
Abstract
Electronic structure calculations were performed to study the role of misfit dislocations on the structure and chemistry of a metal/oxide interface. We found that a chemical imbalance exists at the misfit dislocation which leads to dramatic changes in the point defect content at the interface – stabilizing the structure requires removing as much as 50% of the metal atoms and insertion of a large number of oxygen interstitials. The exact defect composition that stabilizes the interface is sensitive to the external oxygen partial pressure. We relate the preferred defect structure at the interface to a competition between chemical and strain energies as defects are introduced.
Similar content being viewed by others


Introduction
Metal-oxide systems are ubiquitous in technology with applications including microelectronics, magnetoresistance devices1, thermal barriers2, nanocatalysts3 and structural materials for nuclear reactors4. In all these applications the properties of the material are primarily governed by the structure and chemistry of the metal/oxide interface. Metal/oxide interfaces exhibit exotic interfacial structure5 as they are formed by materials with dramatically different properties, crystal symmetry, surface energy and atomic bonding (metallic versus ionic and covalent)6. The structural complexity of a heterointerface such as a metal/oxide interface is a combined effect of the surface structures of the component phases and the unique structural features arising when they form the interface. Oxide surfaces already exhibit a wide range of surface structures to maintain overall charge neutrality, including large deviations in stoichiometry7, faceting, metallization, adsorption of charged species and modification of the electronic structure of the surface8. Once the oxide is joined to the metal, a separate set of structures can form to maintain atomic ordering and alleviate strain across the interface. One such structure is misfit dislocations, formed when two crystals of different inter-planar spacing are joined. In this case, regions at the interface with atom-by-atom matching across the interface (coherent regions) are separated by a region of least coincidence (misfit dislocations). These misfit dislocations have been shown to have important consequences for functional9 and structural properties10 of hetero-interfaces. For example, the work of adhesion of the Ag/MgO interface varies by a factor of four for the interface with and without misfit dislocations11. Hence, to develop a comprehensive understanding of structure-property relationship in metal/oxide systems, it is important to consider misfit dislocations and their coupling to structure and chemistry at metal/oxide interfaces.
Due to the lack of suitable empirical potentials most theoretical calculations of metal/oxide interfaces have relied on density functional theory (DFT), necessitating the study of small systems. Except for a few limited cases12,13, such calculations have focused on fully coherent interfaces14. However, such models provide an inadequate description of many systems that have significant mismatch between the materials because these models do not consider the misfit dislocation structure. In the cases where atomic mismatch between the metal and the oxide were considered12,13, a detailed description of the dislocation core structure was not presented. In order to circumvent the size limitations of DFT, Johansson et al.2 used a combination of DFT and the Peierls-Nabarro model to evaluate the coherent and incoherent contributions to the interfacial energy, respectively, in metal/ceramic systems. Alternatively, analytical approaches such as image charge models6 can be used to examine the properties of metal/oxide interfaces, but these models may not be sufficient to capture the atomic and chemical structure of the misfit dislocation core at metal/oxide interfaces.
In this work, we present electronic structure calculations of the structure of a metal/oxide interface containing misfit dislocations using Fe/Y2O3 as a model system. Fe/Y2O3 is chosen as a surrogate for the more complex Y-Ti-O oxide within the ferritic matrix of nanostructured ferritic alloys (NFAs)4, where the metal/oxide interface is thought to provide most of the radiation resistance observed in this material4,15. The radiation resistance of other nanocomposites is directly proportional to the density of misfit dislocations and their intersections16. If misfit dislocations have a similar role on the radiation resistance in metal/oxide systems, it is critical to understand the structure and chemistry of the misfit dislocations at the metal/oxide interface to understand and predict radiation resistance.
Results
Structure of the ideal interface
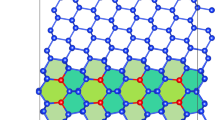
For the electronic structure calculations of the structure and chemistry of the Fe/Y2O3 metal/oxide interface, we created a bi-layer supercell (Fig. 1) following an experimentally17 observed orientation relationship (OR) at the interface of [001]Fe||[100]YO and (010)Fe||(011)YO. Details of the bi-layer structure are presented in the method section. Fig. 2(a) shows the relaxed structure of the initial Fe/Y2O3 interface. In the y direction, there are five atomic columns of iron for every 4 atomic columns of oxygen. The formation of a misfit dislocation with line direction along the x-axis accommodates this lattice mismatch, but leads to a higher Fe/O ratio at the misfit dislocation compared to that in the coherent region. It is notable that in addition to the misfit dislocation with line direction along the x-axis, we also observe a misfit dislocation whose line direction is along the y-axis accommodating the misfit between the two crystals in the x direction. In contrast to the y direction, in the x direction there are eleven columns of iron for every twelve columns of oxygen. Hence, for the misfit dislocation with its line direction along the y direction, the local metal/oxygen ratio is less than 1, resulting in little-to-no chemical imbalance for the metal and thus no strong interaction with defects. As a consequence, this misfit dislocation with its line direction along the y direction is not emphasized in this manuscript. Nonetheless, it can be seen that the chemical imbalance we discuss here is sensitive to the nature of the dislocations i.e. whether the relative extra half plane of the dislocation is on the metal side (dislocation line along x for this interface) or on the oxide side (dislocation line along y).

