





Abstract
The host-selective toxin victorin is produced by Cochliobolus victoriae, the causal agent of victoria blight of oats. Victorin has been shown to bind to the P protein of the glycine decarboxylase complex (GDC) in mitochondria, and induce defense-related responses such as phytoalexin synthesis, extracellular alkalization and programmed cell death. However, evidence demonstrating that the GDC plays a critical role in the onset of cell death is still lacking, and the role of defense-like responses in the pathogenicity has yet to be elucidated. Here, cytofluorimetric analyses, using the fluorescein (VicFluor) or bovine serum albumin–fluorescein derivative of victorin (VicBSA), demonstrated that victorin-induced cell death occurs before these conjugates traverse the plasma membrane. As with native victorin, VicBSA clearly elicits apoptosis-like cell death, production of phytoalexin, extracellular alkalization, and generation of nitric oxide and reactive oxygen intermediates. These results suggest that the initial recognition of victorin takes place on the cell surface, not in mitochondria, and leads to the activation of a battery of victorin-induced responses. Pharmacological studies showed that extracellular alkalization is the essential regulator for both victorin- and VicBSA-induced cellular responses. We propose a model where victorin may kill the host cell by activating an HR-like response, independent of the binding to the GDC, through ion fluxes across the plasma membrane.
Introduction
In many plant–pathogen interactions, plants recognize pathogen-produced avirulence gene products by matching resistance gene products and subsequently evoke a battery of defense responses, including a local and rapid cell death termed hypersensitive response (HR) (Dangl and Jones 2001). In contrast, most pathogens have developed virulence factors that interact with host molecules to cause a spectrum of disease in plants. Of such virulence factors, host-selective toxins (HSTs) produced by certain pathogenic fungi are toxic only to susceptible hosts and hence function as specificity determinants in plant–fungus interactions (Walton 1996). Although approximately 20 HSTs have been identified, the molecular and biochemical basis of disease susceptibility has remained to be elucidated.
The fungus Cochliobolus victoriae, a causal agent of victoria blight of oat (Avena sativa L.), produces an HST, victorin (Wolpert et al. 1985). Victorin is a peptide toxin that is required for the pathogenicity on oat cultivars carrying the dominant Vb allele (Wolpert et al. 1985). The Vb gene has not been separated genetically from Pc-2 (Rines and Luke 1985, Mayama et al. 1995), a resistance gene against certain races of crown rust fungi, Puccinia coronata f. sp. avenae (Meehan and Murphy 1946, Meehan and Murphy 1947), suggesting that both genes may be identical. Therefore, characterization of the Vb gene and the underlying mechanisms of victorin sensitivity may lead to understanding of both disease susceptibility and resistance.
In susceptible oat leaves, a radiolabeled victorin derivative was shown to bind specifically to the P protein component of the glycine decarboxylase complex (GDC) located in the mitochondrial matrix (Wolpert et al. 1994). Victorin strongly inhibits glycine decarboxylase activity (GDA), which plays a pivotal role in the photorespiration cycle, in victorin-sensitive leaf tissues (Navarre and Wolpert 1995). The in vitro binding of victorin to the P protein occurs in both victorin-sensitive and -insensitive oats (Wolpert and Macko 1989), and it requires a mitochondrial permeability transition (MPT) (Curtis and Wolpert 2002), which is elicited by opening of permeability transition pores in the mitochondrial inner membrane, leading to mitochondrial swelling and membrane depolarization (Zamzami et al. 1996, Kroemer et al. 1998). These data suggest that the interaction of victorin with the product of the Vb gene precedes the occurrence of an MPT and the binding of victorin to the GDC. Akimitsu et al. (1993b) have clearly shown, using anti-victorin anti-idiotypic antibodies, that victorin is recognized on the cell surface in victorin-sensitive oats. Therefore, it is unlikely that the Vb gene encodes a subunit of GDC, and it is possible that the inhibition of GDA is not the direct cause of cell death. Further study is required to determine the primary binding site of victorin for the induction of cell death.
Victorin has been shown unambiguously to induce apoptosis-like responses such as DNA laddering and heterochromatin condensation (Navarre and Wolpert 1999, Tada et al. 2001, Yao et al. 2001). Recently, Curtis and Wolpert (2004) demonstrated that victorin elicits the reduction of mitochondrial membrane potential, followed by cell shrinkage without loss of plasma membrane and tonoplast integrity. As reported in animals, victorin-induced apoptosis-like characteristics are mediated by Ca2+ ions, oxidative stress, serine and cysteine proteases and nuclease activities (Navarre and Wolpert 1999, Tada et al. 2001, Yao et al. 2001). Thus, victorin-induced cell death is probably caused by transcriptional and/or metabolic reprogramming of host cells, leading to the activation of a cellular suicide machinery, rather than a necrotic disorganization of the cell.
The most intriguing characteristic of victorin is that it can also serve as a specific elicitor for the induction of phytoalexins, avenanthramides, in oat cultivars carrying the Pc-2 gene (Mayama et al. 1986, Mayama et al. 1995). There are gene dosage effects of Pc-2 on the accumulation of avenanthramides and cell death induced by victorin, suggesting that the Pc-2 gene might control both victorin sensitivity and rust resistance (Mayama et al. 1995). Despite the interest in the toxic nature of victorin, few studies expressed concern about a possible link between its elicitor activity and toxicity. Pharmacological studies indicated that the production of avenanthramide A is mediated by Ca2+ influx, protein kinase and de novo protein synthesis (Tada et al. 2001). Also, victorin has been shown to induce other defense-related responses such as extracellular alkalization (Ullrich and Novacky 1991), K+ efflux (Wheeler and Black 1962) and callose synthesis (Walton and Earle 1985). Taken together with the toxic action of victorin, these results give rise to questions: whether victorin can trigger a general plant defense response, whether the onset of the defense response and cell death is due to side effects of the toxicity caused by the binding of victorin to the GDC, and whether the activation of defense signaling is essential for the virulence function of victorin.
Here we demonstrate that a virulence factor of the filamentous fungus induces a set of defense responses presumably to subvert the host cell. We show that a fluorescein (VicFluor) and bovine serum albumin–fluorescein (VicBSA) conjugate of victorin do not traverse the plasma membrane before they induce cell death. As with native victorin, VicBSA rapidly stimulates the generation of reactive oxygen intermediates (ROIs) and nitric oxide (NO), phytoalexin synthesis, extracellular alkalization and cell death only in victorin-sensitive leaves. These results suggest that victorin is recognized on the cell surface and may elicit an HR-like response independent of the binding of victorin to the GDC. We propose that the victorin-induced defense-like response may be the primary cause of cell death.
