
Abstract
Cerium oxide nanomaterials (nanoceria, CNMs) are receiving increased attention from the research community due to their unique chemical properties, most prominent of which is their ability to alternate between the Ce3+ and Ce4+ oxidation states. While many analytical techniques and methods have been employed to characterize the amounts of Ce3+ and Ce4+ present (Ce3+/Ce4+ ratio) within nanoceria materials, to-date no studies have used multiple complementary analytical tools (orthogonal analysis) with technique-independent oxidation state controls for quantitative determinations of the Ce3+/Ce4+ ratio. Here, we describe the development of analytical methods measuring the oxidation states of nanoceria analytes using technique-independent Ce3+ (CeAlO3:Ge) and Ce4+ (CeO2) control materials, with a particular focus on x-ray photoelectron spectroscopy (XPS) and electron energy loss spectroscopy (EELS) approaches. The developed methods were demonstrated in characterizing a suite of commercial nanoceria products, where the two techniques (XPS and EELS) were found to be in good agreement with respect to Ce3+/Ce4+ ratio. Potential sources of artifacts and discrepancies in the measurement results were also identified and discussed, alongside suggestions for interpreting oxidation state results using the different analytical techniques. The results should be applicable towards producing more consistent and reproducible oxidation state analyses of nanoceria materials.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Cerium oxide (CeO2−x, ceria) nanomaterials (nanoceria, CNMs) are receiving increased attention due to their current and potential use in a vast number of applications, such as chemical mechanical polishing/planarization (CMP) processes, industrial and automotive catalysis, electrochemical devices, agricultural products and medicinal treatments [1–4]. While the performance of nanoceria in these applications depends on many physicochemical properties (e.g. size, shape, surface chemistry), the ability of nanoceria to exist in and cycle between the Ce3+ and Ce4+ oxidation states has been proposed as the key feature behind their unique activity [5–8]. Previous research has suggested that the ratios of these two oxidation states (which is dependent on several factors including their intrinsic physicochemical properties, extrinsic defects or impurities, and surrounding environment) within nanoceria determines the extent of their beneficial properties, such as the ability to function as an oxygen donor for more efficient fuel combustion [9, 10] or reactive oxygen species scavenger for antioxidative therapies [11], but also their potential detrimental effects on biological [12] and environmental systems [13]. As such, more accurate determinations of the Ce3+/Ce4+ ratios could expedite our understanding of nanoceria properties and interactions.
Several analytical techniques have been used to gain insight into the Ce3+/Ce4+ ratios of nanoceria materials, including x-ray photoelectron spectroscopy (XPS) [14–18], electron energy loss spectroscopy (EELS) [19–23], x-ray absorption spectroscopy [24–26], Raman spectroscopy [27–29], and ultraviolet/visible/infrared spectroscopy [30–33]; of these, XPS and EELS are perhaps the most widely used characterization tools [34–37]. However, each of these techniques operates under different fundamental principles [37] with inherent variances and can produce different results on the Ce3+/Ce4+ ratios of nanoceria analytes. These technique-sensitive variances and the lack of technique-independent controls, complicates the ability to compare analytical data generated by different techniques, even in the few cases where multiple techniques were employed in a single study [37, 38]. New approaches in determining the oxidation states of nanoceria, using multiple analytical techniques and technique-independent controls, could help generate more accurate and reproducible measurements of the Ce3+/Ce4+ ratio, thus facilitating a greater understanding of nanoceria properties and interactions [13, 36, 37].
In this work, we describe the development of analysis methods for XPS and EELS using technique-independent Ce3+ (CeAlO3:Ge) and Ce4+ (CeO2) control materials towards quantitatively measuring the oxidation states of nanoceria materials. We discuss the advantages and disadvantages of the techniques themselves, along with different approaches of analysis within each technique. The developed methods were then applied in quantifying the oxidation states of readily available, commercial nanoceria materials, followed by comparison of the results generated by each technique. While our results show that the Ce3+/Ce4+ ratios determined by XPS and EELS are in general agreement, we identify and discuss potential sources of discrepancies between the measurement results, along with recommendations for obtaining and comparing oxidation state results across different analytical techniques.
Experimental
Materials
Bulk cerium (IV) oxide (CeO2, 99.995% Ce) was purchased from Strem Chemicals, Inc. (Newburyport, MA). Cerium carbonate hydrate (Ce2(CO3)3·xH2O, 99.999%) was purchased from Alfa-Aesar (Haverhill, MA). Germanium oxide (GeO2, 99.999%) was purchased from A.D. Mackay, Inc. (Denver, NC). Aluminum oxide (Al2O3, 99.99%) was purchased from Johnson and Matthey (Royston, UK). Four commercial nanoceria products were obtained from four different vendors: a nanopowder comprised of vendor specified 5 nm primary particles, a nanopowder comprised of vendor specified 10 nm primary particles, a nanopowder comprised of vendor specified 25 nm primary particles, and a CMP slurry (primary particle size not specified); henceforth denoted as CNP-5, CNP-10, CNP-25, and CNP-C respectively. Ethanol (EtOH, 200 proof) was purchased from The Warner-Graham Company (Cockeysville, MD). All other materials were used as received without further purification.
