
Abstract
 composite powder has been synthesized via spray pyrolysis of cobalt nitrate-sugar solution at
composite powder has been synthesized via spray pyrolysis of cobalt nitrate-sugar solution at  and assessed for application as anode materials in Li-ion batteries. Microstructural characterization by scanning electron microscopy, transmission electron microscopy, and energy-dispersive X-ray spectroscopy confirm an even distribution of carbon throughout particles, as well as the presence of a carbon-based surface sheath surrounding
and assessed for application as anode materials in Li-ion batteries. Microstructural characterization by scanning electron microscopy, transmission electron microscopy, and energy-dispersive X-ray spectroscopy confirm an even distribution of carbon throughout particles, as well as the presence of a carbon-based surface sheath surrounding  particle agglomerates. Charge-discharge cycling of half-cells indicates a stable reversible discharge capacity above
particle agglomerates. Charge-discharge cycling of half-cells indicates a stable reversible discharge capacity above  . Equivalent circuit modeling of Nyquist plots show the
. Equivalent circuit modeling of Nyquist plots show the  electrode has significant kinetic advantages over non-composite transition metal oxide electrodes.
electrode has significant kinetic advantages over non-composite transition metal oxide electrodes.
Export citation and abstract BibTeX RIS
Poizot et al.1 first reported that electrodes made of nanosized transition metal oxide (MO) particles ( ,
,  ,
,  ,
,  ,
,  , and
, and  ) exhibit reversible capacities up to three times higher than electrodes composed of graphite. The most interesting aspect of this result is that these transition metal oxides do not display the classical hallmarks that have often been used by researchers to identify suitable Li-ion battery materials (i.e., reversible alloying with lithium or the existence of an open crystal framework). Rather, the reaction of these materials with
) exhibit reversible capacities up to three times higher than electrodes composed of graphite. The most interesting aspect of this result is that these transition metal oxides do not display the classical hallmarks that have often been used by researchers to identify suitable Li-ion battery materials (i.e., reversible alloying with lithium or the existence of an open crystal framework). Rather, the reaction of these materials with  involves the thermodynamically unlikely reversible formation and decomposition of
involves the thermodynamically unlikely reversible formation and decomposition of  .2–7 Impetus for this reaction occurs through the formation of highly electrochemically reactive metal nanoparticles during cell discharge
.2–7 Impetus for this reaction occurs through the formation of highly electrochemically reactive metal nanoparticles during cell discharge
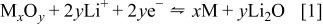
From all of the transition metal oxides studied, the  system offers particular promise due to a reported reversible capacity in excess of
system offers particular promise due to a reported reversible capacity in excess of  .8 Unfortunately, the practical usage of
.8 Unfortunately, the practical usage of  cells in Li-ion batteries has been frustrated by capacity fade and a poor cycle life.9, 10 Part of these problems may be attributed to significant volume changes that occur during lithium uptake and removal (molar volume change ∼100%) which may result in a loss of electrical contact and electrode failure. This situation is analogous to that found in tin-based oxides and alloys that also suffer significant volume changes.11 Another important factor identified as influencing the cyclability of the
cells in Li-ion batteries has been frustrated by capacity fade and a poor cycle life.9, 10 Part of these problems may be attributed to significant volume changes that occur during lithium uptake and removal (molar volume change ∼100%) which may result in a loss of electrical contact and electrode failure. This situation is analogous to that found in tin-based oxides and alloys that also suffer significant volume changes.11 Another important factor identified as influencing the cyclability of the  electrode is the nature and electrochemical behavior of the solid electrolyte interphase (SEI). The SEI begins forming in the first discharge (reduction of MO) as a result of secondary reduction reactions within the electrolyte and has the appearance of a "polymer-like" gel.6 Subsequent charging of the cell will often cause a partial dissolution of the SEI. The exact composition of the SEI obviously varies between systems and electrolytes, but the film is mildly conducting and is composed primarily of lithium-based carbonates and alkylcarbonates. This SEI formation-dissolution phenomenon that is caused by discharge-charge cycling of the cell is particularly influenced by the powder texture (crystallite and particle size, surface area, and surface composition) for each MO system. If precursor powders are too small, the SEI formation process dominates and cycle life of the cell is reduced. Specifically, smaller precursor MO powder particles will form more surface active metal nanoparticles during discharge, which are then available for catalytic reduction of the electrolyte and hence SEI formation. For example, work on nanosized
electrode is the nature and electrochemical behavior of the solid electrolyte interphase (SEI). The SEI begins forming in the first discharge (reduction of MO) as a result of secondary reduction reactions within the electrolyte and has the appearance of a "polymer-like" gel.6 Subsequent charging of the cell will often cause a partial dissolution of the SEI. The exact composition of the SEI obviously varies between systems and electrolytes, but the film is mildly conducting and is composed primarily of lithium-based carbonates and alkylcarbonates. This SEI formation-dissolution phenomenon that is caused by discharge-charge cycling of the cell is particularly influenced by the powder texture (crystallite and particle size, surface area, and surface composition) for each MO system. If precursor powders are too small, the SEI formation process dominates and cycle life of the cell is reduced. Specifically, smaller precursor MO powder particles will form more surface active metal nanoparticles during discharge, which are then available for catalytic reduction of the electrolyte and hence SEI formation. For example, work on nanosized  precursor powders has shown that rapid SEI thickening caused by enhanced electrolyte degradation can occur to a point where Li transfer is significantly impeded and capacity fades.12 Alternatively, work on
precursor powders has shown that rapid SEI thickening caused by enhanced electrolyte degradation can occur to a point where Li transfer is significantly impeded and capacity fades.12 Alternatively, work on  cells that use larger microsized precursor MO powder have accredited an excellent capacity retention to the active formation-dissolution of the SEI.2 Therefore, it is feasible that the SEI may be indirectly engineered by careful selection of the above characteristics to improve the cyclability of the transition MO electrode.
cells that use larger microsized precursor MO powder have accredited an excellent capacity retention to the active formation-dissolution of the SEI.2 Therefore, it is feasible that the SEI may be indirectly engineered by careful selection of the above characteristics to improve the cyclability of the transition MO electrode.
