
Abstract
We developed a layer-by-layer method of preparing PbE (E = S or Se) quantum dot (QD) solar cells using metal halide (PbI2, PbCl2, CdI2, or CdCl2) salts dissolved in dimethylformamide to displace oleate surface ligands and form conductive QD solids. The resulting QD solids have a significant reduction in the carbon content compared to films treated with thiols and organic halides. We find that the PbI2 treatment is the most successful in removing alkyl surface ligands and also replaces most surface bound Cl- with I-. The treatment protocol results in PbS QD films exhibiting a deeper work function and band positions than other ligand exchanges reported previously. The method developed here produces solar cells that perform well even at film thicknesses approaching a micron, indicating improved carrier transport in the QD films. We demonstrate QD solar cells based on PbI2 with power conversion efficiencies above 7%.
Similar content being viewed by others


Introduction
Solution-processed photovoltaics (PV) represent a promising route forward in reducing the cost of solar energy production. Quantum dot (QD) solids are one such solution-processed system currently being researched. In addition to being solution processable, QD solar cells (QDSCs) have a higher limiting single junction power conversion efficiency than is possible using conventional bulk or thin film semiconductors due to enhanced multiple exciton generation (MEG) in the QDs1,2. Recent improvements in QDSC performance and processing ease have resulted from modification of the device architecture, processing of the QD-layers under ambient conditions, improved QD synthetic procedures and surface treatments improving QD passivation3,4,5,6. A critical component of the progress listed above is the incorporation of halides into the QD matrix6,7. Specifically, Cl- anions were shown to improve stability while passivating trap states that lower the minority-carrier lifetime3,4,8. Incorporation of these halide anions has been achieved by using chloride precursors in the QD synthesis3, using post-synthesis solution treatments9,10,11, and, recently, employing ammonium halide salts as the only ligand treatment6,12. However, when using the previously reported halide passivation schemes, organic molecules that are instrumental in delivering the halide anion (e.g. tetrabutylammonium iodide (TBAI), 3-chloropropane-1-thiol, methylammonium iodide (MAI), etc.), leave behind cationic organic residue that could potentially limit device performance. In contrast, here, we demonstrate a procedure that removes nearly all of the organic moieties from the QD solid during device fabrication. The groups of Wang, Talapin and Kovalenko have previously reported solution-phase ligand exchanges using halide ligands but have not applied them to solar cells13,14,15. Our procedure is based upon a layer-by-layer approach demonstrated previously but uses metal halides dissolved in dimethlyformamide (DMF) (rather than thiols in acetonitrile or alcohols) to build-up thick, all-inorganic films by either dip coating or spin coating with PbS or PbSe QDs.
Results
The QD synthesis used in this study follows previous reports where PbE (E = S, Se) QDs are made by cation exchange of CdE QDs with PbCl2/oleylamine4. Of the numerous metal halide materials available, we focus on four metal halides solvated in DMF: PbCl2, PbI2, CdCl2 and CdI2. The chosen metal halides introduce ions (Cl- or I-) that have demonstrated passivation of QD trap states. Metal halides have not previously been used as the sole ligand treatment for QDSCs but rather as a pre- or post-treatment in conjunction with short-chained alkyl thiol ligands. Previous reports indicate soaking QD solids in neat DMF displaces the native oleate ligands derived from oleic acid (OA) and leads to oriented attachment along the (100) facets of the PbE QDs16. Here, we find that the metal-halide:DMF treatment removes Pb-oleate from the QDs while incorporating the metal halide into the film as is discussed below.