Preparation of CeAlO3 and CeAl0.98Ge0.02O3 (CeAlO3:Ge) for use as Ce3+ controls
CeAlO3 and CeAlO3:Ge samples were prepared using traditional mixed oxide synthesis techniques by mixing stoichiometric amounts of Al2O3, Ce2(CO3)3·xH2O, and GeO2 (for the CeAlO3:Ge sample only) in EtOH using zirconia milling media on a vibratory mill for 20 min. The powders were dried and calcined in air at 1200 °C for 1 h in zirconia crucibles to burn-off the carbonate species. The powder was re-ground on a vibratory mill in EtOH with zirconia milling media for 20 min. The powder was dried and mixed with ≈1 mass % organic binder prior to pelletizing using a uniaxial press and steel die. The organic binder was burned out by heating the pellets to 600 °C in air. The pellets were sintered on a platinum foil in a tube furnace for 6 h at 1400 °C at a pO2 of 1.5 × 10−13 MPa achieved by flowing a dry N2/H2 gas mixture. The resultant powders were confirmed to be phase pure by x-ray diffraction (XRD); there were no peaks associated with the precursor phases of CeO2 or Al2O3, and all of the peaks could be assigned to a single phase consistent with the tetragonal I4/mcm perovskite phase of CeAlO3 [39].
X-ray diffraction (XRD)
CNP-C (a CMP slurry) was allowed to dry in a vacuum desiccator until a dry powder was obtained. Resulting powders were ground using an agate mortar and pestle. XRD data were collected using a Panalytical (Almedo, The Netherlands) XPertPro diffractometer utilizing CuKα1 radiation of 1.540 60 Å, 45 kV, 40 mA, with a 0.25° divergence slit from 20° to 130° (2θ) in steps of 0.013°.
Transmission electron microscopy (TEM)
TEM images were taken on a FEI (Hillsboro, OR, USA) Titan 80–300 analytical TEM operated at an accelerating voltage of 300 kV in bright-field mode using a Gatan (Pleasanton, CA) Orius digital camera. Samples were prepared for TEM imaging by suspending the nanoceria powders in EtOH and drop-casting the respective suspensions onto carbon-coated copper TEM grids (Ted Pella Inc., Redding, CA). Particle size analysis was manually performed using Fiji [40] (a distribution of ImageJ [41]) and counting a minimum of 165 particles for each sample.
Scanning transmission electron microscopy (STEM) imaging and EELS
STEM images and EELS data were collected using a probe-corrected FEI (Hillsboro, OR) Titan transmission electron microscope. The instrument was operated at an accelerating voltage of 300 kV and the beam current was approximately 25 pA. For imaging and EELS, the convergence semi-angles were approximately 13.5 mrad and 3.6 mrad, respectively. High-angle annular dark field (HAADF) and low-angle annular dark field (LAADF) images were acquired using a Fischione Model 3000 detector with inner convergence semi-angles of approximately 88 mrad and 28 mrad, respectively. The EELS collection semi-angle was approximately 13.7 mrad and the dispersion was 0.05 eV/ch. A total of 6 to 7 data sets were acquired from different areas in each sample. During the spectral acquisition the beam was rastered over an area of 0.026 μm2. To ensure a precise energy-loss scale the drift tube was iteratively switched between the zero-loss region and the core-loss region as a series of spectra were recorded. During post-processing, spectra were then aligned relative to the zero-loss peak and summed together. Additionally, the drift tube offset was incrementally changed to minimize systematic noise from the charge-coupled device. This process was implemented through a custom script in Digital Micrograph and these concepts have been discussed in-depth elsewhere [42, 43].
CeAlO3:Ge, and bulk CeO2 were used as controls to provide the characteristic Ce3+ and Ce4+ spectra. All samples were prepared by physical deposition of the dry powders onto lacey carbon-coated copper TEM grids (Ted Pella Inc., Redding, CA). Least squares fitting, as employed in EELSMODEL [44], of the control spectra to the commercial samples was used to quantify the EELS data. The mean value is reported along with the standard deviation and number of spectra analyzed. To reduce the effect of differences caused by sample thickness, which can influence the quantitative results of oxidation state [42], the Fourier-ratio deconvolution, as implemented in Digital Micrograph Software (Gatan Inc., Pleasanton, CA), was applied. Backgrounds were removed and deconvolution routines applied to all spectra prior to fitting.
X-ray photoelectron spectroscopy (XPS)
XPS was employed to characterize the distribution of the surface chemical states of cerium, specifically Ce3+ versus Ce4+. To that end, XPS characterization was performed on all 4 commercial ceria containing products and the 2 control powders. The controls were necessary due to the complex nature of the Ce 3d spectra and the propensity for oxidation from Ce2O3 to CeO2. Bulk CeO2 and CeAlO3:Ge were employed as controls to generate representative spectra for Ce4+ and Ce3+, respectively. These powders were both pelleted prior to acquisition of spectra and the CeAlO3:Ge sample was also mechanically polished. The four commercial samples were analyzed in the form they were sold in, specifically as a powder pressed into copper tape (CNP-5, CNP-10, CNP-25) or as a drop cast suspension placed onto a silicon wafer (CNP-C).
XP spectra were acquired on an Axis Ultra DLD XPS system from Kratos Analytical (Manchester, UK) which was maintained at ultra-high vacuum conditions (base pressure of 3 × 10−7 Pa). Spectra were generated using monochromated Al Kα x-rays to achieve photoemission of core level electrons which were acquired along the sample surface normal with 90% of the photoelectrons collected over a 0.94 mm × 2.25 mm area as determined in previous studies [45]. Due to the insulating nature of the various cerium oxide materials, the surface of the samples were neutralized using low energy electrons to compensate for surface charging. Spectra were acquired at 160 eV pass energy with a step size of 1.0 eV for the wide survey spectra and at 40 eV pass energy with a step size of 0.1 eV for the higher resolution elemental regions. For the CeAlO3:Ge control, the use of the ion gun was also necessary for milling the surface with 4 kV Ar+.
