
Abstract
Traditional strengthening ways, such as strain, precipitation and solid-solution, come into effect by pinning the motion of dislocation. Here, through first-principles calculations we report on an extra-electron induced covalent strengthening mechanism, which alters chemical bonding upon the introduction of extra-valence electrons in the matrix of parent materials. It is responsible for the brittle and high-strength properties of Al12W-type compounds featured by the typical fivefold icosahedral cages, which are common for quasicrystals and bulk metallic glasses (BMGs). In combination with this mechanism, we generalize ductile-to-brittle criterion in a universal hyperbolic form by integrating the classical Pettifor's Cauchy pressure with Pugh's modulus ratio for a wide variety of materials with cubic lattices. This study provides compelling evidence to correlate Pugh's modulus ratio with hardness of materials and may have implication for understanding the intrinsic brittleness of quasicrystals and BMGs.
Similar content being viewed by others

Introduction
In the view of electronic structure, good ductile/plastic materials consisting of metallic elements are often characteristic of metallic bonding and the corresponding valence electrons are in a delocalized state1,2. For instance, pure aluminum (FCC Al) is usually soft and lacks strength since Al is regarded as a typical s, p-bonded metal nearly described by a classic free electron gas3. The high-strength brittle materials essentially consist of non-metal elements, i.e., the hardest diamond exhibits a strong directional and covalent bonding framework. Once the covalent bond is broken, new covalent bonds can not be easily and immediately reformed because of a high energy barrier. That's the reason that high-strength brittle materials often resist large stresses with little deformation and break without developing any plastic regime (namely, in a typically brittle nature). However, some ordered intermetallic compounds4 with periodic lattice structures, quasicrystals with ordered but not periodic atomic structures5 and BMGs with completely disordered atomic structures6, despite of metallic constituents in them, also exhibit essentially a common brittle nature at low temperature regime. In fact, it is an interesting issue to understand why those materials are intrinsically brittle although no non-metal elements (i.e., C, N, O, etc) participate in bonds.
Even good ductile/plastic materials can be strengthened by most traditional strengthening methods7, such as strain, solid-solution and dispersed precipitation8,9,10,11, which come into effect by impeding the motion of dislocation12,13. Here, through first-principles calculations, we highlighted a new type of strengthening way, extra-electron induced covalent strengthening, in the icosahedral Al12W-type intermetallic compounds14,15,16,17,18,19, Al12X (X = Cr, Mo, W, Mn, Tc and Re), which have attracted extensive interest20,21,22,23 since the quasicrystals5 are most related to the five-fold icosahedral structure. Although Al and transition metal elements X are both metallic, it has been found that, after introducing proper valence electrons in the center of the icosahedral cage of Al12, the electronic bonding feature is critically transformed into a covalent directional bonding framework from a free electron metallic bonding network, dramatically resulting in a brittle and hard nature in ordered Al12X intermetallic compounds. Certainly, this kind of extra-electron induced covalent strengthening is intrinsically different from traditional ways. Given the fact that icosahedral package is quite common for both quasicrytals5 and metal-metal-based metallic glasses6,24,25,26,27,28,29, this strengthening nature might shed light on the interpretation of their intrinsic brittleness. The analysis on the elastic properties revealed that the classic Pugh's modulus ratio (G/B)30 and Pettifor's Cauchy pressure (C12-C44)31 are well correlated with their ductile-to-brittle transition, also matching the metallic-to-covalent bonding transformation. Furthermore, we extend their correlation to a universal hyperbolic criterion to identify the ductile-to-brittle properties for as large as 332 materials with cubic lattice. Moreover, this unified criterion also provides evidence that Pugh's modulus ratio is closely correlated with hardness of materials as documented in our recently proposed model32,33 of hardness.