Results
VicFluor and VicBSA elicit cell death before their entry into cells
To determine the causative relationship between the timing of victorin-induced cell death and the dynamic distribution of victorin in the cell, we performed cytological analyses using a fluorescein conjugate of victorin (VicFluor) (Curtis and Wolpert 2002), in resistant (Iowa X424) and susceptible plants (Iowa X469). As shown in Fig. 1A, VicFluor induced cell death in a dose- and time-dependent manner in victorin-sensitive leaves. As with native victorin, a high concentration of 0.6 µg ml–1 VicFluor did not affect cell viability by 1 h after treatment, then 95% of the cells died after another 1 h incubation, while a low concentration of 0.05 µg ml–1 VicFluor elicited the production of avenanthramide A without severe cell death (data not shown). These data demonstrate that VicFluor retains the ability to activate the same signaling pathways that are stimulated by native victorin, although VicFluor is less effective than native victorin (Curtis and Wolpert 2002).
We performed quantitative and time course analyses of the fluorescence intensity of VicFluor in mesophyll cells. The morphological changes in the cell were confirmed by detecting the red autofluorescence of chloroplasts, which exhibited an abnormal distribution after cell death (Fig. 1B, arrowhead). While treatment of Iowa X424 leaves with 0.6 µg ml–1 VicFluor did not induce cell death, a small percentage of dead cells (<2%, Fig. 1A, C, asterisk), which might be caused by peeling stress, had about 2-fold higher VicFluor fluorescence compared with that in living cells at 4 h after treatment (Fig. 1C, striped bars). There was no significant increase in intracellular VicFluor fluorescence in living cells during the time course, indicating that VicFluor may not permeate resistant cells before dying or dead cells may lose their membrane integrity. In victorin-sensitive cells, the VicFluor signal increased 2.3-fold at 4 h after treatment compared with the control treatment (30 s) (Fig. 1B, C), demonstrating that dead cells in the two lines displayed equivalent intensity of VicFluor fluorescence. Interestingly, although treatment with VicFluor for 2 h resulted in massive cell death (Fig. 1A), no significant increase in the intracellular VicFluor signal was observed in victorin-sensitive cells by this time (Fig. 1B, C). These data suggest that VicFluor may activate the cell death program in the extracellular space, and enters the cell after the disruption of the plasma membrane integrity.
Although the dramatic increase in the intracellular VicFluor signal was observed after cell death, we could not rule out the possibility that VicFluor entered victorin-sensitive cells below detectable levels before cell death. We produced a victorin-conjugated BSA, which is larger than 66 kDa and hence presumed not to traverse the plasma membrane, using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS), and subsequently the conjugate was coupled with fluorescein (VicBSA) to monitor the localization of VicBSA. VicBSA clearly induced cell death only in victorin-sensitive leaves in a dose- and time-dependent manner (Fig. 2A), whereas a fluorescein–BSA conjugate did not cause any visible cellular damage during the time course (data not shown). Furthermore, we confirmed that VicBSA does not release free victorin sufficient to induce cell death during the treatment period (see Materials and Methods, data not shown). VicBSA had an intense activity for cell death at the concentration of 50 µg ml–1, equivalent to that of 5 ng ml–1 native victorin, which killed >95% of sensitive cells 2 h after incubation. Cytofluorimetric analyses showed that VicBSA was excluded from the cell for at least 4 h after treatment (Fig. 2B, C), and a significant increase in the intracellular VicBSA signal was not detected until 6 h after incubation, although almost 100% of cells treated with 50 µg ml–1 VicBSA died 2 h after treatment (Fig. 2A, B, arrowheads). These results strongly support the hypothesis that victorin is recognized on the cell surface of victorin-sensitive leaves and may stimulate the cell death program independently of the binding to mitochondrial GDC or the other unknown intracellular virulence targets.
Ca2+ and extracellular alkalization are essential signals for victorin-induced cell death
Because victorin has been shown to trigger drastic defense responses (Mayama et al. 1986), it is possible that victorin-induced cell death may occur as a result of immune responses. Thus, we performed pharmacological studies on cell death induced by 5 ng ml–1 victorin and 50 µg ml–1 VicBSA using various inhibitors that affect signal transduction pathways of defense responses and apoptotic cell death (Fig. 3). Given that the interaction of native victorin with the host cell surface leads to cell death, the VicBSA-activated cell death pathway would be blocked by the same inhibitors that prevent victorin-induced cell death. Cell viability was assessed by the fluorescent dye fluorescein diacetate (FDA), which determines membrane integrity. Victorin- and VicBSA-induced cell death was strongly suppressed in the presence of Ca2+ metabolism inhibitors such as the Ca2+ chelator EGTA (10 mM), the Ca2+ channel blocker LaCl3 (1.5 mM) and the Ca2+ competitor ZnSO4 (1 mM) (Fig. 3), suggesting that Ca2+ is a critical regulator of cell death signaling. Changes in the cytosolic Ca2+ concentration are generally followed by the activation of Ca2+-sensing calmodulin, protein kinase and de novo protein synthesis (Chin and Means 2000). However, the calmodulin inhibitor trifluoperazine (500 µM), the protein kinase inhibitors K-252a (2 µM) and staurosporine (500 nM), the protein phosphatase inhibitor okadaic acid (500 nM) and the inhibitor of new protein synthesis cycloheximide (2 µM) did not have any effect on cell death, strongly suggesting that victorin- and VicBSA-induced cell death is controlled by a constitutive machinery independent of phosphorylation-based regulations. The NADPH oxidase inhibitor diphenyleneiodonium chloride (DPI; 50 µM), which is used to block the oxidative burst, did not abolish the induction of cell death. Although the addition of superoxide dismutase (SOD; 150 µg ml–1) and the radical scavenger N-acetylcysteine (NAC; 2 mM) partially reduced victorin-induced cell death 3 h after treatment, the treated cells consequently died by 5 h after treatment (data not shown) as with VicBSA. The cysteine (E-64, 500 µM) and serine protease (aprotinin, 150 µg ml–1) inhibitors did not suppress victorin- and VicBSA-induced cell death. Interestingly, pre-treatment of victorin-sensitive leaves for 2 h with antimycin A (10 µM), which is known to deplete cellular ATP (Atkinson et al. 2004), resulted in significant inhibition of cell death, while pre-treatment for 5 min had no effect on cell death (data not shown), indicating that the cellular ATP content may play an important role in victorin- and VicBSA-induced cell death. The inhibitor profiles of VicBSA completely corresponded to those obtained with native victorin, suggesting that VicBSA and victorin may stimulate the same signal transduction pathway leading to the onset of cell death. Furthermore, these findings led us to hypothesize that active transport of ions (e.g. Ca2+ and H+), which requires ATP as an energy source, might regulate cell death induced by victorin and VicBSA, whereas the protein products of defense-related genes may not be involved in the execution of cell death.