While spectra were analyzed for the Ce 3d, O 1s, and C 1s regions for all samples studied with the addition of the Al 2p and Ge 2p region for the CeAlO3:Ge control results, only the results based on the Ce 3d spectra will be presented. The acquired spectra were processed using the commercially available CasaXPS software (Teignmouth, UK). All spectra were energy corrected by shifting the C(1s) peak maximum binding energy (BE) to 284.6 eV. All Ce 3d spectra were fit with U2 Tougaard background with the second parameter in the cross-section field adjusted so the background intersected the noise between the Ce 3d5/2 and Ce 3d3/2 shakedown features for the Ce3+ spectra and between the Ce 3d5/2 and Ce 3d3/2 shakeup features for the Ce4+ spectra [46]. For further discussion on methodologies for semi-quantitative assessment and corresponding oxidation state distribution associated with the different nanoceria samples based on the controls can be found in the 'Approaches for XPS Analysis of Cerium Oxidation State' section.
Results and discussion
Preparation of CeAlO3 and CeAlO3:Ge for use as Ce3+ controls
The development of our analysis methodology requires stable Ce3+ and Ce4+ materials for use as controls. While bulk CeO2 powder is almost universally used as a Ce4+ control, there is no single Ce3+ control that has been used universally by the research community. As such, one of the goals of this work was to develop a technique-independent Ce3+ control material that could be used across multiple analytical platforms. Towards this end, Ce2O3 would be an ideal Ce3+ control for the orthogonal analysis of oxidation state in nanoceria, yet the preparation of pure Ce2O3 is non-trivial and its air-sensitivity makes it challenging to handle across multiple instruments without complicated and/or cost-prohibitive arrangements [47, 48]. As such, both CeAlO3 and CeAlO3:Ge were identified as potential candidates for use as a Ce3+ control sample due to their stability under ambient conditions, which would enable easier use by a wider research community.
Due to the redox-active nature of the cerium ion and its tendency to oxidize from Ce3+ to Ce4+ when exposed to air, cerium was incorporated into a host lattice to extrinsically reduce the ion to the lower valent state. CeAlO3 was synthesized because it has been shown to successfully stabilize the Ce3+ valence state in the perovskite structure [49]. In order to compositionally design a Ce3+ control in which the Ce3+/Ce4+ ratio is maximized, the CeAlO3 host was doped with a fixed valence acceptor ion Ge4+ on the Al3+ B-site. This type of doping results in a donor type of substitution in an attempt to suppress the concentration of Ce4+.
XRD patterns of the CeAlO3 and CeAlO3:Ge powders are shown in figure 1. The two patterns are essentially identical and also match previous measurements from the literature, illustrating that the two samples are phase pure [39]. XPS analysis revealed CeAlO3:Ge to be more consistent (with respect to peak position, peak shape, and peak intensity) with previous measurements of Ce2O3 samples [46, 50, 51], hence it was chosen over CeAlO3 as the Ce3+ control sample for all subsequent analyses (see XPS section for additional details).

Figure 1. XRD patterns of the CeAlO3 (black) and CeAlO3:Ge (orange) Ce3+ control samples. Both patterns are consistent with tetragonal I4/mcm perovskite CeAlO3.
Download figure:
Standard image High-resolution imageXRD and TEM analysis of the commercial nanoceria products
XRD patterns of the bulk CeO2 control sample and the four commercial nanoceria products are shown in figure S1, which is available online at stacks.iop.org/NANO/30/085703/mmedia. Each pattern is consistent with CeO2 in the cubic fluorite structure. Average particle sizes were calculated from XRD data using the Williamson–Hall method [52]. The integral-breadth of each measured peak (βmeas) and the peaks for a LaB6 (βinst) standard were determined using JADE software (Christchurch, NZ). The peaks for the measured ceria samples as well as the LaB6 standard were fit to Lorentzian line shapes. The integral-breadths were used to determine the volume weighted crystallite sizes (D) and strains (ε) using the Williamson–Hall equation [52]:
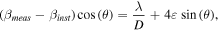
where λ is the incident instrument Cu Kα1 radiation wavelength. The average particle sizes were calculated to be 11.1 ± 0.1 nm, 11.3 ± 0.3 nm, 62.6 ± 1.6 nm, and 61.9 ± 5.8 nm for the commercial nanoceria samples, respectively. On visual inspection of the XRD patterns (figure S1), the large crystallite sizes calculated for CNP-25 and CNP-C are consistent with their respective patterns, which are both quite similar (with respect to peak sharpness) to the bulk CeO2 used as a reference pattern. Likewise, the patterns of CNP-5 and CNP-10 are also quite similar, featuring broader peaks than the other 3 patterns and are hence expected to have smaller calculated crystallite sizes.
