
Abstract
The usefulness of Pt-based nanomaterials for catalysis can be greatly enhanced by coupling morphology engineering to the strategic presence of a second or even third metal. Here we demonstrate the design and preparation of stellated Ag-Pt bimetallic nanoparticles where significant activity difference between the methanol oxidation reaction (MOR) and the oxygen reduction reaction (ORR) may be realized by relegating Ag to the core or by hollowing out the core. In particular the stellated Pt surface, with an abundance of steps, edges, corner atoms and {111} facets, is highly effective for the ORR but is ineffective for MOR. MOR activity is only observed in the presence of a Ag core through electronic coupling to the stellated Pt shell. The bimetallic Ag-Pt stellates therefore demonstrate the feasibility of tuning a Pt surface for two very different structure sensitive catalytic reactions. Stellated bimetallics may therefore be an effective platform for highly tunable catalyst designs.
Similar content being viewed by others


Introduction
Platinum (Pt) nanoparticles are catalytically active for both the anodic (methanol oxidation reaction, MOR) and cathodic (oxygen reduction reaction, ORR) reactions of the direct methanol fuel cell (DMFC)1,2,3. However, they are susceptible to CO poisoning in the MOR and their ORR activity is still limiting the fuel cell performance4,5,6,7,8. Other than the classical approaches of increasing the Pt catalytic performance through particle size reduction, particle shape control and alloying with oxophilic metals9,10,11,12,13, there is increasing interest in combining morphology engineering and the synergistic effects of an adjuvant metal to enhance the Pt catalytic properties11,14,15,16.
The design of electrocatalysts for improved activity, selectivity and resistance to deactivation should be rational and based on a sufficiently good understanding of the reaction mechanism. For example, the importance of the Pt–CO bond in MOR has been well documented17. The chemisorption of CO on Pt involves the donation of lone pair electrons from the filled carbon σ orbital of CO to the empty 5d-orbital of Pt, which is compensated by the back donation of electrons from the Pt dπ to the π* orbitals of CO. A high local density of electrons around Pt is associated with weak chemisorption. With this understanding, Ag2S-Pt composite nanocatalysts have been developed to ameliorate CO deactivation18. The electron transfer from Ag2S to Pt in Ag2S-Pt nanocomposites due to the alignment of energy levels increases the electron density around the Pt sites, causing the weakening of CO chemisorption and hence the increase in MOR activity. In addition, catalytic activity is also affected by the atomic arrangements on specific exposed facets. The enhanced ORR activity of bimetallic Pt-on-Pd heteronanostructures or nanodendrites is an example19,20,21,22. The synthesis of the latter electrocatalysts began with the preparation of truncated Pd cores by solution chemistry method. A dense array of Pt branches was then formed on the Pd cores via the facet-selective growth of Pt.
After a careful review of the literature on recent designs of electrocatalysts for fuel cell applications11,19,20,21,22,23,24,25,26, we hypothesize that the heterogeneous growth of Pt on a metal with different electronegativity may combine the electronic effect in catalysis with the exposure of selective active facets through nanoparticle morphology engineering to increase the diversity in the control of Pt catalytic properties. We will demonstrate this with the design and preparation of stellated Pt nanoparticles with a Ag core (SPNPs-A). The bimetallic stellated nanoparticles could be formed in one-pot by the competing growth of Pt on the twin sites of in-situ formed decahedral Ag seeds. These SPNPs-A could be processed into hollow Pt nanoparticles without changes in size and the stellated morphology (stellated Pt nanoparticles with a hollow interior, or SPNPs-H) by the extraction of the Ag core with a selective etchant, bis(p-sulfonatophenyl)phenylphosphane dihydrate dipotassium salt (BSPP). BSPP works by binding strongly to silver to form water-soluble coordinating compounds. We will also demonstrate that the catalytic activity of these stellated heterogeneous nanoparticles may be varied from MOR to ORR by only altering their internal structure (a Ag or hollow core). Specifically SPNPs-A are highly active for MOR due to presence of the Ag core which alters the Pt activity through the Pt-Ag electronic coupling effects, while the SPNPs-H leverages on the abundance of atomic steps, edges, corner atoms, as well as dominant {111} facets on the stellating geometry to increase the ORR activity of the Pt surface. The stellated morphology offers a convenient platform to construct heterogeneous nanoparticles with very varied catalytic properties quite easily.