There has been a large amount of fundamental studies on the electrochemical performance of transition MOs that have focused on reaction mechanisms,1, 2, 13 the development of possible intermediate phases,14, 15 and the effect of particle size on electrochemical performance.14 However, to enhance the commercial viability of these materials and their application to large scale technology such as electric vehicles, practical improvements must be made in terms of capacity retention and cycle life. To this end, we have synthesized  –carbon (referred to as
–carbon (referred to as  hereafter) composite powders by chemical spray pyrolysis using common sugar (sucrose) as a carbon source. Sucrose is an attractive precursor to synthesize carbon because it is cheap, abundant, and has a high chemical purity. It has been shown that the confinement of MO clusters in a disordered carbon matrix can preserve electrical contact and cushion internal stresses, thereby leading to improved cycle life in a Li-ion cell.16, 17
hereafter) composite powders by chemical spray pyrolysis using common sugar (sucrose) as a carbon source. Sucrose is an attractive precursor to synthesize carbon because it is cheap, abundant, and has a high chemical purity. It has been shown that the confinement of MO clusters in a disordered carbon matrix can preserve electrical contact and cushion internal stresses, thereby leading to improved cycle life in a Li-ion cell.16, 17
Experimental
Composite preparation
Nanostructured  ,
,  , and
, and  powders were prepared in separate experiments using a vertical spray pyrolysis apparatus as described in our previous work.18, 19 The
powders were prepared in separate experiments using a vertical spray pyrolysis apparatus as described in our previous work.18, 19 The  and
and  powders were obtained by spraying aqueous
powders were obtained by spraying aqueous  cobalt or nickel nitrate solutions at ambient temperature through an ultrasonic nozzle at
cobalt or nickel nitrate solutions at ambient temperature through an ultrasonic nozzle at  into an open air
into an open air  quartz tube at
quartz tube at  . For preparation of the
. For preparation of the  powder, the precursor solution contained
powder, the precursor solution contained  sucrose and
sucrose and  cobalt nitrate. The experimental conditions for pyrolysis of this solution were identical to those described for
cobalt nitrate. The experimental conditions for pyrolysis of this solution were identical to those described for  and
and  powders. All final powder products were confirmed by X-ray diffraction (XRD) to be reacted and usable as-received, however powders were dried overnight at
powders. All final powder products were confirmed by X-ray diffraction (XRD) to be reacted and usable as-received, however powders were dried overnight at  in a vacuum oven to evaporate excess moisture prior to electrochemical testing.
in a vacuum oven to evaporate excess moisture prior to electrochemical testing.
Characterization
Analysis of the composition and crystal structure of the powders was conducted using a Philips PW1730 X-ray diffractometer with monochromatized  radiation
radiation  at a scan rate of
at a scan rate of  . The carbon content of the product
. The carbon content of the product  powder was analyzed by thermogravimetric (TG) method performed in static air on a Setaram 92 differential thermal analysis (DTA) apparatus at
powder was analyzed by thermogravimetric (TG) method performed in static air on a Setaram 92 differential thermal analysis (DTA) apparatus at  . An estimate of the crystallite sizes was calculated using the Scherrer equation:
. An estimate of the crystallite sizes was calculated using the Scherrer equation:  , where λ represents the X-ray wavelength, β is the observed full width at half-maximum (fwhm), and
, where λ represents the X-ray wavelength, β is the observed full width at half-maximum (fwhm), and  is the Bragg angle.20 The morphology of the powders was observed by a JEOL JSM-6460A scanning electron microscope (SEM) and JEOL 2010 transmission electron microscope (TEM). The results obtained from TG experiment on
is the Bragg angle.20 The morphology of the powders was observed by a JEOL JSM-6460A scanning electron microscope (SEM) and JEOL 2010 transmission electron microscope (TEM). The results obtained from TG experiment on  powder were compared to the carbon content calculated using an energy dispersive X-ray spectroscopy (EDXS) instrument attached to the abovementioned SEM. The specific surface area of the powders was calculated using the Brunauer, Emmett, Teller (BET) multipoint method with
powder were compared to the carbon content calculated using an energy dispersive X-ray spectroscopy (EDXS) instrument attached to the abovementioned SEM. The specific surface area of the powders was calculated using the Brunauer, Emmett, Teller (BET) multipoint method with  adsorbate at
adsorbate at  using a Quantachrome Nova 1000 Autosorb-1 gas sorption system.21 Powders were weighed, placed in a
using a Quantachrome Nova 1000 Autosorb-1 gas sorption system.21 Powders were weighed, placed in a  chamber, and outgassed at
chamber, and outgassed at  under inert gas flow for
under inert gas flow for  prior to measurement. Particle size distributions were determined using a Malvern Instruments Mastersizer.
prior to measurement. Particle size distributions were determined using a Malvern Instruments Mastersizer.
Electrochemical assessment
Electrochemical measurements were conducted by assembly of standard R2032 type coin cells. In the case of  and
and  powders, working electrodes were constructed by mixing
powders, working electrodes were constructed by mixing  active powder,
active powder,  carbon black (Lexel, 99%), and
carbon black (Lexel, 99%), and  polyvinylidene fluoride (Aldrich, 99%) in a mortar and pestle. For the
polyvinylidene fluoride (Aldrich, 99%) in a mortar and pestle. For the  powder,
powder,  of active material was used with the remaining
of active material was used with the remaining  made up of polyvinylidene fluoride binder. The dry powder mix was blended with N-methylpyrrolidinone to make a slurry, which was then spread uniformly on
made up of polyvinylidene fluoride binder. The dry powder mix was blended with N-methylpyrrolidinone to make a slurry, which was then spread uniformly on  by
by  thick copper foil (99.99%) substrate. The electrodes were dried in a vacuum oven for
thick copper foil (99.99%) substrate. The electrodes were dried in a vacuum oven for  at
at  then cold pressed at
then cold pressed at  in a uniaxial hydraulic press. All electrodes contained no more than
in a uniaxial hydraulic press. All electrodes contained no more than  of active material and were assembled into a test coin cell in a high purity argon-filled glove box (Mbraun, Unilab, USA) where the concentration of
of active material and were assembled into a test coin cell in a high purity argon-filled glove box (Mbraun, Unilab, USA) where the concentration of  and
and  . Battery grade lithium foil was used as both the counter and reference electrodes.