We also acquired TEM images of the commercial nanoceria samples and determined the particle sizes as both mean diameters from differential distributions and median diameters from cumulative distributions. Illustrative TEM images of the four commercial nanoceria products, their differential size distributions, and cumulative undersize distributions are shown in figure 2. Additional TEM images are provided in the supplementary information (SI) as figures S2–S5. Each of the samples are comprised of heavily agglomerated particles, which is not unusual given the high surface energy common to most nanoparticle systems (and exacerbated by decreasing particle size) and the lack of a dispersing agent within the prepared nanoceria suspensions (done to analyze the commercial particles in a manner more similar to the other techniques used in this work) [53]. This heavy agglomeration is especially evident for CNP-5 and CNP-10, which feature primary particles of smaller sizes. While initially difficult to see due to the heavy agglomeration, size analyses of higher magnification images (such as those seen in figure S2 for CNP-5 and figure S3 for CNP-10) reveal these two samples to be comprised of small, rounded nanocrystalline particles with mean particle sizes of 4.6 ± 1.0 nm for CNP-5 (n = 302) and 9.4 ± 2.5 nm for CNP-10 (n = 305). Analysis of the cumulative undersize distributions for these particles results in median diameter (d50) values of 4.6 nm and 9.0 nm respectively, in good agreement with the mean diameters. Individual particles are more easily seen in the images of CNP-25 and CNP-C, where the mean particle sizes were determined to be 23.0 ± 10.1 nm for CNP-25 (n = 311) and 69.1 ± 64.0 nm for CNP-C (n = 166). Here, cumulative distribution analysis results in d50 values of 20.5 nm and 48.3 nm respectively, which are both lower than the respective mean diameter results. CNP-25 appears to be mostly comprised of polyhedral particles, regardless of size (figure S4), with CNP-C having particles of random shape and a wide range of sizes (figure S5).

Figure 2. Illustrative TEM images and respective size distribution plots of CNP-5 (a), (b), CNP-10 (c), (d), CNP-25 (e), (f), and CNP-C (g), (h). Bin widths are as follows: CNP-5, 0.5 nm; CNP-10, 1.0 nm; CNP-25, 5.0 nm; CNP-C, 20.0 nm.
Download figure:
Standard image High-resolution imageFor CNP-5 and CNP-10, the determined mean sizes are in good agreement with the particle sizes reported by the vendors (except for CNP-C (the CMP slurry), in which the primary particle size was not specified by the vendor). Likewise, the determined median values for the CNP-5 and CNP-10 are also in good agreement with the vendor-reported values. The differential distribution graph for CNP-5 features an even distribution of particle sizes centered around the mean value, while the differential distribution graphs for CNP-10 and CNP-25 feature 'tails' of increased particle size that skew the mean diameters slightly above the median diameters. All three of these samples have similarly shaped cumulative distribution curves. For CNP-C however, the mean diameter is much greater than the median diameter, which is unsurprising given the results shown in the size distribution graphs. As mentioned above, CNP-C is comprised of particles having a wide range of sizes and could be described as bi-modal, which roughly half the counted particles falling below 50 nm in diameter, with the other half ranging from 50 nm to nearly 300 nm.
Given the volume weighted nature of the XRD-based crystallite size calculations, particle sizes calculated in this fashion can sometimes greatly differ compared to other analytical techniques [54], especially number weighted techniques such as TEM [55]. Comparing the TEM results with the XRD data shown here highlights this divergence, which is not surprising given the broad size distributions of the samples. For CNP-C, the large particles comprising the sample essentially skew it towards bulk ceria. As such, sizing calculations are often poor for samples with such large grain sizes [56], which hinders meaningful comparison of the XRD and TEM results for this sample. For CNP-25, the XRD data results in a larger particle size than the TEM results. As seen in the TEM images for CNP-25 (figures 2(c) and S4), there are multiple larger (>50 nm) crystallites within the sample, which will skew the XRD result higher due to the volume weighted nature of the calculation. The XRD and TEM results are expected to be more in agreement for the CNP-5 and CNP-10, given their narrower size distributions. For CNP-10, this was the case, with the XRD and TEM measurements in good agreement. However, for CNP-5, the XRD size calculation is larger than that of the TEM results. Unlike the CNP-25 and CNP-C samples, in this case, the discrepancy is more likely associated with the calculation itself. The peaks in the XRD pattern of CNP-5 (figure 1, purple), especially those at higher 2θ values, are sufficiently broad that the peak fitting method used could be less accurate, which could result in error during the calculation. The calculation method used places greater weighting on the higher 2θ peaks when determining the overall average particle size, so the broadness of these high 2θ peaks could exacerbate these peak-fitting inaccuracies. Given the direct observation of the particles, we chose to refer to the TEM-derived mean diameters as the basis for our analyses throughout the rest of the work.
Approaches for EELS analysis of cerium oxidation state
Ce M4,5 EELS spectra from the 2 controls and 4 commercial nanoceria samples are shown in figure 3. The M edge excitations are due to electron transitions between the 3d and 4f states. The 4f state of a Ce4+ ion is unoccupied, while a Ce3+ ion has a single electron. These differences of occupancy are reflected in the electron loss near edge structure (ELNES) of the EEL spectra. The Ce M4,5 edges of the CeAlO3:Ge spectra (figure 3(a)), when compared to the bulk CeO2 spectra (figure 3(b)), are shifted to lower energies, have different relative peak intensities, as well as different edge shapes (e.g., note the loss of the satellite peaks). This characteristic change in ELNES enables least square fitting routines using chemical signatures of control samples suitable for quantification. The oxidation states are reported in table 1 and indicate that the commercial nanoceria samples have varying Ce3+ content. It is also important to note that the EELS measurements were acquired by rastering the beam over relatively large areas (0.026 μm2) across several locations for each sample. As such, the EELs measurements are spatially averaged from numerous particles, with many sizes and crystal orientations analyzed. CNP-25 and CNP-C are nominally the same as the bulk CeO2 sample, while the smaller particles (CNP-5 and CNP-10) have greater Ce3+ content. Besides ELNES fitting routines, multiple methods are available to quantify oxidation state, one of which, is the white-line ratio method [42].