(a) Initial un-relaxed structure of the Fe/Y2O3 interface. In the DFT calculations, atomic relaxation is allowed in all the layers except the top iron layer and bottom yttria layer (as indicated).

(a) Relaxed structure of the Fe/Y2O3 interface. The misfit dislocation is indicated by the dotted lines while the shaded rectangular box indicates the interfacial Fe layer. (b) Vacancy formation energy at Fe sites within the interfacial Fe layer. The red box indicates the dislocation region. It is observed that the vacancy formation energy at certain sites is negative, i.e. a vacancy will form spontaneously at those interfacial iron sites at T = 0 K. (c) Plot of vacancy formation energy vs. excess Bader charge (in terms of electrons) of iron atoms (compared to bulk iron) at the interface within the ideal structure with no point defects.
For the dislocation with line direction along the x-axis the spatial inhomogeneity of the Fe/O ratio leads to a non-uniform displacement of iron atoms within the interfacial Fe layer (Fig. 2(a)) as well as a variation in the iron vacancy formation energy ( ) within the layer (Fig. 2(b)) (Similar spatial inhomogeneity leads to the bulging of the interfacial layer as observed in previous calculations12). Many sites have negative
) within the layer (Fig. 2(b)) (Similar spatial inhomogeneity leads to the bulging of the interfacial layer as observed in previous calculations12). Many sites have negative  and these are primarily concentrated at the dislocation. These unstable sites occur because, at the dislocation, the local stoichiometry (higher Fe/O ratio) provides an insufficient number of oxygen atoms to accept electrons from iron. These iron atoms, which thus cannot donate electrons to the oxygen, are unstable. It can therefore be concluded that in the case of a metal/oxide interface the structure of the misfit dislocations at the interface is not only related to the misfit strain at the interface, but also to the local chemistry across the interface that facilitates, or impedes, electron transfer from the metal to the oxide.
and these are primarily concentrated at the dislocation. These unstable sites occur because, at the dislocation, the local stoichiometry (higher Fe/O ratio) provides an insufficient number of oxygen atoms to accept electrons from iron. These iron atoms, which thus cannot donate electrons to the oxygen, are unstable. It can therefore be concluded that in the case of a metal/oxide interface the structure of the misfit dislocations at the interface is not only related to the misfit strain at the interface, but also to the local chemistry across the interface that facilitates, or impedes, electron transfer from the metal to the oxide.
Another interesting feature observed in our calculations is that the vacancy formation energy of interfacial iron sites within the ideal structure is correlated with the Bader charge18,19 (number of electrons on the iron atoms compared to the iron atoms in unstrained bulk iron) on the interfacial iron atoms as shown in Fig. 2(c). While the formation energy of iron vacancies is a direct measure of the chemical instability of iron atoms at the interface, Fig. 2(c) suggests that the Bader charge on the iron atoms at the interface can also be used as a measure of this instability, which we will exploit later in the manuscript to accelerate the calculations. This result also provides further evidence that the origin of the instability of Fe atoms at the dislocation is related to the ability to donate electrons to the oxygen.
Clearly, the as-constructed interface is structurally unstable. Next, we explore the possibilities of restoring the chemical imbalance at the misfit dislocation using three approaches: introducing iron vacancies (reducing conditions), oxygen interstitials (oxidizing conditions), or a mixture of the two. We note that in examining the interaction of point defects with the interface, we only consider defects on the metal side of the interface; defects in yttria are likely to be much higher in energy (as discussed in the Supplemental information) and are not considered.
Establishing chemical balance by creating interfacial iron vacancies
Fig. 3(a) shows the change in the energy of the system as iron vacancies are introduced at the interface. Iron vacancies were created within the interfacial iron layer at those sites identified to be the most unstable, i.e. having the most negative vacancy formation energies. Starting with the ideal structure, the vacancy formation energies for all Fe positions within the structure were calculated, 1–5 of the most unstable iron atoms were removed and the structure was re-relaxed. This process was repeated until there were no iron sites with a negative vacancy formation energy remaining at the interface (see the Supplemental information for details). The change in energy of the system with removal of n iron atoms at the interface is calculated as follows:

where  is the chemical potential of iron in pure unstrained bulk iron (the cohesive energy of bulk Fe). It is to be noted that for n = 1, ΔE is equal to the vacancy formation energy
is the chemical potential of iron in pure unstrained bulk iron (the cohesive energy of bulk Fe). It is to be noted that for n = 1, ΔE is equal to the vacancy formation energy  at a particular iron site. It can be observed in Fig. 3(a) that the energy of the system decreases continuously until the vacancy concentration at the interface reaches about 0.4 (relative to the initial Fe density within the layer), beyond which the energy of the interface fluctuates until it reaches a minimum at a vacancy concentration of 0.47. This results shows that about half of the iron atoms at the interface must be removed to restore the chemical imbalance and stabilize the interface. This vacancy concentration is significantly higher than that needed to stabilize the misfit dislocation structure in metal/metal systems20 (0.02–0.05), a consequence of the importance of maintaining the chemical balance in metal/oxide systems.
at a particular iron site. It can be observed in Fig. 3(a) that the energy of the system decreases continuously until the vacancy concentration at the interface reaches about 0.4 (relative to the initial Fe density within the layer), beyond which the energy of the interface fluctuates until it reaches a minimum at a vacancy concentration of 0.47. This results shows that about half of the iron atoms at the interface must be removed to restore the chemical imbalance and stabilize the interface. This vacancy concentration is significantly higher than that needed to stabilize the misfit dislocation structure in metal/metal systems20 (0.02–0.05), a consequence of the importance of maintaining the chemical balance in metal/oxide systems.

