
Abstract
The wetting behavior of the Au electrode under molten carbonate fuel cell (MCFC) operating conditions was investigated using a lab-scale cell with optical instrumentation. The general trends which were found were smaller contact angles, (i) under polarization than at open circuit, (ii) under an oxidizing atmosphere than under a reducing atmosphere, (iii) at high temperature rather than at lower temperature, and  in Li-K carbonate than in Li-Na. Contact angles as a function of potential exhibit a dependence which resembles electrocapillary curves in aqueous solution, with the zero-charge (minimum wetting) potential and the open-circuit potential (OCP) being different, especially under a reducing atmosphere. The effect of electrolyte composition on the contact angle is interpreted in terms of activity and surface adsorption of oxides. The effect of polarization on wetting is analyzed from two different viewpoints, near OCP the electrocapillary theory is applied, while at high polarization the effect of migration is invoked. The inferior performance of Li/Na cells, compared to Li/K cells, at temperatures below 600°C, is analyzed from the viewpoint of capillary effects, and it is shown that these effects may explain the observed anode behavior and, at least in part, cathode behavior as well. © 2003 The Electrochemical Society. All rights reserved.
in Li-K carbonate than in Li-Na. Contact angles as a function of potential exhibit a dependence which resembles electrocapillary curves in aqueous solution, with the zero-charge (minimum wetting) potential and the open-circuit potential (OCP) being different, especially under a reducing atmosphere. The effect of electrolyte composition on the contact angle is interpreted in terms of activity and surface adsorption of oxides. The effect of polarization on wetting is analyzed from two different viewpoints, near OCP the electrocapillary theory is applied, while at high polarization the effect of migration is invoked. The inferior performance of Li/Na cells, compared to Li/K cells, at temperatures below 600°C, is analyzed from the viewpoint of capillary effects, and it is shown that these effects may explain the observed anode behavior and, at least in part, cathode behavior as well. © 2003 The Electrochemical Society. All rights reserved.
Export citation and abstract BibTeX RIS
The key technical problems for all types of fuel cells are the optimal design of porous electrodes and the control of the three-phase interface among the electrolyte, the gaseous reactant, and the electrode. In the carbonate fuel cell, which employs a molten carbonate mixture as the electrolyte, the pores of the porous electrodes are partially filled with molten electrolyte. The walls of the pores are completely or partially wetted by the molten electrolyte. When complete wetting occurs, an electrolyte film forms. In the case of partial wetting, the electrolyte forms a meniscus within the pores, and most of the current flow is concentrated in the thin meniscus region. Therefore, the wetting properties of electrode material have a direct relationship on the performance of a porous electrode. A well-performing porous electrode maximizes the active wetted electrode surface (meniscus region) exposed to the gaseous reactant. Besides, wetting influences the efficiency of the internal reforming process and the distribution of the electrolyte in the cell.
In the state-of-the-art (SOA) molten carbonate fuel cell (MCFC) cathode polarization makes the greatest contribution to overall voltage loss in the cell.1 The pore structure of the cathode and the wetting characteristics of the cathode/electrolyte combination are the most important factors determining its performance. These factors also have an effect on electrolyte movement in the long-term operation of the cell. Therefore, the study of wetting behavior of the electrolyte with respect to the active cell components of porous electrodes and matrix is important for the purpose of optimum cell design.
An experimental effort for determining the wetting properties under a variety of conditions has been made by several researchers. However, a consistent explanation of the wetting behavior was not been found in the literature. Such an explanation should take into account the effects of polarization, gas atmosphere, electrolyte composition, and temperature on wetting. It is therefore a considerable challenge. In this study, an attempt is made to present a semiquantitative interpretation based on the physics of adsorption and the fluxes of ionic species during polarization. To lay a basis for this, systematic wetting measurements were made with two different melts (Li-Na and Li-K carbonate eutectic) at three different temperatures, under oxidizing as well as reducing gas atmospheres, and at open circuit as well as under load.
Experimental
For the wetting studies, the experimental equipment used earlier by Matsumura and Selman1 and Mugikura and Selman2 was employed with small modifications. The high-temperature controlled-atmosphere apparatus is a unique observatory that enables a direct view of the wetting process, as illustrated in Fig. 1. The scene is captured by a telescope with charge-coupled device (CCD) camera, sent to a TV screen, and recorded by VCR.
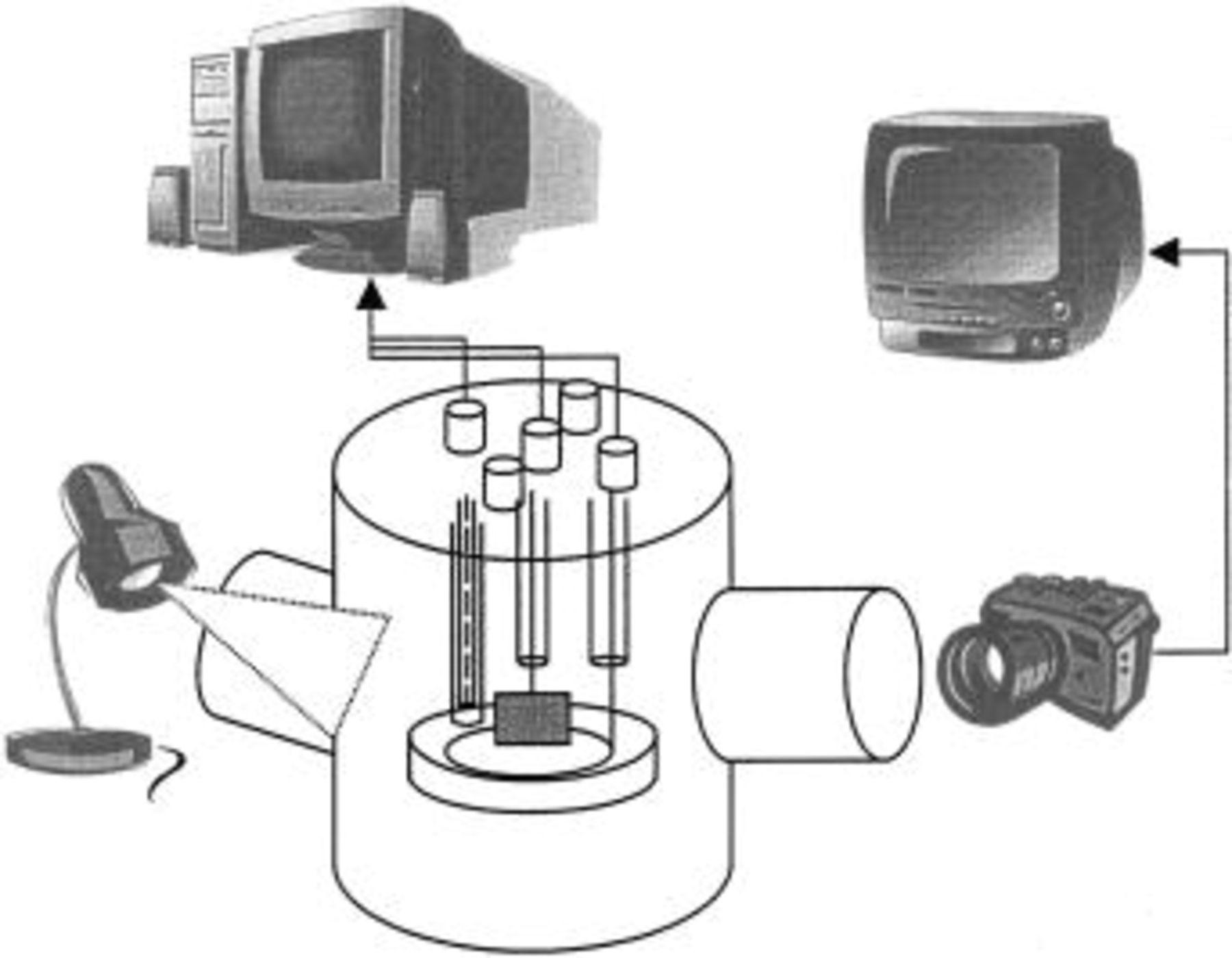
Figure 1. Experimental apparatus display.
Figure 2 shows the three-electrode cell arrangement placed inside the furnace. A thin gold (Au) sheet  was used as the working electrode because Au is extremely corrosion resistant and stable. The wetting behavior can therefore be monitored over a range of anodic as well as cathodic polarization, which is not possible with other metals. For the counter electrode, an Au wire forming a ring was used. The reference electrode was a standard Au
was used as the working electrode because Au is extremely corrosion resistant and stable. The wetting behavior can therefore be monitored over a range of anodic as well as cathodic polarization, which is not possible with other metals. For the counter electrode, an Au wire forming a ring was used. The reference electrode was a standard Au  electrode in a separate compartment. The three electrodes were connected to the electrochemical equipment, Solartron SI 1287 electrochemical interface. The working electrode potential was measured with respect to the reference electrode carrying negligible current.
electrode in a separate compartment. The three electrodes were connected to the electrochemical equipment, Solartron SI 1287 electrochemical interface. The working electrode potential was measured with respect to the reference electrode carrying negligible current.