Results and Discussion
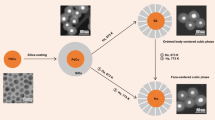
Fig. 1 is a schematic illustration of the synthesis of SPNPs-A by the co-reduction of AgNO3 and K2PtCl4 in oleylamine at elevated temperature. Although both Ag and Pt precursors were heated in one-pot, the Ag precursor was preferentially reduced because the reduction of Ag-oleylamine complexes was kinetically more facile. The fast kinetics resulted in the formation of multiply twinned decahedral Ag nanoparticles which seeded the subsequent deposition of Pt. The twin structure of the Ag seeds, which accounted for many interesting electronic, optical and catalytic properties27,28,29, were essential for the development of the stellated Ag-Pt heterogeneous nanostructure. The anisotropic growth of Pt on the surface of Ag seeds was due to the selective deposition of Pt at high-energy twin boundary sites.

Schematic illustration of the mechanism for the formation of stellated Ag-Pt nanoparticles in the one-pot synthesis.
Transmission electron microscopy (TEM) was used to follow the particle growth process. The results are shown in Fig. 2. The TEM (Fig. 2a) and high-resolution TEM (HRTEM) images (Fig. 2b) show only multiply twinned decahedral Ag nanoparticles with an average diameter of 12.2 nm after reaction for 30 min, although both Ag and Pt precursors were introduced to oleylamine simultaneously. The absence of the Pt component at this stage was confirmed by energy dispersive X-ray spectroscopy (EDX) analysis (Fig. 2c). The Pt signal was only detected by EDX of the nanoparticles recovered from the reaction mixture after 60 min, as shown in Fig. 2f. There were also significant changes in the morphology and the size of the particles in the TEM and HRTEM images (Fig. 2d and 2e). With the deposition of Pt on the in-situ formed Ag nanoparticles, the average diameter was increased to 16.6 nm. Core-shell nanoparticles with a number of Pt humps on the shell were the majority product at this time. At 90 min, the Pt humps developed into short branches on the surface of the core-shell Ag-Pt nanoparticles (Fig. 2g and 2h) and the Pt signal in the EDX spectrum increased accordingly (Fig. 2i). This stellating growth of Pt continued until the Pt precursor in the reaction mixture was completely exhausted (120 min). The final product was SPNPs-A with an average size of 25.2 nm (Fig. 2j and 2k) which is morphologically distinct from the spherical nanoparticles formed in the early stages of the reaction. EDX analysis (Fig. 2l) of the nanoparticles measured the Ag/Pt atomic ratio to be approximately 1.0/1.9, which is in good agreement with the Ag/Pt atomic ratio used in the preparation (1/2). This is suggestion that all of the Ag and Pt precursors had been completely reduced by oleylamine under the prevailing experimental conditions. The completion of reaction was confirmed by extending the reaction time to 180 min, where no further changes in the particle size, morphology and Pt/Ag atomic ratio were detected by TEM (Fig. 2m), HRTEM (Fig. 2n) and EDX (Fig. 2o) respectively.

SPNPs-A collected at different time.
Representative TEM images (a,d,g,j,m), HRTEM images (b,e,h,k,n) and corresponding EDX spectra (c,f,i,l,o) of the stellated Ag-Pt nanoparticles made after the reaction for 30 min (a,b,c), 60 min (d,e,f), 90 min (g,h,i), 120 min (j,k,l) and 180 min (m,n,o), respectively.
The Pt precursor must be present in sufficient quantity to enable the stellated growth of nanoparticles. In the case of an insufficient Pt precursor, for example, for a Ag/Pt atomic ratio of 1/1 or higher, there were no branched structures on the Pt exterior and the products were analogous to those imaged in Fig. 2d (60 min after the co-reduction of Ag and Pt precursors at atomic ratio of 1/2). Core-shell Ag-Pt nanoparticles were formed instead of the stellated Ag-Pt nanoparticles, as shown in Fig. S1 of Supplementary Information (SI).
The distribution of Ag and Pt in the fully-developed stellated nanoparticles was assayed by EDX in high-angle annular dark-field scanning TEM microscopy (HAADF-STEM). The elemental map of a representative nanoparticle in SI Fig. S2a shows uniform distribution of Pt throughout the entire particle (SI Fig. S2c) whereas Ag was concentrated in the core region (SI Fig. S2b). STEM-EDX analysis of three randomly selected nanoparticles confirmed the absence of isolated nanostructures consisting of only Ag or Pt (SI Fig. S3).