(a) Change in energy of the system with the introduction of defects: (a) vacancies; (b) interstitial oxygen; (c, d) mixture of vacancies with twelve and seventeen interstitial oxygen for (c) and (d), respectively. At any stage, the vacancy concentration at the interface was calculated as the ratio of the total number of iron vacancies introduced at the interface with respect to the total number of iron sites present at the interface within the ideal structure. In Fig. 3a the Fe/O ratio was calculated from the number of iron left at the interface at any stage divided by the number of oxygen atoms within the interfacial yttria layer. For the calculation of the Fe/O ratio in Fig. 3b and Fig. 3d both interstitial oxygen and oxygen atoms within the interfacial yttria layer were considered. The capital letters indicate various structures used for subsequent calculations. M is the ideal structure with no defects (vacancy or interstitials) while N and Q are the structures with twelve and seventeen interstitial oxygen (with no vacancies), respectively. In Fig. 3(b) the indicated oxygen chemical potentials are computed for T = 900K. The insets are zoomed-in regions of the curves indicated by the dashed-line boxes. In the insets, points A, B and C represent the vacancy concentration corresponding to the minimum energy structures for the zero, twelve and seventeen-oxygen structures. For the minimum energy structures (A, B and C) all iron vacancy formation energies are positive and any additional iron vacancies increase the energy of the system. In all cases, the associated change in the atomic structure lowers the Fe/O ratio at the interface, restoring the chemical imbalance.
Establishing chemical balance by inserting oxygen interstitials
The second approach for stabilizing the chemical imbalance is to introduce oxygen interstitials as this also decreases the local Fe/O ratio. Fig. 3(b) shows the change in energy of the Fe/Y2O3 system as interstitial oxygen atoms are introduced within the interfacial iron layer under four different partial pressures of oxygen, pO2 (see Supplemental information). The change in energy of the system upon insertion of m oxygen atoms at the interface was calculated as follows:

where μO is the chemical potential of oxygen, taken as the standard state of oxygen in its gaseous state and is related to pO2 (the relationship between μO and pO2 is described in the Supplemental information. The approach used to choose O interstitial sites is also presented in the Supplemental information). Equation 2 further implies that the oxide itself will be hyperstoichiometric and may be a further source of oxygen, a possibility we have not explored here. For pO2 ≤ 2.88 × 10−39 atm (equivalently, μ0 ≥ −8.3 eV at T = 900 K) the energy of the system increases as oxygen interstitials are inserted which implies that the chemical imbalance at the interface can only be established by interfacial iron vacancies. As pO2 is increased, it is thermodynamically preferable to insert a greater amount of oxygen (Fe/O = 1.34 and 1.14 for pO2 = 6.59 × 10−36 atm and pO2 = 2.62 × 10−30 atm, respectively, at the minimum energy interfacial structures). Hence, for pO2 > 2.88 × 10−39 atm, oxygen interstitials are a favorable mechanism for stabilizing the interface. However, it is likely that a combination of the two defect mechanisms is most thermodynamically preferred, which we explore below. It should be noted that these pO2 values are generally less than that needed to form FeO (10−24–10−25 atm) at 900 K (see Supplemental Information), demonstrating that the interface can attract significant amounts of O before the oxidation of bulk Fe occurs. Further, the oxygen partial pressures described here are significantly lower than what can be attained experimentally using ultra high vacuum (UHV) conditions (without the use of a strong oxygen getter like Mg). However, while an external oxygen partial pressure of the order of 10−25 atm may be difficult to achieve, such an equivalently low oxygen chemical potential can exist at the metal/oxide interface buried within a metal/oxide nanocomposite, implying our predicted results under low oxygen partial pressure are still relevant. In addition, for weaker oxide formers such as Ni and Cu, the partial pressures at which the material oxides are much higher and the oxidation at the dislocation might be more directly controllable with external partial pressure of oxygen.
Establishing chemical balance by a combination of defects
To identify the most stable point defect structure for pO2 > 2.88 × 10−39 atm we began with two interfacial structures in which only oxygen interstitials were introduced (N and Q in Fig. 3(b)) and inserted iron vacancies (Fig. 3(c–d)). The process of introducing vacancies is similar to Fig 3(a) (as described in the Supplemental information), with the exception that the starting structures are different. The starting structures in Fig. 3(c) and 3(d) contained 12 and 17 interstitial oxygen atoms, respectively. Compared to the Fe-vacancy loaded structure (A) the minimum energy structures (B and C) with a mixture of oxygen interstitials and iron vacancies have considerably lower energy. Hence, we conclude that for pO2 > 2.88 × 10−39 atm a mixture of oxygen interstitials and iron vacancies stabilizes the interface i.e. the interface is not stabilized by oxygen alone. Finally, it is interesting to note that the final iron vacancy concentration at the interface is roughly 0.5, regardless of the oxygen interstitial content. We discuss the reasons for this below.
