was used to convert calibrated dissolved CH4 concentrations (mg L−1) collected during mud gas logging at Site 6 to dissolved Cl− concentrations.
IsoTubes® collected at Site 2 were shipped to the G.G. Hatch Stable Isotope Laboratory at the University of Ottawa, Canada, where they were stored for 480 d prior to analysis of δ13C-CH4 using a Thermo Finnigan DeltaPlusAdvantage IRMS coupled with a VarioEL III, with an accuracy and precision better than ± 0.2‰. IsoTube® samples collected from Site 7 were shipped to Isobrine Solutions Inc., where they were analyzed within 60 days on a Thermo Finnigan MAT252 IRMS, with an accuracy and precision better than ± 0.5‰. Real-time mud gas δ13C-CH4 values were measured using the LGR CH4 and δ13C analyzer at Sites 5 and 6. A slight enrichment in δ13C-CH4 values measured on Isotube® samples from Site 7 compared to values measured during mud gas isotope logging at Site 6 (+2.9‰ ± 1.6‰; n = 25; Fig. 4) is attributed to experimental error and/or minor secondary microbial reactions (e.g. methane oxidation) during sampling and storage of the Isotube® and core samples33. To test the reliability of the tubes analysis 32 tubes collected at Sites 2, 3, and 4 that were initially run at 480 d after sampling were resubmitted to the University of Ottawa for re-analysis of CH4 and δ13C-CH4 1160 d after collection. Results showed that over the additional ~2.5 a of storage since the initial analysis (480 d), the concentration of CH4 decreased by an average of 13.5% (mean = 13.5 ± 7.0 ppm; n = 32) and the δ13C-CH4 shifted by an average of 0.87‰ (mean = 0.87 ± 1.44‰, n = 32). These data show that long-term storage in Isotubes have a minimal impact on the CH4 concentrations and δ13C-CH4 values.
Additional details on the methods and measured values from Sites 2 and 5 are provided elsewhere16, 33.
Transport Modeling
Simulation of the diffusive transport of the dissolved CH4, Cl− and δ13C-CH4 profiles was based on modelling conducted16 on the shallow (<165 m BTS) profiles at Site 2. The simulations were performed using a commercially available finite element model (CTRAN/W)50. In the current model, we incorporated the deeper profiles through the Pierre Shale Fm obtained from Sites 6 and 7 and the shallow datasets from Sites 2 and 5 (not modeled in ref. 16). The objectives of the modeling were to assess whether the measured profiles can be attributed to diffusion-dominated solute transport and, if so, can the CH4, Cl−, and δ13C-CH4 profiles from each site be explained based on our current understanding of the glacial history of the region (Table S1).
The simulations of CH4, Cl−, 12C-CH4, and 13C-CH4 transport were based on transient one-dimensional diffusive transport:
where n e is the effective porosity, D e is the effective diffusion coefficient (m2 a−1), C is the mass concentration of the solute (g m−3), z is distance (m), and t is time (a). D e in equation (1) was defined according Fick’s first law as:
where J d is the diffusive mass flux rate (g m−2 s−1).
The diffusive transport of 12C-CH4 and 13C-CH4 were determined separately and used to calculate the δ13C-CH4 profile based on Eqn. 3 40:
where \({R}_{({}^{13}C-C{H}_{4})}\) is the isotope ratio of CH4, defined as
where R std is the standard isotopic ratio for Pee Dee Belemnite (PDB; 0.011237) and M is the molecular mass.
The values of D e for Cl− were assumed to be 2.1 × 10−10 and 2.3 × 10−10 m2 s−1 in the till28 and Pierre shale23, respectively. Values of D e for CH4 were assumed to be 2.4 × 10−10 m2 s−1 for both the till and shale51. The n T values were determined on individual core samples. Mean values of n T for the till and shale were 33.2 ± 4.4 and 34.3 ± 2.9, respectively, were used in the modelling. The value of n e for CH4 was assumed to be equal to the total porosity (n T )51. The n e values for Cl− were determined to be n e = n T for the till and n e = 0.7n T for the Pierre Shale Fm16. The value of D e for 12C-CH4 was assumed to be the same as that for CH4 transport in the shale (2.4 × 10−10 m2 s−1). The difference in molecular mass between 12CH4 and 13CH4 results in the D e for 12CH4 exceeding that of 13CH4 by a factor of 1.0195 based on measurements of diffusion in air52. This ratio was assumed to apply to our simulations for aqueous diffusion. As such, the value of D e for 13C-CH4 was 2.35 × 10−10 m2 s−1.