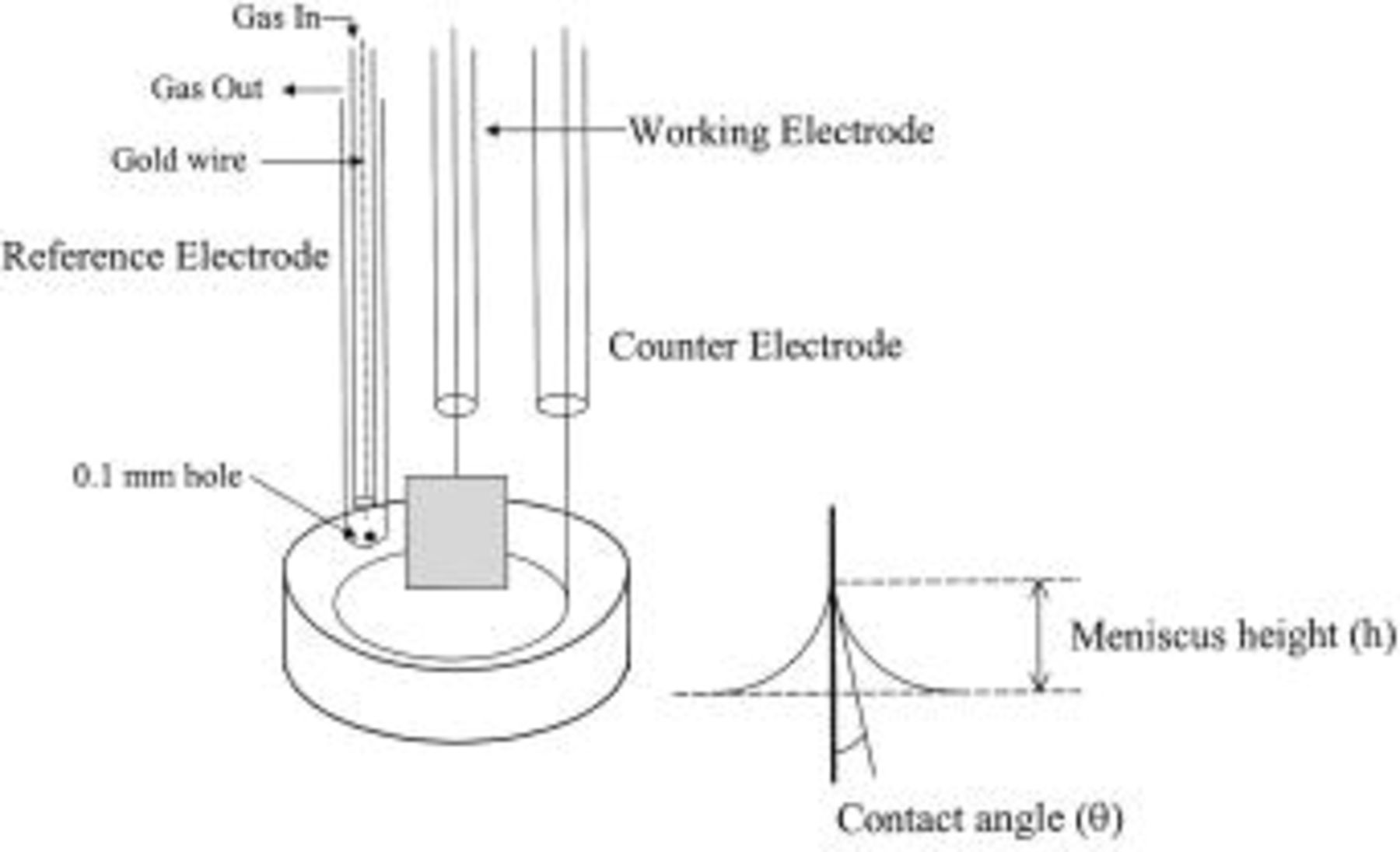
Figure 2. Electrode configuration.
The electrolyte system was an eutectic mixture of lithium and potassium carbonate  or of lithium and sodium carbonate
or of lithium and sodium carbonate  Chemicals of analytical purity were purchased from the Fischer Company. The electrolyte, initially a mixture of solid powders, was placed in a shallow alumina crucible situated in the middle of the furnace. The quantities of solid powders were chosen such that, upon heating to the operating temperature, the melt would fill the crucible entirely so that the meniscus could be photographed from an almost perpendicular angle. The preparation procedure of the electrolyte mixture is summarized in Table I. As the table indicates, the carbonates were further purified prior to use to remove traces of oxide and hydroxide by passing
Chemicals of analytical purity were purchased from the Fischer Company. The electrolyte, initially a mixture of solid powders, was placed in a shallow alumina crucible situated in the middle of the furnace. The quantities of solid powders were chosen such that, upon heating to the operating temperature, the melt would fill the crucible entirely so that the meniscus could be photographed from an almost perpendicular angle. The preparation procedure of the electrolyte mixture is summarized in Table I. As the table indicates, the carbonates were further purified prior to use to remove traces of oxide and hydroxide by passing  through the melt for 24 h.
through the melt for 24 h.
Table I.
| Preparation procedure of electrolyte system. | |
|---|---|
| 1. Heat carbonate powders to 400°C gradually. | |
2. Maintain at this temperature for over 12 h and supply 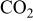 gas simultaneously. gas simultaneously. | |
| 3. Increase the temperature to 550°C and observe melting. | |
| 4. Pump to vacuum using a mechanical pump and keep at near vacuum for 3 h to remove impurities and traces of vapor. | |
| 5. Raise the temperature to 600°C. | |
6. Keep this temperature for 24 h under 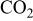 for carbonate purification. for carbonate purification. | |
7. Raise the temperature to 650°C under 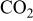 atmosphere. atmosphere. | |
| 8. Dip electrodes into molten electrolyte. |
Before assembling the experimental setup, every component contacting the electrolyte was cleaned by ultrasonication in a microcleaning solution, an organic-removing agent  and mixed acid
and mixed acid  The surface of the working electrode, in particular, was polished and cleaned thoroughly to enable a precise and reproducible measurement of meniscus height.
The surface of the working electrode, in particular, was polished and cleaned thoroughly to enable a precise and reproducible measurement of meniscus height.
Three types of gas atmosphere were used, pure  oxidizing atmosphere
oxidizing atmosphere  and reducing atmosphere
and reducing atmosphere  The gases were purchased mixed from Praxair. Even in an inert gas stream contacting molten carbonate
The gases were purchased mixed from Praxair. Even in an inert gas stream contacting molten carbonate  is always present, since carbonate dissociates in the absence of
is always present, since carbonate dissociates in the absence of  forming oxide ions and
forming oxide ions and 
Most of the experiments were carried out at 650°C, the operating temperature of MCFC. Measurements at 600 and 700°C were also made to investigate the temperature dependence of wetting.
Results
The wetting characteristics of carbonate melts at a solid electrode material are expressed as a contact angle, which depends on the gas composition, pressure, temperature, and the polarization. For the determination of contact angles in molten carbonate, researchers have mainly used two experimental methods, namely, the sessile drop method and the meniscus rise method. In this work, the meniscus rise method is used. The meniscus height is measured with the instrumentation described above, and the contact angle is calculated from the meniscus height using liquid/gas interfacial energy data available in the literature.
Measurements were made of the height to which the carbonate melt meniscus rises at the flat gold sheet that serves as a working electrode. The sheet electrode is first immersed. When it is pulled up slowly, a meniscus forms and its height reaches a maximum (steady) value after some time. This is referred to as the meniscus height (h). The meniscus height at a solid/liquid/gas interface is sensitive to the radius of curvature of the solid/liquid interface, so that it has been found to depend on surface roughness of the electrode. Therefore, a well-defined preparation procedure must be scrupulously followed, as described earlier.
Figures 3 and 4 indicate the changes of meniscus height with applied potential in three different gas environments, for Li-K and Li-Na electrolyte, respectively. In both electrolytes, and for each gas composition, the meniscus height shows a behavior that roughly resembles the typical electrocapillary curve known for mercury electrodes in aqueous solution. The dependence of meniscus height on potential is roughly parabolic. The lowest points of the various curves shown in Fig. 3 and 4 are markedly different as to potential and meniscus height, and the rates of increase of meniscus height with potential also differ widely. The reproducibility of the meniscus height measurement was checked throughout the experiment and the error bars in Fig. 3 and 4 indicate the range observed.
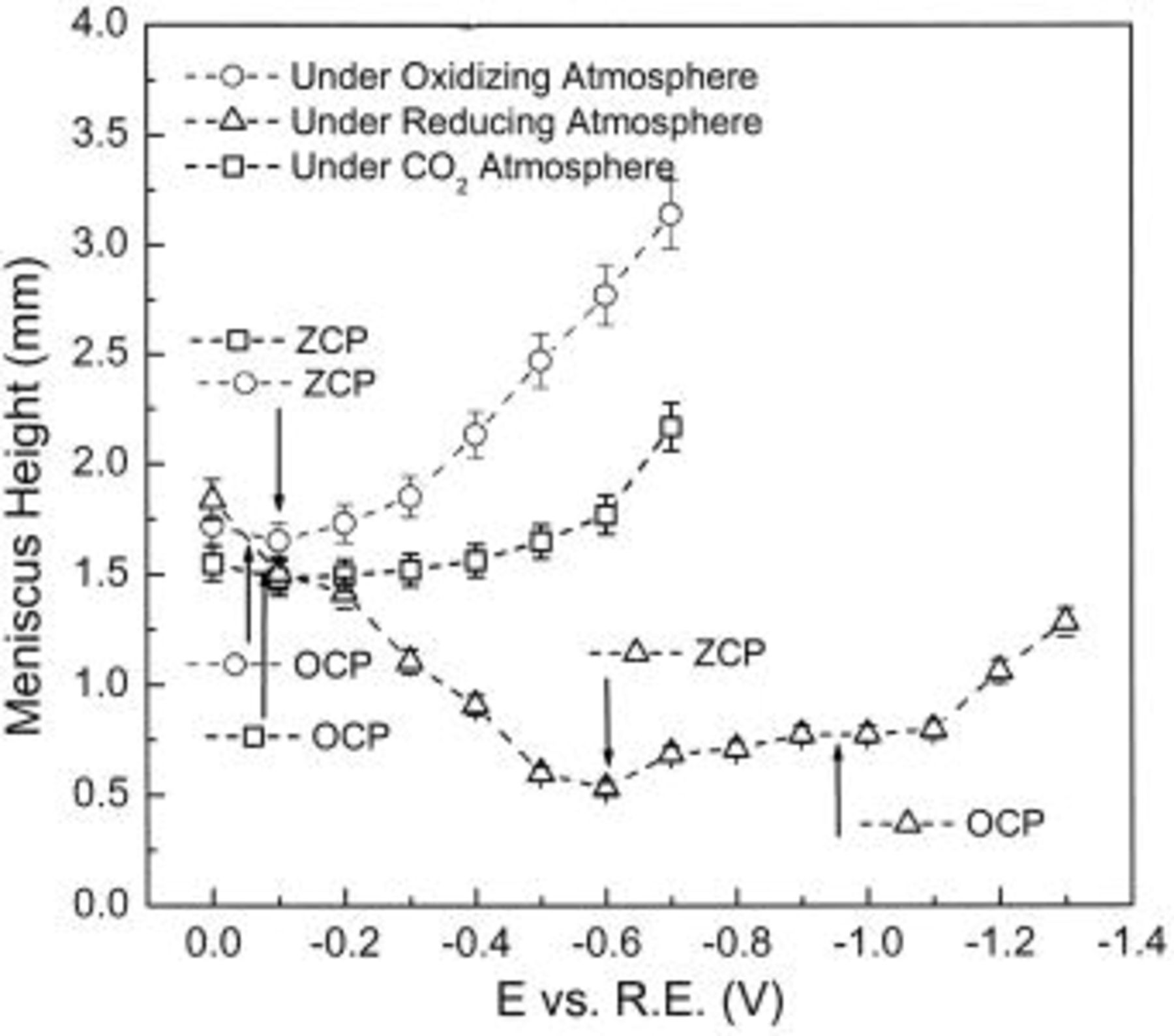
Figure 3. Meniscus height measurement in Li-K melt at 650°C and 1 atm.