The HRTEM images sampled from isolated stellated particles (SI Fig. S2d–i) show that each particle had a solid body and a number of branches which interrupted the surface into disjoint intervals. These HRTEM images also indicate well-developed crystallinity. The lattice fringes in the branches of the stellated particles associated well with the Pt {111} planes. In the body region, the superposition of the diffraction patterns of Ag and Pt made it difficult to differentiate between Ag and Pt in the HRTEM images. It is noteworthy to mention that the lattice planes in the Pt branches were not parallel to those in the body of the stellated nanostructure and the angles between them also varied from particle to particle. It is therefore deduced that there was no epitaxial growth between the branch and the body of SPNPs-A and the growth of Pt branches occurred in different orientations. This result is significantly different from previous reports on the seed-mediated synthesis of Pt-on-Pd nanostructures where the {111} planes of Pt were parallel to the {111} planes of Pd due to the very good lattice matching between Pt and Pd19,20,21.
The twinned structure of the Ag seeds provides an expeditious means to modify the internal structure of SPNPs-A. Multiply-twinned Ag nanoparticles are inherently unstable; known to be slowly etched by dissolved O2 and Cl− dissociated from the Pt precursor (K2PtCl4) and influence strongly on the continuity and compactness of the overlaid Pt shell30,31. The Ag+ released from the O2/Cl− etching of twinned Ag seeds diffuses out through the discontinuous Pt shell due to the prevailing Ag+ concentration gradient between the core of the Ag-Pt nanoparticles and the surrounding solution. This diffusion has been developed into a general protocol for the fabrication of noble metal nanoparticles with a hollow or cage-bell structure31. A variation of this protocol was used in this work to completely remove the Ag core from SPNPs-A, resulting in hollow Pt nanoparticles with an intact stellation morphology (SPNPs-H, or stellated Pt nanoparticles with a hollow interior). The disappearance of the Ag signal in the EDX analysis of SPNPs-A after the BSPP treatment evidenced the complete elimination of the Ag core from SPNPs-A (Fig. 3d). The voidage in the core region formed by the removal of Ag by BSPP could be discerned by the strong contrast between central and surface regions in the TEM and HRTEM images of Fig. 3a, b and c. The particle size and the stellation morphology were practically unchanged by the BSPP treatment. Hence the BSPP treatment was highly selective and did not cause the collapse of the SPNPs-A structure.

SPNPs-H. TEM (a), HRTEM (b,c) and corresponding EDX spectrum (d) of stellated Pt nanoparticles with a hollow interior prepared from the stellated Ag-Pt bimetallic nanoparticles.
Both SPNPs-A and SPNPs-H were loaded on Vulcan carbon and tested for electrocatalytic activity for the methanol oxidation reaction (MOR) and the oxygen reduction reaction (ORR) at room temperature). As shown by the representative TEM and HRTEM images in SI Fig. S4, SPNPs-A and SPNPs-H could be dispersed very well on the carbon support by conventional means and the stellated morphology of the nanoparticles was intact. The loading of the stellated particles on carbon was fixed at 20 wt% of Pt in order to be comparable to the commercial catalyst (E-TEK Pt/C, 20 wt% Pt nanoparticles (3.2 nm) on Vulcan XC-72 carbon support) which was used to benchmark the catalyst performance.
The electrochemical active surface areas (ECSAs) of SPNPs-A, SPNPs-H and the E-TEK Pt/C catalysts were measured by cyclic voltammetry (Fig. 4a). The ECSAs normalized by the mass of Pt are shown in Fig. 4b. The ECSAs of SPNPs-A and SPNPs-H were 40.5 m2 gPt−1 and 50.6 m2 gPt−1 respectively and were 55.6% and 69.5% of the ECSA of the E-TEK Pt/C catalyst (72.8 m2 gPt−1). The lower ECSAs of stellated nanoparticles are most likely due to the presence of residual impurities adsorbed on the surface and the larger sizes compared with Pt nanoparticles (~3 nm) in commercial catalysts. Fig. 4b also shows that the removal of the Ag core increased the Pt ECSA by 20% (10.1 m2 gPt−1), suggesting that the ECSA of the stellated particles was mostly contributed by the Pt branches. The curvature of the spatially separated Pt branches suggests an abundance of atomic steps, edges and corner atoms, which are known to increase the Pt catalytic activity22,32,33,34.