Dip coating QDs allows for a controlled thickness of a compact film with appropriate surface coverage17. In Fig. 1A, we show the increase in absorption of PbS QD films with increasing number of deposition cycles while preserving the first exciton feature originating from the individual QD size. In Fig. 1B, we show baseline-corrected Fourier-Transform Infrared (FTIR) spectra of dropcast films capped with the native oleate ligand (black lines) and the corresponding spectra after ligand treatment with various metal halide salts in DMF (red lines). Based on the ratios of the largest absorbance feature at 2925 cm−1 (corresponding to the νa (–CH2) mode), the iodide salts remove more Pb-oleate than their respective chloride salts (i.e. CdI2 removes more than CdCl2) and the lead salts remove more than the cadmium salts (i.e PbI2 is more effective than CdI2). This trend is deduced using a ratio of the absorbance at 2925 cm−1 i.e. [post-soak]/[pre-metal halide soak]; 26% oleate remains after treatment with CdCl2, 14% after CdI2 treatment, 5.1% after PbCl2 treatment and 1.4% after PbI2 treatment. However, some of the residual organics from DMF are still present after rinsing, as indicated by the peak near 1640 cm−1. The generality of the concept is shown by using each of the compounds in Fig. 1B, but focusing on PbI2 in DMF as a treatment to prepare QD solar cells since it is most effective at removing the oleate.

A) Absorption spectra calculated by measuring the transmission and reflection spectra of dip coated PbS QD films on glass as a function of the number of dip cycles using PbI2. Inset: photograph of PbS QD films with variable thickness controlled by the number of dip cycles given in the legend. B) Fourier-Transform Infrared (FTIR) spectra of 1.3 eV bandgap PbS QDs dropcast from hexane (black traces) and then soaked for 2 hours in 10 mM metal halide in N,N-dimethylformamide (DMF) (red traces). The peak at 1640 cm−1 is attributed to residual DMF that can be removed with heating and/or placing the film under vacuum.
To further detail the composition and properties of the QD films treated with PbI2 (PbSPbI2), we examined the atomic concentrations and energy levels using x-ray photoelectron spectroscopy (XPS) and compare to that of other ligand-exchanged QD films. We fabricate films using iodine-containing ligands: TBAI and PbI2, as well as the sulfur-containing ligands: MPA and NH4SCN. Both NH4SCN and MPA have carbon signatures greater than 20%. Comparing QD films treated with TBAI to those treated with PbI2 in Table 1, we find that the percentage of carbon present in the film is greatly reduced (from 26.7% to 2.5%) when using the PbI2 treatment. Interestingly, the MPA and NH4SCN do not displace the Cl present in the QDs (Cl added during the ion exchange reaction via PbCl2/oleylamine4), whereas after treating QD films with TBAI or PbI2, Cl is not detected by XPS. This demonstrates the strong bonding character of iodine to the surface of lead chalcogenide quantum dots and may be responsible for the lessened sensitivity to oxygen in PbS and PbSe devices found here and previously6.
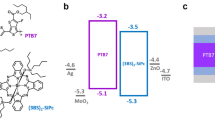
Researchers have demonstrated a link between the stoichiometry in ionic QDs and majority carrier type in QD films18,19. For instance, the Pb:E ratio decreased with the addition of chalcogens from ligands like MPA or NH4SCN resulting in p-type QD films18,19,20. Changes in the stoichiometry would therefore alter the Fermi level position within the bandgap. The XPS spectra can be used to determine the work function (ϕ = difference between Fermi energy and vacuum level) and the onset of emitted electrons from VB states relative to the Fermi energy (EF – EVB onset). Here we find that treating the QDs with MPA or NH4SCN decreases the Pb:S ratio compared to the I- treated films and the separation between the onset of the valence band (VB) states and the Fermi level is also smaller, consistent with previous reports21. In addition, the I- treatments lead to a deeper VB (i.e. larger energy difference between vacuum and the VB onset) than the sulfur-containing ligand treatments as shown in Fig. 2. With total cation:anion ratio equal to unity, the lower Pb:S ratios for the MPA and NH4SCN treatments compared to the I- ligand treatment support the conclusion that the MPA and NH4SCN treatments lead to more p-type films than the I- ligand treatments22.

Summary of photoelectron spectroscopy results of 1.3 eV bandgap PbS QDs with various surface treatments.
The EF - EVB onset and ϕ of PbS QDs changes with PbI2, TBAI, PbCl2, NH4SCN, MPA, CdCl2 and CdI2 surface treatments/ligand exchange.