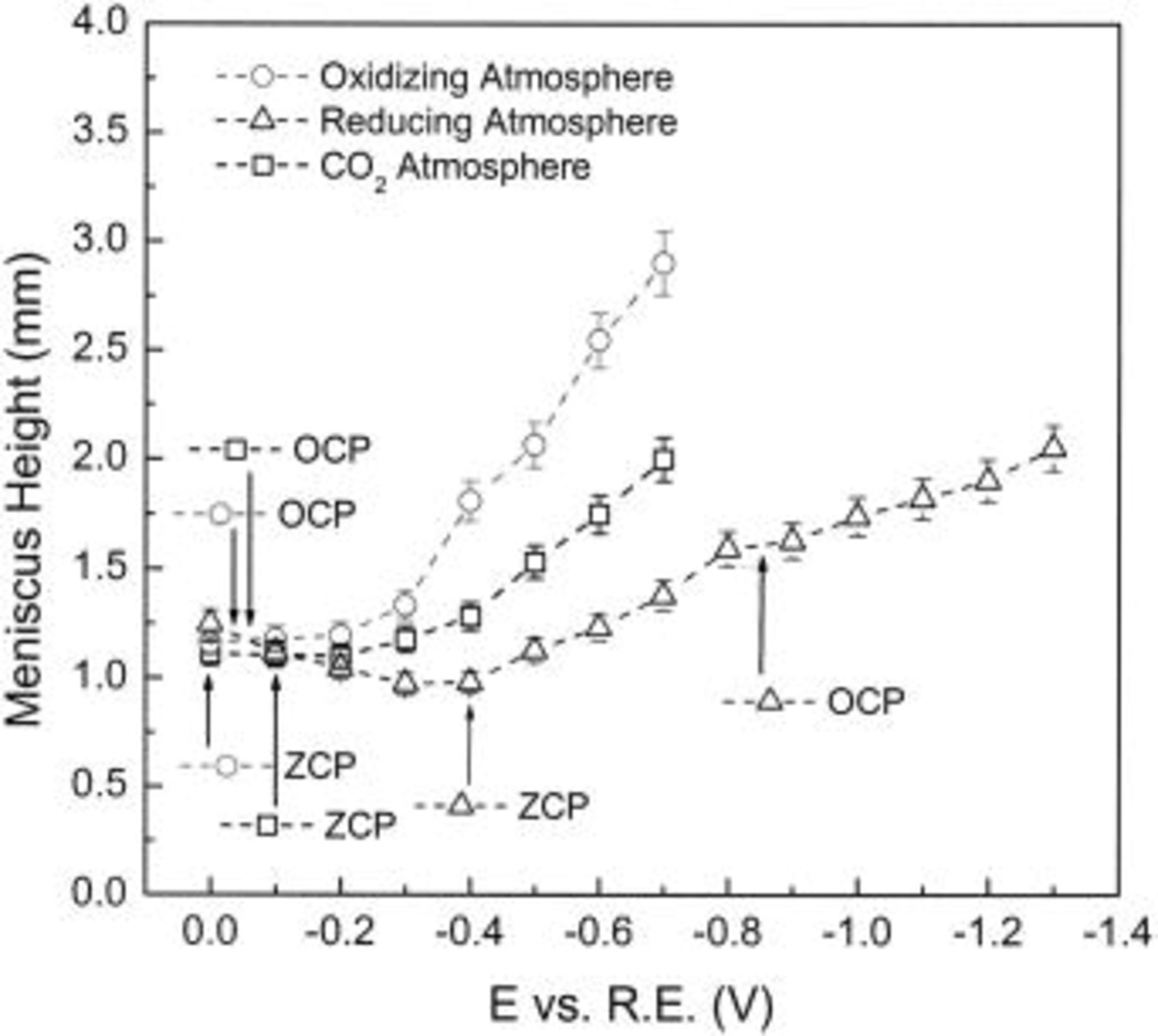
Figure 4. Meniscus height measurement in Li-Na melt at 650°C and 1 atm.
The theory of interfacial equilibrium thermodynamics makes use of interfacial tension as an intensive variable expressing the surface energy per unit area of interface between two phases. The surface energy between a liquid phase and a gas phase,  may be determined by measuring the surface shape and applying a static force balance between gravity and surface force. For example, from the measured height of a meniscus at a solid-liquid-gas interface, together with the contact angle (optically determined), the liquid-gas surface tension can be determined by the Neumann equation3 (Eq. 1)
may be determined by measuring the surface shape and applying a static force balance between gravity and surface force. For example, from the measured height of a meniscus at a solid-liquid-gas interface, together with the contact angle (optically determined), the liquid-gas surface tension can be determined by the Neumann equation3 (Eq. 1)
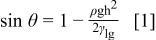
Here, ρ is the density, g the gravitational acceleration, and  the liquid/gas interfacial tension (N/m). Conversely, if
the liquid/gas interfacial tension (N/m). Conversely, if  is known, the contact angle can be calculated. This equation is applied in the present work. The contact angle can be calculated from the meniscus height using well-established values of
is known, the contact angle can be calculated. This equation is applied in the present work. The contact angle can be calculated from the meniscus height using well-established values of  known from the literature.
known from the literature.
Figure 5 shows the contact angle values calculated from the meniscus height values in Fig. 3 and 4. Clearly, the highest values of the contact angle in Fig. 5 correspond to the lowest values of meniscus height in Fig. 3 and 4.
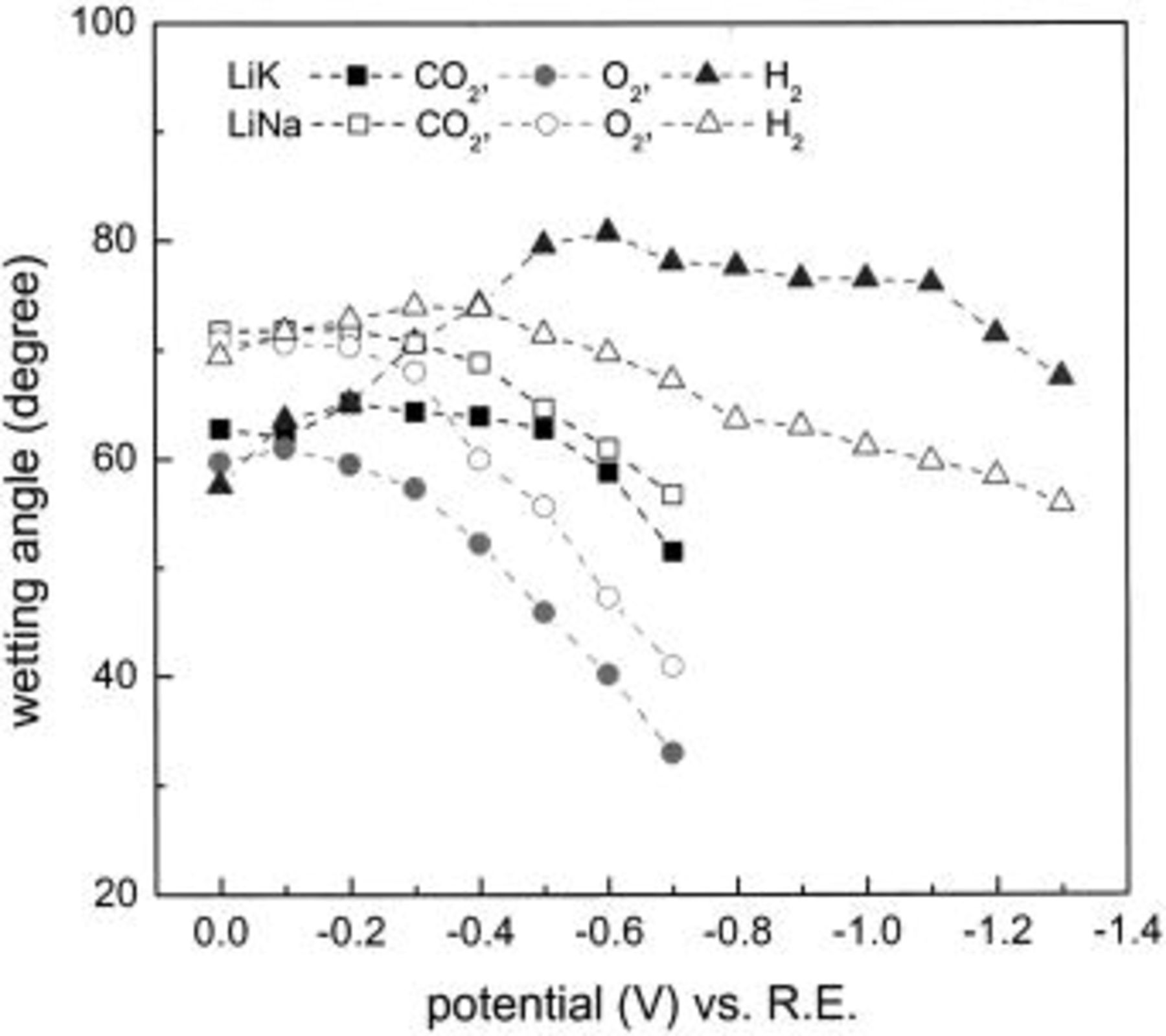
Figure 5. Contact angle in Li-K and Li-Na melts at 650°C and 1 atm.
Analysis and Discussion
Zero-charge potential and open-circuit potential (OCP).—
The minimum of each quasiparabolic curve (lowest meniscus height, i.e., lowest wetting tendency and greatest contact angle) occurs at a potential which does not coincide with the OCP except, approximately, in the case of pure  atmosphere. The minimum-height potential corresponds to the zero-charge potential (ZCP) of conventional electrocapillary curves, therefore the notation ZCP is adopted here for this potential. In an oxidant environment the ZCP of both Li-K and Li-Na electrolyte is not very different from the OCP, but in a reducing environment, the ZCP is much more positive than the OCP, especially for the Li-Na electrolyte
atmosphere. The minimum-height potential corresponds to the zero-charge potential (ZCP) of conventional electrocapillary curves, therefore the notation ZCP is adopted here for this potential. In an oxidant environment the ZCP of both Li-K and Li-Na electrolyte is not very different from the OCP, but in a reducing environment, the ZCP is much more positive than the OCP, especially for the Li-Na electrolyte  Only in pure
Only in pure  are the ZCP and apparent OCP value not much different. In 100% pure
are the ZCP and apparent OCP value not much different. In 100% pure  the OCP is actually not well-defined thermodynamically. But the very low partial pressure of oxygen established during the potential measurements (perhaps by small anodic and cathodic potential excursions) establish an effective OCP which practically coincides with the ZCP.
the OCP is actually not well-defined thermodynamically. But the very low partial pressure of oxygen established during the potential measurements (perhaps by small anodic and cathodic potential excursions) establish an effective OCP which practically coincides with the ZCP.