Electrochemical measurements.
Cyclic voltammograms of SPNPs-A, SPNPs-H and E-TEK Pt/C in argon-purged HClO4 (0.1 M) at 50 mV/s (a); Histogram of the ECSAs for SPNPs-A, SPNPs-H and E-TEK Pt/C (b); Cyclic voltammograms of SPNPs-A, SPNPs-H and E-TEK Pt/C in argon-purged HClO4 (0.1 M) with 1 M methanol at 20 mV/s (c); Histogram of peak current densities of SPNPs-A, SPNPs-H and E-TEK Pt/C due to MOR in the forward and reverse scans (d); ORR polarization curves for SPNPs-A, SPNPs-H and E-TEK Pt/C catalysts in an O2-saturated HClO4 solution (0.1 M) at 20 mV/s and a rotating speed of 1600 rpm (e); Histogram of half-wave potentials of SPNPs-A, SPNPs-H and E-TEK Pt/C in the ORR polarization curves (f).
Besides the stellated morphology, an important feature in SPNPs-A affecting catalytic activity is the electronic coupling between the Ag core and the Pt shell with stellates. The difference in the electronegativity of Ag and Pt (1.83 and 2.28 respectively) implies that electrons may be drawn from Ag to Pt in SPNPs-A. XPS analysis was therefore used to measure the electron transfer, in any, in SPNPs-A. The binding energies of the Pt 4f7/2 and 4f5/2 peaks of SPNPs-A were shifted appreciably to lower values compared with the same peaks in the E-TEK Pt/C catalyst and SPNPs-H (SI Fig. S5a). This is indication of electron transfer from Ag to Pt in the SPNPs-A. The electron-donation from Ag to Pt in SPNPs-A also resulted in the shift of the Ag 3d peak to more positive values than that in monometallic Ag, as shown in SI Fig. S5b. The electron donation from Ag to Pt in SPNPs-A could result in a substantial increase in the local electron density around the Pt sites, which has been shown to reduce the irreversible adsorption of CO, an intermediate product in the MOR and catalyst poison, from inhibiting the catalytically active sites18.
SI Fig. S6 shows the CO stripping voltammograms of SPNPs-A, SPNPs-H and commercial Pt/C after the working electrode has been held at −0.15 V for 30 min in CO saturated 0.1 M HClO4. The CO stripping peaks of the SPNPs-A shifted to a more negative potential as compared to SPNPs-H and commercial Pt/C, thus indicating a more facile CO removal and hence, an improved CO tolerance in practice. The ease of CO removal over SPNPs-A relative to SPNPs-H and commercial Pt/C reflected the effectiveness of electron coupling between the Ag and Pt in the stellated Ag-Pt nanoparticles.
The voltammograms for MOR and the polarization curves for ORR of SPNPs-A, SPNPs-H and the E-TEK Pt/C catalyst are given in Fig. 4c and 4e respectively. The current densities in the voltammograms and polarization curves were normalized by the ECSA of Pt. For MOR, the peak current densities associated with methanol oxidation in the forward and reverse scans are summarized in Fig. 4d. The comparison of current densities shows that SPNPs-A had higher specific activity than SPNPs-H and the E-TEK Pt/C catalyst. Because of the identical size and morphology of SPNPs-A and SPNPs-H, the higher catalytic activity of SPNPs-A for MOR could only be attributed to the presence of Ag and the possibility of electronic coupling between Ag and Pt. Furthermore Fig. 4d also shows that after the removal of the Ag core from SPNPs-A, the resultant SPNPs-H showed comparable MOR activity as the E-TEK Pt/C catalysts in MOR, suggesting that the stellated morphology contributed very limitedly, if any, to the activity of Pt for methanol oxidation.