The XPS results of the VB onset and ϕ show that the ligand can dictate the Fermi level position within the bandgap and can control the overall band positions relative to vacuum21,23,24. Our results with the TBAI, MPA and NH4SCN ligand treatments agree with those of Brown et al.21 and we find (Fig. 2) that the PbI2 ligand exchanged PbS QD film has the lowest lying VB onset and ϕ of all of the ligands studied here and in previous work with PbS QD films. Furthermore, treating the PbS QDs with CdCl2 or CdI2 yields a shallower valence band onset. Control over both the band positions and majority carrier type within QD solids enables deliberate engineering of the energetics within a device.
We then fabricated PbE QDSCs with the structure shown in Fig. 3A. The processing details and characteristics of devices made for this study are summarized in Table 2. The devices are fabricated in air using a layer-by-layer coating process. Both dip coating and spin coating yield nominally the same results with details given in Table 2 and the Methods section. Both of these deposition protocols allow for conformal films with well-controlled thickness. As mentioned above, a unique feature of QD solids is the ability to control the absolute energy levels by applying different ligands. This effect has been attributed to ligand-induced surface dipoles21. Such control allows the energetics within a device to be engineered by using multiple surface treatments during the QD deposition to create bilayer (or in principle, more complex) structures of QD solids. For example, Semonin et al. demonstrated increased performance in PbSe solar cells by stacking ethanedithiol- (EDT) and hydrazine-treated layers1. Other combinations using TBAI and EDT or tetramethylammoinium hydroxide have been used to enhance carrier collection resulting in improved device performance6,25. In Fig. 3B, we compare devices with only a PbSPbI2 layer to those with bilayer structures where the PbSPbI2 layer is followed by either PbSMPA or PbSNH4SCN layers and find that the bilayer structure can greatly improve the current density-voltage (J-V) characteristics of the device. Although PbS QDs treated with the inorganic SCN- ligand have been reported to be more conductive in QD films than the organic MPA ligand26,27, we find that the QD devices presented here function more efficiently with PbSPbI2/PbSMPA than PbSPbI2/PbSNH4SCN.

A) Schematic representation of the device structure superimposed on a false-color scanning electron microscope image for a completed PbSPbI2/PbSMPA device. Scale bar is 500 nm. B) Current-voltage characteristics using only a PbI2 treatment shows low FF (blue trace) but using a secondary layer treated with MPA (red trace) and with the inorganic SCN- ligand (gold trace) aids in band alignment yielding improved FF and PCE. C) Current-voltage characteristics of devices incorporating the four metal halides discussed above are shown. Using CdCl2 as opposed to PbI2 improves the VOC to over 615 mV. D) External quantum efficiency (EQE) curves for PbS QDSCs with PbI2 and CdCl2 ligand treatments (PbSMPA is the back layer as shown in panel A). E) Current-voltage characteristics of air-fabricated PbSePbI2 QDSC. The inset shows the external quantum efficiency for the device.
Adopting the PbSPbI2/PbSMPA bilayer structure, we then compare PV devices using each of the metal halides discussed. Figure 3A shows a scanning electron microscopy (SEM) image of a completed PbSPbI2/PbSMPA device indicating highly uniform QD deposition throughout the device. The difference in contrast shown in the SEM for the PbI2- vs. MPA-capped QDs indicates that the layers remain distinct with likely different material density or perhaps conductivity. While we have optimized the device fabrication conditions for the PbI2 treatment, we note that each of the metal halides results in functioning devices and each affect the PV performance in unique ways. For example, in Fig. 3C, we show that devices fabricated using CdCl2 have an improved open circuit voltage over those fabricated from PbI2-treated QDs and reach a PCE of 5.6%. The spectral response of a CdCl2-treated device (Fig. 3D) exhibits a 100-nm blue shift in the wavelength of the first exciton feature that is likely due to a surface ion exchange which reduces the size of the PbS core and increases the bandgap28. Metal halide treatments can also be used to fabricate PbSe QDSCs under ambient conditions (PbSe is generally more prone to oxidation than PbS). The J-V characteristics of a 1.3 eV bandgap PbSe QDs device are shown in Fig. 3E with the inset showing the external quantum efficiency (EQE) of the device with >70% response throughout most of the visible spectrum.