The ZCP represents the condition under which the double-layer charge at the electrode is zero, regardless of current passage or not. Figure 6 illustrates schematically the double-layer structure and the charge profile in the double layer. In agreement with earlier authors,2 the oxide ion  is specifically adsorbed at the electrode and occupies the inner plane (inner Helmholtz plane, IHP) of the electric double layer. At the ZCP, the negative charge of this inner layer is compensated by the outer plane, which is occupied mainly by alkali cations and carbonate ions in such a ratio as to produce a net positive charge. In molten salts the diffuse double layer is believed to be nonexistent or unimportant, so the double layer is overall more compact than that in aqueous solutions. The resulting structure has a potential that is fairly close to OCP in the case of oxidizing atmosphere (and, as noted, in pure
is specifically adsorbed at the electrode and occupies the inner plane (inner Helmholtz plane, IHP) of the electric double layer. At the ZCP, the negative charge of this inner layer is compensated by the outer plane, which is occupied mainly by alkali cations and carbonate ions in such a ratio as to produce a net positive charge. In molten salts the diffuse double layer is believed to be nonexistent or unimportant, so the double layer is overall more compact than that in aqueous solutions. The resulting structure has a potential that is fairly close to OCP in the case of oxidizing atmosphere (and, as noted, in pure  .
.
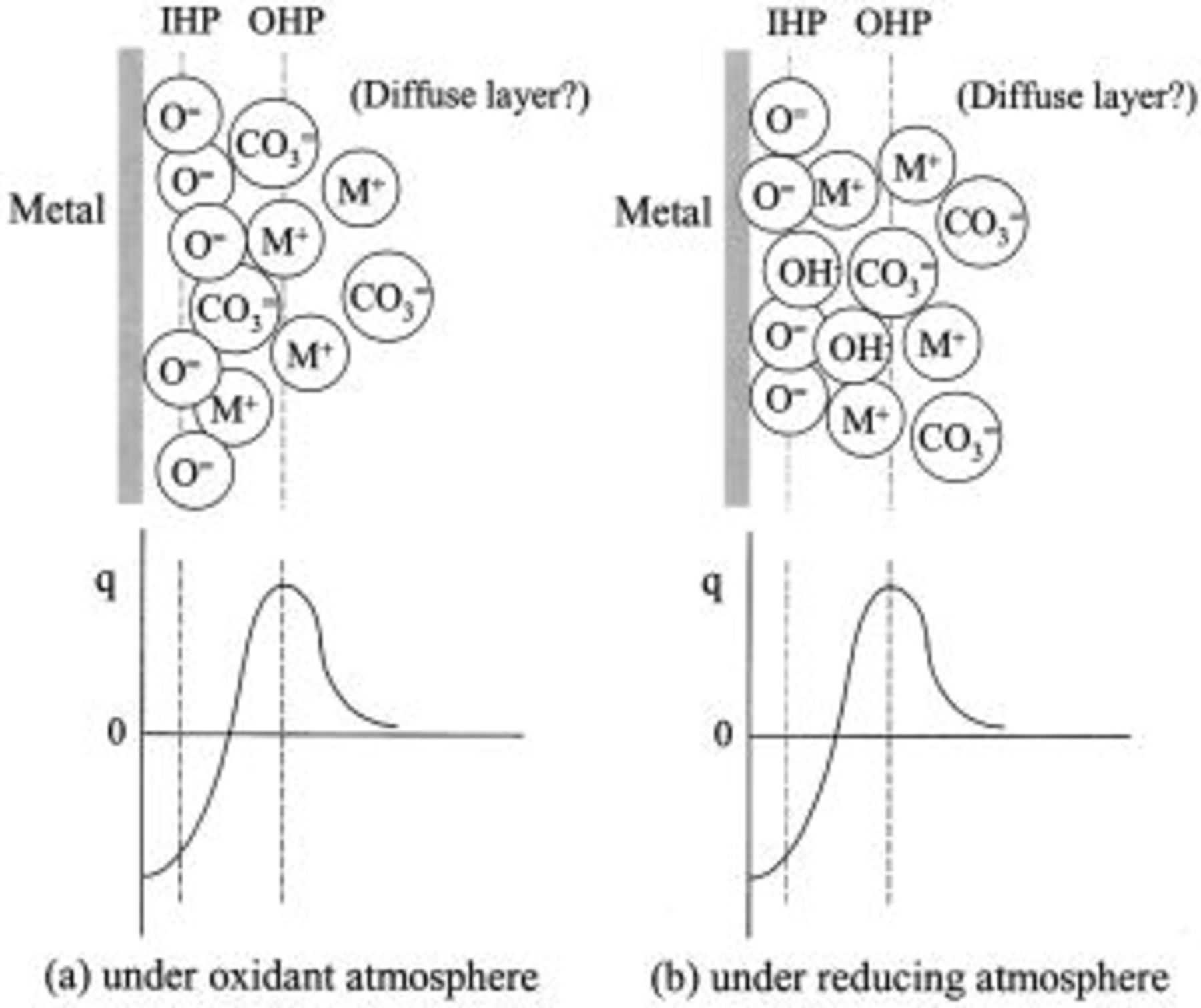
Figure 6. Electric double-layer structure at a metal electrode in molten carbonate, and charge profile in the double layer.  indicates alkali ions
indicates alkali ions 
Assuming that we may apply equilibrium thermodynamics to these characteristic potentials (see the discussion in the next subsection), the closeness of OCP and ZCP values under oxidant atmosphere implies that the activity of the adsorbed oxide ions is not too different from that in the bulk melt. This seems to hold for both Li-K melt and Li-Na melt, although the Li-Na melt is more basic, i.e., it has a much greater oxide ion activity than Li-K. However, in reducing atmosphere the ZCP is 350 mV (in Li-K) to 450 mV (in Li-Na) more positive than the OCP. This suggests that the surface oxide ion activity at the electrode is much smaller than the bulk oxide activity at OCP. One reason for this may be the presence of  ions in a much larger concentration than
ions in a much larger concentration than  ions. The
ions. The  ions are produced by anodic polarization, even slightly, under
ions are produced by anodic polarization, even slightly, under  atmosphere. The difference between ZCP and OCP is larger for the Li-Na electrolyte than for the Li-K electrolyte, which is probably related to the much larger bulk oxide activity in Li-Na compared to Li-K eutectic, at an approximately equal extent of hydrolysis.
atmosphere. The difference between ZCP and OCP is larger for the Li-Na electrolyte than for the Li-K electrolyte, which is probably related to the much larger bulk oxide activity in Li-Na compared to Li-K eutectic, at an approximately equal extent of hydrolysis.
Faradaic effect on meniscus rise upon polarization.—
With either Li-K or Li-Na electrolyte under oxidant atmosphere, the meniscus height at OCP is, within the reproducibility of the measurements, lower than when the metal surface is polarized in the cathodic direction (and also in the anodic direction, although the data in Fig. 3 and 4 do not show this clearly). The polarized surface pulls up the meniscus above its open-circuit height, i.e., it is apparently more easily wetted by the electrolyte. However, this increased wetting results not only from surface and gravity forces at the meniscus in static balance, but also from pressure gradients exerted by bulk liquid movement in the meniscus region due to diffusion and migration of ions. Strictly speaking, equilibrium thermodynamics does not apply to the meniscus rise at any potential except the OCP. Nevertheless, interfacial thermodynamics are useful for a first-approximation analysis, and some key thermodynamic relationships therefore are introduced and checked in the following subsection.
The effect of polarization under pure  is somewhat similar to that under oxidant atmosphere, with the difference that the meniscus height first decreases slightly (in Li-K) or stays constant (in Li-Na) before increasing upon cathodic polarization beyond 200 mV. The rate of increase of meniscus height with polarization is slower than under the oxidant.
is somewhat similar to that under oxidant atmosphere, with the difference that the meniscus height first decreases slightly (in Li-K) or stays constant (in Li-Na) before increasing upon cathodic polarization beyond 200 mV. The rate of increase of meniscus height with polarization is slower than under the oxidant.
Under a reducing atmosphere the meniscus height upon anodic polarization decreases (very weakly for Li-K, but strongly for Li-Na), and upon cathodic polarization increases (for Li-K as well as Li-Na).
Figure 7 shows a schematic of the electrode reaction and transport processes taking place in the meniscus region during current passage, i.e., away from OCP. In contrast to Fig. 6 (which is on a nanometer scale), Figure 7 (on a micrometer to nanometer scale) assumes a faradaic process is taking place and shows the associated fluxes of reactant and product species, including  ions. A net transport of
ions. A net transport of  ions takes place by combined diffusion and migration to or from the high end of the meniscus, where the electrode reaction is concentrated. Because of the requirement of melt electroneutrality, the net
ions takes place by combined diffusion and migration to or from the high end of the meniscus, where the electrode reaction is concentrated. Because of the requirement of melt electroneutrality, the net  flux causes a migration of alkali ions to or from the meniscus tip. The net flux of alkali ions in the meniscus is necessarily kept at zero by a compensating flux due to the alkali ion gradient in the meniscus.
flux causes a migration of alkali ions to or from the meniscus tip. The net flux of alkali ions in the meniscus is necessarily kept at zero by a compensating flux due to the alkali ion gradient in the meniscus.