The activity trend of the three catalysts for ORR was quite different from that for MOR. The low ORR activity of SPNPs-A is a strong contrast to its high MOR activity (Fig. 4e). As summarized in Fig. 4f, the half-wave potential of SPNPs-A was 470 mV and was 77% and 87% of the half-wave potentials of SPNPs-H and E-TEK Pt/C respectively. The current density at the half-wave potential of SPNPs-A (2.15 mA cm−2) was also lower than that of SPNPs-H (2.70 mA cm−2) and E-TEK Pt/C (2.48 mA cm−2). The report of Watanabe and co-workers provided some possible understanding of the poor ORR activity of SPNPs-A35. The report discussed the alloying effect of Fe, Ni or Co with Pt, where increase in ORR activity was observed. As Fe, Ni and Co have more 5d vacancies than Pt, they would withdraw electrons from Pt and consequently increase the 5d vacancies in Pt. This increased the electron exchange between O2 and Pt to promote oxygen reduction through enhanced O2 adsorption. However, for SPNPs-A, the electron donation from Ag to Pt decreased the 5d vacancies in Pt. Thus, O2 adsorption was weakened on SPNPs-A and the ORR activity was decreased because of inadequate activation of the O2 molecules.
On the other hand, SPNPs-H was more active than SPNPs-A and the E-TEK Pt/C catalyst for ORR. This is consistent with previous observations of high catalytic activities of Pd-Pt dendrites and Pt-on-Pd structures for oxygen reduction at room temperature19,20. Since SPNPs-A and SPNPs-H had identical size and morphology, we are led to conclude that the high ORR catalytic activity of SPNPs-H was derived from their stellated structure without the compensating effect of a Ag core in the case of SPNPs-A. An abundance of atomic steps, edges and corner atoms in the high curvature stellated structure and exposed {111} facets in SPNPs-H accounted for a heightened activity for ORR. Thus, catalytic activity may be varied from MOR to ORR, two very different structure sensitive reactions, by controlling the composition of the stellated structure.
In summary, stellated Pt nanoparticles with a Ag core were produced by a one-pot method. These stellated nanoparticles could be further processed into hollow Pt nanoparticles without changes in the stellation morphology by a highly selective Ag extraction procedure. In comparison with a reference E-TEK Pt/C catalyst, the stellated Pt nanoparticles with Ag or hollow core were more active than E-TEK Pt/C in MOR and ORR respectively. The electronic coupling between Ag and Pt was a major contributor to the enhancement of MOR activity whereas the Pt stellates, with abundant atomic steps, edges and corner atoms as well as exposed {111} facets, were principally responsible for the high activity of stellated particles in ORR. The synthesis of stellated nanoparticles is relatively simply and yet stellation appears to provide an effective platform for producing heterogeneous structured nanomaterials with very different catalytic performance. The technique may be extended to explore the stellating growth of Pt and other catalytic metals on multiply twinned polyhedral metal seeds other than the Ag seeds. This could result in some interesting examples with technological importance.
Methods
General materials
Potassium tetrachloroplatinate(II) (K2PtCl4, 98%), silver nitrate (AgNO3, 99%), aqueous HClO4 solution (70%, ACS reagent) and Nafion 117 solution (5% in a mixture of lower aliphatic alcohols and water) from Aladdin Reagents, bis(p-sulfonatophenyl)phenylphosphane dihydrate dipotassium salt (BSPP, 97%) from Strem Chemicals, oleylamine (70%, technical grade) from Sigma-Aldrich, methanol (99%), ethanol (99.5%) and toluene (99.5%) from Beijing Chemical Works and Vulcan XC-72 carbon powders (XC-72C, BET surface area = 250 m2 g−1 and average particle size = 40 ~ 50 nm) from Cabot Corporation, were used as received. All glassware and Teflon-coated magnetic stir bars were cleaned with aqua regia, followed by copious washing with de-ionized water before drying in an oven.
Synthesis of stellated Pt nanoparticles with Ag cores (SPNPs-A)
An one-pot approach was used for the synthesis of bimetallic Ag-Pt nanoparticles with stellated morphologies. In brief, 34 mg of AgNO3 (0.2 mmol) and 166 mg of K2PtCl4 (0.4 mmol) were added to 20 ml of oleylamine in a three-necked flask equipped with a condenser and a stir bar. The solution was brought to and kept at 165°C for 2 h under flowing Ar or N2 for the reduction of Ag+ and Pt2+ by oleylamine. The Ag and Pt precursor molar ratio was controlled at 1/2. After reaction, the SPNPs-A were purified by precipitation with methanol, centrifugation, washing with methanol and re-dispersed in 20 ml of toluene.