A previous study reported that change in the plasma membrane permeability is the early toxic action of HSTs, and thus electrolyte leakage is thought to be an indicator of cell death (Walton 1996). However, the ion leakage is also a physiological response associated with both the defense response (Mackey et al. 2002) and toxin-induced cell death. Thus, to confirm the assumption that stimulation of ion transport is the critical checkpoint to victorin- and VicBSA-induced cell death, we monitored the effects of various inhibitors on the kinetics of extracellular alkalization induced by 5 ng ml–1 victorin and 50 µg ml–1 VicBSA (Fig. 4). As we expected, pre-treatment with antimycin A for 2 h strongly reduced extracellular alkalization, which was evident by 30 min after victorin or VicBSA treatment, while a shorter pre-treatment period of 5 min did not suppress the alkalization (data not shown). Addition of LaCl3 also completely inhibited extracellular alkalization during the time course. In contrast, the chemical agents that did not reduce cell death, such as cycloheximide, K-252a and E-64, failed to block the rapid increase in extracellular pH. It is important to note that both antimycin A and LaCl3 abrogated the alkalization induced by water treatment, while the other chemicals could not, demonstrating the potential ability of the two reagents to suppress the active ion movement. These data indicate that the ion fluxes across the plasma membrane may be a critical upstream signal in the cell death pathway.
Victorin and VicBSA serve as specific elicitors
Mayama et al. (1986) demonstrated that low concentrations of victorin, unlike some abiotic and biotic elicitors, stimulate the biosynthesis of phytoalexins, avenanthramides, in primary leaves of susceptible cultivar Iowa X469, suggesting that victorin functions as a specific elicitor, not as a general toxin which simply damages cells. To investigate the potential ability of VicBSA as a victorin derivative, we determined whether VicBSA treatment resulted in the induction of avenanthramide A. Low concentrations of VicBSA (0.5–2 µg ml–1) stimulated the accumulation of avenanthramide A in victorin-sensitive leaves 24 h after treatment in which >60% of the treated cells were alive (data not shown), but had no significant effect in victorin-insensitive leaves (Fig. 5). At higher concentrations (>5 µg ml–1), the production of avenanthramide A was dramatically decreased to the basal level, presumably because almost all the treated cells died within 12 h, at which time little accumulation of avenanthramides was observed even in leaves treated with a low concentration of VicBSA (data not shown).
The oxidative burst and the generation of NO are the most prominent defense responses highly associated with the HR in oat plants (Tada et al. 2004). We determined the kinetics of ROI and NO generation by monitoring the fluorescence of 2′,7′,-dichlorofluorescein (DCF) and 4,5-diaminofluorescein (DAF), respectively. The lower epidermis of primary leaves was peeled off and floated on a test solution containing DCF or DAF, with the peeled surfaces in contact with the liquid. After treatment with 5 ng ml–1 victorin (closed symbols) and 50 µg ml–1 VicBSA (open symbols), significant production of ROIs and NO was detected by 90 min (Fig. 6), when VicBSA was restricted to the extracellular space of susceptible cells (Fig. 2). At 3 h after victorin treatment, the fluorescence intensity of DCF and DAF in toxin-sensitive leaves (circles) reached 4.0-fold and 3.3-fold higher than that detected in the toxin-insensitive cultivar Iowa X424 (squares), respectively. VicBSA elicited changes in the kinetics of ROI and NO production similar to those induced by native victorin. The increase in the fluorescence of DCF and DAF in a test solution was continuously observed even after the 2 h time point at which most of the exposed cells died (Fig. 2A, 6). These results are consistent with our previous observation that although the cells most proximal to the peeled side died 2 h after victorin treatment, the other layers remained intact (Yao et al. 2001), suggesting that the generation of ROIs and NO could be elicited in the remaining viable cells. These results indicate that victorin and VicBSA stimulate a set of defense responses typically incited during a gene-for-gene type resistance.
VicBSA induces apoptosis-like cell death in susceptible cells
In previous studies, we showed that victorin causes the cell to undergo apoptosis-like cell death characterized by DNA laddering and heterochromatin condensation in the nuclei (Tada et al. 2001, Yao et al. 2001, Yao et al. 2002). Thus we assessed whether VicBSA activates the same signal cascade that is elicited by victorin. The genomic DNA extracted from victorin-sensitive leaves treated with 50 µg ml–1 VicBSA exhibited clear DNA laddering from 4 h after treatment, at which time VicBSA is excluded from the susceptible cell, but was not observed in toxin-insensitive leaves through the incubation period (Fig. 7A). Ultrastructural observation showed that heterochromatin had began to aggregate into large masses in victorin-sensitive leaves treated with 50 µg ml–1 VicBSA for 2 h (Fig. 7C), while heterochromatin in the control leaves was evenly scattered within the nucleus (Fig. 7B). Although the plasma membrane shrunk away from the cell wall at 2 h after treatment (Fig. 7C, arrowheads), other organelles remained morphologically intact as observed in the control cells. As reported for native victorin (Yao et al. 2001), further condensed chromatin was observed in the cells adjacent to the VicBSA-exposed layer in which almost all cells collapsed 4 h after treatment (Fig. 7D). We conclude that VicBSA also stimulates apoptosis-like cell death in victorin-sensitive leaves via a cell surface mediator.
Discussion
Although the primary function of phytotoxic compounds is thought to be to induce cell death and/or to suppress defense response in host plants, we propose here an alternative function of phytotoxic compound to trigger the plant defense machinery for pathogenicity. Our data have demonstrated that victorin serves as a specific elicitor in oat plants carrying the Vb/Pc-2 gene, as initially suggested by Mayama et al. (1986). Victorin triggers a wide range of defense responses such as programmed cell death (PCD) (Tada et al. 2001, Yao et al. 2001), an oxidative burst, NO generation and the activation of defense-related genes responsible for phytoalexin synthesis, PR protein accumulation and detoxification of ROI stress (S. Betsuyaku, H. Nakayashiki, Y. Tosa and S. Mayama, unpublished ). Recently, we demonstrated that two genes encoding hydroxycinnamoyl-CoA:hydroxyanthranilate N-hydroxycinnamoyltransferase and S-adenosyl-l-methionine:trans-caffeoyl-CoA 3-O-methyltransferase, which catalyze the biosynthesis of avenanthramides, are highly up-regulated in response to victorin (Yang et al. 2004). Considering that defense responses evoked by a pathogen-encoded avirulence gene product often accompany PCD (Lam et al. 2001), it is possible that victorin carries out its virulence function by activating the innate immune system.