. Battery grade lithium foil was used as both the counter and reference electrodes.  2400 was used as a separator membrane soaked in
2400 was used as a separator membrane soaked in 
 dissolved in a 1:1 (vol:vol) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) electrolyte (Merck KgaA, Germany). Charge-discharge cycles of the half-cells were measured between 0.02 and
dissolved in a 1:1 (vol:vol) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) electrolyte (Merck KgaA, Germany). Charge-discharge cycles of the half-cells were measured between 0.02 and  vs lithium at the C/30 rate using a Neware battery cycler in galvanostatic mode. Specific capacity was calculated based on the mass of active material in the electrode and all electrochemical testing was carried out at ambient temperature
vs lithium at the C/30 rate using a Neware battery cycler in galvanostatic mode. Specific capacity was calculated based on the mass of active material in the electrode and all electrochemical testing was carried out at ambient temperature  . Electrochemical impedance spectroscopy (EIS) measurements were conducted using a CH Instruments electrochemical workstation (CHI 660A). The impedance spectra were recorded potentiostatically by applying an ac voltage of
. Electrochemical impedance spectroscopy (EIS) measurements were conducted using a CH Instruments electrochemical workstation (CHI 660A). The impedance spectra were recorded potentiostatically by applying an ac voltage of  at various open-circuit voltages (OCVs) on the tenth discharge. Simulation of impedance spectra with an equivalent circuit was performed over identical frequency range using Zview computer software (Version 2.8, Scribner Associates, USA). The program fits the parameters of the equivalent circuit of each of the electrodes using the complex nonlinear least squares (CNLS) method, which provides simultaneous fits for each parameter. Note that the electrodes used for the impedance analysis had a uniform film thickness in the
at various open-circuit voltages (OCVs) on the tenth discharge. Simulation of impedance spectra with an equivalent circuit was performed over identical frequency range using Zview computer software (Version 2.8, Scribner Associates, USA). The program fits the parameters of the equivalent circuit of each of the electrodes using the complex nonlinear least squares (CNLS) method, which provides simultaneous fits for each parameter. Note that the electrodes used for the impedance analysis had a uniform film thickness in the  range to allow a valid comparison of impedance values.
range to allow a valid comparison of impedance values.
Results and Discussion
Figure 1 shows the XRD patterns for the sprayed  ,
,  , and
, and  powders. The diffraction patterns seen in Fig. 1a are indexed to the cubic
powders. The diffraction patterns seen in Fig. 1a are indexed to the cubic  phase with
phase with  space group (JPDD ICDD reference 65-3103). Crystallite sizes for the
space group (JPDD ICDD reference 65-3103). Crystallite sizes for the  and
and  powders were calculated using the Scherrer equation at 10.2 and 12.0 nm, respectively. The swollen nature of the
powders were calculated using the Scherrer equation at 10.2 and 12.0 nm, respectively. The swollen nature of the  graph at low scattering angle
graph at low scattering angle  is indicative of disordered carbon.22, 23 The diffraction pattern in Fig. 1b shows Bragg reflections indexed according to pure phase
is indicative of disordered carbon.22, 23 The diffraction pattern in Fig. 1b shows Bragg reflections indexed according to pure phase  bunsenite compound with cubic crystal structure and
bunsenite compound with cubic crystal structure and  space group (JPDD ICDD reference 01-1239). The broadened nature of the three main diffraction peaks at 37.4, 43.5, and 62.6° [(111), (200), and (220) reflections, respectively] suggests that the powder has a nanocrystalline morphology, which is confirmed by a calculated crystallite size of
space group (JPDD ICDD reference 01-1239). The broadened nature of the three main diffraction peaks at 37.4, 43.5, and 62.6° [(111), (200), and (220) reflections, respectively] suggests that the powder has a nanocrystalline morphology, which is confirmed by a calculated crystallite size of  . Dark field TEM analysis (data not shown) supports that the crystallite sizes for all powders are of the magnitude calculated empirically.
. Dark field TEM analysis (data not shown) supports that the crystallite sizes for all powders are of the magnitude calculated empirically.