Figure 3. Ce M4,5 EEL spectra from the CeAlO3:Ge (a), bulk CeO2 (b), CNP-5 (c), CNP-10 (d), CNP-25 (e), and CNP-C (f). Calculated contributions from Ce3+ (blue) and Ce4+ (red) to each of the nanoceria sample spectra are highlighted. The background has been subtracted using a power-law curve fit to the spectra preceding the Ce edge. Note the change in the edge structure between the Ce3+ and Ce4+ standards.
Download figure:
Standard image High-resolution imageTable 1. Quantified EELS data from both quantification methods for each sample listing the Ce3+ content (mean ± standard deviation). N represents the number of data sets that were acquired from different rastered areas in each sample (see the experimental section for more detail).
| % Ce3+ | |||
|---|---|---|---|
| Sample | Fitting | White-line | N |
| CNP-5 | 15.6 ± 2.4 | 10.7 ± 3.6 | 6 |
| CNP-10 | 7.1 ± 1.8 | 3.3 ± 0.6 | 6 |
| CNP-25 | 2.9 ± 1.3 | 1.0 ± 2.4 | 7 |
| CNP-C | 2.1 ± 1.0 | 1.1 ± 1.7 | 6 |
The white-line ratio is commonly implemented to correlate the ratio of integrated intensity for two core loss peaks—for cerium compounds the ratio of the M5 to M4 edges is used—to a formal oxidation state [42, 57, 58]. Here, we subtracted the background using a power-law fit and then applied a Fourier-ratio deconvolution, as implemented in Digital Micrograph, to reduce differences caused by sample thickness. The white-line ratio was then calculated by taking the second derivation of the spectra and integrating under the positive portion of the curve. This procedure was implemented in Digital Micrograph using a publicly available script written by David Mitchell [59]. To correlate the white-line ratio values from the commercial nanoceria materials to a formal oxidation state, linear interpolation using the white-line ratio values from the Ce3+ and Ce4+ controls was applied.
The values calculated from the white-line ratio analysis are also found in table 1. The white-line ratio derived values follow the same qualitative trend as the values determined by least squares fitting but give lower values for all the commercial nanoceria samples. To understand this discrepancy, the white-line ratios of synthetic spectra were measured. The synthetic spectra are based on linear combinations of background removed and Fourier deconvoluted Ce3+ and Ce4+ control sample spectra. The integrated intensity from 875 to 910 eV, the region encompassing the near edge structure of the Ce M4,5 edges, of both the control sample spectra were equalized. The Ce3+ content for each of the simulated spectra was then quantified using the procedure described in the prior paragraph. The results have been plotted in figure 4, where the black line represents the actual Ce3+ content and the red points represent the quantified value of Ce3+ using the white-line ratio method. The method used to correlate white-line ratio provides a nonlinear response. When the Ce3+ content is less than ≈28%, this implementation produces a positive error and when the Ce3+ content is above ≈28%, it produces a negative error. This means that using linear interpolation with the Ce3+ and Ce4+ controls acting as endpoints appears to be an invalid method to correlate the white-ratio values to formal oxidation states. Similar nonlinear behavior has been reported in transition metal oxide systems and accordingly more complex equations are then used to correlate white-line ratio values to formal oxidation state [42, 60]. Such an approach could be applied to the cerium-oxide system when quantifying the white-line ratio. We make two notes on this analysis. First, differences in methods of background subtraction and peak integration will influence the white-line ratio values and it may be possible that certain data processing routines will better allow for linear interpolation between Ce3+ and Ce4+ controls. Second, synthetic data was generated and analyzed, therefore collecting and analyzing experimental data from controls with oxidation states between +3 and +4 would be useful in confirming this finding.

Figure 4. Quantified values of Ce3+ content (red points) from synthetic spectra with differing amounts of Ce3+ content. The white-line ratio method produces results (red points) that deviate from a linear relationship (black line).
Download figure:
Standard image High-resolution imageADF-STEM imaging analysis of cerium oxidation state
ADF-STEM images of the commercial particles are presented in figures S6 and S7, while ADF-STEM images of the control samples are presented in figures S8 and S9 (SI). When a sample is orientated to a zone axis, a condition which supports strong electron channeling of the incident electron probe, the contrast is sensitive to local changes of the crystal lattice (i.e., defects). In ceria it has previously been reported that static displacements introduced upon the reduction of CeO2 to CeO2−x will generate contrast in annular dark field STEM images [61]. An example of this phenomenon with corresponding EELS data is also shown in figure 5. Pairs of LAADF- and HAADF-STEM images act to highlight regions of the samples where the electron scattering distribution has changed due to lattice distortions. Representative pairs of images are shown in figures 5 and 6. Differences in contrast are apparent at the surface terminations, the LAADF intensity increases while the HAADF intensity decreases. These regions are approximately 0.5–1 nm wide. This contrast suggests these surface regions are reduced relative to the surrounding areas (and confirmed through EELS analysis as seen in figure 5). If the reduced regions are in part associated with free surfaces this would indicate why the Ce3+ content tracks inversely to particle diameter.

Figure 5. LAADF—(a) and HAADF-STEM (b) images of CNP-25. EELS spectra from the denoted regions are inset in (a). Regions near the surface (red, green) show greater Ce3+ character compared to bulk CeO2 (purple). Also note the contrast differences of the twin boundary, which runs through the center of the particle, which also shows its having greater Ce3+ character.