We propose a model in which victorin is recognized on the cell surface triggering apoptosis-like cell death and defense responses independently of the binding to the GDC (Fig. 8) as originally demonstrated by Akimitsu et al. (Akimitsu et al. 1992, Akimitsu et al. 1993a, Akimitsu et al. 1993b). We provide two lines of evidence for this proposal: first, VicFluor, a fluorescein conjugate of victorin, does not accumulate in victorin-sensitive cells until 4 h after treatment, as shown by Curtis and Wolpert (2004), although most of the cells are dead by 2 h (Fig. 1). VicFluor also serves as a potent elicitor leading to the production of avenanthramide A, ROIs and NO (Y. Tada, K. Kusaka, Y. Tosa and S. Mayama, unpublished), and has been shown to accumulate in the mitochondria after the induction of cell shrinkage (Curtis and Wolpert 2004). Secondly, VicBSA, a macromolecular conjugate in which victorin and fluorescein are covalently attached to BSA, clearly induces cell death 2 h after treatment without entering cells until 6 h (Fig. 2). Interestingly, low concentrations of VicBSA elicit the production of avenanthramide A in victorin-sensitive leaves (Fig. 5). Furthermore, extracellular alkalization and the generation of ROIs and NO are rapidly induced in response to VicBSA, before it accumulates in the cell, with similar kinetics to those detected with native victorin. Thus, VicBSA and VicFluor most probably attack extracellular component(s) to incite nearly all, or all, victorin-induced responses. These indicate that the protein product of the Vb gene could be involved in the recognition of victorin on the cell surface, including the plasma membrane and cell wall, and that although the binding of victorin to the mitochondrial GDC is initially suggested to play a central role in victorin’s mode of action (Wolpert et al. 1994, Navarre and Wolpert 1995), the P protein might be the primary site of action neither for acute cell death nor for defense responses as recently proposed by Wolpert et al. (2002).
Akimitsu et al. (Akimitsu et al. 1992, Akimitsu et al. 1993a) showed, using anti-victorin polyclonal antibodies, that victorin binds to 65 and 45 kDa proteins in purified plasma membrane of both susceptible and resistant cultivars. Furthermore, the authors demonstrated that anti-victorin anti-idiotypic antibodies elicit callose synthesis in protoplasts from susceptible leaves as induced by native victorin (Akimitsu et al. 1993b). They suggested that the primary site of action of victorin exists on the surface of the plasma membrane, since the antibodies are speculated to be too large to enter cells. The current study further supports these results and raises the possibility that this recognition can lead to a battery of victorin-induced responses. Actually, our unpublished data (R. Saito, S. Ryu, K. Kusaka, Y. Tada, H. Nakayashiki, Y. Tosa and S. Mayama) demonstrated that victorin does interact with a novel mediator(s) extracted from the extracellular space of both susceptible and resistant leaves.
Despite the data showing that the cell surface molecule(s) may be a key mediator of victorin-induced responses, we cannot rule out the possibility that the inactivation of GDA plays a major role in the pathogenicity of C. victoriae. Navarre and Wolpert (1999) demonstrated that victorin causes a senescence-like response in victorin-sensitive leaves characterized by a proteolytic cleavage of the Rubisco large subunit (LSU) as well as chlorophyll loss. The binding of victorin to the GDC appears to be prerequisite for LSU cleavage because pyridoxal phosphate, which competes with victorin for binding to the P-protein, prevents Rubisco cleavage, indicating that the inhibition of GDA may facilitate plant destruction by abrogating the photorespiratory cycle. In addition, the application of the GDC inhibitor aminoacetonitrile results in apoptosis-like cell death (Yao et al. 2002) and disease symptoms similar to those induced by native victorin (Y. Tada, S. Hata, Y. Tosa and S. Mayama, unpublished). Therefore, although the inhibition of GDA by victorin seems to play a partial role, if any, in the onset of cell death and defense responses, it may contribute to symptom development and provide subsequent benefit for the pathogen in terms of feeding nutrients and colonizing host plants.
We propose that active transport of ions, which requires ATP as an energy source, is the critical regulator of physiological and morphological changes induced by victorin. This hypothesis largely comes from the observations as follows. (i) ATP depletion by pre-treatment with antimycin A for 2 h results in the strong inhibition of cell death (Fig. 3), whereas a shorter pre-treatment period of 5 min with antimycin A does not negate cell death. The significant reduction of cellular ATP levels occurs in oat suspension cells by 2 h after antimycin A treatment but not by 5 min (M. Sakamoto, H. Nakayashiki, Y. Tosa and S. Mayama, unpublished). Curtis and Wolpert (2002) demonstrated that the mitochondrial uncoupler FCCP, which inhibits ATP synthesis and leads to the hydrolysis of cellular ATP, prevents VicFluor binding to the P protein and DNA laddering. The authors discussed that an MPT may be required for the access of victorin to the mitochondrial matrix. However, there is an alternative possibility that FCCP may rescue cell death through the reduction of ATP levels as in leaves treated with antimycin A. (ii) Among inhibitors of signal transduction pathways, only reagents, other than antimycin A, that affect divalent cation metabolism can strongly suppress victorin-induced cell death (Fig. 3). Administration of inhibitors that perturb the activity of protein kinase, phosphatase, calmodulin, proteases, ROI generation and protein synthesis has no suppressive effect on cell death in a long-term treatment. Considering our previous studies showing that cysteine and serine proteases, protein kinases and ROIs play important roles in the induction of DNA laddering or heterochromatin condensation (Tada et al. 2001, Yao et al. 2001, Yao et al. 2002), the current data may suggest that the commitment to cell death precedes ROI generation, and activation of protein kinases, cysteine and serine proteases which lead to the late characteristics of apoptosis. These facts do not contradict the hypothesis that the ion fluxes across the plasma membrane could be the primary cause of cell death. Supporting this assumption, (iii) all reagents including antimycin A that negate victorin-induced cell death remarkably inhibit the increase in extracellular pH, a hallmark of the activation of ion transport (Fig. 4). We suggest that extracellular alkalization is an upstream signal responsible for the cell death program and is not induced as a result of cell death because only these chemicals can shut down the transient alkalization, which might be activated by peeling stress, in leaves treated with water, showing the predominant function as inhibitors of ion transport systems.
Several lines of evidence suggest that the stimulation of ion fluxes is tightly associated with the onset of cell death and resistance response. The syringolide elicitors from Pseudomonas syringae isolates elicit the HR (Keen and Buzzell 1991) accompanied by extracellular alkalization, K+ efflux and Ca2+ influx (Atkinson et al. 1996) in soybean cultivars that carry the complementary resistance gene Rpg4. As in the case of victorin-treated leaves, the blockage of Ca2+ influx by LaCl3 significantly suppresses the increase in extracellular pH and K+ efflux, demonstrating their important roles in a the syringolide-activated signaling pathway (Atkinson et al. 1996). Furthermore, the non-HR response of parsley elicited by an oligopeptide elicitor Pep-13 of Phytophthora sojae also requires a sustained increase in free Ca2+ and extracellular alkalization as components of the signal cascade (Nurnberger et al. 1994, Blume et al. 2000). Intriguingly, the expression of the proton pump bacterio-opsin (bO) from Halobacterium halobium in various plants results in the induction of HR cell death, systemic resistance and genes encoding PR proteins and PAL in the absence of pathogen (Mittler et al. 1995). These responses are caused by passive proton leakage through the bO channel, indicating that the loss of ion homeostasis may be the cause of the HR (Pontier et al. 2002). Here we show that the continual exposure of toxin-sensitive cells to victorin leads to a sustained increase in extracellular pH until cell death occurs (Fig. 4), exhibiting the possible role of the abnormal ion balance across the plasma membrane in cell death.