One-dimensional transport modelling was undertaken in two phases. Phase 1 defined the distributions of CH4, Cl−, and δ13C-CH4 across the shale prior to glaciation, and Phase 2 assessed the impact of Sutherland and Saskatoon Gr glacial periods on the final (present-day) profiles for CH4, Cl−, and δ13C-CH4 at Sites 2 and 5. Details of the methodologies used in each phase of modelling are presented in Supplemental Information.
The relative root mean square error (RRSME)53 was used to compare the simulated and measured concentration profiles for each scenario and determine the best-fit model results (data not presented).
References
Boyer, C., Clark, B., Jochen, V., Lewis, R. & Miller, C. K. Shale gas: A global resource. Oilfield Rev. 23, 28–39 (2011).
US Energy Information Administration (US EIA). Annual Energy Outlook 2013, with Projections to 2040. United States Department of Energy: Washington, DC (2013).
Vengosh, A., Jackson, R. B., Warner, N., Darrah, T. H. & Kondash, A. A Critical Review of the Risks to Water Resources from Unconventional Shale Gas Development and Hydraulic Fracturing in the United States. Environ. Sci. Technol. Lett. 48(15), 8334–8348 (2014).
Jackson, R. E., Gorody, A. W., Mayer, B., Ryan, M. C. & Van Stempvoort, D. R. Groundwater protection and unconventional gas extraction: The critical need for field-based hydrogeological research. Groundwater. 51(4), 488–510, doi:10.1111/gwat.12074 (2013).
Kargbo, D. M., Wilhelm, R. G. & Campbell, D. J. Natural gas plays in the Marcellus Shale: challenges and potential opportunities. Environ. Sci. Technol. 44(15), 5679–5684 (2010).
Osborn, S., Vengosh, A., Warner, N. R. & Jackson, R. B. Methane contamination of drinking water accompanying gas-well drilling and hydraulic fracturing. Proc. Natl. Acad. Sci. USA 108, 8172–8176 (2011).
Vidic, R. D., Brantley, S. L., Vandenbossche, J. M., Yoxtheimer, D. & Abad, J. D. Impact of Shale Gas on Regional Water Quality. Science 340(6132), doi:10.1126/science.1235009 (2013).
Shurr, G. W. & Ridgley, J. L. Unconventional shallow biogenic gas systems. AAPG Bull. 86(11), 1939–1969 (2002).
Schmoker, J. W. & Hester, T. C. Organic Carbon in Bakken Formation, United States Portion of Williston Basin. AAPG Bulletin 67(12), 2165–2174 (1983).
Meissner, F. F. Guidebook to Geology and Horizontal Drilling of the Bakken Formation. pp 19–42 (Montana Geological Society, 1991).
Muscio, G. P. A., Horsfield, B. & Welte, D. H. Occurrence of thermogenic gas in the immature zone – implications from the Bakken in-source reservoir system. Org. Geochem. 22(3–5), 461–476 (1994).
Fenton, M. M., Schreiner, B. T., Nielsen, E. & Pawlowicz, J. G. Chapter 26 – Quaternary Geology of the Western Plains. In: Geological Atlas of the Western Canadian Sedimentary Basin. G. D. Mossop and I. Shetsen (comp.), Canadian Society of Petroleium Geologists and Alberta Research Council, http://ags.aer.ca/publications/chapter-26-quaternary-geology-of-the-western-plains [April 27, 2017].
Christiansen, E. A. Pleistocene stratigraphy of the Saskatoon Area, Saskatchewan, Canada: an update. Can. J. Earth Sci. 29, 1767–1778 (1992).
Bilodeau, D. & Potter, G. Weyburn Core Hole Project: Coring, Drilling and Instrumentation, Report prepared for Cenovus Energy Inc. by SNC Lavalin (2012)
Hendry, M. J., Richman, B. & Wassenaar, L. I. Correcting for Methane Interferences on δ2H and δ18O Measurements in Pore Water Using H2O(liquid)-H2O(vapour) Equilibration Laser Spectroscopy. Anal. Chem. 83, 5789–5796 (2011).
Hendry, M. J., Barbour, S. L., Schmeling, E. E., Mundle, S. O. C. & Huang, M. Fate and transport of dissolved methane and ethane in the cretaceous shales in the Williston Basin, Canada. Water Resour. Res. 52(8), 6440–6450 (2016a).
Hendry, M. J. & Wassenaar, L. I. Implications of transport of δD in porewaters for groundwater flow and the timing of geologic events in a thick aquitard system. Water Resour. Res. 35(6), 1751–1760 (1999).
Hendry, M. J., Wassenaar, L. I. & Kotzer, T. Chloride and chlorine isotopes (36Cl and δ37Cl) as tracers of solute migration in a thick, clay-rich aquitard system. Water Resour. Res. 36(1), 385–296 (2000).