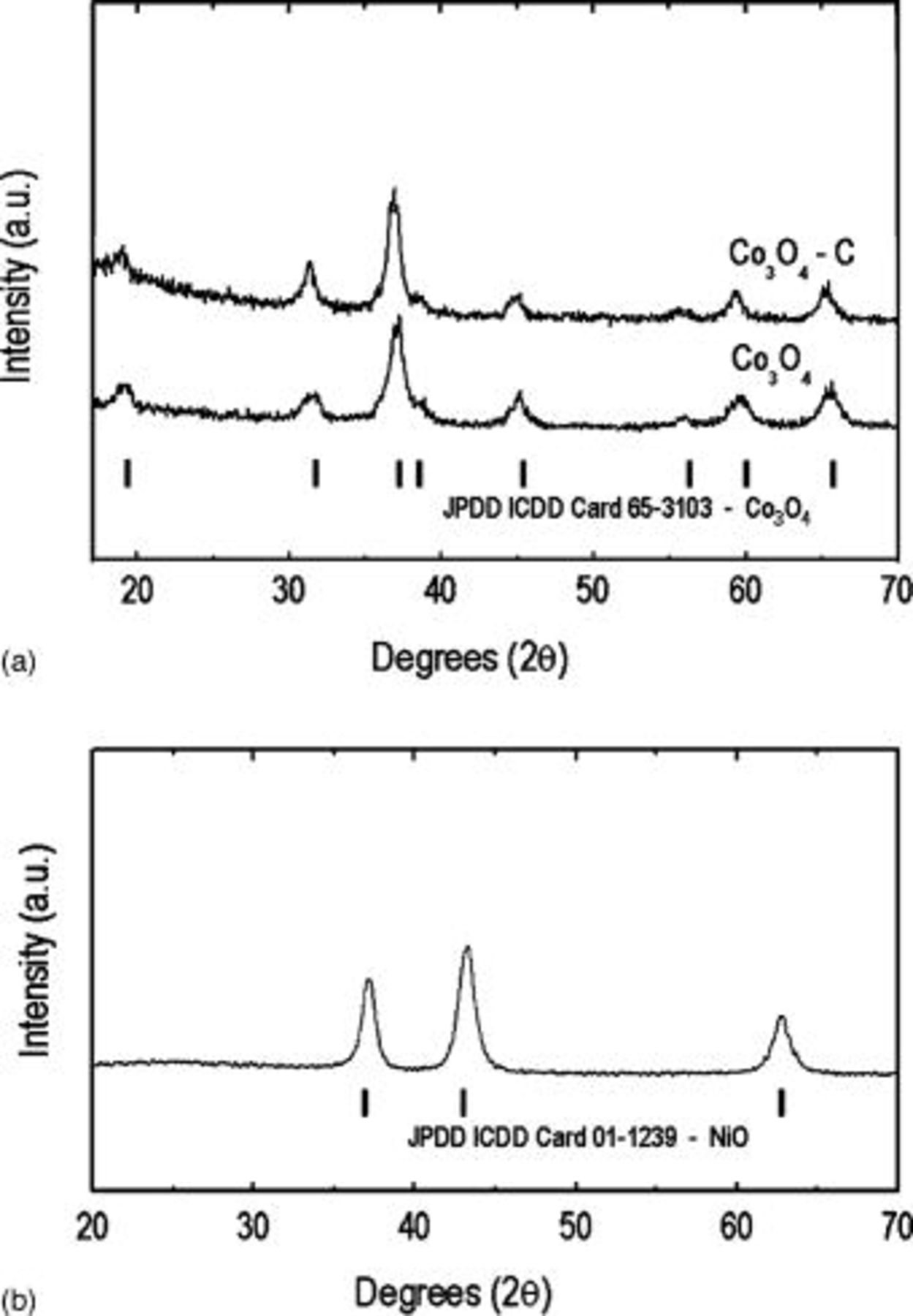
Figure 1. XRD patterns of (a)  ,
,  and (b)
and (b)  powders.
powders.
The actual quantity of carbon in the  composite powder was calculated by TG analysis at
composite powder was calculated by TG analysis at  and EDXS area analysis at
and EDXS area analysis at  . The close agreement of these values confirms that the C content of the composite to be in the
. The close agreement of these values confirms that the C content of the composite to be in the  range. It is expected that a large fraction of the carbon in the precursor solution was also lost to the atmosphere as
range. It is expected that a large fraction of the carbon in the precursor solution was also lost to the atmosphere as  during pyrolysis, and can be explained by considering the behavior of sucrose upon heating. When sucrose is hydrolyzed, it first decomposes into its two smaller monosaccharide isostructural constituents, namely, glucose and fructose (Reaction 2).24 Slow heating initiates a series of up to 25 sequential reactions occur whereby different classes of compounds are formed including aldehydes, ketones, carboxylic acids.25 However, regardless of heating rate there are two main exothermic reactions of decomposition that occur before
during pyrolysis, and can be explained by considering the behavior of sucrose upon heating. When sucrose is hydrolyzed, it first decomposes into its two smaller monosaccharide isostructural constituents, namely, glucose and fructose (Reaction 2).24 Slow heating initiates a series of up to 25 sequential reactions occur whereby different classes of compounds are formed including aldehydes, ketones, carboxylic acids.25 However, regardless of heating rate there are two main exothermic reactions of decomposition that occur before  and involve the formation of large quantities of
and involve the formation of large quantities of  ,
,  (Reaction 3), and carbon (Reaction 4)
(Reaction 3), and carbon (Reaction 4)
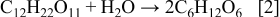
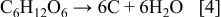
Figure 2 shows SEM and TEM images of the as-prepared  powder at different magnifications. Figure 2a shows a low magnification SEM of the morphology of the
powder at different magnifications. Figure 2a shows a low magnification SEM of the morphology of the  powder. The majority of particles measure less than
powder. The majority of particles measure less than  and consist of a mixture of partially and fully developed hollow spheres, with the average wall thickness of
and consist of a mixture of partially and fully developed hollow spheres, with the average wall thickness of  . EDXS analysis indicates that carbon is evenly distributed across the surface of
. EDXS analysis indicates that carbon is evenly distributed across the surface of  particles and also throughout the wall thickness. The TEM image in Fig. 2b shows that each particle visible in Fig. 2a is actually an agglomeration of sub-micrometer
particles and also throughout the wall thickness. The TEM image in Fig. 2b shows that each particle visible in Fig. 2a is actually an agglomeration of sub-micrometer  particles coated in a thin C-rich sheath, with the composition of the surface layer being confirmed by selected area electron diffraction (SAED). It is thought that this type of morphology may form as a result of the highly reactive particles coalescing within the reaction chamber, moments after being sprayed. The thickness of the C sheath is
particles coated in a thin C-rich sheath, with the composition of the surface layer being confirmed by selected area electron diffraction (SAED). It is thought that this type of morphology may form as a result of the highly reactive particles coalescing within the reaction chamber, moments after being sprayed. The thickness of the C sheath is  and can be seen in Fig. 2c coating grains containing a fine distribution of disordered carbon (black spots
and can be seen in Fig. 2c coating grains containing a fine distribution of disordered carbon (black spots  ) and domains of crystalline
) and domains of crystalline  .
.