Unlike other HSTs, the current hypothesis is that victorin induces disease susceptibility by activating host defense responses. This proposal may seem to contradict a general concept that toxins abrogate resistance to pathogens. However, although high concentrations of victorin up-regulate a set of defense-related genes (Yang et al. 2004, S. Betsuyaku, H. Nakayashiki, Y. Tosa and S. Mayama, unpublished), the production level of avenanthramides is very low (Mayama et al. 1995). Our unpublished data (Y. Takata, Y. Kimura, H. Nakayashiki, Y. Tosa and S. Mayama) demonstrated that the antifungal protein chitinase, which is highly induced by low concentrations of victorin, also does not accumulate in response to toxic concentrations of victorin. It is plausible that the effective production of such antimicrobial compounds would take longer than the execution of cell death and the expression of the defense-related genes. Furthermore, avenanthramides have been shown not to inhibit the fungal growth effectively. Thus, C. victoriae may synchronously elicit HR-like cell death at and around the infection site and exploit the dead cells for the pathogenicity, avoiding the accumulation of PR proteins and other antifungal compounds. The necrotrophic pathogen Botrytis cinerea has been shown to elicit an HR in Arabidopsis to utilize dead cells for successful colonization, while the Arabidopsis dnd1 (defense, no death) mutant, loss-of-HR phenotype, significantly prevents fungal growth of B. cinerea (Govrin and Levine 2000). In addition, the suppression of PCD by transgenic expression of the baculovirus p35 gene, a specific inhibitor of apoptogenic protease (caspase), protects tomato plants from disease symptoms induced by Alternaria alternata and P. syringae pv. tomato (Lincoln et al. 2002). Together with our data, these suggest that PCD during the HR and pathogenicity does not necessarily retard fungal growth especially in plant–necrotroph interactions, and that certain necrotrophic pathogens have evolved their tools to trigger cell death by modification of plant intrinsic cell death signaling.
In mammalian cells, effectors from pathogenic bacteria often mimic host molecules and interact with host cell surface receptors to reduce phagocytic activity and the development of immunity (Knodler et al. 2001). We demonstrated that victorin elicits an HR-like response in Vb/Pc-2 oats, possibly for pathological cell death, suggesting that victorin might mimic a specific elicitor against Pc-2 to stimulate cell death signaling, which is normally triggered during defense against the crown rust fungus. Although we cannot exclude the possibility that the Vb gene differs from the Pc-2 gene, our data suggest an alternative strategy of the necrotrophic pathogen to invade plants.
Materials and Methods
Plant materials
Seven-day-old primary leaves of oat (A. sativa L.) cultivars Iowa X424 and Iowa X469 were used in this study. Iowa X469 carrying the Vb/Pc-2 gene is sensitive to the host-selective toxin victorin C produced by C. victoriae. Iowa X424 is a near isogenic line of Iowa X469 that does not carry the Vb/Pc-2 gene and hence is insensitive to victorin. Seeds were dipped in water for 12 h, sown on vermiculite, and grown in a growth chamber at 20°C under illumination for 16 h daily with 10,000 lux fluorescent light.
Victorin C and chemical treatments
The host-selective toxin victorin C was kindly provided by Dr. Wolpert (Oregon State University, Corvallis, OR, USA) and was applied at a concentration of 5 ng ml–1. E-64d, aprotinin, cycloheximide, EGTA, LaCl3, ZnSO4, trifluoperazine, SOD and NAC were obtained from Nacalai Tesque (Kyoto, Japan). Antimycin A, staurosporine, DPI and okadaic acid were obtained from Sigma (St Louis, MO, USA). K-252a was purchased from Alomone Labs (Jerusalem, Israel). SOD and NAC were used as ROI scavengers at concentrations of 150 µg ml–1 and 2 mM, respectively. EGTA, a calcium chelator, ZnSO4, a calcium antagonist, and LaCl3, a calcium channel blocker, were used at 10, 1 and 1.5 mM, respectively. K-252a and staurosporine were used as protein kinase inhibitors at concentrations of 2 µM and 500 nM, respectively. Okadaic acid was used as a protein phosphatase inhibitor at 500 nM. E-64d was used as a cell-permeable cysteine protease inhibitor at 500 µM. Aprotinin was used as a serine protease inhibitor at 150 µg ml–1. Cycloheximide was used as an inhibitor of de novo protein synthesis at 2 µM. DPI was used as an NADPH oxidase inhibitor at a concentration of 50 µM. Antimycin A was used as a mitochondrial complex III inhibitor at 10 µM. The effectiveness of inhibitors and an enzyme used in this experiment has already been demonstrated in our previous studies (Tada et al. 2001, Kusaka et al. 2004, Tada et al. 2004). The lower epidermis of primary oat leaves was peeled off, and 5 cm segments, taken at 1–6 cm from the leaf tip, were floated on 3 ml of testing solutions with the exposed mesophyll surface in contact with the liquid, and kept at 20°C under continuous light for the indicated times.
Cytofluorimetric analysis
An IX70 (Olympus, Tokyo, Japan) fluorescent microscope equipped with a MicroMax 12-bit CCD camera system (Princeton Instruments, Inc., Trenton, NJ, USA) and an optical filter changer (Lambda 10–2, Sutter Instruments, Novato, CA, USA) was used. A xenon lamp is coupled with a monochromator (Polychrome II, TILL Photonics, Planegg, Germany) to provide the excitation light source. ROIs generated from oat leaves were measured by monitoring the fluorescence of DCF (Molecular Probes, Eugene, OR, USA). NO accumulation was measured by the fluorescence of DAF (Daiichi Kagaku, Tokyo, Japan). The epidermis was peeled from 7-day-old leaves, and 1 cm×5 segments were incubated with the test solutions containing 100 µM DCF or 50 µM DAF for the indicated times. The test solutions were observed using 488 and 515 nm with excitation at 520 and 495 nm emission wavelengths, respectively. Images from the microscope were acquired, and the fluorescence intensity values were analyzed by MetaMorph software (Universal Imaging Corporation, Downingtown, PA, USA). All fluorescence intensity values would be in the range between 0 and 4,095, and experimental points represent the mean ± SD of three replicates of the same experiment.
