
Abstract
 and
and  magic angle spinning nuclear magnetic resonance (NMR) allows for the identification and quantification of LiF in the solid electrolyte interphase (SEI) at both electrodes in lithium-ion rechargeable batteries. An estimate of the percent of lithium not reintercalated into the cathode after numerous cycles is also obtained. In the cells presented, more LiF formed on the surface of the anode compared to its respective cathode. Positive correlations have been established for the amount of LiF formed on the cathode with both the number of cycles and the % Li loss from the cathode. © 2005 The Electrochemical Society. All rights reserved.
magic angle spinning nuclear magnetic resonance (NMR) allows for the identification and quantification of LiF in the solid electrolyte interphase (SEI) at both electrodes in lithium-ion rechargeable batteries. An estimate of the percent of lithium not reintercalated into the cathode after numerous cycles is also obtained. In the cells presented, more LiF formed on the surface of the anode compared to its respective cathode. Positive correlations have been established for the amount of LiF formed on the cathode with both the number of cycles and the % Li loss from the cathode. © 2005 The Electrochemical Society. All rights reserved.
Export citation and abstract BibTeX RIS
At the heart of the portable technology race is the constant need for a better rechargeable battery. Increasing the calendar life of Li-ion batteries is also a vital goal in different aerospace and medical technologies. One of many obstacles to long life of these batteries is the growth of an electrically insulating layer, termed the solid electrolyte interphase (SEI), on the battery electrodes.1 2 The SEI, a product of the reaction between the electrode materials and the electrolyte components, forms immediately upon battery assembly. The SEI serves to protect the electrodes, forming a passivating layer, which prevents further reaction between the electrolyte and electrodes. However, with successive charge cycles, the continual deposition of this layer is believed to play a role in the gradual reduction of capacity for these batteries. This contribution to cycling limitation is a result of increasing ionic impedance of the SEI with increasing thickness,2 preventing the lithium ions from intercalating into the electrodes, and a reduction of the number of lithium ions available for insertion as they are irreversibly incorporated into the SEI.3
Experimental techniques, such as Fourier transform infrared (FTIR),4 electrical impedance spectroscopy (EIS),5
6 and X-ray photoelectron spectroscopy (XPS),7
8 have been used to analyze SEI layer formation qualitatively.  LiOH,
LiOH,  LiF, polyolephines, and semi-carbonates have been identified. While XPS is semi-quantitative, depth profiling is usually achieved by Argon sputtering, where the efficiency of sputtering is dependent upon the type of material, and concerns exist regarding surface reactions and material decomposition during experimentation.7 Nuclear magnetic resonance (NMR), on the other hand, is inherently quantitative and nondestructive. NMR has been used extensively to characterize the
LiF, polyolephines, and semi-carbonates have been identified. While XPS is semi-quantitative, depth profiling is usually achieved by Argon sputtering, where the efficiency of sputtering is dependent upon the type of material, and concerns exist regarding surface reactions and material decomposition during experimentation.7 Nuclear magnetic resonance (NMR), on the other hand, is inherently quantitative and nondestructive. NMR has been used extensively to characterize the  cathode9
10
11
12
13
14 and carbonaceous anode15
16
17 materials. Recently, Ota et al. have used
cathode9
10
11
12
13
14 and carbonaceous anode15
16
17 materials. Recently, Ota et al. have used  and
and  liquid state NMR to study surface film formation on nickel substrates.18 Only a few papers to date have specifically addressed the issue of SEI formation with solid state NMR on cycled electrodes.17
19
20 Wang et al. have performed
liquid state NMR to study surface film formation on nickel substrates.18 Only a few papers to date have specifically addressed the issue of SEI formation with solid state NMR on cycled electrodes.17
19
20 Wang et al. have performed  static NMR on cycled
static NMR on cycled  cathodes and graphite anodes,20 and were able to compare the amount of SEI formation in cells stored at partial charge and elevated temperature. Menetrier et al. investigated a series of mixtures of
cathodes and graphite anodes,20 and were able to compare the amount of SEI formation in cells stored at partial charge and elevated temperature. Menetrier et al. investigated a series of mixtures of  and active material to shed light on the effect of proximity of
and active material to shed light on the effect of proximity of  in the SEI layer to the active material.21 Recently, Song et al. used ex situ FTIR spectroscopy with the attenuated total reflection technique to investigate surface film formation on