Download figure:
Standard image High-resolution image
Figure 6. LAADF—(top row) and HAADF-STEM (middle row) images of CNP-C (a), CNP-25 (b), and CNP-10 (c). Intensity profiles (bottom row) taken from the boxed region, as indicated in (a), illustrate that at exposed surfaces a contrast difference can be noted; the LAADF signal (red dashed line) becomes more intense, conversely, the HAADF (black solid line) becomes less intense. This contrast is due to local displacements of the lattice and can be associated with relatively higher concentration of reduced ceria.
Download figure:
Standard image High-resolution imagePreviously reported STEM-EELS measurements have yielded similar observations that the surface and near surface regions of nanoceria are reduced [21, 23, 62]. The process of using STEM-EELS spectrum imaging to map the spatial distribution of reduced cerium oxide in samples can be quantitative. By comparison, the imaging used here to identify the reduced regions is not quantitative and is susceptible to interpretation challenges. The benefit of this imaging technique compared to STEM-EELS spectrum imaging is a reduction in electron dose and dose rate necessary to acquire data with comparable spatial sampling. This is important because energetic electrons reduce ceria potentially distorting results [19], and judicious control of dose rate can help to limit this artifact [63]. Interpreting the contrast of the LAADF and HAADF images as a proxy for reduction requires care because changes to contrast are not necessarily limited to those introduced by the static displacements associated with the reduction of CeO2. Extended defects can also modulate the scattering distribution. For example, an aggregate of CeO2 nanocrystals, where grain boundaries are present, may introduce additional contrast (i.e., originating from grain boundary strains) into the image thus convoluting the interpretation. Since we are observing the materials in projection, there is also a lower size limit suitable for this type of analysis. In the case when the material with lattice distortions localizes near surfaces, as the particle size decreases, the fraction of distorted and undistorted lattice becomes similar and the propagation and scattering of electrons no longer markedly changes with position. As such, a contrast change would not be observed near the surface relative to the core. Data from CNP-5 is not included in figure 6 because there is no significant change in the angular distribution of scattered electrons between the core and surface of the particle and thus image contrast near surface terminations becomes negligible. Therefore, using contrast in the ADF-STEM images as a proxy to identify regions of reduced CeO2 requires careful consideration and experimental design to ensure that the contrast is interpreted appropriately. More extensive discussion of the ADF-STEM methodology can be found elsewhere in the literature [61, 63].
Approaches for XPS analysis of cerium oxidation state
CeO2 and CeAlO3:Ge control samples were characterized by XPS to further characterize the distribution of surface oxidation states in commercial nanoceria samples with respect to cerium. Control samples were essential for the interpretation of the acquired spectra from commercial sources, which are shown in figure 7. Figure 7(c) presents the data for the Ce 3d region for the Ce4+ chemical state with the characteristic 6 peaks associated with CeO2: two traditional photoelectron peaks (labeled 'a' and 'd'), each with a 'so-called' shakeup (labeled 'c' and 'f') and shakedown (labeled 'b' and 'e') structures built into the line shape due to previously described final state effects associated with charge transfer [64, 65]. Due to the rapid surface oxidation of Ce2O3 to CeO2, we were forced to employ a Ce3+ control with hetero atoms (Al and Ge) specifically employed to inhibit the oxidation of the cerium to Ce4+. To that end, figures 7(a) and (b) presents the Ce 3d spectra for CeAlO3:Ge (the control for the Ce3+ chemical state) before and after argon ion sputtering, respectively. The Ce3+ oxidation state was characterized by four peaks, two traditional photoelectron peaks (labeled 'a' and 'd') and two peaks (labeled 'b' and 'e') also attributed to final state effects [65]. Employing an Ar+ ion beam for 7 min to clean the surface yielded a spectrum that closely resembled previously observed results as demonstrated in figure 7(b) [46, 50, 51]. The clearest difference between the spectra in figures 7(a) and (b) is the peak to peak separation between the dominant photoelectron peak and the additional transition at lower binding energies. Indeed, the peak to peak separation for the Ar+ sputtered control in figure 7(b) was 4.4 eV, which was comparable to literature references of ≈4.2 eV [46] as opposed to the shoulder shifted by 3.6 eV for the native CeAlO3 Ce 3d [5] component. For this reason, the argon sputtered example was chosen as the control spectrum despite acknowledged concerns of any effects occurring from Ar+ sputtering, such as ion beam mixing [15].

Figure 7. XP spectra of CeAlO3:Ge (a), sputtered CeAlO3:Ge (b), and CeO2 (c). Peaks 'a' and 'd' are the primary Ce 3d3/2 and Ce 3d5/2 photoelectron peaks, respectively. Peaks 'b' and 'e' are the respective Ce 3d3/2 and Ce 3d5/2 shakedown peaks, while peaks 'c' and f' are the respective Ce 3d3/2 and Ce 3d5/2 shakeup peaks. Spectra (b) and (c) were ultimately used as reference spectra for Ce3+ and Ce4+, respectively.
Download figure:
Standard image High-resolution imageAs previously stated, the purpose of this study was to identify the distribution of different cerium oxidation states in commercial nanoceria products. This is commonly accomplished by deconvolution of the spectral envelop with several curves varying in shape (e.g. Gaussian/Lorentzian fits), full width at half maximum parameter, and position parameters, to name a few, a process more commonly known as peak fitting. Perhaps a less utilized peak fitting approach employs the spectral line shapes of known controls as fits to the experimental spectra. This approach is useful when controls are available for the different oxidation states for which one is probing.