Results
Comparison of lattice structures between FCC Al and Al12X
A series of Al12X (X = Cr, Mo, W, Mn, Tc and Re) compounds14,15,16,18,20 crystallize in the Al12W-type structure with the space group of  (No. 204) with X at the 2a site and with Al at the 24g site. As illustrated in Fig. 1, the Al12W-type structure is closely correlated with the FCC Al phase. In order to conveniently understand their correlation, Al atoms can be categorized into three classes Al1, Al2 and Al3 in the 2×2×2 FCC supercell (Fig. 1a). It has been noted that the Al1 and Al2 atoms correspond to the X and Al atoms in the Al12W-type structure, respectively (Fig. 1b). Their distinction lies in two aspects: (i) the Al12W-type structure lacks of Al3 atoms; (ii) in FCC Al each Al has twelve nearest-neighbor Al atoms to form an Al12 cuboctahedron whereas in the Al12W-type structure each X atom is surrounded by an icosahedron of twelve Al atoms (called Al12-icosahedron). In other words, the Al12-icosahedron can be considered as a distorted version of the Al12-cuboctahedron because of the removal of the Al3 atoms in FCC Al. The optimized structural parameters of Al12W-type compounds are in good agreement with available experimental data.
(No. 204) with X at the 2a site and with Al at the 24g site. As illustrated in Fig. 1, the Al12W-type structure is closely correlated with the FCC Al phase. In order to conveniently understand their correlation, Al atoms can be categorized into three classes Al1, Al2 and Al3 in the 2×2×2 FCC supercell (Fig. 1a). It has been noted that the Al1 and Al2 atoms correspond to the X and Al atoms in the Al12W-type structure, respectively (Fig. 1b). Their distinction lies in two aspects: (i) the Al12W-type structure lacks of Al3 atoms; (ii) in FCC Al each Al has twelve nearest-neighbor Al atoms to form an Al12 cuboctahedron whereas in the Al12W-type structure each X atom is surrounded by an icosahedron of twelve Al atoms (called Al12-icosahedron). In other words, the Al12-icosahedron can be considered as a distorted version of the Al12-cuboctahedron because of the removal of the Al3 atoms in FCC Al. The optimized structural parameters of Al12W-type compounds are in good agreement with available experimental data.

Comparison of the lattice structures between FCC Al and Al12X.
(a), Supercell (2×2×2) of FCC Al.Here, Al atoms are classified into three types: Al1, Al2 and Al3. (b), Unit cell of Al12X. The small and large balls denote aluminum and X atoms, respectively. The Al12W-type structure is closely correlated with the FCC supercell when Al1 atom is replaced by a valence electron rich transition metal element X and the Al3 atoms have been removed.
Electronic structures and chemical bonds
Although the FCC structure of Al is closely related to that of Al12W-type compounds, their electronic structures differ highly as evidenced in Fig. 2a and 2b. FCC Al exhibits a nearly free electron feature because its profile of the density of states can be described well through the classic free electronic theory. However, for Al12Re the appearance of a typical pseudogap at the Fermi level originated from the strong hybridization between Re d-orbital and Al s, p-orbitals18,20 evidences a significant deviation from the free electron feature. In fact, the similar pseudogap feature due to sp-d hybridization has been extensively observed in many other Al-based transition metal aluminides, such as Al3Ti and Al3V which were revealed to exhibit covalent bonding feature34,35.

Extra-electron induced covalent strengthening.
(a)-(d), Total DFT electronic densities of states of FCC-Al, Al12W-type Al12X (X = Re, □ and He). □ denotes that X is replaced by a vacancy. Here, Al12□ and Al12He are artificial and unstable, as evidenced by their positive enthalpies of formation (Supplementary Table S1). Their DOS profiles are compared with those obtained using the classic free electron model. e and f, Section contour maps of the difference of charge densities for e the Al-Al covalent bonds connecting the nearest-neighboring icosahedra as illustrated by the (020) plane and f the intra Al-Re covalent bonds within the Al12 icosahedron in the (0y0) plane of Al12Re. Similar results have been observed for all other Al12X (X = Cr, Mo, W, Mn and Tc), but are not shown here. The red and blue isovalues correspond to the charge accumulations and depletions, respectively. g, The comparison of calculated bulk moduli (B in GPa), Young moduli (E in GPa), shear moduli (G in GPa) and Cauchy pressure C12-C44 as well as Pugh'smodulus ratio of G/B (right side) in the series of Al12□, FCC Al and Al12X (X = Cr, Mo, W, Mn, Tc and Re) (details refer to Supplementary Table S3 which summarizes all elastic data used here).