Evidence of chemical balance being established at the interface
Fig. 4(a–c) show planar views of the atoms at the interface (iron atoms, oxygen in yttria and oxygen interstitials) for the starting structures in Fig. 3 (labeled M, N and Q) while Fig. 4(d–f) present the corresponding minimum energy structures (labeled A, B and C). In other words, these figures represent the structures of the interface before (M, N, Q) and after (A, B, C) minimizing the energy versus vacancy content for different initial interstitial oxygen content. For the case when the chemical imbalance is restored by vacancies only (Figs. 4(a) and 4(d)), most of the vacancies reside in the misfit dislocation region of the interface (Fig. 4(d)). Most of the remaining iron vacancies correlate with the structural oxygen vacancies in the bixbyite structure. Figs. 4b and 4c indicate that the original oxygen interstitials tend to reside at the misfit dislocations while Figs. 4e and 4f show that the Fe vacancies also prefer the misfit dislocation region. Hence, these figures reveal that both types of defects (iron vacancies and oxygen interstitials) are preferentially created at the misfit dislocation, indicating that the chemical imbalance at the misfit dislocation is the driver for the defect behavior at the interface.

(a–c) Planar view of the interfacial iron atoms (green), interfacial oxygen atoms in yttria (red) and oxygen interstitials (dark red) corresponding to the structures M, N and Q in Fig. 3; (d–f) Planar view of the interfacial iron atoms, interfacial oxygen atoms and oxygen interstitials corresponding to the minimum energy structures A, B and C in Fig. 3. These represent the structures before (M, N, Q) and after (A, B, C) minimizing the energy versus vacancy content for different oxygen content. The blue box represents the dislocation region. (g) Spatial variation of local Fe/O ratio at the interface at and away from the misfit dislocation for four different structures, namely the ideal structure (M), the structure with seventeen oxygen (Q) and the structures corresponding to the minimum energy structures A and C in Fig. 3, which are structure M and Q after minimizing the energy versus vacancy content. The local Fe/O ratio was measured based on the number of Fe and O atoms within five rectangular regions in the y-direction. The width of each rectangular region is equal to the cell size in the y-direction divided by the number of columns of iron (i.e. five).
This effect is quantified in Fig. 4(g) in which the spatial variation in the local Fe/O ratio between the dislocation and coherent region is presented. The local Fe/O ratio in the ideal structure is higher at the misfit dislocation than in the coherent region and, to a lesser extent, at the misfit dislocation in the structure containing only oxygen interstitials. However, once vacancies are introduced (A in Fig. 3a and Fig. 4d and C in Fig. 3d and Fig. 4f) the Fe/O ratio at the dislocation is significantly reduced such that it is less than in the neighboring coherent region, further confirming that the chemical imbalance imposed by the misfit dislocation is one important factor for determining the ultimate defect structure at the interface. For both structures with vacancies, the Fe/O ratio at the dislocation is less than 1, suggesting a tendency to form an iron-oxide (FeOm with m > 1) at the dislocation. This result indicates that the nucleation of any iron oxide phase at the interface would likely to occur first at the misfit dislocation and only later in the remainder of the interface.
Possibility of the formation of iron oxides at the interface
In this section we discuss the increased thermodynamic propensity to form iron oxide at the misfit dislocation due to the creation of point defects in order to establish chemical stability at the misfit dislocation. Details of the thermodynamic analysis to predict the formation of oxide at the misfit dislocation are presented in the Supplemental information. It was found that due to the creation of point defects the formation of any iron-oxide phases at the interface at low pO2 will likely initiate at the misfit dislocation, resulting in a complex interfacial structure in which new phases form at the dislocation while the coherent regions remain relatively intact. Further, as the pO2 is increased, the misfit dislocation will still tend to attract more oxygen than the surrounding coherent regions of the interface, possibly leading to a non-uniform distribution of oxide phases between the coherent and the misfit dislocation regions. While the prediction of a specific oxide phase is beyond our current capabilities (due to cell size limitations), secondary oxide phases have been observed at metal/oxide interfaces both theoretically and experimentally1,21,22. In principle, one could assume a given FeOx interfacial phase and orientation relationship and explicitly examine the Fe/FeOx and FeOx/Y2O3 interfaces separately, but without further experimental guidance on both the nature (stoichiometry) of such an FeOx phase and the orientation relationship, it would be very difficult to perform calculations on a reasonable interfacial structure. Further, spectroscopic measurements by Gegner et al23 suggested that it is possible to have excess oxygen residing at misfit dislocations in metal/oxide interfaces, which agrees with our prediction. Finally, we observed that the vacancy concentration for the minimum energy structures, between 0.47–0.50, is largely independent of the interstitial oxygen content (structures A, B and C in Fig. 3 with numbers of interstitial oxygen of 0, 12 and 17, respectively). These results suggest that the free volume created by Fe vacancies available at misfit dislocations is independent of the oxygen content at the dislocation, thus providing a possible explanation for the experimental observation23 of hydrogen trapping at metal/oxide interfaces, irrespective of oxygen content.