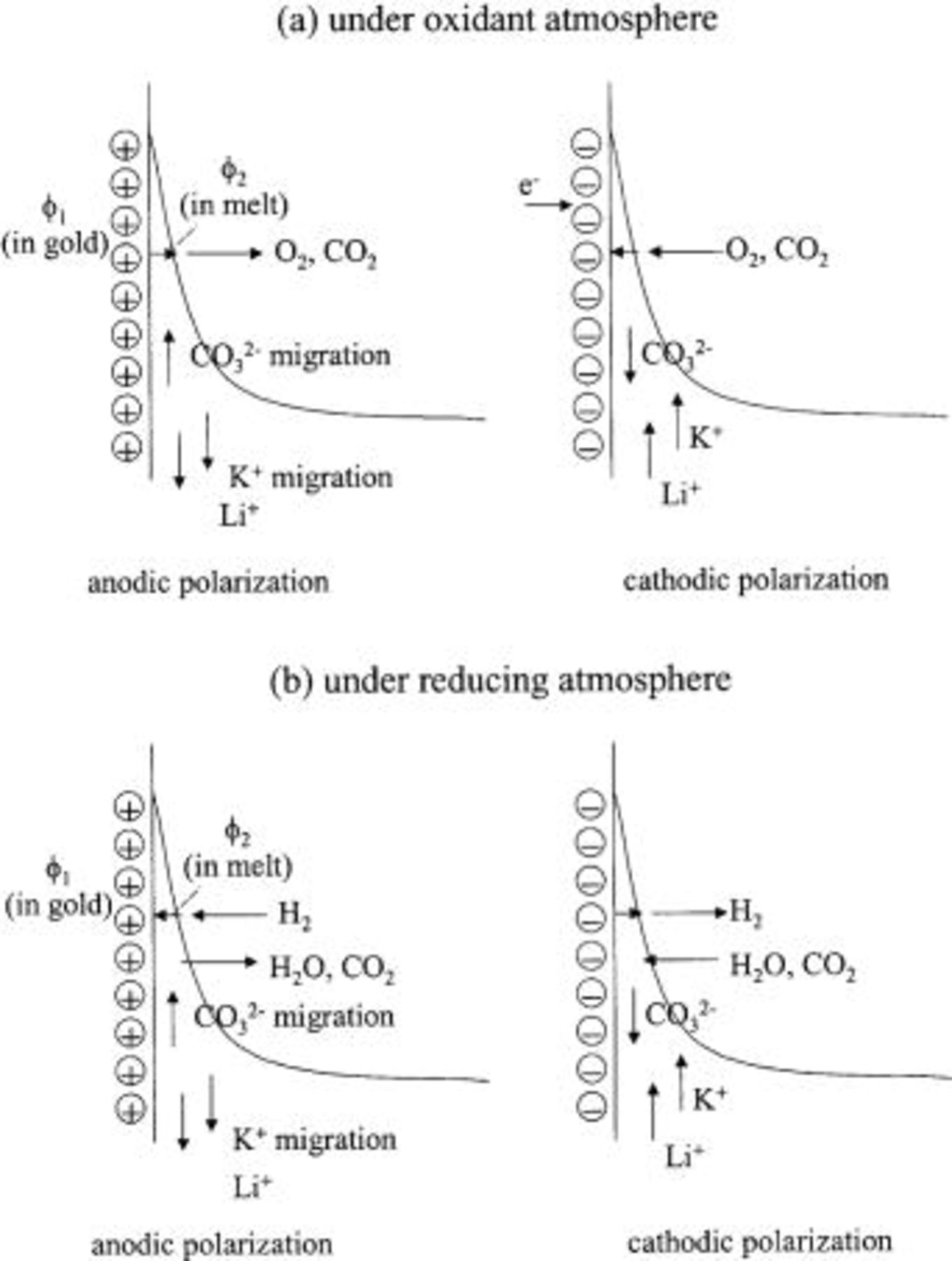
Figure 7. Schematic of electrode and transport processes in the meniscus at a MCFC electrode.
The combined fluxes of alkali and carbonate cations yield a net flow toward, or away from, the tip of the meniscus. This flow is driven by a pressure gradient, which results from the purely capillary (interfacial) forces acting at the tip of the meniscus, as well as the hydrostatic (gravity) force. (The latter is always negative, i.e., downward pointing.) The net flow resulting from the total pressure gradient together with the mass change in the tip of the meniscus due to generation or consumption of carbonate, changes the shape of the meniscus from that which would prevail in the presence of electrocapillary forces only. The result, depending on whether polarization is anodic or cathodic, is a lengthening or shortening, respectively, of the meniscus, relative to the height corresponding to a purely electrocapillary curve.
Figures 3 and 4 confirm this qualitative analysis of the faradaic polarization effect. They show meniscus height curves which are roughly superpositions of an approximately parabolic curve, symmetric with respect to ZCP, and a linear effect near OCP which raises the meniscus when the potential is positive with respect to OCP (anodic polarization) and depresses it below OCP (cathodic polarization). Figure 8 shows schematically the concept of the superimposed electrocapillary and faradaic effects. The superposition is most clearly recognized in the case of reducing atmosphere, but it is also apparent for the cathodic side of oxidizing and pure  atmosphere. However, Fig. 3 and 4 suggest also that (i) the polarization effect causes a relatively small height correction compared to the pure electrocapillary meniscus height and (ii) the polarization effect increases with polarization only over a small potential range near the OCP, and weakens farther away from OCP. The reasons for this second, rather complicated feature of the faradaic effect are not obvious, and further insight may have to await a quantitative modeling analysis of wetting behavior under polarization.
atmosphere. However, Fig. 3 and 4 suggest also that (i) the polarization effect causes a relatively small height correction compared to the pure electrocapillary meniscus height and (ii) the polarization effect increases with polarization only over a small potential range near the OCP, and weakens farther away from OCP. The reasons for this second, rather complicated feature of the faradaic effect are not obvious, and further insight may have to await a quantitative modeling analysis of wetting behavior under polarization.
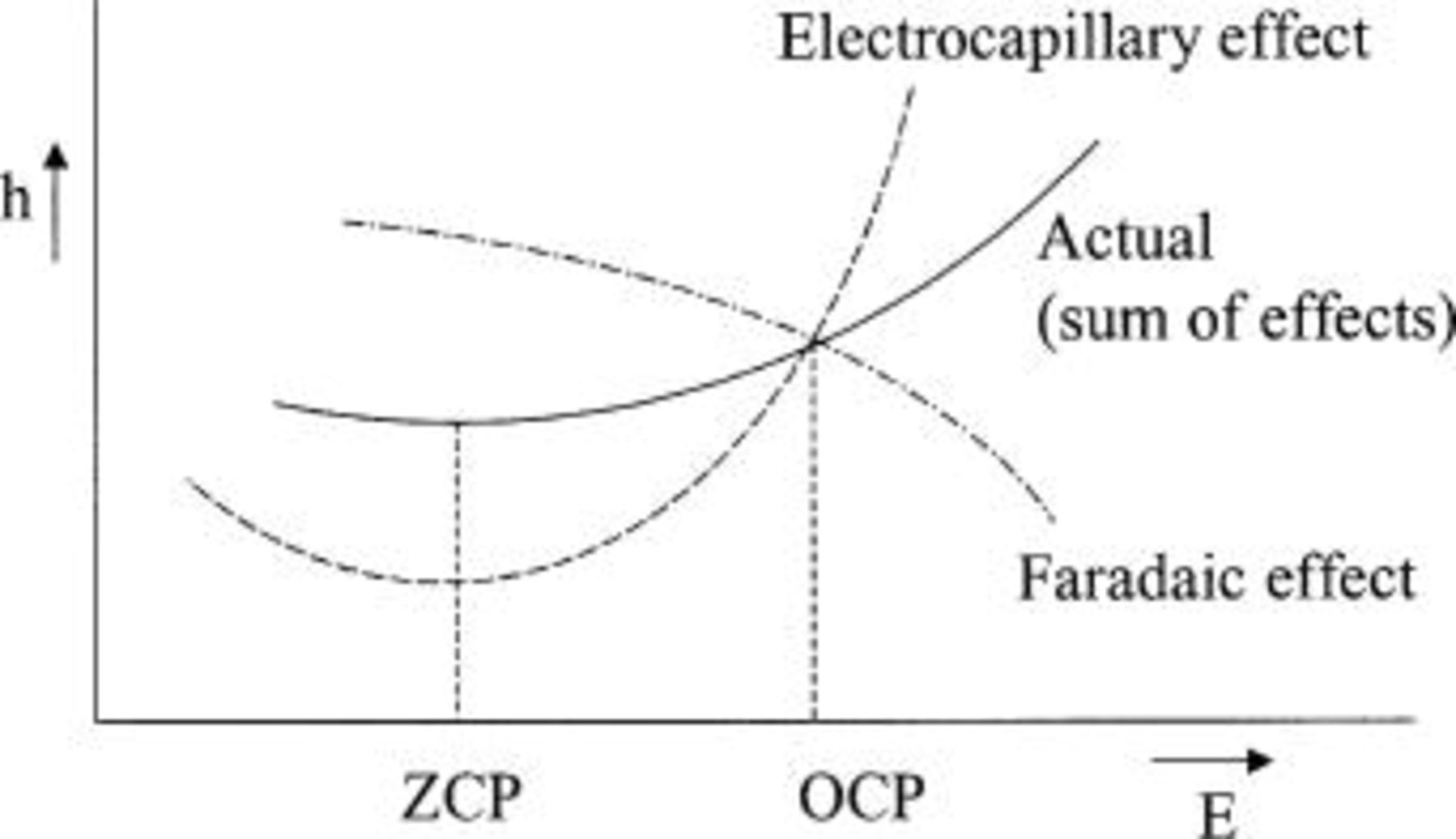
Figure 8. Schematic of the superposition of electrocapillary and faradaic effects on meniscus height.
Thermodynamic analysis of wetting effects.—
As mentioned earlier, the contact angle θ values determined from h beyond the immediate vicinity of OCP are apparent, or effective, values, i.e., strictly speaking they have no thermodynamic significance. However, as explained in the previous subsection, polarization effects do not overwhelm electrocapillary meniscus response. Therefore, a more thorough thermodynamic analysis is undertaken here to check some of the preliminary conclusions drawn from inspection of the meniscus height curves (Figs. 3 and 4).
The three interfacial tensions (actually, surface energies), 
 and
and  are related to each other by fundamental force balances. The relationship between the interfacial tensions and the contact angle is given by the Young-Dupre equation,4 i.e.
are related to each other by fundamental force balances. The relationship between the interfacial tensions and the contact angle is given by the Young-Dupre equation,4 i.e.
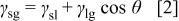
The quantity  is relatively easy to measure, and its values for various carbonate melts are available. On the other hand, the surface energy (or interfacial tension) between a solid and a gas phase, or between a solid and a liquid phase is much more difficult to determine.
is relatively easy to measure, and its values for various carbonate melts are available. On the other hand, the surface energy (or interfacial tension) between a solid and a gas phase, or between a solid and a liquid phase is much more difficult to determine.