In order to further understand the nature of the chemical bonding in Al12Re, we calculated the charge density differences of two planes as shown in Fig. 2e and 2f, (i) between two nearest neighboring Al12Re-icosahedra and (ii) between the sublattices of Re and Al12. The former is to check the inter-icosahedron bonding feature (corresponding to inter-Al-Al bonds as illustrated by a dashed line in Fig. 1b, whereas the latter is designed to show the intra-Al-Re bonding feature within each Al12Re icosahedron. It has been noted that apparent covalent Al-Al bonds can be confirmed due to the strong charge accumulations (c.f., Fig. 2e). From Fig. 1b, each Al atom has four nearest neighbor Al atoms which are equivalently located in the two neighbor Al12-icosahedra. Interestingly, the similar covalent bonds between metallic atoms have been also reported not only in some intermetallic compounds (Al3Ti and Al3V)34,35 but also in the Re2C compound (Re-Re covalent bond36) according to Mulliken overlap population analysis as shown in Ref. [37]. In addition, Figure 2f compiles the charge density of the (020) plane which shows a directional Re-Al bond within each Al12-icosahedron. The significant charge accumulation along all Re-Al bonds can be visualized, representing their covalent feature. Re atoms occupy the center of each Al12-icosahedron and their nearest neighbors being twelve Al atoms are arranged in the form of a nearly perfect icosahedron. Hence, the twelve Re-Al covalent bonds are constructed in totally twelve different directions with a three-dimensional framework.
Extra-electron induced covalent strengthening
Furthermore, we analyzed a series of other Al12X compounds as mentioned above. All of them exhibit a very similar electronic structure with the formation of a typical pseudogap (not shown here) and a covalent bonding framework. In terms of the chemical bonding nature of Al12W-type compounds, it would be naturally expected that they should have stronger mechanical properties than FCC Al due to the chemical bonding transformation from a metallic FCC-Al to a covalent Al12X. As expected, Fig. 2g evidences a dramatic increase of the elastic properties (in particular, shear (G) and Young (E) moduli) after the addition of X. Interestingly, it has been found that their mechanical performances are closely related to the valence electron number imposed by X. If no valence electrons are imposed by a vacancy (□) or an inert He atom, or a metallic element with a valence number smaller than 3 (i.e., Al) at X, their chemical bonding frameworks still remain metallic. From Fig. 2c for Al12□ and 2d for Al12He, their DOS profiles are similar to that of FCC Al. Although many peaks appear due to the reduced symmetry, the disappearance of the typical pesudogap is consistent with the lack of sp-d hybridization. In addition, it has been noted that their profiles can be further described by the classic free electron theory, evidencing the metallic bonding framework in those artificial compounds. Therefore, there is no doubt that their elastic properties (shear (G), Young (E) and bulk moduli (B)) are highly similar to those of FCC Al. However, when X is replaced by Cr, Mo, W, Mn, Tc, Re the valence electrons number of which exceeds that of Al, the abruptly increased mechanical properties are consistent with the formation of strong covalent bonding. From FCC Al (Al12□, Al12He and Al12Al) to isoelectronic Al12X (X = Cr, Mo and W), their E and G are abruptly increased by more than 100%. These values are even increased much heavier in the series of isoelectronic Al12X (X = Mn, Tc, Re) which have one more valence electron introduced in each Al12-icosahedron than the cases of X = Cr, Mo and W. Hence, we have defined this feature from metallic bonding to covalent bonding transformation in combination with the mechanical strengthening depending on the introduction of valence electrons of a critical number larger than three imposed by X in Al12X as an extra-electron induced covalent strengthening mechanism.