Competing energetic contributions leading to the formation of defects
It is surprising to observe that the vacancy concentration for the minimum energy structures is largely independent of the interstitial oxygen content, as indicated in the previous section. Intuitively, one might expect that an increase in oxygen content would result in a reduction in the number of iron vacancies required to restore the chemical imbalance. That is, if the driving force for defect segregation to the misfit dislocation is the formation of iron-oxygen bonds, either removing iron or introducing oxygen should satisfy that driving force. However, adding oxygen not only provides the needed chemical stability, but also strains the metal/oxide interface, resulting in two competing effects, one that reduces the energy of the system and another that, concurrently, increases the energy. The system relieves the strain by forming iron vacancies. Thus, the formation of iron vacancies has at least two driving forces: (i) restoring the chemical imbalance (which decreases with oxygen content); (ii) relieving the strain (which increases with oxygen content). In the following section, the combined effect of these two competing energetic contributions is examined.
Local energetic contributions
Generally, the stability of an interface is governed by competing energetic contributions. In the case of a metal/oxide interface with misfit dislocations, we show in this section that the structure of the interface is determined by the competition between chemical and strain energy. Fig. 5(a–b) shows that iron atoms (position p and c within the ideal structure) that are initially very unstable (very negative  ) exhibit increased stability with the introduction of oxygen in the neighborhood, which helps to restore the local chemical imbalance. Although interstitial oxygen strains the lattice locally, the reduction in chemical energy is more than the increase in strain energy, resulting in stabilization of these iron atoms. The opposite is true for an iron atom (position f) that was initially relatively stable. Iron atoms at position q, d and e, which were relatively more stable compared to position p, exhibit slightly increased instability with the introduction of oxygen in the neighborhood. In this case, the strain energy dominates with the introduction of oxygen. Fig. 5 indicates the local and complex competition that occurs between the two energetic contributions, namely, chemical and strain energy with the introduction of interstitial oxygen.
) exhibit increased stability with the introduction of oxygen in the neighborhood, which helps to restore the local chemical imbalance. Although interstitial oxygen strains the lattice locally, the reduction in chemical energy is more than the increase in strain energy, resulting in stabilization of these iron atoms. The opposite is true for an iron atom (position f) that was initially relatively stable. Iron atoms at position q, d and e, which were relatively more stable compared to position p, exhibit slightly increased instability with the introduction of oxygen in the neighborhood. In this case, the strain energy dominates with the introduction of oxygen. Fig. 5 indicates the local and complex competition that occurs between the two energetic contributions, namely, chemical and strain energy with the introduction of interstitial oxygen.

(a) Planar view of interfacial Fe atoms within the ideal structure. The blue box represents the dislocation region. (b) Calculated vacancy formation energy before and after an oxygen atom is introduced at an interstitial position within the interface of the ideal structure to quantitatively analyze the energetic contributions of oxygen interstitials to the Fe vacancy formation energies at different locations within the interface plane. We also found that the introduction of more than one oxygen atom (not shown) in the neighborhood of any iron atom destabilizes that iron atom, implying that the strain energy dominates when multiple interstitial oxygen atoms reside in the neighborhood of an iron atom.
Energetic contribution throughout the interface
The competition between the two energetic contributions is also evident even when the combined effect of all the interfacial iron atoms is considered. As established in Fig. 2(c), the excess Bader charge on an interfacial iron atom is a signature of the chemical instability of the atom. One might then expect that, with the introduction of oxygen interstitials, the chemical instability at the interface might be restored as the insertion of oxygen interstitials moves the Fe/O ratio at the interface closer to unity (Fig. 3 (b)). Fig. 6 shows that the number of iron atoms at the interface with excess Bader charge decreases as interstitial oxygen atoms are introduced into the interfacial iron layer (blue curve). Thus, with increasing interstitial oxygen content the chemical imbalance at the interface is indeed restored. However, inserting oxygen interstitials also introduces strain into the system. The average Voronoi volume of the iron atoms at the interface is a measure of the average free volume available at the interface24,25. A decrease in the Voronoi volume correlates with an increase in the strain energy of the system. The red points in Fig. 6 show that the average Voronoi volume of the iron atoms at the interface decreases continuously with the increasing interstitial oxygen content. Thus, introducing interstitial oxygen reduces the Voronoi volume, indicating that the strain energy of the system has increased with higher oxygen content. Fig. 6 illustrates the competition between the two energetic contributions, namely the chemical and strain energy, with the introduction of oxygen interstitials even when the combined effect of all the interfacial iron atoms is considered. This competition is much more complex at the local level as shown in Fig. 5. The complexity can be further understood from the fact that the vacancy formation energy for some of the iron atoms presented in Fig. 5 differs depending on the order in which the oxygen interstitials and iron vacancies are inserted/created at the interface. Nonetheless, the main purpose in presenting Fig. 5 and Fig. 6 is to demonstrate that the point defect structure associated with misfit dislocation is determined by two competing energetic components, the strain and chemical energy.