in the SEI layer to the active material.21 Recently, Song et al. used ex situ FTIR spectroscopy with the attenuated total reflection technique to investigate surface film formation on  cathode; however they were not able to detect LiF as one of the decomposition products of
cathode; however they were not able to detect LiF as one of the decomposition products of  due to LiF's invisibility in the mid-IR (700 to
due to LiF's invisibility in the mid-IR (700 to  range.22
range.22
Menetrier et al. have reported the  NMR spectra for
NMR spectra for  at different states of charge;14
at different states of charge;14  where
where  has a chemical shift of
has a chemical shift of  During the initial stage of charging, a new resonance appeared at around 60 ppm, which coexisted with the resonance due to
During the initial stage of charging, a new resonance appeared at around 60 ppm, which coexisted with the resonance due to  until 25 to 30% of the lithium have been removed. The new resonance was ascribed to the presence of a second phase,
until 25 to 30% of the lithium have been removed. The new resonance was ascribed to the presence of a second phase,  caused by the insulator to metal transition that occurs in
caused by the insulator to metal transition that occurs in  upon Li deintercalation. Upon further charging, the resonance at around 60 ppm shifts toward more positive frequencies until it reaches approximately 100 ppm when
upon Li deintercalation. Upon further charging, the resonance at around 60 ppm shifts toward more positive frequencies until it reaches approximately 100 ppm when  in
in  Here we show that we can use this information to follow the state of charge of
Here we show that we can use this information to follow the state of charge of  in
in  full cells as a function of cycle number, and the fraction of Li lost to side reactions (which include the formation of the SEI on the cathode and anode and any Li that remains stuck in the anode). In addition,
full cells as a function of cycle number, and the fraction of Li lost to side reactions (which include the formation of the SEI on the cathode and anode and any Li that remains stuck in the anode). In addition,  NMR is employed to determine the LiF content.
NMR is employed to determine the LiF content.
Experimental
For all cells in this study, the cathode was commercial  mixed with poly(vinylidene fluoride) (PVDF) and graphite, and the anode was graphitic mesocarbon microbeads (MCMB) mixed with Super P and PVDF. The electrolyte used in this study was
mixed with poly(vinylidene fluoride) (PVDF) and graphite, and the anode was graphitic mesocarbon microbeads (MCMB) mixed with Super P and PVDF. The electrolyte used in this study was  dissolved into a mixture of ethylene carbonate (EC), propylencarbonate (PC), ethyl methyl carbonate (EMC), and diethyl carbonate (DEC). Cell A was cycled at 60°C from 3.0 to 3.9 V. Cells B-F were cycled from 3.0 to 4.1 V at room temperature with a C/2 charge rate and a 2/3C discharge rate to 40% depth of discharge (DOD). All cells were removed from cycling at failure. The criteria for failure in this study was defined as the point when the cell reached 3.0 V upon discharge without delivery of 40% of its original capacity. The cells were then disassembled at the 3.0 V point at failure. For NMR, the cathode and anode material were scraped from the foil backing inside an Argon dry box and packed into 2.5 mm rotors. Lithium loss in the cathode was determined by chemical shift comparison instead of
dissolved into a mixture of ethylene carbonate (EC), propylencarbonate (PC), ethyl methyl carbonate (EMC), and diethyl carbonate (DEC). Cell A was cycled at 60°C from 3.0 to 3.9 V. Cells B-F were cycled from 3.0 to 4.1 V at room temperature with a C/2 charge rate and a 2/3C discharge rate to 40% depth of discharge (DOD). All cells were removed from cycling at failure. The criteria for failure in this study was defined as the point when the cell reached 3.0 V upon discharge without delivery of 40% of its original capacity. The cells were then disassembled at the 3.0 V point at failure. For NMR, the cathode and anode material were scraped from the foil backing inside an Argon dry box and packed into 2.5 mm rotors. Lithium loss in the cathode was determined by chemical shift comparison instead of  spin counting. The extra unknown amounts of the different Lithium environments formed in the SEI in cycled cells makes spin counting problematic and leads to larger errors in estimation. It is important to note that the Li loss determined electrochemically and by NMR may differ. Electrochemically, any particles which have been isolated will result in capacitance loss even though intercalated lithium is still present. Alternatively, NMR will be sensitive to all particles isolated or not in the active material.
spin counting. The extra unknown amounts of the different Lithium environments formed in the SEI in cycled cells makes spin counting problematic and leads to larger errors in estimation. It is important to note that the Li loss determined electrochemically and by NMR may differ. Electrochemically, any particles which have been isolated will result in capacitance loss even though intercalated lithium is still present. Alternatively, NMR will be sensitive to all particles isolated or not in the active material.