A universal ductile-to-brittle criterion
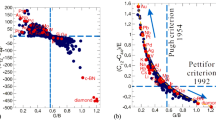
Interestingly, this mechanism is further supported by both classical criteria of Cauchy pressure C12-C44 (as proposed by Pettifor in 199231) and of Pugh's modulus ratio G/B (as proposed by Pugh30). From Fig. 2g, for both FCC Al and Al12□ their C12-C44 remains nearly the same positive value, implying the metallic bonding framework in terms of Pettifor's suggestion31. In addition, their G/B values also meet Pugh's criterion when G/B is smaller than 0.571, in agreement with a ductile property. However, when X is replaced by transition metal elements (X = Cr, Mo, W, Mn, Tc and Re) their C12-C44 values are all positive, revealing a directional (covalent) bonding framework from Pettifor's criterion of Cauchy pressure and their G/B values are all larger than 0.571, suggesting their brittle mechanical properties based on Pugh's criterion of modulus ratio. In order to further assess the influence of the Pugh's modulus ratio (G/B) and Cauchy pressure (C12-C44) on the mechanical properties of cubic materials, we also plotted in Fig. 3a C12-C44 against G/B for these Al12X. Unexpectedly, Figure 3a shows a nearly linear relationship, building a nice connection between the classic Pugh's modulus ratio and the classic Cauchy pressure. Because the artificial compound of Al12□ shows an electronic structure and metallic bonding feature similar to FCC Al, we see that it locates closest to FCC Al in the upper left corner. However, Al12X compounds are dramatically moved to the lower right corner mainly due to the presence of directional covalent bonding, resulting in a brittle behavior. In particular, Al12X (X = Mn, Tc and Re) locates in the lower right corner. In this sense, there is no doubt that they should be the most brittle and strongest materials among those Al12X aluminides collected here.

A universal ductile-to-brittle criterion.
(a), Nearly linear correlation between C12-C44 and G/B for those Al12X aluminides. (b), The correlation in a is further extended to a large scale data collected for 332 compounds (571 group data sets; Supplementary Tables S2 and S3) from literature. (c), A renormalized hyperbolic correlation derived by dividing Young modulus E from (C12-C44) for all the summarized data of b. The horizontal line of C12-C44 denotes the critical zero Cauchy pressure defined by Pettifor31, whereas the vertical line of G/B = 0.571 corresponds to critical Pugh's modulus ratio defined by Pugh30. (d), The relation between Cauchy pressure and the experimental ultimate intensile strength (UTS)38 for selected solid phases with cubic lattice of some pure elements. For details, see text.
When a wide variety of samples (in total, 571 group data sets for 332 compounds collected from literature) are included for comparison, their correlation does not remain linearly any more, rather revealed a highly scattered distribution in Fig. 3b. Strikingly, when Cauchy pressure C12-C44 is renormalized by multiplying with a factor of  (E – Young modulus), all those data in Fig. 3b can be, unexpectedly, uniformed as a beautiful hyperbola (Fig. 3c). The most spectacular fact is that diamond, the hardest known of highest strength material, locates at the lowest right corner with the largest G/B ratio and the lowest
(E – Young modulus), all those data in Fig. 3b can be, unexpectedly, uniformed as a beautiful hyperbola (Fig. 3c). The most spectacular fact is that diamond, the hardest known of highest strength material, locates at the lowest right corner with the largest G/B ratio and the lowest  . In contrast, the most ductile and plastic Au exhibits the most positive (
. In contrast, the most ductile and plastic Au exhibits the most positive ( ) and the lowest Pugh's modulus ratio. This fact demonstrated that this hyperbolic relationship can be unified as a rule to identify the intrinsic strength and ductility of cubic materials. If we use Pugh's modulus G/B as a factor of strength, as demonstrated by ultimate tensile strength (UTS) experimentally measured for some pure elemental solids with cubic lattice in Fig. 3d and the revised Cauchy pressure
) and the lowest Pugh's modulus ratio. This fact demonstrated that this hyperbolic relationship can be unified as a rule to identify the intrinsic strength and ductility of cubic materials. If we use Pugh's modulus G/B as a factor of strength, as demonstrated by ultimate tensile strength (UTS) experimentally measured for some pure elemental solids with cubic lattice in Fig. 3d and the revised Cauchy pressure  as a factor of ductility, the hyperbolic correlation exactly shows a well-known fact for materials7,8. Namely, the high-strength materials, in general, lack of good ductility, whereas the ductile materials do not exhibit good strength.
as a factor of ductility, the hyperbolic correlation exactly shows a well-known fact for materials7,8. Namely, the high-strength materials, in general, lack of good ductility, whereas the ductile materials do not exhibit good strength.