We test the thickness dependence of the PbS absorber layer by producing devices composed of 4, 6, 7, 8 and 10 sequential spin coating steps. After each spin, the film is treated by soaking in 10 mM PbI2 in DMF for 3 minutes. The last two coatings were treated by 10% MPA in methanol rather than PbI2. In Fig. 4A, we plot the open-circuit voltage (VOC), short-circuit current (JSC), fill factor (FF) and PCE as a function of the total QD layer thickness. The solid symbols represent the average of 6 devices and the hollow symbols represent the champion device for each film thickness. Current-voltage characteristics of the devices are shown in Fig. 4B with the best device reaching a PCE of 7.25% which corresponds to a thickness of 500 nm. Figure 4C shows the EQE response of the devices as a function of thickness and indicates a general trend of increasing spectral response for lower energy photons (i.e. photons with wavelength between 600 and 1200 nm). We also determined that the internal quantum efficiency (IQE) increases in this same manner with the thickest cell showing a flat response of 80–85% (Fig. 4D). Ideally, the IQE should be independent of the cell thickness unless there is high carrier recombination at the back interface. For the device with a PbS QD thickness of 740 nm, the IQE is roughly 80% for all photons absorbed in the QD layer (i.e. photon energy above the bandgap of the PbS QDs and below the absorption of the glass/ITO substrate). Electron transport is sufficient to extract 80% of carriers generated in the device, indicated by the IQE and flat spectrum, despite being significantly thicker than the highest efficiency reported PbS QDSC6. The PbI2 treatment described here is, therefore, very promising for improving the overall efficiency of QDSCs as the JSC only begins to drop as the film thickness approaches 740 nm.

A) VOC, JSC, FF and PCE plotted as a function of device thickness for spincoated PbI2-treated devices. Solid symbols represent the average of 6 devices and hollow symbols are the best devices. B) J-V curves of cells shown in panel A with the best cell reaching a PCE of 7.25% with 500 nm thickness of the QD layer. C) EQE response of cells showing improved response of longer wavelength light as the cell absorbs more light. D) Internal quantum efficiency (IQE) determined by dividing the EQE by the absorption. The color coding is consistent in panels B-D and annotated in the legend of panel B.
To conclude, we present metal halide treated films of PbE yielding high efficiency devices. This inorganic ligand treatment allows for relatively thick films (~600 nm) to be incorporated into devices while still maintaining good transport (i.e. high current) in the device. The XPS results highlight the control over the PbS QD absorber layer by choice of ligand. We have shown with XPS that different chemical treatments affect the QD surface and subsequently, how these surface treatments directly control the energy levels of the QD absorber layer. Additionally, XPS and FTIR analysis confirmed that the metal halide exchange lessens the residual organic elements in the film. Furthermore, using PbS QDs as the low bandgap cell in tandem configurations where better collection of the near infrared photons is needed is now more feasible as collection efficiency throughout nearly the entire spectrum exceeds 50% with an absorber thickness >700 nm.
Methods
The QDs were synthesized following a previously published procedure4. For PbSe, CdSe was first synthesized following a modified version of the procedure published by Pu et al.29 to obtain ~5 nm, monodisperse CdSe. The CdSe was then chemically converted to PbSe through a cation exchange reaction by mixing 0.834 g PbCl2 with 10 mL oleylamine (OLA), degassing and heating to 140°C for 30 min. The mixture was then heated to 190°C and 2 mL of CdSe (100 mg/mL, in ODE) is injected. The reaction was left at 180°C for 30 seconds then quenched with a water bath. As the reaction cools, 10 mL hexane and 8 mL OA are injected at 70°C and 30°C, respectively. The reaction was allowed to cool and the QDs were washed by precipitation-redispersion with ethanol and hexane. The final dispersion was centrifuged to remove any excess chloride salts and filtered through a 0.2 μm Nylon filter.
PbS was synthesized by the cation exchange of CdS. CdS was synthesized following the procedure published by Zhang et al.30. The cation exchange follows that of the CdSe, except the precursors are cooled to 90°C before the injection of CdS (150 mg/mL in toluene) and the reaction runs for 60 seconds. The product was washed and filtered in the same way as described above.