If the solid/gas interfacial tension (or surface energy), expressed in J/cm2 and the liquid/gas interfacial tension are not significantly affected by the applied potential, the change in contact angle directly reflects the change in the solid/liquid interfacial tension (energy) under polarization, as follows2
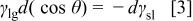
The surface tension of an electrode in contact with a solution at thermodynamic equilibrium (open-circuit potential, OCP) depends on the metal-solution potential difference (dE). The equation describing this dependence is called the electrocapillary equation or Lippmann equation5

where  is surface charge density (C/cm2);
is surface charge density (C/cm2);  the surface concentration (mol/cm2), and
the surface concentration (mol/cm2), and  the electrochemical potential (J/mol). Thus, assuming equilibrium conditions, thermodynamics relates the change of electrode potential (dE) to the change in the interfacial tension between electrode and electrolyte
the electrochemical potential (J/mol). Thus, assuming equilibrium conditions, thermodynamics relates the change of electrode potential (dE) to the change in the interfacial tension between electrode and electrolyte  Equation 4 implies that, by measuring the variation of interfacial tension with composition at constant potential, the surface concentration of species i can be obtained from the derivative of interfacial tension with respect to bulk concentration of i. Also, by measuring the variation of interfacial tension with potential, E, at constant composition, the surface charge
Equation 4 implies that, by measuring the variation of interfacial tension with composition at constant potential, the surface concentration of species i can be obtained from the derivative of interfacial tension with respect to bulk concentration of i. Also, by measuring the variation of interfacial tension with potential, E, at constant composition, the surface charge  can be obtained from the derivative of interfacial tension with respect to E.
can be obtained from the derivative of interfacial tension with respect to E.
According to the Lippmann equation, the interfacial tension decreases with increasing (i.e., more positive) potential if the net surface charge,  is positive (specific adsorption of cations, e.g., alkali ions in molten carbonate). The reason is that polarization draws counterons (in the above cases, respectively, anions or cations) into the inner plane (IHP) of the electric double layer. These counteract the interfacial energy buildup represented by
is positive (specific adsorption of cations, e.g., alkali ions in molten carbonate). The reason is that polarization draws counterons (in the above cases, respectively, anions or cations) into the inner plane (IHP) of the electric double layer. These counteract the interfacial energy buildup represented by  The decrease of interfacial tension upon (nonfaradaic) polarization is reflected as a decrease in θ, i.e., an increase in meniscus height, h. Note also that in accordance with the Gibbs adsorption isotherms the interfacial tension must decrease with increasing interfacial excess concentration of either cations or anions.
The decrease of interfacial tension upon (nonfaradaic) polarization is reflected as a decrease in θ, i.e., an increase in meniscus height, h. Note also that in accordance with the Gibbs adsorption isotherms the interfacial tension must decrease with increasing interfacial excess concentration of either cations or anions.
The  behavior predicted by Eq. 4 agrees with the observations for wetting under a reducing atmosphere, both in the Li-K and Li-Na melt, and for anodic as well as cathodic polarization. (In Li-K melt under anodic polarization, the weak response suggests
behavior predicted by Eq. 4 agrees with the observations for wetting under a reducing atmosphere, both in the Li-K and Li-Na melt, and for anodic as well as cathodic polarization. (In Li-K melt under anodic polarization, the weak response suggests  Therefore, the assumption that small anions such as
Therefore, the assumption that small anions such as  and
and  are specifically adsorbed under reducing gas atmosphere seems to be correct.
are specifically adsorbed under reducing gas atmosphere seems to be correct.
On the other hand, under oxidant atmosphere, the parabolic response to polarization seems to indicate that  is <0 upon cathodic polarization and >0 upon anodic polarization. In both cases, the meniscus height increases upon polarization.
is <0 upon cathodic polarization and >0 upon anodic polarization. In both cases, the meniscus height increases upon polarization.
As discussed earlier, this thermodynamic argument does not properly account for the behavior of meniscus height, h, with polarization, E, beyond the immediate vicinity of the OCP. That there is a qualitative agreement with the thermodynamically predicted trend may be explained by the assumption, inferred earlier from inspection of the meniscus height vs. E curves, that the thermodynamic forces may be quantitatively stronger than the microscale hydrodynamic forces generated by diffusion and migration, so their effect is dominant. However, confirmation of this assumption requires further modeling of the faradaic process at the meniscus to fit the curves of Fig. 3 and 4.
Effect of gas environment.—
Changes in wetting tendency (θ in Eq. 1) reflect changes in adsorption of particular charged and uncharged species as well as potential (Eq. 4). This adsorption determines the nature and net charge of the electric double layer, which in turn determines the contact angle. In molten carbonates,  ions have been considered to be the main (preferentially adsorbed) charged species in the inner layer (so-called inner Helmholtz plane) of the electric double layer at the electrode.2 Under anodic polarization, the specific adsorption of
ions have been considered to be the main (preferentially adsorbed) charged species in the inner layer (so-called inner Helmholtz plane) of the electric double layer at the electrode.2 Under anodic polarization, the specific adsorption of  ions at the electrode
ions at the electrode  increases, which leads to a decrease in interface energy
increases, which leads to a decrease in interface energy  Consequently, this results in the increase in meniscus height.
Consequently, this results in the increase in meniscus height.
As observed earlier, in an oxidant atmosphere wettability near OCP is better than in reducing or pure  gas atmospheres. Figures 3 and 4 illustrate that this holds practically over the entire range of applied potentials. The better wettability in oxidant atmosphere is reasonable since an oxidant atmosphere generates a comparatively large bulk concentration of oxide ions, via carbonate dissociation and complex oxide formation. This enhances surface adsorption of oxide ions (simple or complex) which is believed to enhance wetting.
gas atmospheres. Figures 3 and 4 illustrate that this holds practically over the entire range of applied potentials. The better wettability in oxidant atmosphere is reasonable since an oxidant atmosphere generates a comparatively large bulk concentration of oxide ions, via carbonate dissociation and complex oxide formation. This enhances surface adsorption of oxide ions (simple or complex) which is believed to enhance wetting.
Based on the schematic of Fig. 6, an argument can be made that, under a reducing atmosphere, the transport of reacting species to and from the meniscus area (reaction zone) reinforces the basic thermodynamic condition derived in the previous section, i.e.,  for both anodic and cathodic polarization. On the other hand, under an oxidant atmosphere, the transport of reacting and product species (complex oxide species such as
for both anodic and cathodic polarization. On the other hand, under an oxidant atmosphere, the transport of reacting and product species (complex oxide species such as  or
or  on the anodic side, and
on the anodic side, and  on the reduced side) appears to reinforce the reversal of charge that occurs near the ZCP/OCP.
on the reduced side) appears to reinforce the reversal of charge that occurs near the ZCP/OCP.
Additionally, an oxide layer formed on the electrode surface in an oxidant environment can improve the wettability. Therefore, NiO, the SOA cathode material of MCFC technology, shows a contact angle close to 0°. Porous cathodes made of NiO or other oxides may be assumed to be covered by an electrolyte film of a certain thickness (usually assumed to be 0.1 to 1 μm, but actually never measured).
Effect of electrolyte composition.—
The typical Li-K electrolyte is now being replaced with Li-Na, attractive for its lower values for ionic resistance, evaporation loss, and NiO solubility.6 Figure 9 compares the two electrolyte systems at eutectic composition under oxidizing atmosphere. Wetting by Li-K carbonate is better than by Li-Na, at least at 650°C.
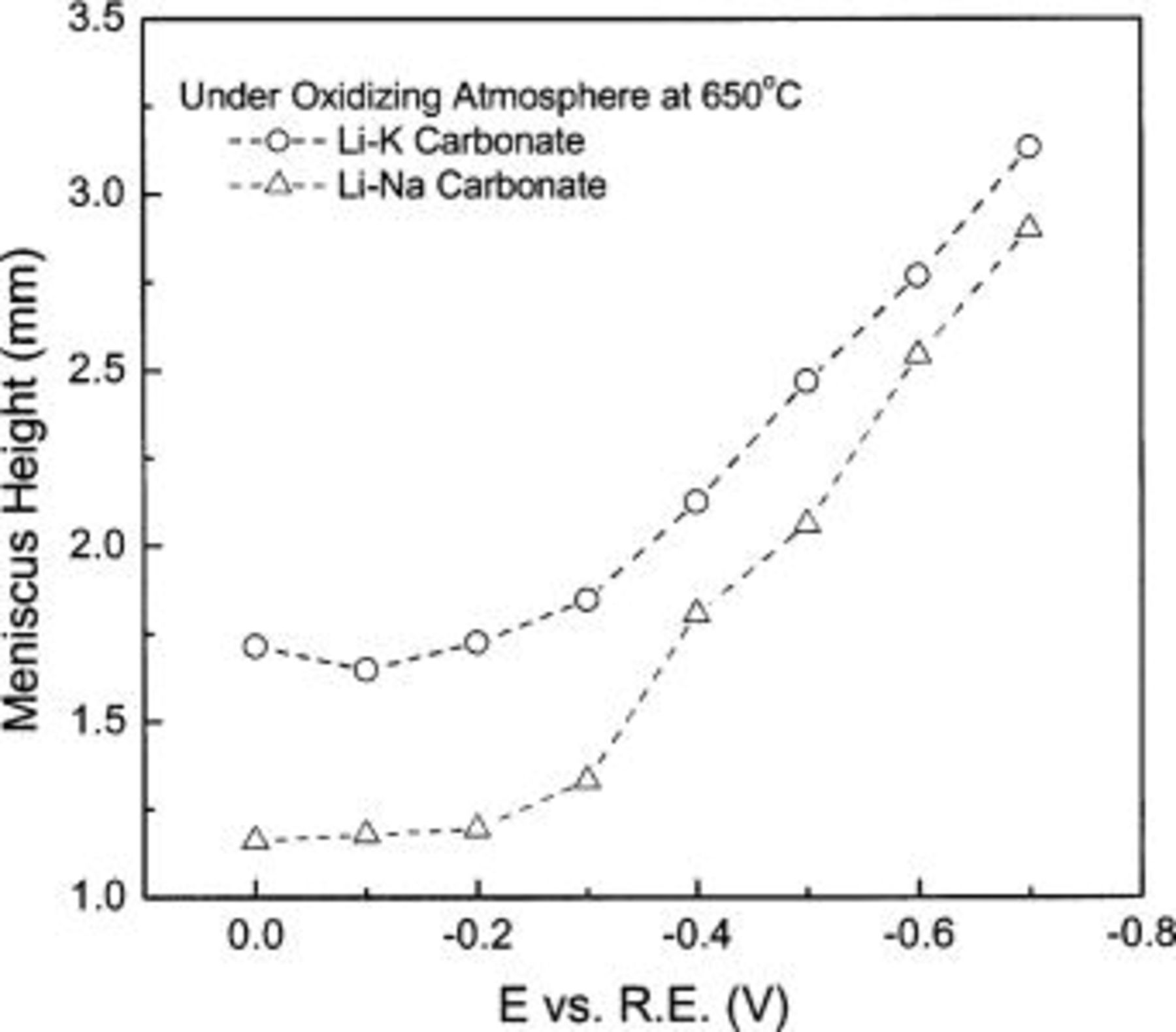
Figure 9. Meniscus height comparison in Li-K and Li-Na melts.