The application of ductile-to-brittle criterion to hardness of materials
From Fig. 3c it can be seen that Pugh's modulus ratio (k = G/B) seems to mirror the hardness of materials. For instance, the hardest diamond has a largest k of 1.2 and the second hardest cubic-BN has a k of about 1.0, just smaller than that of diamond. Therefore, it is our aim to further check a wide variety of hard materials by comparing their experimental Vickers hardness (Hv) as a function of k in Fig. 4a. The resulting trend seems to show a good correlation, namely, hardness increases with increasing k, despite of some scattering data available. Given this fact that the Poisson's ratio39 (ν) is reversely proportional to k, accordingly, hardness certainly exhibits a decreasing tendency as ν increases. This fact provides the compelling evidence to validate our recently proposed hardness model32,33, Hv = 2(k2G)0.585−3. It indicates that the hardness not only correlates with shear modulus as observed by Teter40, but also with bulk modulus as observed by Gilman41. Our work combines those aspects32 that were previously argued strongly, and, most importantly, is capable to correctly reproduce the hardness of a wide variety of hard materials including all known superhard materials as illustrated in Fig. 4b.

The application of ductile-to-brittle criterion to hardness of materials.
(a), Correlation between Experimental Vickers hardness (Hv) and its Pugh's modulus ratio (k = G/B) for hard materials. (b), Experimental Vickers hardness as a function of a product (k2G) between the squared Pugh's modulus ratio (k2) and shear modulus G33,34. Circles correspond to hard materials with cubic structure, whereas solid squares denote non-cubic-lattice hard materials (Supplementary Table S4).
Discussion
We have systematically investigated a series of Al12X (X = Cr, Mo, W, Mn, Tc and Re) intermetallic compounds, revealing their brittle and high-strength mechanical properties. This class of compounds attracts our interest because their structures are characteristic of the ordered icosahedral units which are composed of Al12 cages with a centered transition metal X. This unique five-fold icosahedral feature is similar to the basic structural unit of some typical quasicrystals and BMGs. Interestingly, we have established a direct structural connection between Al12X and FCC Al and the latter is, apparently, a typical metal whose electronic structure can be described well within free-electron gas model. We found that the introduction of extra-valence electrons (typically, larger than three) imposed by X induces a covalent bonding framework in Al12X. These covalent bonds are not only for all the intra-Al-X bonds within each icosahedron but also for the inter-Al-Al bonds between any two neighboring icosahedra. Undoubtedly, the occurrence of the covalent bonding framework results in the typical brittle and high-strength properties of Al12X. We have then proposed a new strengthening way, called extra-electron induced covalent strengthening. Our findings extend the classical strengthening ways of metal from a mechanical viewpoint (typically, pinning dislocations by solid-solution, precipitation and stress) to an electronic viewpoint by modifying chemical bonds in the local or whole materials. We believe that the extra-electron induced covalent strengthening may originally exist in materials but perhaps, was neglected in some classical ways because one used to focus on dislocation effect from mechanical aspects. Therefore, we cannot rule out the possibility that, in some solid-solution strengthening cases, the solution addition may have an effect on modifying the local chemical bonds which lift up the activation energy barrier of dislocations, thereby taking action of strengthening effect. In addition, this new way would also have some potential applications to accomplish the whole or local strengthening effects (i.e., on the surface strengthening) through the introduction of extra electrons provided by proper alloying addition.