The FTIR absorbance measurements were taken on a Thermo-Nicolet 6700 FT-IR spectrometer in transmission mode with a resolution of 4 cm−1. Clean Si plates were used for background measurements and films of OA-capped QDs were drop cast onto the Si plates for the oleate-capped measurements. The samples were then submerged in 10 mM metal halide in DMF solutions for 2 hours and rinsed with acetonitrile. These metal halide treated samples were then measured and spectra with sloping baselines were baseline-corrected.
The XPS measurements were performed on a Physical Electronics, Inc. 5600 ESCA instrument, which has been discussed in detail previously31. Briefly, the radiation is produced by a monochromatic Al (Kα) source centered at 1486.6 eV. The VB spectra were taken with a step size of 0.05 eV and a pass energy of 5.85 eV. The electron binding energy scale was calibrated using the Fermi edge of cleaned metallic substrates (Au, Mo, Cu, and/or Ag), giving the spectra an uncertainty of ±0.05 eV. We verify that charging during the photoemission experiments is insignificant by measuring the X-ray power dependence of various spectral features (core levels, VBMs, and/or secondary electron cutoffs). We find the VB onset by determining the intersection between the baseline and a linear fit to the main VB feature32.
Solar cell fabrication consisted of dip coating or spin-coating on patterned ITO-coated glass slides from Thin Film Devices where we first deposited a TiO2 layer with a sol-gel method. TiO2 sol-gel was prepared in air by mixing 5 mL anhydrous ethanol, 2 drops hydrochloric acid and 125 μL DI water. This mixture was stirred while 375 μL titanium ethoxide is added drop-wise to ensure that no precipitates form. This yielded a clear liquid that was stirred for 48 hours with the headspace of the vial filled with nitrogen. It was then stored in a freezer until needed. The ITO/glass substrates were cleaned vigorously with ethanol and UV-ozone treated before depositing TiO2. Within 10 min of UV-ozone treatment, 70 μL TiO2 sol-gel was spun at 1400 RPM for 30 sec. The TiO2 is wiped off the ITO contact pads using ethanol and the films are dried at 115°C then annealed at 450°C for 30 min. The films are stored in air and sit in air for at least 1 day before use. For dip coating, immersing the substrates into a ~15 mg/mL solution of QDs in hexane and smoothly removing them leaves a thin film of QDs as discussed previously17. Dipping this film into the 10 mM metal halide/DMF solution for 30–60 seconds renders the QD layer insoluble in hexane and allows for thick films to be built up layer-by-layer (where the term “layer” does not imply a monolayer of QDs, but rather one coating of QDs). A post-ligand treatment with neat acetonitrile (ACN) was necessary to remove the DMF because the residual DMF does not dry rapidly. It should be noted that the metal halides discussed here are not appreciably soluble in ACN making ACN a poor choice of solvent for the ligand exchange. A mixture of 20 vol.% DMF/ACN solvated the PbI2 and devices made in this fashion performed nearly as well as those with PbI2 in DMF for the ligand treatment solvent (Table 2). Typical dip coated devices used 10–15 layers of PbI2-treated QDs followed by 3–4 layers treated with 10% MPA in methanol (MeOH) or alternatively a 10 mM solution in MeOH was used for the NH4SCN treatment. Spin coated devices used a variable number of layers for the PbI2-treatment as discussed in the manuscript with 2 layers of MPA-capped QDs in each case. The QDs were dispersed in octane at a concentration of 40 mg/mL and spun at 1000 rpm for 45 s before being immersed in 10 mM metal halide solution for 3 minutes and rinsed with ACN. The last 2 cycles of QDs were treated with 10% MPA in MeOH by dipping the device into a MPA/MeOH solution, rinsing twice with MeOH and drying with nitrogen. All devices presented here were fabricated at room temperature (~23.9–26.7°C) and relative humidity that fluctuates between 16–20%. A MoOx/Al back contact was then thermally evaporated as discussed by Gao et al.33.
Device testing was carried out using Newport solar simulators adjusted by measuring a calibrated Si photodiode reference to match the AM1.5 spectrum. Some devices were tested in glovebox atmosphere while others were tested in air; details annotated in Table 2. Device area is 0.11 cm2 but an aperture of 0.059 cm2 was used to define the active illuminated area. Spectral response measurements were performed on an Oriel IQE-200 system.
Additional information
Competing financial interests: The authors declare no competing financial interests.