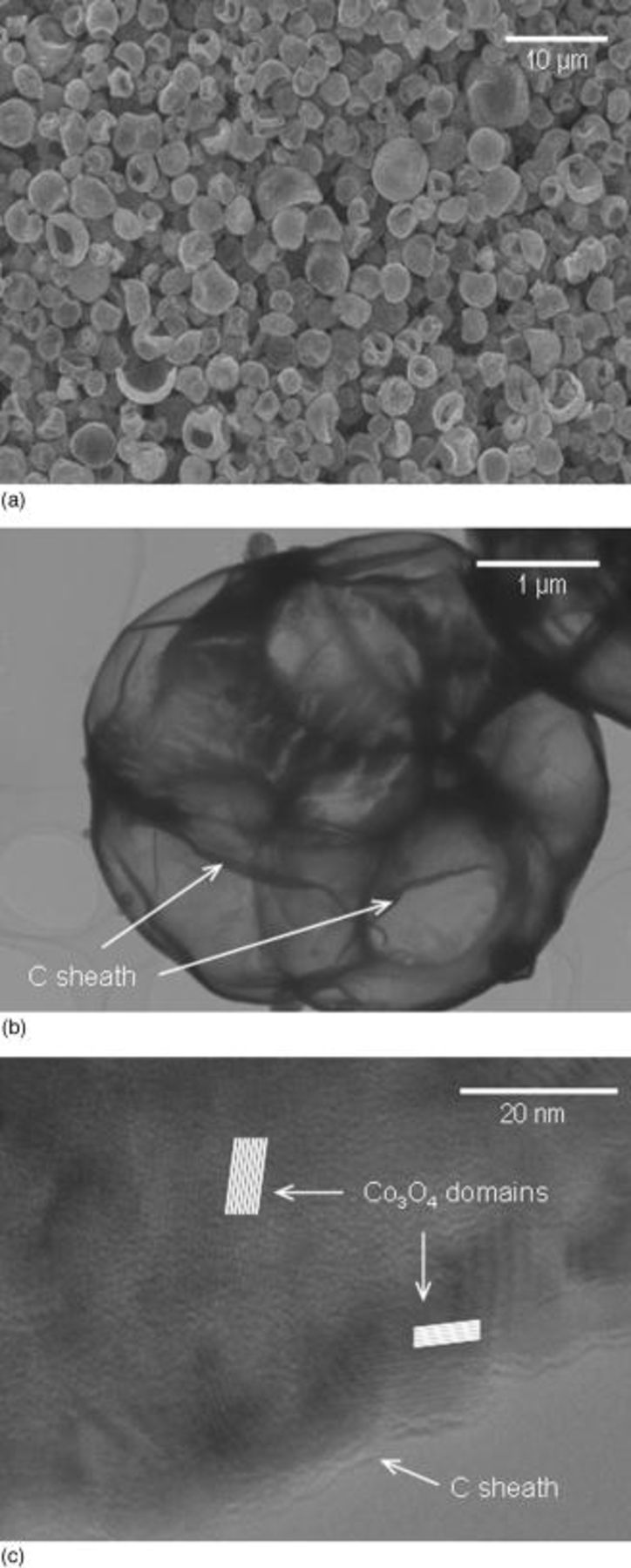
Figure 2. Images of  powder showing (a) a low magnification SEM view, (b) TEM of the nanoporous carbon sheath coated on a
powder showing (a) a low magnification SEM view, (b) TEM of the nanoporous carbon sheath coated on a  particle, (c) and a TEM image of finely distributed disordered C and crystalline
particle, (c) and a TEM image of finely distributed disordered C and crystalline  domains within an individual particle.
domains within an individual particle.
Figure 3 shows the particle size distribution and BET surface areas of  ,
,  , and
, and  powders. All powders show a narrow Gaussian distribution from 2 to
powders. All powders show a narrow Gaussian distribution from 2 to  that supports SEM observations. The surface area of powders in decreasing order was
that supports SEM observations. The surface area of powders in decreasing order was 
 ,
, 
 , and
, and 
 . The higher surface area measured in
. The higher surface area measured in  is explained by the additional gases given off during processing that have resulted in an aerated powder product. Small angle X-ray scattering (SAX) experiments conducted in other works26 have indicated that pyrolyzed carbons contain a mix of open and closed nanopores that can significantly increase the powder surface area. In these studies, a maximum surface area was achieved at spray temperatures
is explained by the additional gases given off during processing that have resulted in an aerated powder product. Small angle X-ray scattering (SAX) experiments conducted in other works26 have indicated that pyrolyzed carbons contain a mix of open and closed nanopores that can significantly increase the powder surface area. In these studies, a maximum surface area was achieved at spray temperatures  , which corresponds well with the processing temperature used in our present study. From an electrochemical performance viewpoint, the higher surface area measured in
, which corresponds well with the processing temperature used in our present study. From an electrochemical performance viewpoint, the higher surface area measured in  powder implies that more electrolyte could be exposed to the particle surface. It is well documented that although this may produce initial improvements in the cell capacity, the electrode capacity can rapidly fade upon cycling due to the accelerated degradation of the electrolyte and concomitant formation of a thick SEI layer that surrounds active particles.3, 12 However, as seen in Fig. 4, the
powder implies that more electrolyte could be exposed to the particle surface. It is well documented that although this may produce initial improvements in the cell capacity, the electrode capacity can rapidly fade upon cycling due to the accelerated degradation of the electrolyte and concomitant formation of a thick SEI layer that surrounds active particles.3, 12 However, as seen in Fig. 4, the  electrode demonstrated superior electrochemical performance compared to
electrode demonstrated superior electrochemical performance compared to  and
and  electrodes. In Fig. 4a, the
electrodes. In Fig. 4a, the  electrode maintains a capacity
electrode maintains a capacity  for 50 cycles, whereas
for 50 cycles, whereas  and
and  electrodes show a steady decay to 300 and
electrodes show a steady decay to 300 and  respectively. Indeed, the
respectively. Indeed, the  electrode also demonstrates better capacity retention than
electrode also demonstrates better capacity retention than  electrodes that were prepared by thermal decomposition of a cobalt octacarbonyl compound and reported by our group elsewhere.9, 10 The first and fiftieth cycle discharge curves for electrodes made from
electrodes that were prepared by thermal decomposition of a cobalt octacarbonyl compound and reported by our group elsewhere.9, 10 The first and fiftieth cycle discharge curves for electrodes made from  ,
,  , and
, and  powders is shown in Fig. 4b. Here we see the voltage plateaus that were present for the noncomposite
powders is shown in Fig. 4b. Here we see the voltage plateaus that were present for the noncomposite  (at
(at  ) and
) and  (at
(at  ) during the first cycle, almost disappear in the fiftiethth cycle. Conversely for the
) during the first cycle, almost disappear in the fiftiethth cycle. Conversely for the  electrode, the voltage plateau that was responsible for the high capacity achieved in early cycling is preserved in the fiftieth cycle. We believe that part of the reason for the improved cyclability may be attributed to carbon coating on the surface of
electrode, the voltage plateau that was responsible for the high capacity achieved in early cycling is preserved in the fiftieth cycle. We believe that part of the reason for the improved cyclability may be attributed to carbon coating on the surface of  crystals. We expect that this coating could simultaneously shield electrolytes from advanced decomposition caused by exposure to Co nanoparticles, enhance the overall electronic conductivity of the electrode, and also provide a cushion effect to accommodate volume expansion and contraction during discharge-charge cycling.11 Furthermore, any volume changes upon cycling may also be partially accommodated by the fine distribution of disordered carbon present throughout the bulk powder.