From the ductile FCC Al to the brittle and high-strength Al12X, the transition of electronic chemical bonding from metallic to covalent framework is consistent with the enhanced elastic mechanical properties interpreted well by empirical Pettifor's criterion of Cauchy pressure (C12-C44)31 and Pugh's modulus ratio (G/B)30. As mentioned above, Pettifor highlighted a critical zero Cauchy pressure (C12-C44) to separate the metallic and covalent (directional) bonding framework and Pugh also yielded a critical value of G/B = 0.571 as a boundary between ductile and brittle properties. We have shown the interplay between these two empirical criteria proposed by Pettifor and Pugh can be unified as an intrinsic ductile-to-brittle criterion in a universal hyperbolic correlation (see Fig. 3c), by fitting a wide range of materials with cubic lattices collected from 332 compounds in the total 557 group data set. As a consequence of the generalization of their criteria, this new form of the hyperbolic relation indeed uncovers there is no so-called critical separated boundaries between the ductile and brittle properties of materials. In particular, as shown in Fig. 3c, the crossing point between the hyperbolic curve and the vertical dashed line of the critical value (G/B = 0.571) defined by Pugh30 exactly corresponds to the critical zero value of C12-C44 proposed by Pettifor31. This fact evidences the intrinsic coherency of these two classical criteria. It still needs to be emphasized that, although mechanical properties related with permanent deformation of materials are highly complex, the currently unified criterion seems to provide the simple and start-up applications to identify roughly the intrinsic ductile-to-brittle property. Given the fact that our collected data for those 332 compounds all correspond to room temperature for experiments and to sufficiently low temperature (i.e., absolute zero) for ab initio calculations, this empirical correlation should be thus limited to the low-temperature scale of materials. In addition, this unified criterion also unveil the substantial evidence that Vickers hardness of materials is correlated with Pugh's modulus ratio32,33, as successfully applied to CrB442 and WB3/WB443 as well as a series of superhard phases of cold-compressed graphite44.
Methods
First-principles calculations were performed using the Vienna ab initio Simulation Package (VASP)45 with the ion-electron interaction described by the projector augmented wavepotential (PAW)46. The energy cutoff for the plane-wave expansion of eigenfunctions was set to 500 eV. We used the generalized gradient approximation (GGA) based on the Perdew-Burke-Ernzerhof (PBE) scheme47 for the exchange-correlation functional. Optimization of structural parameters was achieved by minimizing forces and stress tensors. Highly converged results were obtained utilizing a dense 13×13×13  -point grid for the Brillouin zone integration. The independent elastic constants of the Al12W-type icosahedral compounds were derived from the total energies as a function of lattice strains48. These strain energies were fitted to third-order polynomials from which the elastic constants at the equilibrium structures were calculated.
-point grid for the Brillouin zone integration. The independent elastic constants of the Al12W-type icosahedral compounds were derived from the total energies as a function of lattice strains48. These strain energies were fitted to third-order polynomials from which the elastic constants at the equilibrium structures were calculated.
References
Clugston, M. & Flemming, R. Advanced Chemistry (Oxford Univ. Press, Oxford, 2008).
Cottrell, A. Introduction to the Modern Theory of Metals (Institute of Metals, London, 1988).
Nakashima, P. N. H., Smith, A. E., Etheridge, J. & Muddle, B. C. The bonding electron density in aluminum. Science 331, 1583–1586 (2011).
Cahn, R. W., Liu, C. T. & Sauthoff, G. Ordered Intermetallics – Phyiscal Metallurgy and Mechanical Behavior (Kluwer Academic Press, 1992).
Dubois, J. M. Useful Quasicrystals (World Scientific Publishing, 2005).
Inoue, A. Stabilization of metallic supercooled liquid and bulk amorphous alloys. Acta Mater. 48, 279–306 (2000).
Donald, P. The strengthening of metals (Reinhold Publishing Corporation, New York, 1964).
Ritchie, R. O. The conflicts between strength and toughness. Nature Mater. 10, 817–822 (2011).
Valiev, R. Z. Nanostructuring of metals by severe plastic defromation for advanced properties. Nature Mater. 3, 511–516 (2004).
Sato, J. et al. Cobalt-based high-temperature alloys. Science 312, 90–91 (2006).
Hirata, A. et al. Atomic structure of nanoclusters in oxide-dispersion-strengthened steels. Nature Mater. 10, 922–926 (2011).