Plot of average Voronoi volume of interfacial iron atoms (left) and the number of interfacial iron atoms with excess Bader charge (right) as oxygen interstitials are inserted. The Bader charge is measured relative to bulk iron. The ideal structure with no defects has eighteen iron atoms with an excess Bader charge. The number is reduced to two upon the introduction of 12 oxygen interstitials (Fe/O = 1.14).
Summary and Conclusions
To summarize, using electronic structure calculations, we have determined how misfit dislocations influence the local atomic structure and chemistry of a model metal/oxide interface. We discovered that a chemical imbalance could exist at the misfit dislocation, which leads to dramatic changes in the point defect content at the interface, with as much as 50% of the metal atoms removed and significant oxygen interstitials inserted to stabilize the structure. Further, the exact composition of point defects that stabilize the metal/oxide interface is dependent on the oxygen content. It is notable that irrespective of the oxygen content at the interface it was observed that most of the iron atoms at the dislocation are removed in the minimum energy structures. This effectively moves the position of the misfit dislocation approximately one atomic layer into the iron bulk and away from the interface. While an accurate estimation of the preferred position of the dislocation cannot be performed with the current cell size, this result is consistent with experiments. Such ‘stand-off’ dislocations are commonly observed experimentally in metal/oxide interfaces (Nb/Al2O3, Cu/MnO, Ag/NiO, Nb/MgO6 and FeAl/Y2O317) where the dislocation core lies a few atomic layers away from the interface within the metal and the metal is coherent with the oxide. This dislocation structure is a consequence of the strong driving force for every iron atom to bond directly with an oxygen atom, pushing the system to form a coherent interface at the expense of moving the misfit dislocation away from the interface. Finally, we relate the preferred defect structure at the interface to a competition between chemical and strain energies as defects are introduced.
Taken together, our results have significant implications for designing novel metal/oxide interfaces. The results obtained in this work provide two design parameters, dislocation density and pO2 or, more generally, the oxygen content at the interface, to control the structure and chemistry of metal/oxide interfaces. For example, alloying elements with strong oxide forming tendencies (greater than the matrix) will tend to segregate to the misfit dislocation in the presence of oxygen, the local concentration of which can, in principle, be controlled by varying pO2 or the availability of oxygen at the interface (for example, by changing the oxide used to form the interface). Thus, the concentration of alloying elements at the interface will depend on both the misfit dislocation density and pO2 and will vary spatially at the interface between the dislocation and the coherent regions. This coupling of structure and dopant segregation could enable the control of various properties in metal/oxide systems that depend on the metal/oxide interface, including interfacial adhesion and strength as well as ionic and electronic mobility, which has important implications in designing new metal/oxide interfaces for numerous applications, including thermal barrier coatings, fast ion conductors and nanostructured ferritic alloys. We expect that these new insights into the role of the misfit dislocation structure on the structure and chemistry of the interface will extend to other metal/ceramic systems (metal/carbide and metal/nitride) as they have similar structural characteristics due to chemical imbalance at metal/oxide interfaces caused by misfit dislocations12,26. However, in some cases (e.g. metal/carbide) other factors, such as covalency, may also be important.
It is to be noted that the details of the reconstruction will vary between different metal/oxide interfaces. For example, in the present metal/oxide system we predict that about fifty percent of the iron atoms have to be removed to establish the chemical balance. For an arbitrary metal/oxide interface the fraction of metal vacancies to be created will depend on several factors:
-
i
Propensity to form oxide by the metal: For example, keeping everything else constant, if we change the metal from Fe to Cr (i.e. an Cr/yttria interface) it is possible that the fraction of vacancies will be more than 50 percent. This is because Cr has a stronger oxide-forming tendency compared to Fe. Hence, the extent of chemical imbalance on the metal side might be higher in Cr/yttria interface compared to the present Fe/yttria interface.
-
ii
Nature of the misfit dislocation: As previously discussed the extent of chemical imbalance depends on the nature of the extra half plane of the misfit dislocation i.e. if the misfit dislocation has excess metal or oxygen planes. For example, if the extra half plane is on the oxygen sub-lattice (instead on the metal side as described here) there is excess oxygen at the misfit dislocation compared to metal, the extent of the chemical imbalance will be nominal and, at such misfit dislocations, there will not be as large of a driving force for chemical restructuring.
-
iii
Orientation relationship (OR): For the same metal and oxide combination the extent of reconstruction will be different for different ORs. This is because different ORs have different metal/oxygen ratios at the interface and hence different degrees of chemical imbalance.
Overall, this work significantly shifts the conventional understanding of misfit dislocations at metal/oxide interfaces. We expect that almost all metal/oxide interfaces will contain large numbers of structural defects around the misfit dislocations; the exact concentration of which will likely depend on the relative propensity of the metal to form oxide and the nature of the misfit dislocation i.e. if the misfit dislocation has excess metal or oxygen planes. The insights from this work will play a critical role in how future metal/oxide interfaces in applications from nuclear energy to nanocatalysis to nanoelectronics should be modeled and understood.