crystals. We expect that this coating could simultaneously shield electrolytes from advanced decomposition caused by exposure to Co nanoparticles, enhance the overall electronic conductivity of the electrode, and also provide a cushion effect to accommodate volume expansion and contraction during discharge-charge cycling.11 Furthermore, any volume changes upon cycling may also be partially accommodated by the fine distribution of disordered carbon present throughout the bulk powder.
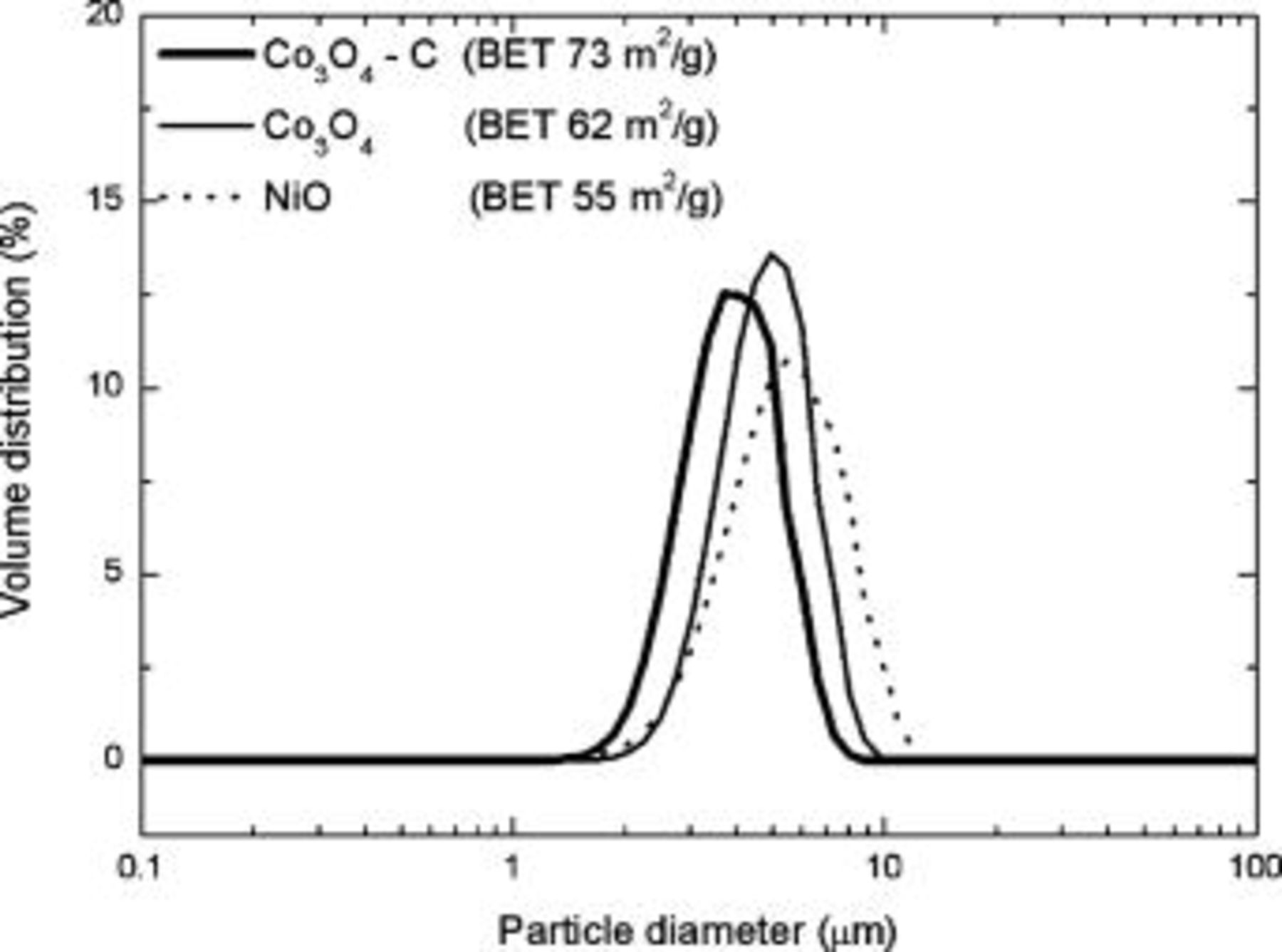
Figure 3. Particle size distribution and BET specific surface areas of  ,
,  , and
, and  powders.
powders.