The difference in wettability may be explained in two ways. First, considering the correlation of interfacial properties with interfacial structure at the molecular level, the wetting tendency may increase with increasing ionic radius of the alkali ions present. This is supported by the fact that surface tension decreases with increasing cation radius. Table II compares the physical properties of various pure carbonate melts.4
Table II.
| Physical properties of pure carbonate melts. | ||||||
|---|---|---|---|---|---|---|
| Composition | Cation radius(pm) | MP(K) | Density(kg/m3) | Surface tension(mN/m) | ||
| 873 K | 973 K | 873 K | 973 K | |||
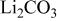 | 60 | 993 | 1884 | 1843 | 249.2 | 245.1 |
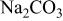 | 95 | 1124 | 2081 | 2038 | 224.9 | 219.8 |
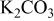 | 133 | 1164 | 2033 | 1988 | 188.4 | 181.9 |
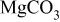 | 65 | - | - | - | - | - |
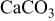 | 99 | 1098 | - | - | - | - |
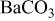 | 135 | 1573 | - | - | - | - |
Second, considering the relationship of wetting properties to the nature of the species that are assumed to be specifically adsorbed (oxide and hydroxide ions, as discussed earlier), the basicity of the carbonate melt, defined as  may have a strong effect on the wetting characteristics of the melt. From the dissociation reaction of the carbonate
may have a strong effect on the wetting characteristics of the melt. From the dissociation reaction of the carbonate
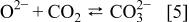
the equilibrium constant for this reaction is defined as
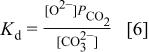
The dissociation constant  is dependent on the cation of the melt. For alkali cations, the dissociation constant decreases from lithium to potassium and for earth alkali cations, the constant increases from magnesium to barium.7 Table III shows the dissociation constant
is dependent on the cation of the melt. For alkali cations, the dissociation constant decreases from lithium to potassium and for earth alkali cations, the constant increases from magnesium to barium.7 Table III shows the dissociation constant  and the hydrolysis constant
and the hydrolysis constant  of carbonate melts at 823, 923, and 1023 K.8
of carbonate melts at 823, 923, and 1023 K.8
Table III.
| Dissociation constant and hydrolysis constant of carbonate melts. | ||||||
|---|---|---|---|---|---|---|
| Composition |
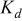 |
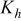 | ||||
| 823 K | 923 K | 1023 K | 823 K | 923 K | 1023 K | |
| Li |
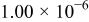 |
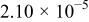 |
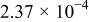 |
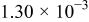 | ||
| Li-Naa |
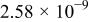 |
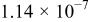 |
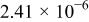 |
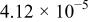 |
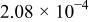 |
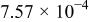 |
| Li-Na-Kb |
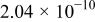 |
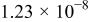 |
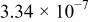 |
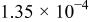 | ||
| Li-Kc |
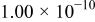 |
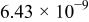 |
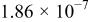 |
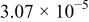 |
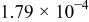 |
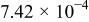 |
| Na-K |
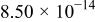 |
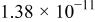 |
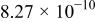 |
 | ||
a 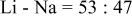 | ||||||
b 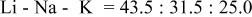 | ||||||
c 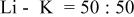
| ||||||
The Li-Na melt has a greater dissociation constant and a higher oxide ion concentration and is considered more basic (according to the Lux-Flood acid/base concept) than the Li-K melt. However, the effective basicity of the melt can also be influenced by gas composition.
From the experimental observation that the more basic carbonate melt, Li-Na, exhibits poorer wettability, the addition of acidity-enhancing components into the electrolyte should improve the wetting. This may be useful in the formulation of an optimal electrolyte composition for the next-generation electrolyte system.
Basicity is also dependent on temperature. As temperature is increased, the melt becomes more basic with a greater dissociation constant (Table III). This suggests that wetting may be affected by temperature via the basicity.
Effect of temperature.—
The temperature dependence of wetting was investigated for Li-Na carbonate by meniscus height measurements at 600 and 700, as well as 650°C, as shown in Fig. 10. The meniscus height increases with temperature, because as shown in Table II, the  interfacial energy (surface tension) decreases with increasing temperature. As a result, the wettability of the electrode is enhanced. This is analytically understood from the surface analogue of the Gibbs-Duhem relation
interfacial energy (surface tension) decreases with increasing temperature. As a result, the wettability of the electrode is enhanced. This is analytically understood from the surface analogue of the Gibbs-Duhem relation
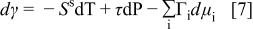
where  is the surface entropy per square centimeter of surface and τ is thickness of the surface (excess concentration) layer.5
9 In addition, Eq. 7 predicts that, for a constant thickness of the interfacial layer, wetting will be affected negatively by pressurization. This may also have practical implications for MCFC operation.
is the surface entropy per square centimeter of surface and τ is thickness of the surface (excess concentration) layer.5
9 In addition, Eq. 7 predicts that, for a constant thickness of the interfacial layer, wetting will be affected negatively by pressurization. This may also have practical implications for MCFC operation.
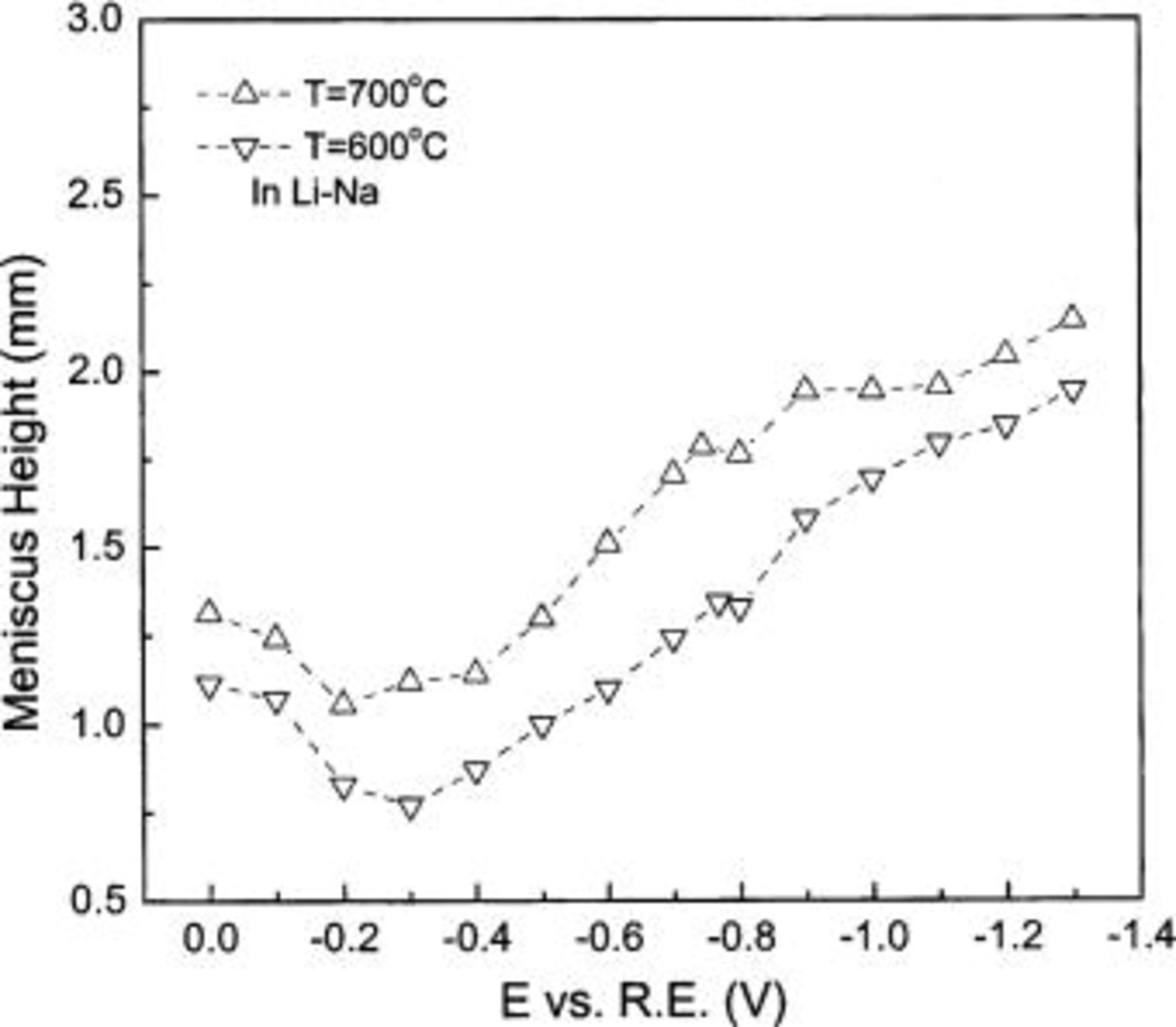
Figure 10. Temperature dependence in Li-Na under reducing atmosphere.
If the above explanation for the effect of temperature on wetting is correct, it introduces a contradiction with our previous inference that increased basicity is the cause of poor wetting. That inference was arrived at by comparing the wetting characteristics of Li-K (good wetting) and Li-Na melt (poor wetting). Since an elevated temperature makes carbonate melts more basic and the melts at higher temperature (700°C) show better wetting behavior than at 600°C, this contradicts our previous inference about basic melts being more poorly wetting. It is clearly not appropriate to explain the effect of electrolyte composition and temperature on wetting tendency exclusively by reference to the basicity of the melt. Basicity is, anyway, a bulk melt property, while wetting behavior is influenced by interfacial concentrations. The latter may not be proportional to bulk concentrations, as might be expected (for dilute solutes) if no specific adsorption occurs.