Methodology
Calculations of all interface structures and properties were performed using density functional theory as implemented in the Vienna ab-initio Simulation Package (VASP)27,28. In these calculations the projector augmented wave29 method was utilized with a plane wave cut-off of 300 eV. All the calculations were spin polarized and the Perdew–Burke–Ernzerhof30 parameterization of the generalized gradient approximation (GGA) was used for the exchange–correlation functional. The PAW potentials were generated using the electronic configurations: 3p6 3d7 4s1 for iron, 4s2 4p6 4d1 5s2 for yttrium and 2s2 2p4 for oxygen. Further, the Monkhorst and Pack scheme was utilized for Brillion-Zone sampling with a 1x1x1 k-point mesh. The maximum forces on every relaxed atom were converged to 0.05 eV/Angstrom for all the calculations except for limited calculations involving re-relaxation of the supercell with additional oxygen interstitials and/or vacancies, where the maximum forces on atoms were converged to 0.15 eV/Angstrom.
The bi-layer supercell for the DFT calculations consisted of three layers each of iron (a total of 165 atoms with 55 atoms in each layer) and yttria (a total of 180 atoms with Y:O = 2:3). Iron has the BCC crystal structure while yttria is a bixbyite31, a fluorite derivative structure with one-fourth of the oxygen sites vacant and ordered. Compared to a 3D particle within Fe, this bi-layer cell geometry simplifies the analysis of the structure while retaining the essential features of the system. The top iron and bottom yttria layer are in contact with a vacuum layer with a thickness of 7.25 Å. The experimentally observed OR involves a non-polar (110) termination of Y2O3, indicating that no special considerations are necessary to eliminate spurious surface dipoles. Yttria layers are strained by −1.1% and −4.6% in the in-plane x and y direction respectively, to match the periodic unit cell parameters required to fit and integral number of iron atoms. We expect that this introduced strain, necessary to keep the simulation cell to a tractable size, primarily changes the misfit dislocation spacing as compared to the fully relaxed case and not the basic properties of the misfit dislocations themselves. Furthermore, straining the yttria layers allows us to use bulk iron as the reference for calculating the chemical potential of iron. In any case, the overall reconstruction of the metal/oxide interface and the structure of the misfit dislocation studied in the current work has its origin in the chemical imbalance caused by excess metal compared to oxygen at the dislocation and our results will be relatively insensitive to whether the misfit strain is assigned to yttria or to the metal or shared between the two. In our calculations the top and bottom layer of iron and yttria were kept frozen to replicate the bulk crystal and the interface is assumed to be atomically flat. We also performed additional calculations of the iron/yttria interface in which the top-most iron layer was allowed to relax (not shown). We found that the iron atoms undergo bending or “warping” at the misfit dislocation, an effect observed in electronic structure calculations of metal/ceramic interfaces (e.g. metal/oxide, metal/nitride) with misfit dislocations by other authors12,26. Such warping of the metallic layers originates from the chemical imbalance (Fe/O >1) at the misfit dislocation.
Finally to ensure that our results are not an artifact of the various constraints such as system size, plane wave energy cut-off in our calculations, we have examined the influence of those constraints. We found that the central physical conclusions reached above are not artifacts of the limitations of our calculation procedure (see Supplemental information). In the Supplemental information we also present the validity of using Bader charge as the guiding parameter to introduce defects.
References
Meyerheim, H. L. et al. Geometrical and compositional structure at metal-oxide interfaces: MgO on Fe(001). Phys. Rev. Lett. 87, 076102 (2001).
Johansson, S. A. E., Christensen, M. & Wahnstrom, G. Interface energy of semicoherent metal-ceramic interfaces. Phys. Rev. Letts. 95, 226108 (2005).
Yanhui, W. & Diyong, W. The experimental research for production of hydrogen from n-octane through partially oxidizing and steam reforming method. Int. J. Hydrogen Energy 26, 795–800 (2001).
Odette, G. R., Alinger, M. J. & Wirth, B. D. Recent developments in irradiation-resistant steels. Annu. Rev. Mater. Res. 38, 471–503 (2008).
Rosenhahn, A., Schneider, J., Becker, C. & Wandelt, K. Oxidation of Ni3Al (111) at 600, 800 and 1050 K investigated by scanning tunneling microscopy. J. Vac. Sci. Technol. A 18, 1923–1927 (2000).
Ernst, F. Metal-oxide interfaces. Mater. Sci. Eng. A R14, 97–156 (1995).
Noguera, C. Physics and chemistry at oxide Surfaces (Cambridge Univ. Press, Cambridge, 1996).
Goniakowski, J., Finocchi, F. & Noguera, C. Polarity of oxide surfaces and nanostructures. Rep. Prog. Phys. 71, 016501 (2008).
Li, Y. L. et al. Influence of interfacial dislocations on hysteresis loops of ferroelectric films. J. Appl Phys. 104, 104110 (2008).
Misra, A., Hirth, J. P. & Kung, H. Single-dislocation-based strengthening mechanisms in nanoscale metallic multilayers. Phil. Mag. A, 82, 2935–2951 (2002).
Hong, T., Smith, J. R. & Srolovitz, D. J. Theory of metal- ceramic adhesion. Acta Metal. Mater. 43, 2721–2730 (1994).
Benedek, R. et al. First principles simulation of a ceramic/metal interface with misfit. Phys. Rev. Lett. 84, 3362–3365 (2000).