Synthesis of stellated Pt nanoparticles with hollow interiors (SPNPs-H)
To remove the Ag component from the interior region of stellated Ag-Pt nanoparticles, 10 ml of SPNPs-A solution was diluted with toluene to 50 ml and then mixed with 50 ml of water containing 250 mg of BSPP. The molar ratio of BSPP/Ag in the mixture was calculated to be ~5/1. The mixture was aged for 48 h under vigorous stirring at room temperature to completely remove the Ag from the core region of SPNPs-A. The mixture was left to stand and the upper toluene phase was collected (nominally 4 mM of Pt).
Particle characterizations
TEM, HRTEM and STEM were performed on the JEOL JEM-2100 and FEI Tecnai G2 F20 electron microscope operating at 200 kV with a supplied software for automated electron tomography. For the TEM measurements, a drop of the nanoparticle solution was dispensed onto a 3-mm carbon-coated copper grid. Excessive solution was removed by an absorbent paper and the sample was dried under vacuum at room temperature. An EDX analyzer attached to the TEM operating in the scanning transmission electron microscopy (STEM) mode was used to analyze the chemical compositions of the synthesized nanoparticles. UV-visible spectra of the reaction system at different time during the synthesis of stellated Ag-Pt bimetallic nanoparticles were collected on a Hitachi U-3900 spectrophotometer. X-ray photoelectron spectroscopy (XPS) was conducted on a VG ESCALAB MKII spectrometer. Powder XRD patterns were recorded on a Rigaku D/Max-3B diffractometer, using Cu Kα radiation (λ = 1.54056 Å). Samples for XPS and XRD analyses were concentrated from the toluene solution of nanoparticles to 0.5 mL using flowing N2. 10 ml of methanol was then added to precipitate the nanoparticles, which were recovered by centrifugation, washed with methanol several times and then dried at room temperature in vacuum.
Electrochemical measurements
Electrochemical measurements were carried out in a standard three-electrode cell connected to a Bio-logic VMP3 (with EC-lab software version 9.56) potentiostat. A leak-free Ag/AgCl (saturated with KCl) electrode was used as the reference electrode. The counter electrode was a platinum mesh (1 × 1 cm2) attached to a platinum wire.
For the loading of the catalyst on Vulcan XC-72 carbon support, a calculated amount of carbon powder was added to the toluene solution of SPNPs-A or SPNPs-H. After stirring the mixture for 24 h, the stellated nanoparticles/C (20 wt% Pt on carbon support) was collected by centrifugation, washed thrice with methanol and then dried at room temperature in vacuum.
The working electrode was a thin layer of Nafion-impregnated catalyst cast on a vitreous carbon disk. This electrode was prepared by ultrasonically dispersing 10 mg of the stellated nanoparticles/C in 10 ml of aqueous solution containing 4 ml ethanol and 0.1 ml of Nafion solution. A calculated volume of the ink was dispensed onto the 5 mm glassy carbon disk electrode to produce a nominal catalyst loading of 20 μg cm−2 (Pt basis). The carbon electrode was then dried in a stream of warm air at 70°C for 1 h.
The room temperature cyclic voltammograms of SPNPs-A and SPNPs-H in argon-purged HClO4 (0.1 M) were recorded between −0.2 V and 1 V at 50 mV·s−1 and used for the determination of electrochemically active surface area (ECSA) of Pt. The catalyst performance in room-temperature methanol oxidation reaction (MOR) was also measured by cyclic voltammetry. For these measurements the potential window of 0 V to 1 V was scanned at 20 mV·s−1 until a stable response was obtained, before recording the voltammograms. The electrolyte was methanol (1 M) in perchloric acid (0.1 M). For each catalyst (SPNPs-A and SPNPs-H), current density was normalized by its ECSA to obtain the specific activities.
The catalyst performance in room-temperature oxygen reduction reaction (ORR) was evaluated in HClO4 electrolyte solution (0.1 M) using a glass carbon rotating disk electrode (RDE) at a rotation speed of 1600 rpm. Negative-going linear sweep voltammograms were recorded from 0.8 V to 0 V at 20 mV·s−1 at room temperature in the presence of bubbling ultra-pure oxygen to maintain a saturated oxygen atmosphere near the working electrode. In the ORR polarization curve, the current density was also normalized in reference to the ECSA to obtain the specific activities.
References
Steele, B. C. H. & Heinzel, A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001).