References
Semonin, O. E. et al. Peak external photocurrent quantum efficiency exceeding 100% via MEG in a quantum dot solar cell. Science 334, 1530–1533 (2011).
Semonin, O. E., Luther, J. M. & Beard, M. C. Quantum dots for next-generation photovoltaics. Mater. Today 15, 508–515 (2012).
Zhang, J., Gao, J., Miller, E. M., Luther, J. M. & Beard, M. C. Diffusion-controlled synthesis of PbS and PbSe quantum dots with in situ halide passivation for quantum dot solar cells. ACS Nano 8, 614–622 (2013).
Zhang, J. et al. PbSe quantum dot solar cells with more than 6% efficiency fabricated in ambient atmosphere. Nano Lett. 14, 6010–6015 (2014).
Kramer, I. J. & Sargent, E. H. The architecture of colloidal quantum dot solar cells: Materials to devices. Chem. Rev. 114, 863–882 (2013).
Chuang, C.-H. M., Brown, P. R., Bulović, V. & Bawendi, M. G. Improved performance and stability in quantum dot solar cells through band alignment engineering. Nat. Mater. 13, 796–801 (2014).
Tang, J. et al. Colloidal-quantum-dot photovoltaics using atomic-ligand passivation. Nat. Mater. 10, 765–771 (2011).
Bae, W. K. et al. Highly effective surface passivation of PbSe quantum dots through reaction with molecular chlorine. J. Am. Chem. Soc. 134, 20160–20168 (2012).
Zhitomirsky, D. et al. Engineering colloidal quantum dot solids within and beyond the mobility-invariant regime. Nat. Commun. 5, 3803 (2014).
Ip, A. H. et al. Hybrid passivated colloidal quantum dot solids. Nat. Nanotechnol. 7, 577–582 (2012).
Crisp, R. W. et al. Nanocrystal grain growth and device architectures for high-efficiency CdTe ink-based photovoltaics. ACS Nano 8, 9063–9072 (2014).
Ning, Z., Dong, H., Zhang, Q., Voznyy, O. & Sargent, E. H. Solar cells based on inks of n-type colloidal quantum dots. ACS Nano 8, 10321–10327 (2014).
Niu, G. et al. Inorganic halogen ligands in quantum dots: I-, Br-, Cl- and film fabrication through electrophoretic deposition. PCCP. 15, 19595–19600 (2013).
Dirin, D. N. et al. Lead halide perovskites and other metal halide complexes as inorganic capping ligands for colloidal nanocrystals. J. Am. Chem. Soc. 136, 6550–6553 (2014).
Zhang, H., Jang, J., Liu, W. & Talapin, D. V. Colloidal nanocrystals with inorganic halide, pseudohalide and halometallate ligands. ACS Nano 8, 7359–7369 (2014).
Baumgardner, W. J., Whitham, K. & Hanrath, T. Confined-but-connected quantum solids via controlled ligand displacement. Nano Lett. 13, 3225–3231 (2013).
Luther, J. M. et al. Structural, optical and electrical properties of self-assembled films of PbSe nanocrystals treated with 1,2-ethanedithiol. ACS Nano 2, 271–280 (2008).
Luther, J. M. & Pietryga, J. M. Stoichiometry control in quantum dots: A viable analog to impurity doping of bulk materials. ACS Nano 7, 1845–1849 (2013).
Oh, S. J. et al. Designing high-performance PbS and PbSe nanocrystal electronic devices through stepwise, post-synthesis, colloidal atomic layer deposition. Nano Lett. 14, 1559–1566 (2014).
Oh, S. J. et al. Stoichiometric control of lead chalcogenide nanocrystal solids to enhance their electronic and optoelectronic device performance. ACS Nano 7, 2413–2421 (2013).
Brown, P. R. et al. Energy level modification in lead sulfide quantum dot thin films through ligand exchange. ACS Nano 8, 5863–5872 (2014).
Choi, H., Ko, J.-H., Kim, Y.-H. & Jeong, S. Steric-hindrance-driven shape transition in PbS quantum dots: Understanding size-dependent stability. J. Am. Chem. Soc. 135, 5278–5281 (2013).