VicFluor was prepared as described by Curtis and Wolpert (2002). A 100 µg aliquot of victorin was incubated with 1 mg of 5-(and 6)-carboxyfluorescein, succinimidyl ester (NHS–fluorescein, Pierce, Rockford, IL, USA) for 4 h at 25°C in 1 ml of reaction buffer A (0.1 M MOPS-KOH, pH 7.2), and purified as described previously (Mayama et al. 1986). VicFluor and chloroplast autofluorescence were detected with excitation at 488 nm during 3,000 and 100 ms exposure times, respectively, and the intensity of VicFluor fluorescence was estimated by MetaMorph software.
VicBSA was produced by using EDC (Pierce) and NHS (Pierce). A 600 µg aliquot of BSA was incubated with 11.5 mg of NHS and 75 mg of EDC for 1 h at 25°C in 1 ml of reaction buffer B (0.1 M MES-KOH, pH 6.0, 0.5 M NaCl) and then 1.4 µl of 2- mercaptoethanol was added to quench the EDC. The reaction mixture was concentrated 5-fold with Microcon 30 kD (Amicon, Beverly, MA, USA), followed by the addition of 100 µl of victorin and 200 µl of 1 M sodium bicarbonate. After incubation at 25°C for 12 h, 10 µl of stop solution (0.5 M ethanolamine, 0.5 M NaCl, pH 8.3) was added to the reaction mixture, and the BSA conjugate of victorin was eluted on a PD-10 column (Amersham Biosciences) with 3.5 ml of MilliQ. After concentration of the conjugate to 450 µl using Microcon 30 kD, 50 µl of 1 M sodium bicarbonate and 100 µl of 1 mg ml–1 NHS–fluorescein were added and incubated at 25°C for 4 h. VicBSA was purified on the PD-10 column with 3.5 ml of MilliQ and concentrated to 2.5 ml by Microcon 30 kD. The purification procedure was repeated four times, and then we confirmed that the flow-through fraction of Microcon 30 kD did not have toxic activity. The VicBSA solution was recovered 2 h after treatment and dialyzed by Microcon 30 kD. The flow-through fraction did not induce cell death in susceptible leaves, and free victorin was not detected in the fraction by HPLC.
Cell death
The epidermis-peeled leaf segments treated with 5 ng ml–1 victorin and the indicated chemicals for various intervals were incubated with 0.01% FDA for 5 min at room temperature. After washing with distilled water, viable cells were monitored with a fluorescence microscope with excitation at 495 nm using an Olympus IX70 fluorescent microscope. The number of dead cells was expressed as a percentage of the total cells counted [(total cell number – viable cell number]/total cell number×100]. The results were confirmed in three independent experiments. The estimation of viable cells and the detection of VicFluor or VicBSA localization were performed after preparing independent samples.
Detection of pH changes
Detection of pH changes was carried out using a pH meter F-22 (Horiba, Kyoto, Japan) according to the time course. The epidermis-peeled leaf segments (0.45 g) were floated on the 5 ml solution containing 5 ng ml–1 victorin or 50 µg ml–1 VicBSA and the indicated reagents. Recovered testing solution was used for pH measurement.
DNA extraction and analysis
Oat leaves (0.3 g) were ground into a fine powder using liquid nitrogen. Each sample was incubated for 30 min at 65°C in 600 µl of 2% (w/v) CTAB solution [100 mM Tris–HCl, pH 8.0, 1.4 M NaCl, 20 mM EDTA and 2% {w/v) cetyltrimethyl ammonium bromide], then mixed with an equal volume of chloroform : isoamyl alcohol (24 : 1, v/v). After gently shaking for 5 min, the mixture was centrifuged for 15 min at 10,000×g. Total DNA was precipitated by the addition of 800 µl of 1% (w/v) CTAB solution [50 mM Tris–HCl, pH 8.0, 10 mM EDTA and 1% (w/v) cetyltrimethyl ammonium bromide] to the supernatant. After centrifugation for 10 min at 8,000×g, the pellet was dissolved in 500 µl of 1 M CsCl, and subjected to alcohol precipitation with a 2-fold volume of 100% ethanol. DNA was recovered by centrifugation for 10 min at 10,000×g, followed by washing with 70% ethanol, and dissolved in 500 µl of Tris-EDTA buffer containing 40 µg ml–1 RNase. After incubation at 37°C for 30 min, phenol extraction and ethanol precipitation were performed to recover DNA. Samples were run on a 2% (w/v) agarose gel and stained with 0.5 µg ml–1 ethidium bromide.
Electron microscopic analysis
The treated tissues were cut into ∼1–2 mm2 pieces. Then, the samples were pre-fixed in 2.5% (v/v) glutaraldehyde and 2% (v/v) paraformaldehyde with 1/15 M phosphate (PBS) buffer (pH 7.2). After three washes with PBS buffer, the samples were post-fixed with 1% (w/v) osmium tetroxide in PBS for 1 h and then dehydrated. For electron microscopic observation, samples were embedded in Epon 812 resin (Nisshin EM, Tokyo, Japan) and blocks were cut with a Histo diamond knife (Diatome, Bienne, Switzerland) on a Reichert-Nissei Ultracut (Leica AG, Austria) to obtain ultrathin sections (90 nm), which were then collected on copper grids (200 mesh). Sections were examined and photographed using a Hitachi-7100 (Hitachi, Tokyo, Japan) transmission electron microscope at an accelerating voltage of 75 kV.
Acknowledgments
We are grateful to Dr. T. J. Wolpert (Oregon State University, Corvallis, OR, USA) for kindly providing victorin C. This work was supported in part by a Grant-in-Aid for Scientific Research (No. 12052215) from the Ministry of Education, Science, Sports and Culture of Japan.
These authors contributed equally to this work.
Present address: Duke University, Department of Biology, LSRC Building, Research Drive, Box 91000, Durham, NC 27708-1000, USA.
Present address: Max Planck Institute for Plant Breeding Research, Carl-von-Linné-Weg 10, D-50829 Köln, Germany.
Present address: Hiroshima University, Department of Applied Bioscience, 1-4-4 Kagamiyama, Higashi-Hiroshima, Hiroshima, 739-8528 Japan.