Perry, M. L. & Fuller, T. F. A historical perspective of fuel cell technology in the 20th century. J. Electrochem. Soc. 149, S59–S67 (2002).
Gasteiger, H. A., Kocha, S. S., Sompalli, B. & Wagner, F. T. Activity benchmarks and requirements for Pt, Pt-alloy and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 56, 9–35 (2005).
Parsons, R. & VanderNoot, T. The oxidation of small organic molecules: A survey of recent fuel cell related research. J. Electroanal. Chem. 257, 9–45 (1988).
Antolini, E., Lopes, T. & Gonzalez, E. R. An overview of platinum-based catalysts as methanol-resistant oxygen reduction materials for direct methanol fuel cells. J. Alloys Compd. 461, 253–262 (2008).
Marković, N. M. & Ross, P. N. Surface science studies of model fuel cells electrocatalysts. Surf. Sci. Rep. 45, 117–229 (2002).
de Brujin, F. A., Dam, V. A. T. & Janssen, G. J. M. Review: Durability and degradation issues of PEM fuel cell components. Fuel Cells 8, 3–22 (2008).
Chen, W., Kim, J., Sun, S. & Chen, S. Electrocatalytic reduction of oxygen by FePt alloy nanoparticles. J. Phys. Chem. C 112, 3891–3898 (2008).
Wang, C., Daimon, H., Onodera, T., Koda, T. & Sun, S. A. General approach to the size- and shape-controlled synthesis of platinum nanoparticles and their catalytic reduction of oxygen. Angew. Chem. Int. ed. 47, 3588–3591 (2008).
Chen, J., Lim, B., Lee, E. P. & Xia, Y. Shape controlled synthesis of platinum nanocrystals for catalytic and electrocatalytic applications. Nano Today 4, 81–95 (2009).
Peng, Z. & Yang, H. Designer platinum nanoparticles: Control of shape, composition in alloy, nanostructure and electrocatalytic property. Nano Today 4, 143–164 (2009).
Kang, Y. & Murray, C. B. Synthesis and electrocatalytic properties of cubic Mn-Pt nanocrystals (Nanocubes). J. Am. Chem. Soc. 132, 7568–7569 (2010).
Chen, A. & Holt-Hindle, P. Platinum-based nanostructured materials: Synthesis, properties and applications. Chem. Rev. 110, 3767–3804 (2010).
Cozzoli, P. D., Pellegrino, T. & Manna, L. Synthesis, properties and perspectives of hybrid nanocrystal structures. Chem. Soc. Rev. 35, 1195–1208 (2006).
Zhang, J., Sasaki, K. & Adzic, R. R. Stabilization of platinum oxygen-reduction electrocatalysts using gold clusters. Science 315, 220–222 (2007).
Zhang, L.-F., Zhong, S.-L. & Xu, A.-W. Highly branched concave Au/Pd bimetallic nanocrystals with superior electrocatalytic activity and highly efficient SERS enhancement. Angew. Chem. Int. Ed. 52, 645–649 (2013).
Nilsson, A. & Pettersson, L. G. M. [Absorbate electronic structure and bonding on metal surfaces]. Chemical bonds at surfaces and interfaces [Nilsson, A., Pettersson, L. G. M. & Nørskov, J. K. (eds.)] [78–138] (Elsevier, 2008).
Yang, J. & Ying, J. Y. Nanocomposites of Ag2S and noble metals. Angew. Chem. Int. Ed. 50, 4637–4643 (2011).
Lim, B. et al. Pd-Pt bimetallic nanodendrites with high activity for oxygen reduction. Science 324, 1302–1305 (2009).
Peng, Z. & Yang, H. Synthesis and oxygen reduction electrocatalytic property of Pt-on-Pd bimetallic heteronanostructures. J. Am. Chem. Soc. 131, 7542–7543 (2009).
Guo, S., Dong, S. & Wang, E. Three-dimensional Pt-on-Pd bimetallic nanodendrites supported on graphene nanosheet: Facile synthesis and used as an advanced nanoelectrocatalyst for methanol oxidation. ACS Nano 4, 547–555 (2010).
Wang, L., Nemoto, Y. & Yamauchi, Y. Direct synthesis of spatially-controlled Pt-on-Pd bimetallic nanodendrites with superior electrocatalytic activity. J. Am. Chem. Soc. 133, 9674–9677 (2011).