The two approaches (deconvolution of the spectral envelop versus fitting spectral line shapes of known controls) both have strengths and weaknesses. For example, peak fitting with several Gaussian/Lorentzian curves can be a useful approach when carefully performed on traditional photoelectric transitions composed of one, or two well separated, component(s) (most s, p and d block elements) to separate and distinguish between different oxidation states. Ideally, the parameters for these curves are determined by a combination of experimental and literature values based on the different oxidation states that are observed. However, due to the final state effects [64, 65] present in the nanoceria samples, it becomes much more challenging to compare spectra. How the peak fitting model is 'constrained' can greatly impact the calculated distribution of oxidation states, but it is important to have reasonable constraints on peaks to retain any scientific meaning when working with spectra that contain 2, much less 10 components. In the case of using control spectra to fit the line shape, it can be challenging to find an appropriate control material where the cerium is 100% one oxidation state. In the event one gets close to a pure sample in one oxidation state as the first control, one can develop a model line shape by subtracting out the minority oxidation state from the second control, which we refer to as a 'modified control'.
Figure 8 illustrates the impact deconvolution models and parameters have when evaluating XP spectra to determine oxidation state distribution in CNP-C (the CMP slurry). The approaches observed in figure 8 (from bottom to top) are: (1) Control spectra (CeAlO3:Ge and CeO2) used as is; (2) Ce3+ control spectra and modified Ce4+ control spectra; (3) fitting with each peak maximum position constrained to a range of 1.1 eV; and (4) peak fitting with positions opened to a range of 2.5 eV or greater.

Figure 8. Different models for deconvoluting the Ce 3d spectral envelop using CNP-C. Ce3+ percentages calculated by each method are shown. Residual standard deviations (STDs) of the fits, from top to bottom, were 7.33, 5.62, 2.04 and 2.00. Peaks associated with Ce4+ are shown in shades of red, while peaks associated with Ce3+ are shown in shades of blue.
Download figure:
Standard image High-resolution imageWhile one might expect to obtain a better fit from a combination of peaks, in this case, the better fits were obtained by using the raw control spectra as our representations of Ce3+ and Ce4+, as demonstrated by the improved residual STDs (caption of figure 8). It is also clear that both peak fitted samples resulted in significantly higher Ce3+ contributions. Furthermore, the calculated amount of Ce3+ measured by peak fitting varied by as much as a factor of two, ranging from 18.3% to 38.9% depending upon if the peak maximums were energetically constrained vs unconstrained, respectively. On the other hand, fitting with control spectra resulted in roughly 2.3% attributed to Ce3+ oxides. This suggests that even in the constrained peak fitting, there is likely an overestimate of the amount of Ce3+ due to the design of the software to minimize the residuals. In the last example, the modified Ce4+ control, where we assume that the control utilized had some minor Ce3+ component results in a value of 13.5% for Ce3+ contributions. This was performed to demonstrate that there may be some contributions from Ce3+ when studying cerium oxide specimens, highlighting the difficulty in obtaining a pure Ce4+ control material.
Ultimately, the choice was made to use the fitting protocol used in the 'control' conditions in figure 8. The benefits to this was that it was (a) the least arbitrary choice, with distribution of oxidation states determined by unmodified controls; and (b) using control spectra generally led to better fits. One potential negative was the challenge in obtaining pure controls, which meant there was the potential for Ce3+ in the CeO2 control, which could skew the results.
Line shapes representing the contributions of Ce3+ and Ce4+, consistent with the fits shown in figures 7(b) and (c), respectively, were employed to qualitatively demonstrate the distribution of oxidation states for the commercial samples (figure 9). It is important to note that for each sample, multiple spots were characterized. However, due to a known (and observed) impact of x-rays on transforming CeO2 from Ce4+ to Ce3+, it was decided to only report the analysis of the first measurement for each specimen. Another important point is that the distribution of oxidation states can change with time and storage conditions [31, 66], so the measurements used were of the first sample made as close to the acquisition of the samples as possible. Qualitatively, for both CNP-5 and CNP-10, the samples did increase in Ce3+ contribution with increased x-ray flux (data not shown), supporting the decision to only use measurement data from the first spot for each sample [15]. The measurements revealed that CNP-5 had the largest component of Ce3+ at 18.4%, followed by CNP-10 with 9.0% Ce3+, with CNP-25 and CNP-C having very little contribution from Ce3+ (none detected (ND) and 2.3% respectively).

Figure 9. XP spectra of CNP-5 (a), CNP-10 (b), CNP-25 (c), and CNP-C (d). Calculated contributions from Ce4+ (red) and Ce3+ (blue) to each spectrum are highlighted and vertically offset to improve visualization of the raw (black) and fitted (magenta) spectra.
Download figure:
Standard image High-resolution imageComparison of EELS and XPS analyses of commercial nanoceria materials
Previous research has suggested that the Ce3+/Ce4+ ratios of nanoceria materials correlate with the primary particle size of the nanoceria material in question [67–70]. Smaller particles tend to feature more point defects than their larger counterparts. In the ceria crystal structure, these defects often arise as oxygen vacancies, which results in an increased concentration of Ce3+ [71–73]. As such, the aforementioned particle sizes (from TEM-derived mean diameters) were correlated with XPS and EELS measurements of the nanoceria products' oxidation states to investigate this trend. The XPS and EELS results were also compared to each other to evaluate the similarities and differences between the two techniques, including fundamental principles that might complicate comparison of results. A graphical comparison of the Ce3+ content in the commercial nanoceria products determined by both techniques is presented as figure 10.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Percentage Ce3+ content of the commercial samples as determined by XPS (blue) and EELS (red) with respect to TEM-determined average particle size. Single measurement results are reported for XPS analysis and are meant to reflect the numerical values shown in figure 9. Full numerical results are shown in table S1 (located in the SI).