Stumpp, C. & Hendry, M. J. Spatial and temporal dynamics of water flow and solute transport in a heterogeneous glacial till: The application of high-resolution profiles of d18O and d2H in pore waters. J. Hydrol. 438–439, 203–214 (2012).
Hendry, M. J., Cherry, J. A. & Wallick, E. I. Origin and distribution of sulfate in a fractured till in southern Alberta, Canada. Water Resour. Res. 22(1), 45–61 (1986).
van Stempvoort, D. R., Hendry, M. J., Schoneau, J. & Krouse, H. R. Sources and dynamics of sulfur in weathered till, Western Glacial Plains of North America. Chem. Geol. 111, 35–56 (1994).
Humez, P. et al. Redox controls on methane formation, migration and fate in shallow aquifers. Hydrol. Earth Syst. Sci. 20, 2759–2777 (2016).
Hendry, M. J. & Harrington, G. Comparing vertical profiles of natural tracers in the Williston Basin to estimate the onset of deep aquifer activation. Water Resour. Res. 50(8), 6496–6506 (2014).
Smith, L. A. Characterizing The Hydrogeologic Properties of Thick Claystone Aquitards in the Williston Basin Using Grouted-In Pressure Transducers, PhD thesis, University of Saskatchewan (2016).
Keller, C. K., van der Kamp, G. & Cherry, J. A. Fracture permeability and groundwater flow in clayey till near Saskatoon, Saskatchewan. Can. Geotech. J. 23, 229–240 (1986).
Shaw, J. & Hendry, M. J. Hydrogeology of a thick clay till and Cretaceous clay sequence, Saskatchewan, Canada. Can. Geotech. J. 35(6), 1041–1052 (1998).
Remenda, V. H., Cherry, J. A. & Edwards, T. W. D. Isotopic composition of old ground water from Lake Agassiz: implications for Late Pleistocene climate. Science 266(5193), 1975 (1994).
Hendry, M. J., Barbour, S. L., Novakowski, K. & Wassenaar, L. I. Palaeo-hydrogeology of the Cretaceous sediments of the Williston Basin using stable isotopes of water. Water Resour. Res. 49(8), 4580–4592 (2013).
Gimmi, T., Waber, H. N., Gautschi, A. & Rübel, A. Stable water isotopes in pore water of Jurassic argillaceous rocks as tracers for solute transport over large spatial and temporal scales. Water Resour. Res. 43(4), W04410, doi:10.1029/2005WR004774 (2007).
Mazurek, M. et al. Natural tracer profiles across argillaceous formations. Appl. Geochem. 2011, doi:10.1016/j. apgeochem.2011.03.124 (2011).
Patriarche, D., Michelot, J., Ledoux, E. & Savoye, S. Diffusion as the main process for mass transport in very low water content argillites: 1. Chloride as a natural tracer for mass transport – Diffusion coefficient and concentration measurements in interstitial water. Water Resour. Res. 40(1), W01516, doi:10.1029/2003WR002600 (2004).
Rübel, A. R., Sonntag, C., Lippmann, J., Pearson, F. J. & Gautschi, A. Solute transport in formations of very low permeability: profiles of stable isotope and dissolved noble gas contents of pore water in the Opalinus Clay, Mont Terri, Switzerland. Geochim. Cosmochim. Acta 66(8), 1311–1321 (2002).
Hendry, M. J., Barbour, S. L., Schmeling, E. E. & Mundle, S. O. C. Measuring concentrations of dissolved methane and ethane and the 13C of methane in shale and till. Groundwater, doi:10.1111/gwat.12445 (2016b).
Bernard, B. B., Brooks, J. M. & Sackett, W. M. Natural gas seepage in the Gulf of Mexico. Earth and Planet. Sci. Lett. 31(1), 48–54 (1976).
Schoell, M. The hydrogen and carbon isotopic composition of methane from natural gases of various origins. Geochim. Cosmochim. Acta 44(5), 649–661 (1980).
Whiticar, M. J. Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 161(1–3), 291–314 (1999).
Mayer, K. U., Kunimaru, T., Nakata, K. & Hasegawa, T. Can argillaceous formations isolate nuclear waste? Insights from isotopic, noble gas, and geochemical profiles. Geofluids. doi:10.1111/gfl.12132 (2015).
Prinzhofer, A. A. & Pernaton, E. Isotopically light methane in natural gas: bacterial imprint or diffusive fractionation? Chem. Geol. 142(3–4), 193–200 (1997).
Prinzhofer, A. & Battani, A. Gas isotopes tracing: an important tool for hydrocarbons exploration. Oil Gas Sci. Technol. 58(2), 299–311 (2003).