Benedek, R., Seidman, D. N., Minkoff, M., Yang, M. & Alavi, A. Atomic and electronic structure and interatomic potentials at a polar ceramic/metal interface: MgO/Cu. Phys. Rev. B 60, 16094–16102 (1999).
Sinnott, S. B. & Dickey, E. C. Ceramic/metal interface structures and their relationship to atomic- and meso-scale properties. Mater. Sci. Eng. R 43, 1–59 (2003).
Odette, G. R. & Hoelzer, D. T. Irradiation-tolerant nanostructured ferritic alloys: Transforming helium from a liability to an asset. JOM, 62, 84–92 (2010).
Kashinath, A., Misra, A. & Demkowicz, M. J. Stable storage of helium in nanoscale platelets at semicoherent interfaces. Phys. Rev. Letts. 110, 086101 (2013).
Inkson, B. J. & Threadgill, P. L. Y2O3 morphology in an oxide dispersion strengthened FeAl alloy prepared by mechanical alloying. Mat. Res. Soc. Symp. Proc. 460, 767 (1997).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 254–360 (2006).
Bader, R. F. W. Atoms in molecules - A quantum theory (Oxford University Press, New York, 1990).
Liu, X. Y., Hoagland, R. G., Demkowicz, M. J., Nastasi, M. & Misra, A. The influence of lattice misfit on the atomic structures and defect energetics of face centered cubic–body centered cubic interfaces. J. Eng. Mater. Tech. 134, 021012 (2012).
Luches, P. et al. Iron oxidation, interfacial expansion and buckling at the Fe/NiO(001) interface. Phys. Rev. Lett. 96, 106106 (2006).
Pipel, E., Woltersdorf, J., Gegner, J. & Kirchheim, R. Evidence of oxygen segregation at Ag/MgO interfaces. Acta Mater. 48, 2571–2578 (2000).
Gegner, J., Horz, G. & Kirchheim, R. Segregation of oxygen at metal/oxide- interfaces. Interface Sci. 5, 231–243 (1997).
Erhart, P. A first-principles study of helium storage in oxides and at oxide–iron interfaces. J. Appl. Phys. 111, 113502 (2012)
Rycroft, C. H., Grest, G. S., Landry, J. W. & Bazant, M. Z. Analysis of granular flow in a pebble-bed nuclear reactor. Phys. Rev. E 74, 021306 (2006).
Liu, L. M., Wang, S. Q. & Ye, H. Q. Atomic and electronic structures of the lattice mismatched metal–ceramic interface. J. Phys.: Condens. Matter 16, 5781–5790 (2004)
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Blochl, P. E. Projector Augmented-Wave Method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Xu, Y. N., Gu, Z. Q. & Ching, W. Y. Electronic, structural and optical properties of crystalline yttria. Phys. Rev. B 23, 14993–15000 (1997).
Acknowledgements
S.C. was primarily supported by the LDRD Director's postdoctoral fellowship at Los Alamos National Laboratory (LANL). S.C. further acknowledges support by the Center for Materials at Irradiation and Mechanical Extremes (CMIME), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number 2008LANL1026, which supported the combined vacancy-oxygen calculations and subsequent analysis. B.P.U., who constructed initial interfacial geometries and assisted with analysis and interpretation of the calculations, was supported by CMIME. D.M., who assisted with analysis, editing and converted chemical potentials to partial pressures, was supported by Nuclear Energy University Program (NEUP) award 10-392 and the National Science Foundation under Grant No. 110564. We thank Dr. Amit Misra and Dr. Christopher Taylor from LANL for useful comments on this manuscript. This research used resources provided by the LANL Institutional Computing Program, which is supported by the U.S. Department of Energy National Nuclear Security Administration under Contract No. DE-AC52-06NA25396. Further, some of the initial sets of calculations were also performed using Environmental Molecular Sciences Laboratory, a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
Author information
Authors and Affiliations
Contributions
S.C. performed all the electronic structure calculations. B.P.U. set up the initial supercell structure. S.C. and B.P.U. analyzed the data. D.M. provided inputs in relating oxygen chemical potential and partial pressure of oxygen. S.C. prepared the initial draft of the manuscript, while all the authors have contributed to the discussion of results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Choudhury, S., Morgan, D. & Uberuaga, B. Massive Interfacial Reconstruction at Misfit Dislocations in Metal/Oxide Interfaces. Sci Rep 4, 6533 (2014). https://doi.org/10.1038/srep06533
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06533
This article is cited by
-
Vacancy assisted He-interstitial clustering and their elemental interaction at fcc-bcc semicoherent metallic interface
Scientific Reports (2018)
-
Asymmetric interaction of point defects and heterophase interfaces in ZrN/TaN multilayered nanofilms
Scientific Reports (2017)
-
Epitaxial Fe Thin Films on {100} Y2Ti2O7: Model Interfaces for Nano-Oxide Dispersion Strengthened Steels
Metallurgical and Materials Transactions A (2017)
-
Energetics of vacancy segregation to [100] symmetric tilt grain boundaries in bcc tungsten
Scientific Reports (2016)
-
Structural modifications due to interface chemistry at metal-nitride interfaces
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