Download figure:
Standard image High-resolution image{kind=link}
In general, an inverse correlation between particle size and Ce3+ content is confirmed by our measurements (figure 10). For example, CNP-5 is comprised of the smallest particles and is thus expected to have the most Ce3+ of all the nanoceria products analyzed. The Ce3+ content of CNP-5, as determined by EELS, is almost double that determined for CNP-10, where the particle sizes are larger. Likewise, the Ce3+ content is expected to be low for the samples featuring even larger particles (CNP-25 and CNP-C); consistent with the present results. Qualitatively, the XPS and EELS findings are consistent.
Comparing the XPS and EELS results was one of the primary motivations of this work due to both techniques being among the principal techniques for determining the oxidation states of cerium oxide systems yet having distinct differences in their principles of operation. By using technique-independent controls and analyzing the commercial samples under 'identical' conditions, we sought to identify potential artifacts and discrepancies that can arise when performing these measurements (see earlier discussions) towards enabling improved comparison of results across techniques. As described previously, both XPS and EELS calculations of the Ce3+ content followed the trend of increasing Ce3+ content with decreasing particle size. While the absolute values of these measurements are in general agreement, the differences highlight the challenges of using either technique in isolation to provide a 'definitive' result [3]. While at first glance, the XPS determined Ce3+ values were (except for CNP-25, where no Ce3+ was found in XPS analysis) only slightly higher than those provided by EELS, it is important to remember that the XPS values are representative of one point only and may be different due to many sources of unquantifiable error. As indicated earlier, another source includes the presence of a small amount of Ce3+ in the bulk CeO2 (Ce4+) control, as well as the propensity of Ce3+ to oxidize with increased time from manufacturing date, suggesting the Ce3+ content in the commercial samples are likely to be slightly underestimated by different and unknown amounts depending on storage conditions. Another example includes the differences in sample area analyzed between the two techniques, with the sampling area of XPS being orders of magnitude greater than that of EELS and providing a more 'global' measurement of the samples. Although the EELS measurements are averaged from multiple sample locations where we rastered the electron beam over relatively large sample regions, which should result in an averaged oxidation state from numerous particles (e.g. each EELS raster area of CNP-5 should sample hundreds of particles), it is still smaller than the area analyzed by XPS. As such, there is the possibility (however unlikely) of sampling bias compared to a more 'global' technique such as XPS. it is possible for the EELS determined Ce3+ content to vary depending on the area of analysis. For example, our data from both EELS and ADF-STEM analyses show (in agreement with previous reports) [21, 38] that Ce3+ content tends to be increased at surface/edge regions of individual particles relative to the particle center (see figure 5(a)) or in particles where defect are present, such as the twin boundary of the particle also shown in figure 5(a). In addition, both XPS and EELS have the capacity for reduction of Ce4+ to Ce3+ in cerium oxide samples [14, 19]. This phenomenon is dependent on both the beam energy dosage and rate of dosage, as described in the EELS discussion above for electrons [63], but also applicable to x-ray photons [14], and can induce differences in the Ce3+ content determined by each technique. While not comprehensive, the observations described here serve as examples of the inherent sources of variation originating from the analytical technique utilized, which should be considered when performing said analyses and comparing to results determined by a different technique or different method within a given technique. Based on our findings and supplemented by those found in the literature, we propose the following steps when pursing oxidation state studies of cerium oxide materials, especially in the context of comparing new findings with previous literature reports: (1) control materials (for ceria, Ce3+ and Ce4+) should be obtained and calibrated to the analytical technique to be utilized; (2) if possible, the same control material should be used for each analytical technique to be utilized; (3) technique-specific variables (e.g. sampling area, energetics of analytical probe) should be identified and considered to limit artifacts that might impact the determination of oxidation state; (4) sample-specific variables (e.g. sample size/shape, sample uniformity, sample matrix, sample aging) should also be identified and accounted for when measuring oxidation state.
Conclusions
In summary, we have successfully employed air-stable and technique-independent Ce3+ and Ce4+ control materials to develop new methods towards more accurate and reproducible oxidation state measurements of nanoceria materials. The advantages and disadvantages of the developed approaches were discussed in comparison with prevailing methods, with our results highlighting the impact of the method used for quantifying oxidation state, even within a single analytical technique. The methodologies were also demonstrated in characterizing several commercially available nanoceria products, where the oxidation state results determined by XPS and EELS were in good agreement, although multiple sources of variance were identified. These potential variances were discussed alongside recommendations designed to assist the research community in obtaining more robust oxidation state measurements on nanoceria and facilitate improved comparison of results between analytical approaches.
NIST disclaimer
Certain commercial equipment, instruments, and materials are identified in this paper to specify an experimental procedure as completely as possible. In no case does the identification of particular equipment or materials imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments, or equipment are necessarily the best available for the purpose.
Acknowledgments
Two of the authors, CMS and RAM, acknowledge funding and support from the National Academy of Sciences- National Research Council Postdoctoral Research Associateship Program. We thank Alline F Myers of the Center for Nanoscale Science and Technology (CNST) at NIST for her assistance in initial investigative measurements and for providing technical support in TEM imaging.