Figure 4. Comparison of electrochemical performance for  ,
,  , and
, and  electrodes showing discharge capacity over (a) 50 cycles and the (b) first and fiftieth cycle discharge curves.
electrodes showing discharge capacity over (a) 50 cycles and the (b) first and fiftieth cycle discharge curves.
EIS was used to further elucidate the nature of the cyclability improvement demonstrated in the  electrode, by collecting Nyquist plots at various predetermined voltages (2.0, 1.6, 1.2, 1.0, 0.8, 0.4, and
electrode, by collecting Nyquist plots at various predetermined voltages (2.0, 1.6, 1.2, 1.0, 0.8, 0.4, and  ) on the tenth discharge cycle. Impedance spectra were recorded by applying an ac voltage of
) on the tenth discharge cycle. Impedance spectra were recorded by applying an ac voltage of  amplitude over the frequency range
amplitude over the frequency range  to
to  , after a
, after a  rest at each potential to attain equilibrium. EIS is a powerful technique for Li-ion electrode kinetic analysis due to the information that can be deduced for each reaction process; concerning that of the electrolyte, passivation layer(s), charge transfer, and
rest at each potential to attain equilibrium. EIS is a powerful technique for Li-ion electrode kinetic analysis due to the information that can be deduced for each reaction process; concerning that of the electrolyte, passivation layer(s), charge transfer, and  diffusion. The impedance spectra on 10th discharge for
diffusion. The impedance spectra on 10th discharge for  and
and  electrodes at various OCV are presented in Fig. 5. For all measurements, the overall shape of Nyquist plots show the characteristic depressed semicircle in the high and middle-frequency range and an extended tail at
electrodes at various OCV are presented in Fig. 5. For all measurements, the overall shape of Nyquist plots show the characteristic depressed semicircle in the high and middle-frequency range and an extended tail at  to the
to the  axis. The semicircle is often considered to be the superposition of two individual semicircles, even though the exact separation of these is not well developed in our case. Usually, a small, high-frequency semicircle is identified that corresponds to the formation of a passivated film (SEI) and creates impedance at the surface of the working electrode particles in contact with the organic electrolyte.27–29 The resistance
axis. The semicircle is often considered to be the superposition of two individual semicircles, even though the exact separation of these is not well developed in our case. Usually, a small, high-frequency semicircle is identified that corresponds to the formation of a passivated film (SEI) and creates impedance at the surface of the working electrode particles in contact with the organic electrolyte.27–29 The resistance  and capacitance
and capacitance  values associated with the migration of
values associated with the migration of  through the SEI are described by Eq. 5, 6
through the SEI are described by Eq. 5, 6

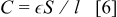
where  is the electrode surface area and
is the electrode surface area and  is the thickness, ρ is the resistivity, and
is the thickness, ρ is the resistivity, and  the permittivity of the SEI layer.30 However, because only one semicircle is distinguishable, the exact contribution to impedance of this component is difficult to quantify.31 The semicircle that dominates at intermediate frequency represents the charge transfer impedance
the permittivity of the SEI layer.30 However, because only one semicircle is distinguishable, the exact contribution to impedance of this component is difficult to quantify.31 The semicircle that dominates at intermediate frequency represents the charge transfer impedance  through the electrode–electrolyte interface, and is considered to account for a large proportion of the overall kinetic impedance of the cell. From Fig. 5a and 5b, the size of this semicircle in both electrodes reduced considerably at voltages lower than
through the electrode–electrolyte interface, and is considered to account for a large proportion of the overall kinetic impedance of the cell. From Fig. 5a and 5b, the size of this semicircle in both electrodes reduced considerably at voltages lower than  since the primary reaction plateau for
since the primary reaction plateau for  occurs at just below this value. The sloping line at
occurs at just below this value. The sloping line at  to the
to the  axis found at low frequency is known as Warburg impedance and is representative of diffusion of
axis found at low frequency is known as Warburg impedance and is representative of diffusion of  within the bulk anode.32 The high frequency intercept at the
within the bulk anode.32 The high frequency intercept at the  axis corresponds primarily to the electrolyte resistance
axis corresponds primarily to the electrolyte resistance  , however small contributions may exist from the wire resistance of the system and solid-solid contact.
, however small contributions may exist from the wire resistance of the system and solid-solid contact.