A clue to the properties influencing the temperature dependence of wetting is provided by the temperature dependence of the surface tension (interfacial tension). According to the Young-Dupre equation (Eq. 2), the molecular forces that affect the interfacial tension(s) should determine, to a considerable extent, the contact angle. The interfacial excess concentrations, e.g., of adsorbed  ions, play a leading role in determining the interfacial tension(s). They decrease tension(s) with temperature, thereby improving the wetting properties, but that increased bulk concentration (or activity) of adsorbed species (such as
ions, play a leading role in determining the interfacial tension(s). They decrease tension(s) with temperature, thereby improving the wetting properties, but that increased bulk concentration (or activity) of adsorbed species (such as  increases the interfacial tension, which decreases the wetting tendency. Between 650 and 700°C the direct temperature effect on interfacial concentration (and tension) seems to be dominant over the indirect effect on the latter via the increased bulk basicity. But below 650°C, the indirect effect may be dominant.
increases the interfacial tension, which decreases the wetting tendency. Between 650 and 700°C the direct temperature effect on interfacial concentration (and tension) seems to be dominant over the indirect effect on the latter via the increased bulk basicity. But below 650°C, the indirect effect may be dominant.
Effect on electrode performance.—
Literature reports confirm that the performance of MCFC cells with Li-Na and Li-K electrolyte diverge drastically at low temperature. Sishtla et al.6 reported that a Li/Na cell showed a lower performance than a Li/K cell below 600°C and ascribed this to increased cathode polarization. One particular reason for increased cathode polarization could be a reduction in electrolyte-wetted pore area of the cathode. It has been suggested that the greater temperature sensitivity of the Li/Na melt with respect to wetting of the electrode material, compared to the Li/K melt, may cause the cathode to become liquid-filled (drowned) to a greater extent in the Li/Na melt than in the Li/K melt, as the operating temperature is lowered to 600°C or below. It is therefore relevant to explore whether the results obtained in this project support this explanation, and to compare them with results obtained by other investigators who addressed this question, for example, Suski et al.10 and Kawase et al.11
The work of Suski et al.10 merits particular attention because, besides reporting contact angles of Li/K and Li/Na carbonate on Au, they reported also, for the first time, contact angles on Ni (anode material) and on NiO (cathode material), in their respective gas environments. They found that the wettability of the Au electrode in a  atmosphere decreases significantly when switching from Li/K melt to Li/Na melt (i.e., the contact angle increases when replacing Li/K by Li/Na melt). This is in agreement with our results. Suski et al.10 found the same trend for Ni (anode) as for Au under reducing atmosphere. Kawase et al.11 also found contact angles at Ni under reducing atmosphere to be higher for Li-Na than for Li-K, to an even larger extent than Suski et al.10 Thus, there is general agreement, among these two references and the present work, that wettability under reducing conditions, both at Ni and at Au, is less in Li/Na than in Li/K electrolyte.
atmosphere decreases significantly when switching from Li/K melt to Li/Na melt (i.e., the contact angle increases when replacing Li/K by Li/Na melt). This is in agreement with our results. Suski et al.10 found the same trend for Ni (anode) as for Au under reducing atmosphere. Kawase et al.11 also found contact angles at Ni under reducing atmosphere to be higher for Li-Na than for Li-K, to an even larger extent than Suski et al.10 Thus, there is general agreement, among these two references and the present work, that wettability under reducing conditions, both at Ni and at Au, is less in Li/Na than in Li/K electrolyte.
Similarly under reducing conditions Suski et al.10 found that the temperature gradient of the contact angle of Li/Na electrolyte at Au (approximately −0.12°/K) is more negative than that of Li/K electrolyte, and they reported the same trend for the temperature coefficient of the contact angle at Ni (anode), although its value (approximately −0.03°/K in Li/Na melt) is much smaller than that at Au. Interestingly, Kawase et al.11 report, for the effect of the electrolyte on the contact angle at Ni under reducing conditions, the opposite trend, i.e., Li-Na is better wetted than Li-K. Moreover, the value of the temperature coefficient, in Li/Na as well as in the Li/K electrolyte, is much more negative than that found by Suski et al.10 (approximately −0.34°/K in Li-Na and −0.50°/K in Li/K). In the present work the temperature coefficient of the contact angle at Au in Li/Na electrolyte under a reducing atmosphere is found to be approximately −0.10°/K, which is close to that reported by Suski et al.).10 Overall, there appears to be general agreement that the temperature coefficient of the contact angle under reducing conditions is negative, regardless of electrolyte composition. There is also agreement between the work of Suski et al.10 and the present results, that the temperature coefficient under reducing conditions is more negative in Li-Na than in Li/K electrolyte.
Although these trends under reducing conditions (greater θ and more negative  in Li/Na than in Li/K electrolyte) seem well-supported by the data from three sources, we note a significant spread in the measured values of θ among these sources, as illustrated in Table IV by data for 600 and 650°C. As discussed below, this impacts on the certainty with which conclusions may be drawn about the role of capillary effects in the low-temperature performance degradation observed with Li/Na electrolyte. It also suggests that the data are not reproducible and numerous enough to conclude with certainty that the metal (Au, Ni) has any effect on the wetting properties.
in Li/Na than in Li/K electrolyte) seem well-supported by the data from three sources, we note a significant spread in the measured values of θ among these sources, as illustrated in Table IV by data for 600 and 650°C. As discussed below, this impacts on the certainty with which conclusions may be drawn about the role of capillary effects in the low-temperature performance degradation observed with Li/Na electrolyte. It also suggests that the data are not reproducible and numerous enough to conclude with certainty that the metal (Au, Ni) has any effect on the wetting properties.
Table IV.
| Contact angle values. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Temp (°C)Material | 600 | 650 | ||||||
| Au | Ni | NiO | Au | Ni | NiO | |||
| Suski et al.10 | Li/K |
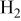 | 38 | 16 | 36 | 15 | ||
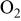 | 33 | 26 | 30 | 25 | ||||
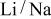 |
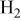 | 46 | 23 | 40 | 21.5 | |||
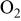 | 42 | 27 | 40 | 26 | ||||
| Kawase et al.11 | Li/K |
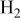 | 73 | 45 | ||||
| Li/Na |
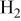 | 91 | 74 | |||||
| Hong | Li/Na |
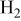 | 63 | 57 | ||||
Because cell performance and capillary properties of the electrolyte in an MCFC are linked by fixed-volume capillary equilibrium,4 one must consider also the data reported (in the literature) for contact angles under oxidizing atmosphere. According to Suski et al.10 the NiO cathode wettability is hardly affected by the electrolyte composition. From the data in Table IV, it can be seen that there is a much stronger electrolyte effect at Au than at NiO, and this may be related to the role of the metal (Au inert, NiO active in dynamic oxide-carbonate equilibrium). The temperature effect on the contact angle is also weak (approximately −0.02°/K) and barely different for Li/Na than for Li/K. Overall, based on the single source available,10 it appears that both temperature and cationic composition have a weak effect on the contact angle at NiO.
With the reservations imposed by the spread in θ values illustrated by Table IV, one may conclude that the following fairly represents the overall changes in capillary properties when comparing the performance of a Li/Na cell with that of a Li/K cell. In the Li/Na cell subjected to temperatures decreasing below 600°C, the greater contact angle of Li/Na electrolyte at the anode as well as its more strongly negative  of the anode (compared to the Li-K electrolyte) lead to a sharply increased contact angle. This is combined with a relatively unaffected contact angle at the cathode.
of the anode (compared to the Li-K electrolyte) lead to a sharply increased contact angle. This is combined with a relatively unaffected contact angle at the cathode.
This would easily explain at least part of the electrode polarization data reported by Sishtla et al.6 (Fig. 4) where anode polarization in Li/Na is always higher than in Li/K between 575-675°C. The increased resistance due to poor electrolyte fill of the anode is compatible with the above-reported decrease in wettability of the Ni anode (and probably any metal) in Li/Na electrolyte. The cathode polarization behavior reported by Sishtla et al.6 is more difficult to explain merely by changes in capillary properties (mainly of the anode). When comparing Li/Na and Li/K cathode polarization, an inversion is noticed around 600°C. Below 600°C polarization is higher in Li/Na electrolyte, whereas above this temperature it is higher in Li/K. One would expect that the capillary displacement of electrolyte from anode to cathode caused by lowering the temperature would lead to a monotonic increase in cathode polarization (if the cell is loaded for optimal operation at 650°C). This would be more pronounced for Li/Na than for Li/K electrolyte.
It appears therefore that causes other than differences in wetting tendency must be invoked to explain the observed cathode behavior in the Li/Na electrolyte. These may be related, for example, to changes in the mechanism of oxygen reduction with temperature. Thus, one may speculate that the peroxide ions which dominate the oxygen reduction mechanism in Li/Na electrolyte decrease less steeply in concentration with decreasing temperature, compared to the superoxide ions in Li/K electrolyte. But below 600°C this effect is overwhelmed by the flooding of the porous cathode and consequent decrease in available reaction area.
This review of available data and analysis of low-temperature performance makes clear that the accuracy and reproducibility of wettability data must be improved to develop a better understanding of electrode behavior at low temperature. This must go hand in hand with investigations focused more narrowly on the mechanism and electrode kinetics. Nevertheless, from the available data some useful conclusions may be drawn.
Conclusions
Using a gold electrode in molten carbonate, the dependence of meniscus height on polarization, gas composition, electrolyte melt composition, and temperature was examined. Plausible explanations of how these parameters affect the wetting behavior have been put forward.
Under all of the gas atmospheres, meniscus height was observed to depend on applied potential in the manner of an electrocapillary curve, with increase of meniscus height in both the cathodic and anodic direction away from the zero-charge potential (ZCP). Meniscus behavior upon cathodic polarization under oxidizing atmosphere was found to depend more strongly on polarization than that upon anodic polarization or under a reducing atmosphere. Better wetting under oxidant atmosphere was ascribed to specific adsorption of oxide ions on the electrode surface. Better wettability of the Li-K carbonate compared to the Li-Na melt appears to correlate with the difference in interfacial tension, rather than the bulk melt basicity, though both are affected by differences in cationic radius of the respective melts. The wetting tendency increased with increasing temperature, which may be explained with the aid of a form of the Gibbs-Duhem equation. In conclusion, as the Young-Dupre equation suggests, changes in wetting tendency (contact angle) cannot be considered separate from changes in interfacial energy (tension), for which temperature and bulk melt composition appear to provide independent influences.
The data obtained in this work support, at least in part, an explanation of the inferior performance of MCFC cells with Li/Na electrolyte at low (<600°C) temperature, compared with Li/K cells. They agree fairly well with the results of other investigators indicating a greater contact angle and more negative temperature dependence of the contact angle in Li/Na, compared to Li/K, electrolyte, at least for the nickel anode. If the capillary properties of the NiO cathode are only weakly affected by changes of electrolyte and temperature, which is suggested by the literature, then a fixed-volume capillary equilibrium model can explain the observed degradation of cell performance below 600°C.
Illinois Institute of Technology assisted in meeting the publication costs of this article.