Jasieniak, J., Califano, M. & Watkins, S. E. Size-dependent valence and conduction band-edge energies of semiconductor nanocrystals. ACS Nano 5, 5888–5902 (2011).
Munro, A. M., Zacher, B., Graham, A. & Armstrong, N. R. Photoemission spectroscopy of tethered CdSe nanocrystals: Shifts in ionization potential and local vacuum level as a function of nanocrystal capping ligand. ACS Appl. Mater. Interfaces 2, 863–869 (2010).
Zhitomirsky, D. et al. N-type colloidal-quantum-dot solids for photovoltaics. Adv. Mater. 24, 6181–6185 (2012).
Fafarman, A. T. et al. Thiocyanate-capped nanocrystal colloids: Vibrational reporter of surface chemistry and solution-based route to enhanced coupling in nanocrystal solids. J. Am. Chem. Soc. 133, 15753–15761 (2011).
Jeong, K. S. et al. Enhanced mobility-lifetime products in PbS colloidal quantum dot photovoltaics. ACS Nano 6, 89–99 (2011).
Pietryga, J. M. et al. Utilizing the lability of lead selenide to produce heterostructured nanocrystals with bright, stable infrared emission. J. Am. Chem. Soc. 130, 4879–4885 (2008).
Pu, C. et al. Highly reactive, flexible yet green Se precursor for metal selenide nanocrystals: Se-octadecene suspension (Se-Sus). Nano Res. 6, 652–670 (2013).
Zhang, H., Hyun, B.-R., Wise, F. W. & Robinson, R. D. A generic method for rational scalable synthesis of monodisperse metal sulfide nanocrystals. Nano Lett. 12, 5856–5860 (2012).
Perkins, C. L. & Hasoon, F. S. Surfactant-assisted growth of CdS thin films for photovoltaic applications. J. Vac. Sci. Technol. A. 24, 497–504 (2006).
Kraut, E. A., Grant, R. W., Waldrop, J. R. & Kowalczyk, S. P. Semiconductor core-level to valence-band maximum binding-energy differences: Precise determination by x-ray photoelectron spectroscopy. Phys. Rev. B. 28, 1965–1977 (1983).
Gao, J. et al. N-type transition metal oxide as a hole extraction layer in PbS quantum dot solar cells. Nano Lett. 11, 3263–3266 (2011).
Acknowledgements
The development of the quantum dot synthesis and characterization in this work are supported by the U.S. Department of Energy Office of Science, Office of Basic Energy Sciences Energy Frontier Research Centers program within the Center for Advanced Solar Photophysics through contract number DE-AC36-08GO28308. The device work presented here is supported by the U.S. Department of Energy (DOE) SunShot program under Award No. DE-EE0005312. EMM acknowledges funding through the NREL Directors Fellowship program.
Author information
Authors and Affiliations
Contributions
R.C. developed the PbI2 ligand exchange, synthesized PbS QDs, fabricated and characterized PbS devices and materials. D.K. performed and analyzed the FTIR experiments. A.M. synthesized PbSe QDs, fabricated and measured PbSe devices. E.M. performed and analyzed photoelectron experiments. J.Z. optimized the QD synthesis. M.B. and J.L. conceived the experiments, analyzed the results and oversaw the project. All authors aided in preparing and editing the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Crisp, R., Kroupa, D., Marshall, A. et al. Metal Halide Solid-State Surface Treatment for High Efficiency PbS and PbSe QD Solar Cells. Sci Rep 5, 9945 (2015). https://doi.org/10.1038/srep09945
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09945
This article is cited by
-
Dry Transfer Printed Hole Transport Layer for Hysteresis-Free Colloidal Quantum Dot Solar Cells
International Journal of Precision Engineering and Manufacturing-Green Technology (2024)
-
How to get high-efficiency lead chalcogenide quantum dot solar cells?
Science China Physics, Mechanics & Astronomy (2023)
-
Solution-processable integrated CMOS circuits based on colloidal CuInSe2 quantum dots
Nature Communications (2020)
-
High efficiency perovskite quantum dot solar cells with charge separating heterostructure
Nature Communications (2019)
-
Tuning colloidal quantum dot band edge positions through solution-phase surface chemistry modification
Nature Communications (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