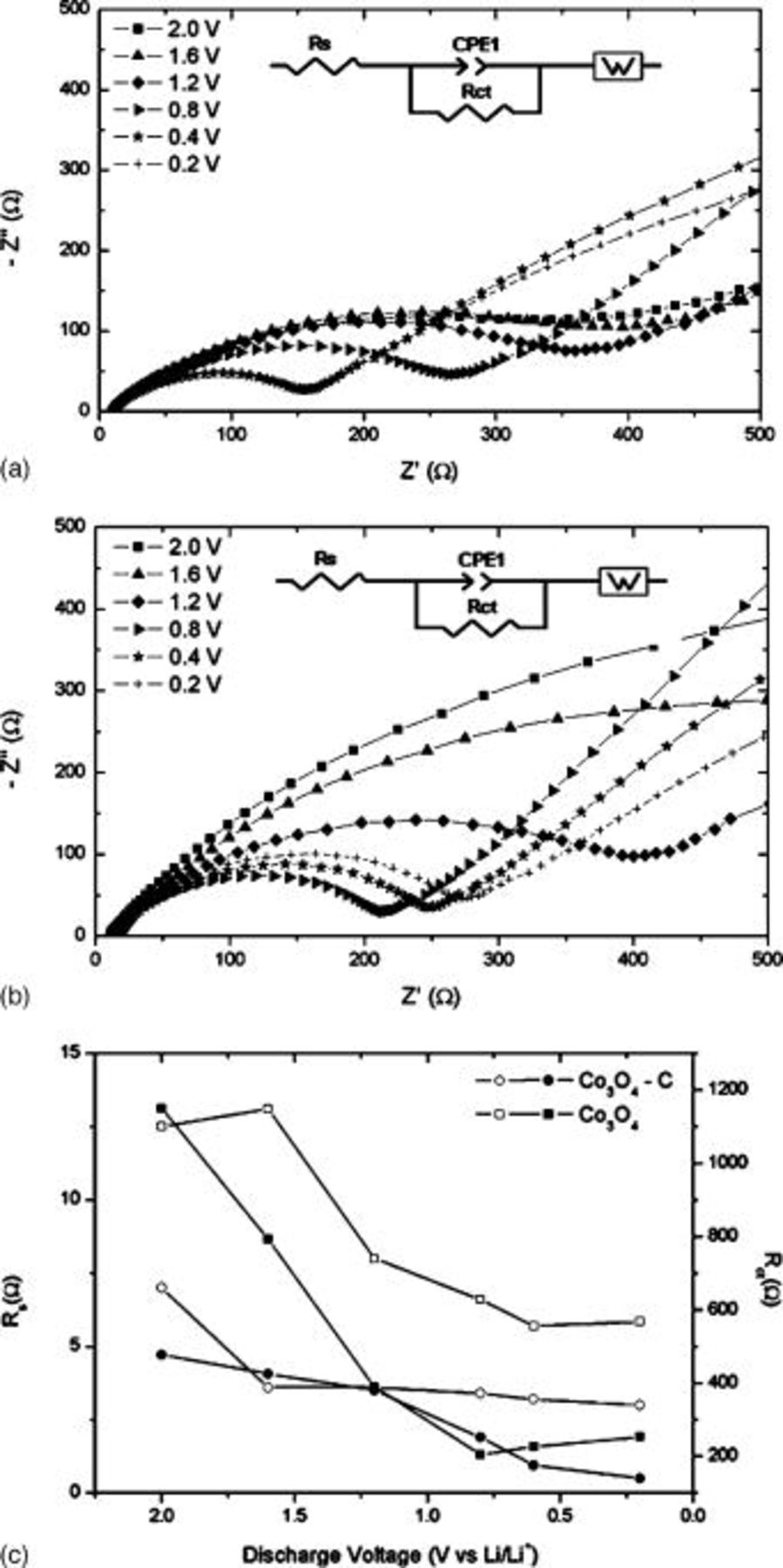
Figure 5. Nyquist plots of (a)  and (b)
and (b)  electrodes and (c) scatter plots of various equivalent circuit parameters vs discharge voltage.
electrodes and (c) scatter plots of various equivalent circuit parameters vs discharge voltage.  is represented by open symbols and
is represented by open symbols and  by closed symbols.
by closed symbols.
To obtain quantitative results, an equivalent circuit (insets of Fig. 5a and 5b) was used to fit the Nyquist plots where  =solution resistance,
=solution resistance,  =charge transfer resistance, and W is the
=charge transfer resistance, and W is the  diffusion resistance. A constant phase element
diffusion resistance. A constant phase element  was used to account for the complexity of the double layer effect associated with charge transfer resistance. The fit result parameters are summarized in Fig. 5c where we see
was used to account for the complexity of the double layer effect associated with charge transfer resistance. The fit result parameters are summarized in Fig. 5c where we see  values for the
values for the  electrode are lower than those of the
electrode are lower than those of the  electrode. The values of
electrode. The values of  at various potentials also show the same trend, except at 0.8 and
at various potentials also show the same trend, except at 0.8 and  where the values are similar. We attribute the generally lower trend of values of
where the values are similar. We attribute the generally lower trend of values of  and
and  for the
for the  electrode to the improvement of electronic conductivity due to the formation of a disordered carbon sheath. Moreover, the sheath that coats
electrode to the improvement of electronic conductivity due to the formation of a disordered carbon sheath. Moreover, the sheath that coats  crystals in the
crystals in the  composite could also induce the formation of a high quality stable SEI layer, thereby contributing to the excellent cycling performance.27
composite could also induce the formation of a high quality stable SEI layer, thereby contributing to the excellent cycling performance.27
Conclusions
We have produced nanocrystalline  composite powder by spray pyrolysis technique that has a narrow Gaussian type particle size distribution
composite powder by spray pyrolysis technique that has a narrow Gaussian type particle size distribution  and high specific surface area
and high specific surface area  . Preliminary electrochemical testing of
. Preliminary electrochemical testing of  electrodes indicates a high reversible capacity of
electrodes indicates a high reversible capacity of  , whereas the noncomposite
, whereas the noncomposite  and
and  electrodes show a steady decay upon cycling to less than 40% of their original capacity. By synthesizing
electrodes show a steady decay upon cycling to less than 40% of their original capacity. By synthesizing  powders we have overcome rapid capacity fade often associated with finely divided
powders we have overcome rapid capacity fade often associated with finely divided  powder. The formation of a C sheath on the particle surface can potentially shield the catalytic action of the Co nanoparticles on electrolyte reduction and the retard development of a thick SEI that can impede charge transfer. In addition, we expect that the disordered C matrix throughout the bulk powder and the carbon sheath assist to cushion the volume expansion associated with the reversible formation of Co nanoparticles. It can be assumed that these results are not limited to the
powder. The formation of a C sheath on the particle surface can potentially shield the catalytic action of the Co nanoparticles on electrolyte reduction and the retard development of a thick SEI that can impede charge transfer. In addition, we expect that the disordered C matrix throughout the bulk powder and the carbon sheath assist to cushion the volume expansion associated with the reversible formation of Co nanoparticles. It can be assumed that these results are not limited to the  system but also to other promising transition MO–C composite powders.
system but also to other promising transition MO–C composite powders.
Acknowledgments
The authors acknowledge financial support provided by the Australian Research Council (ARC) through ARC linkage project LP0453766 and the Centre of Excellence program.
The University of Wollongong assisted in meeting the publication costs of this article.