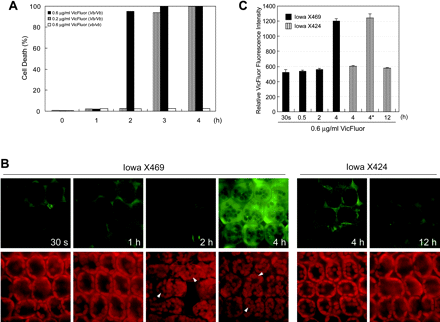
Fig. 1 VicFluor permeates victorin-sensitive cells after cell death. (A) The kinetics of VicFluor-induced cell death. Epidermis-peeled victorin-sensitive (Iowa X469, Vb/Vb) and insensitive leaves (Iowa X424, vb/vb) were incubated with VicFluor at the indicated concentrations for 1–4 h. Living and dead cells were observed by FDA staining at the indicated time points, and dead cells were expressed as a percentage of the total cells counted. Data are means (n = 2). (B) Cytofluorimetric analyses of VicFluor distribution. The localization of VicFluor fluorescence (upper panels) and chloroplasts (lower panels) in victorin-sensitive and -insensitive leaves was monitored in the same field by a fluorescence microscope coupled with a CCD camera. Epidermis-peeled leaf segments were treated with 0.6 µg ml–1 VicFluor for the indicated time and viewed with excitation of 488 nm. The images are representative of at least four experiments. Note that dead cells demonstrated by arrowheads show the abnormal distribution of the chloroplasts. (C) Changes in the intensity of intracellular VicFluor fluorescence. Epidermis-peeled leaf segments of Iowa X469 (black bars) and Iowa X424 (striped bars) were treated with 0.6 µg ml–1 VicFluor. The VicFluor signal in 20 independent mesophyll cells was monitored with excitation of 488 nm and evaluated by MetaMorph software. Data represent the mean value and SD of three independent experiments. Note that an asterisk shows the fluorescence value obtained from dead cells (<2%) in toxin-insensitive leaves.
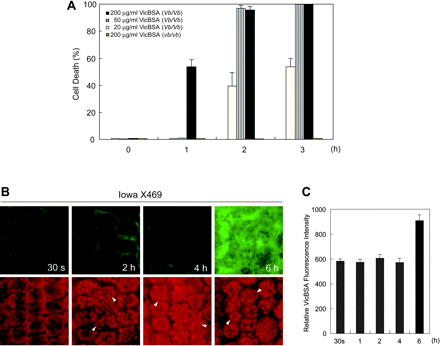
Fig. 2 A bovine serum albumin–fluorescein conjugate of victorin (VicBSA) induces cell death in victorin-sensitive leaves. (A) The kinetics of VicBSA-induced cell death. Epidermis-peeled leaf segments of victorin-sensitive (Iowa X469, Vb/Vb) and -insensitive cultivars (Iowa X424, vb/vb) were incubated with VicBSA at the indicated concentrations for 1–3 h. Living cells were observed by FDA staining, and dead cells were expressed as percentages of the total cells counted. Data represent the mean value and SD of three independent experiments. (B) Distribution of VicBSA in victorin-sensitive cells. The localization of VicBSA fluorescence (upper panels) and chloroplasts (lower panels) in Iowa X469 was monitored in the same field by a fluorescence microscope coupled with a CCD camera. Epidermis-peeled leaves were treated with 50 µg ml–1 VicBSA for the indicated time, and the fluorescence was detected at 488 nm. The images are representative of at least three experiments. Note that dead cells demonstrated by arrowheads show an abnormal distribution of the chloroplasts. (C) Changes in the intensity of the intracellular VicBSA signal. Epidermis-peeled victorin-sensitive leaves were treated with 50 µg ml–1 VicBSA for the indicated time. The intensity of VicBSA fluorescence in 20 independent mesophyll cells was monitored with excitation of 488 nm and evaluated by MetaMorph software. Data represent the mean value and SD of three independent experiments.
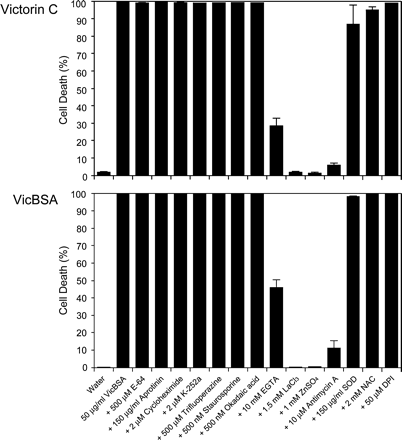
Fig. 3 Effect of inhibitors of signal transduction pathways on victorin- and VicBSA-induced cell death. Epidermis-peeled victorin-sensitive leaves were treated with 5 ng ml–1 victorin C or 50 µg ml–1 VicBSA plus the indicated reagents for 3 h. Living cells were estimated by FDA staining, and dead cells were expressed as a percentage of the total cells counted. Data represent the mean value and SD of three independent experiments.
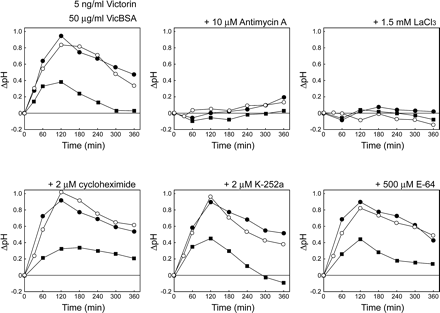
Fig. 4 Victorin- and VicBSA-induced cell death is associated with extracellular alkalization. Epidermis-peeled victorin-sensitive leaves were incubated with 5 ng ml–1 victorin C (closed circle) or 50 µg mL–1 VicBSA (open circle) plus the indicated chemicals or with inhibitors alone (square). The kinetics of pH changes in test solutions were monitored by pH meter.
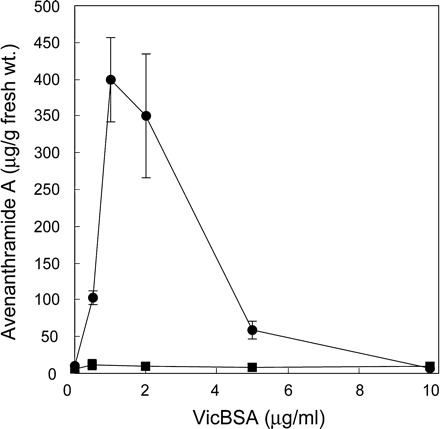
Fig. 5 VicBSA elicits the production of avenanthramide (A) Peeled leaf segments of victorin-sensitive (circle) and insensitive cultivars (square) were treated with VicBSA at the indicated concentrations. Avenanthramide A in test solutions was estimated 24 h after treatment by HPLC. Data represent the mean ± SD from three experiments.