Zhou, S., Mcllwrath, K., Jackson, G. & Eichhorn, B. Enhanced CO tolerance for hydrogen activation in Au-Pt dendritic heteroaggregate nanostructures. J. Am. Chem. Soc. 128, 1780–1781 (2006).
Lee, H., Habas, S. E., Somorjai, G. A. & Yang, P. Localized Pd overgrowth on cubic Pt nanocrystals for enhanced electrocatalytic oxidation of formic acid. J. Am. Chem. Soc. 130, 5406–5407 (2008).
Peng, Z. & Yang, H. PtAu bimetallic heteronanostructures made by post-synthesis modification of Pt-on-Au nanoparticles. Nano Res. 2, 406–415 (2009).
Wang, F. et al. Heteroepitaxial growth of high-index-faceted palladium nanoshells and their catalytic performance. J. Am. Chem. Soc. 133, 1106–1111 (2011).
Wang, Z. L. Transmission electron microscopy of shape-controlled nanocrystals and their assemblies. J. Phys. Chem. B 104, 1153–1175 (2000).
Tang, Y. & Ouyang, M. Tailoring properties and functionalities of metal nanoparticles through crystallinity engineering. Nat. Mater. 6, 754–759 (2007).
Brodersen, S. H., Grønbjerg, U., Hvolbæk, B. & Schiøtz, J. Understanding the catalytic activity of gold nanoparticles through multi-scale simulations. J. Catal. 284, 34–41 (2011).
Wiley, B., Herricks, T., Sun, Y. & Xia, Y. Polyol synthesis of silver nanoparticles: Use of chloride and oxygen to promote the formation of single-crystal, truncated cubes and tetrahedrons. Nano Lett. 4, 1733–1739 (2004).
Liu, H. et al. Hollow and cage-bell structured nanomaterials of noble metals. J. Am. Chem. Soc. 134, 11602–11610 (2012).
Mahmoud, M. A., Tabor, C. E., El-Sayed, M. A., Ding, Y. & Wang, Z. L. A new catalytically active colloidal platinum nanocatalyst: The multiarmed nanostar single crystal. J. Am. Chem. Soc. 130, 4590–4591 (2008).
Lee, S. W. et al. Roles of surface steps on Pt nanoparticles in electro-oxidation of carbon monoxide and methanol. J. Am. Chem. Soc. 131, 15669–15677 (2009).
Liu, L., Pippel, E., Scholz, R. & Gösele, U. Nanoporous Pt-Co alloy nanowires: Fabrication, characterization and electrocatalytic properties. Nano Lett. 9, 4352–4358 (2009).
Toda, T., Igarashi, H., Uchida, H. & Watanabe, M. Enhancement of the electroreduction of oxygen on Pt alloys with Fe, Ni and Co. J. Electrochem. Soc. 146, 3750–3756 (1999).
Acknowledgements
Financial support from the 100 Talents Program of the Chinese Academy of Sciences, National Natural Science Foundation of China (No.: 21173226, 21376247) and State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences (MPCS-2012-A-11, MPCS-2011-D-08, MPCS-2010-C-02) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
H.L., F.Y. and H.C. performed the materials synthesis, characterization and electrochemical measurements. Q.Y. contributed to the TEM characterization. J.Y., J.X. and J.Y.L. supervised the project. J.Y. wrote the main manuscript text and all authors participated in the review of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Liu, H., Ye, F., Yao, Q. et al. Stellated Ag-Pt bimetallic nanoparticles: An effective platform for catalytic activity tuning. Sci Rep 4, 3969 (2014). https://doi.org/10.1038/srep03969
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03969
This article is cited by
-
Ag/g-C3N4 nanosheets as a progressive support of Pt catalyst for improved electrocatalytic oxidation of methanol
Journal of Materials Science (2024)
-
Surface controllable anchoring of Cu onto nanostructured PtNi for efficient electrochemical hydrogen evolution from seawater
Science China Materials (2023)
-
A high-efficiency electrochemical sensor of dopamine based on WS2 nanosheets decorated with dandelion-like platinum–silver nanoparticles
Journal of Materials Science: Materials in Electronics (2022)
-
Pt3Ag alloy wavy nanowires as highly effective electrocatalysts for ethanol oxidation reaction
Nano Research (2020)
-
Amino-acid- and peptide-directed synthesis of chiral plasmonic gold nanoparticles
Nature (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