Olson, G. B. & Cohen, M. in Dislocations in Solids Vol. 7 (ed. Nabarro, F. R. N.) Ch. 37, 295–407 (North-Holland, 1986).
Lu, K. The future of metals. Science 328, 319–320 (2010).
Adam, J. & Rich, J. B. The crystal structure of WAl12, MoAl12 and (Mn,Cr)Al12 . Acta Crystallogr. 7, 813–816 (1954).
Barlock, J. G. & Mondolfo, L. F. Structure of some aluminium-iron-magnesium-manganese-silicon alloys. Z. Metallk. 66, 605–611 (1975).
Darby, J. B., Downey, J. W. & Norton, L. J. Intermediate phases in the technetium-aluminum and technetium-silicon systems. J. Less-Common Metals 8, 15–19 (1965).
Bennett, L. H., Cahn, J. W., Schaefer, R. J., Rubinstein, M. & Stauss, G. H. Icosahedral symmetry versus local icosahedral environments in Al-Mn alloys from NMR. Nature 326, 372–373 (1987).
Kirihara, K. et al. Covalent bonds in AlMnSi icosahedral quasicrystalline approximant. Phys. Rev. Lett. 85, 3468–3471 (2000).
Tao, X. M. et al. First-principles investigations of elastic, electronic and thermodynamic properties of Al12X (X = Mo, W and Re). Intermetallics 24, 15–21 (2012).
Laissardiere, G. T., Manhm, D. N. & Mayou, D. Electronic structure of complex Hume-Rothery phases and quasicrystals in transition metal aluminides. Prog. Mater. Sci. 50, 679–788 (2005).
Stadnik, Z. M. et al. Electronic structure of quasicrystals studied by ultrahigh-energy-resolution photomission spectroscopy. Phys. Rev. B 55, 10938–10951 (1997).
Singh, A., Ranganathan, A. & Bendersky, L. A. Quasicrystalline phases and their approximants in Al-Mn-Zn alloys. Acta Mater. 45, 5327–5336 (1997).
Nozawa, K. & Ishii, Y. Structure-induced covalent bonding in Al-Li Compounds. Phys. Rev. Lett. 104, 226406 (2010).
Chen, M. W. Mechanical behavior of metallic glasses: microscopic understanding of strength and ductility. Annu. Rev. Mater. Res. 38, 445–469 (2008).
Chen, M. W., Dutta, I., Zhang, T., Inuoe, I. & Sakurai, T. Kinetic evidence for the structural similarity between a supercooled liquid and an icosahedral phase in Zr65Al7.5Ni10Cu12.5Ag5 bulk metallic glass. Appl. Phys. Lett. 79, 42–44 (2001).
Luo, W. K. et al. Icosahedral short-range order in amorphous alloys. Phys. Rev. Lett. 92, 145502 (2004).
Yuan, C. C., Xiang, J. F., Xi, X. K. & Wang, W. H. NMR signature of evolution of ductile-to-brittle transition in bulk metallic glasses. Phys. Rev. Lett. 107, 236403 (2011).
Hirata, A. et al. Direct observation of local atomic order in a metallic glass. Nature Mater 10, 28–33 (2011).
Miracle, D. B. A physical model for metallic glass structures: an introduction and update. JOM 64, 846–855 (2012).
Pugh, S. F. Relation between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 45, 823–843 (1954).
Pettifor, D. G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 8, 345–349 (1992).
Chen, X.-Q., Niu, H. Y., Li, D. Z. & Li, Y. Y. Intrinsic correlation between hardness and elasticity in polycrystalline materials and bulk metallic glasses. Intermetallics 19, 1275–1281 (2011).
Chen, X.-Q., Niu, H. Y., Franchini, C., Li, D. Z. & Li, Y. Y. Hardness of T-carbon: Density functional theory calculations. Phys. Rev. B 84, 121405(R) (2011).
Krajci, M. & Hafner, J. Covalent bonding and bandgap formation in intermetallic compounds: a case study for Al3V. J. Phys: Condens. Matter. 14, 1865–1879 (2002).