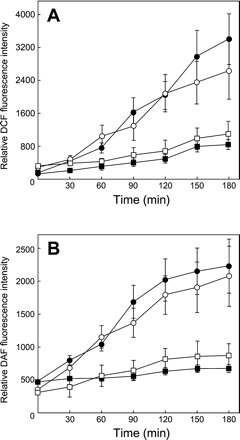
Fig. 6 Victorin and VicBSA induce the generation of reactive oxygen intermediates (ROIs, A) and nitric oxide (NO, B) in victorin-sensitive leaves. Primary leaves of sensitive cultivar Iowa X469 (circle) and insensitive cultivar Iowa X424 (square) were treated with 5 ng ml–1 victorin C (closed symbols) or 50 µg ml–1 VicBSA (open symbols) for the indicated time. The time course of ROI and NO accumulation was monitored by the fluorescence of dichlorofluorescein (DCF) and diaminofluorescein (DAF), respectively. Points and bars represent the mean value and SD of three independent experiments.
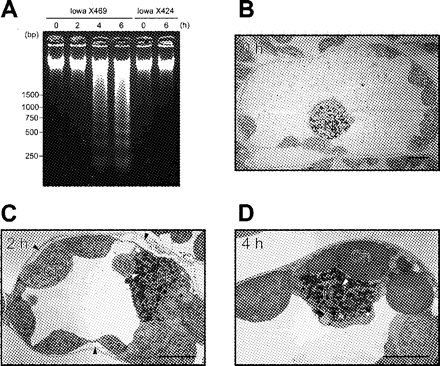
Fig. 7 VicBSA induces apoptotic responses in victorin-sensitive leaves. (A) Time course of VicBSA-induced DNA laddering. Peeled leaf segments of victorin-sensitive (Iowa X469) and -insensitive cultivars (Iowa X424) were treated with 50 µg ml–1 VicBSA for the indicated time. DNA was analyzed by ethidium bromide staining on a 2% (w/v) agarose gel. (B–D) Ultrastructural changes in VicBSA-treated cells. Peeled victorin-sensitive leaves were incubated with 50 µg ml–1 VicBSA for the indicated time. Open arrowheads show localized condensation of heterochromatin, and closed arrowheads indicate plasmolysis. N, nucleus; Ch, chloroplast. Bar in (F) to (H) = 5 µm.
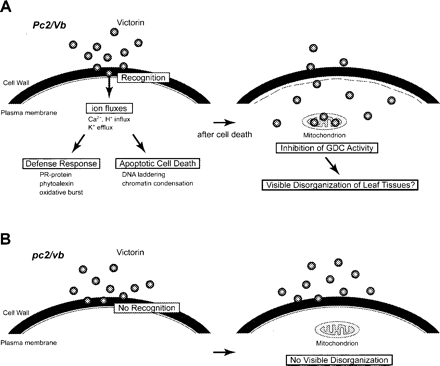
Fig. 8 The current model of victorin-induced cellular responses. Victorin-sensitive cells. (A) Victorin may interact with an extracellular mediator(s) in Vb/Pc-2 oats and stimulates ion fluxes across the plasma membrane, followed by the activation of defense responses and rapid cell death. After cell death, victorin may permeate cells and is distributed in the mitochondria, inducing a senescence-like response (Navarre and Wolpert 1999). (B) Victorin-insensitive cells. Victorin neither induces cell death nor enters the resistant cells.
Abbreviations
- DAF
4,5-diaminofluorescein
- DCF
2′,7′,-dichlorofluorescein
- DPI
diphenyleneiodonium chloride
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- FDA
fluorescein diacetate
- GDA
glycine decarboxylase activity
- GDC
glycine decarboxylase complex
- HST
host-selective toxin
- MPT
mitochondrial permeability transition
- NAC
N-acetylcysteine
- NHS
N-hydroxysuccinimide
- PCD
programmed cell death
- ROI
reactive oxygen intermediate
- SOD
superoxide dismutase
- VicBSA
bovine serum albumin–fluorescein derivative of victorin
- VicFluor
fluorescein derivative of victorin
References
Akimitsu, K., Hart, L.P. and Walton, J.D. (
Akimitsu, K., Hart, L.P. and Walton, J.D. (
Akimitsu, K., Hart, L.P., Walton, J.D. and Hollingsworth, R. (
Atkinson, S.J., Hosford, M.A. and Molitoris, B.A. (
Atkinson, M.M., Midland, S.L., Sims, J.J. and Keen, N.T. (
Blume, B., Nurnberger, T., Nass, N. and Scheel, D. (
Chin, D. and Means, A.R. (
Curtis, M.J. and Wolpert, T.J. (
Curtis, M.J. and Wolpert, T.J. (
Dangl, J.L. and Jones, J.D. (
Govrin, E.M. and Levine, A. (
Keen, N.T. and Buzzell, R.I. (
Knodler, L.A., Celli, J. and Finlay, B.B. (
Kroemer, G., Dallaporta, B. and Resche-Rigon, M. (
Kusaka, K., Tada, Y., Shigemi, T., Sakamoto, M., Nakayashiki, H., Tosa, Y. and Mayama, S. (
Lam, E., Kato, N. and Lawton, M. (
Lincoln, J.E., Richael, C., Overduin, B., Smith, K., Bostock, R. and Gilchrist, D.G. (
Mackey, D., Holt, BF 3rd, Wiig, A. and Dangl, J.L. (
Mayama, S., Bordin, A.P.A., Morikawa, T., Tanpo, H. and Kato, H. (
Mayama, S., Tani, T., Midland, S.L., Sims, J.J. and Keen, N.T. (
Meehan, F. and Murphy, H.C. (
Mittler, R., Shulaev, V. and Lam, E. (
Navarre, D.A. and Wolpert, T.J. (
Navarre, D.A. and Wolpert, T.J. (
Nurnberger, T., Nennstiel, D., Jabs, T., Sacks, W.R., Hahlbrock, K. and Scheel, D. (
Pontier, D., Mittler, R. and Lam, E. (
Rines, H.W. and Luke, H.H. (
Tada, Y., Hata, S., Takata, Y., Nakayashiki, H., Tosa, Y. and Mayama, S. (
Tada, Y., Mori, T., Shinogi, T., Yao, N., Takahashi, S., Betsuyaku, S., Sakamoto, M., Park, P., Nakayashiki, H., Tosa, Y. and Mayama, S. (
Ullrich, C.I. and Novacky, A.J. (
Walton, J.D. and Earle, E.D. (
Wolpert, T.J., Dunkle, L.D. and Ciuffetti, L.M. (
Wolpert, T.J. and Macko, V. (
Wolpert, T.J., Macko, V., Acklin, W., Jaun, B., Seibl, J., Meili, J. and Arigoni, D. (
Wolpert, T.J., Navarre, D.A., Moore, D.L. and Macko, V. (
Yang, Q., Trinh, H.X., Imai, S., Ishihara, A., Zhang, L., Nakayashiki, H., Tosa, Y. and Mayama, S. (
Yao, N., Tada, Y., Park, P., Nakayashiki, H., Tosa, Y. and Mayama, S. (
Yao, N., Tada, Y., Sakamoto, M., Nakayashiki, H., Park, P., Tosa, Y. and Mayama, S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}