Zhang, T. & Krooss, B. M. Experimental investigation on the carbon isotope fractionation of methane during gas migration by diffusion through sedimentary rocks at elevated temperature and pressure. Geochim. Cosmoschim. Acta 65(16), 2723–2742 (2001).
Fuex, A. N. Experimental evidence against an appreciable isotopic fractionation of methane during migration. Org. Geochem. 12, 725–732 (1980).
Cheung, K., Klassen, P., Mayer, B., Goodarzi, F. & Aravena, R. Major ion and isotope geochemistry of fluids and gases from coalbed methane and shallow groundwater wells in Alberta, Canada. Appl. Geochem. 25(9), 1307–1329 (2010).
Tilley, B. & Muehlenbachs, K. Gas maturity and alteration systematics across the Western Canada Sedimentary Basin from four mud gas isotope depth profiles. Org. Geochem. 37(12), 1857–1868 (2006).
Tilley, B. & Muehlenbachs, K. Isotopically determined Mannville group gas families. In: Let it Flow – Flow of Ideas, Hydrocarbons and Business, Proceedings of the 2007 CSPG CSEG Convention (2007).
Humez, P. et al. Occurrence and origin of methane in groundwater in Alberta (Canada): Gas geochemical and isotopic approaches. Sci. Total Environ. 541, 1253–1268 (2016).
Darrah, T. H., Vengosh, A., Jackson, R. B., Warner, N. R. & Poreda, R. J. Noble gases identify the mechanisms of fugitive gas contamination in drinking-water wells overlying the Marcellus and Barnett Shales. PNAS 111(39), 14076–14081 (2014).
McIntosh, J. C., Grasby, S. E., Hamilton, S. M. & Osborn, S. G. Origin, distribution and hydrogeochemical controls on methane occurences in shallow aquifers, southwestern Ontario, Canada. Appl. Geochem. 50, 37–52 (2014).
Jackson, R. E. & Heagle, D. J. Sampling domestic farm wells for baseline groundwater quality and fugitive gas. Hydrogeol. J. 24(2), 269–272 (2016).
Kampbell, D. H. & Vandegrift, S. A. Analysis of dissolved methane, ethane, and ethylene in ground water by a standard gas chromatographic technique. J. Chromatogr. Sci. 36, 253–256 (1998).
Geo-Slope International Ltd. Contaminant Modeling with CTRAN/W, An Engineering Methodology, (3rd eds) pp 1–89 (GEO-SLOPE International, Ltd, 2008).
Jacops, E., Volckaert, G., Maes, N., Weetjens, E. & Govaerts, J. Determination of gas diffusion coefficients in saturated porous media: He and CH4 diffusion in Boom Clay. Appl. Clay. Sci. 83‒84, 217–223 (2013).
Marrero, T. R. & Mason, E. A. Gas diffusion coefficients. J. Phys. Chem. Ref. Data 1, 3–118 (1972).
Huang, M., Gallichand, J., Dang, T. & Shao, M. Evaluation of EPIC soil water and yield components in the gully region of Loess Plateau, China. J. Agric. Sci. 144, 339–348 (2006).
Young, H. R., Rongyu, L. & Kuroda, M. Silicification in Mississippian Lodgepole Formation, northeastern flank of Williston Basin, Manitoba, Canada. J. Earth Sci 23(1), 1–18 (2012).
Acknowledgements
We acknowledge financial support from Cenovus Energy and a Senior NSERC-IRC (grant 184573) to MJH. We would also like to thank C. Sandau (Chemistry Matters), B. Rostron (Isobrine Solutions), and S. Arkadakskiy for their input and expertise in the development of the geochemical sampling programs for Sites 6 and 7.
Author information
Authors and Affiliations
Contributions
M.J.H. conceived the idea of defining the fate and transport of gases through the shale, identified the sampling parameters required, and wrote the first draft of the paper. E.S. conducted all field and much of the lab work and did most of the data conversions. S.L.B., M.J.H., and M.H. collaborated on the conceptual basis for the numerical modeling. M.H. performed the numerical modeling, and SOCM conducted field work and refined the interpretation of isotopic data and put it in a basin context. All authors edited and commented on the manuscript and contributed to the final text.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hendry, M.J., Schmeling, E.E., Barbour, S.L. et al. Fate and Transport of Shale-derived, Biogenic Methane. Sci Rep 7, 4881 (2017). https://doi.org/10.1038/s41598-017-05103-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05103-8
This article is cited by
-
Diffusion mechanism of deep shale gas and its carbon isotope fractionation: a combined simulated and mathematical analysis
Geosciences Journal (2023)
-
Methods and benefits of measuring non-hydrocarbon gases from surface casing vents
International Journal of Environmental Science and Technology (2023)
-
Modeling of methane migration from gas wellbores into shallow groundwater at basin scale
Environmental Earth Sciences (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