Jahnatek, M., Krajci, M. & Hafner, J. Interatomic bonding, elastic properties and ideal strength of transition metal aluminids: A case study for Al3(V, Ti). Phys. Rev. B 71, 024101 (2005).
Zhao, Z. S. et al. Bulk Re2C: crystal structure, hardness and ultra-incompressibility. Crystal Growth & Design 10, 5024–5026 (2012).
He, J. L., Wu, E. D., Wang, H. T., Liu, R. P. & Tian, Y. J. Ionicities of boron-boron bonds in B12 Icosahedra. Phys. Rev. Lett. 94, 015504 (2005).
Gale, W. F. & Totemeier, T. C. Smithells Metals Reference Book 8th Edn (Elsevier Butterworth-Heinemann Ltd., Oxford, UK, 2004).
Greaves, G. N., Greer, A. L., Lakes, R. S. & Rouxel, T. Poisson's ratio and modern materials. Nature Mater. 10, 823–837 (2011).
Teter, D. M. Computational alchemy: The search for new superhard materials. MRS Bull. 23, 22–27 (1998).
Gilman, J. J. Hardness-a Strength Miroprobe, Chapter 4 of The Science of Hardness Testing and Its research Applications (Westbrook, J. H. & Conrad, H. Eds. American Society of Metal, Metal Park, Ohio, 1973).
Niu, H. Y. et al. Structure, bonding and possible superhardnss of CrB4 . Phys. Rev. B 85, 144116 (2012).
Liang, Y. C. et al. An unexpected softening from WB3 to WB4 . EPL 98, 66004 (2012).
Niu, H. Y. et al. Families of superhard crystalline carbon allotropes constructed via cold compression of graphite and nanotubes. Phys. Rev. Lett. 108, 135501 (2012).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Beckstein, O., Klepeis, J. E., Hart, G. L. W. & Pankratov, O. First-principles elastic constants and electronic structure of α-Pt2Si and PtSi. Phys. Rev. B 63, 134112 (2001).
Acknowledgements
We thank Zhefeng Zhang, Jian Xu, Zhaoping Lu, Cesare Franchini, Peter Franz Rogl, Raimund Podloucky, K.-M. Hou, Zhenyu Zhang, Aleksey N. Kolmogorov, Yongjun Tian for their valuable help and discussions. This work was supported by the “Hundred Talents Project” of the Chinese Academy of Sciences and from NSFC of China (Grand Numbers: 51074151 and 51174188). The authors also acknowledge the computational resources from the Supercomputing Center (including its Shenyang Branch in the IMR) and the Vienna Scientific Cluster as well as the National Supercomputing Center in Tianjin (TH-1A system). Dedicated to Professors, Wei Ke and Yiyi Li, a couple of scientists, on the occasion of their 80th birthday.
Author information
Authors and Affiliations
Contributions
X.-Q.C., D. Z. L. and Y. Y. L. designed and coordinated the overall study and X.-Q.C. wrote the paper. H.Y.N. and X.-Q.C. performed theoretical calculations and H.Y.N. collected all data with the help from P.T.L., W.W.X. and X. Y. C. All contributed to the discussion of the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Online supplementary Materials
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Niu, H., Chen, XQ., Liu, P. et al. Extra-electron induced covalent strengthening and generalization of intrinsic ductile-to-brittle criterion. Sci Rep 2, 718 (2012). https://doi.org/10.1038/srep00718
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00718
This article is cited by
-
Structural, electronic, elastic, optical and thermoelectric properties of ASiCl3 (A = Li, Rb and Cs) chloroperovskites: a DFT study
Optical and Quantum Electronics (2024)
-
Elastic, optoelectronic and photocatalytic properties of semiconducting CsNbO3: first principles insights
Scientific Reports (2023)
-
Design of refractory multi-principal-element alloys for high-temperature applications
npj Computational Materials (2023)
-
The stability, mechanical, electronic, and thermal features of the new superhard double transition-metal mono-nitrides and mono-carbides compounds
Indian Journal of Physics (2023)
-
Electronic band structure, mechanical and optical characteristics of new lead-free halide perovskites for solar cell applications based on DFT computation
Bulletin of Materials Science (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