The  MAS spectra was acquired at 272.1 MHz using a Bruker 700 MHz system. Data was collected using a single π/4 pulse, 10 s delays between scans, and a 25 kHz spinning speed.
MAS spectra was acquired at 272.1 MHz using a Bruker 700 MHz system. Data was collected using a single π/4 pulse, 10 s delays between scans, and a 25 kHz spinning speed.  MAS spectra were acquired at 564.6 MHz using a Bruker 600 MHz system. Data was collected using a single π/2 pulse, 10 s delays between scans, and spinning speeds of 25 to 35 kHz.
MAS spectra were acquired at 564.6 MHz using a Bruker 600 MHz system. Data was collected using a single π/2 pulse, 10 s delays between scans, and spinning speeds of 25 to 35 kHz.  and
and  chemical shifts are quoted relative to the external references 1M LiCl (in
chemical shifts are quoted relative to the external references 1M LiCl (in  and
and  respectively, both set to 0 ppm.
respectively, both set to 0 ppm.
 integrated intensities were determined by using spectral simulation and the presence of PVDF binder as a fluorine reference. The LiF content was then calculated and normalized to allow comparisons between cathodes and corresponding anodes using equation 1. In this study, only calculations using equation 1 for unrinsed cells are reported.
integrated intensities were determined by using spectral simulation and the presence of PVDF binder as a fluorine reference. The LiF content was then calculated and normalized to allow comparisons between cathodes and corresponding anodes using equation 1. In this study, only calculations using equation 1 for unrinsed cells are reported.
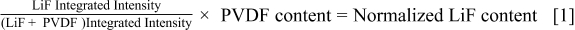
Results and Discussion
The first focus of this study was to build a chemical shift library for  inorganic compounds using the high field NMR 700 MHz spectrometer. Several spectra are displayed in Fig. 1. Because diamagnetic lithium has a very small chemical shift range
inorganic compounds using the high field NMR 700 MHz spectrometer. Several spectra are displayed in Fig. 1. Because diamagnetic lithium has a very small chemical shift range  the high field was necessary to gain enough resolution to separate the different compounds that may be present in the SEI.
the high field was necessary to gain enough resolution to separate the different compounds that may be present in the SEI.

Figure 1.
 chemical shift spectra of various inorganic compounds that maybe present in a SEI layer.
chemical shift spectra of various inorganic compounds that maybe present in a SEI layer.
Figure 2 shows the  MAS NMR spectrum (at 272.1 MHz) of a cell cycled at 60°C for 3000 cycles from 3.0 to 3.9 V (cell A). Two resonances are seen at 60 (strong) and 0.3 ppm (weak), which are assigned to
MAS NMR spectrum (at 272.1 MHz) of a cell cycled at 60°C for 3000 cycles from 3.0 to 3.9 V (cell A). Two resonances are seen at 60 (strong) and 0.3 ppm (weak), which are assigned to  and to the SEI,
and to the SEI,  and residual
and residual  respectively. Unfortunately, the 0.3 ppm resonance lacks sufficient resolution to allow the individual components in the SEI layer to be distinguished. The broadening is ascribed to the anisotropic magnetic susceptibility of the carbon used to fabricate the electrodes and the presence of
respectively. Unfortunately, the 0.3 ppm resonance lacks sufficient resolution to allow the individual components in the SEI layer to be distinguished. The broadening is ascribed to the anisotropic magnetic susceptibility of the carbon used to fabricate the electrodes and the presence of 
 The overall shift of the resonance suggests that
The overall shift of the resonance suggests that  or
or  may be present but no further analysis is possible. Based on the frequency of the more intense resonance at 60 ppm, and the earlier work by Menetrier et al. ,14 an estimate for the Li loss from the cathode of approximately 25% can be determined. As a result, the cathode composition at the time of disassembly is approximately
may be present but no further analysis is possible. Based on the frequency of the more intense resonance at 60 ppm, and the earlier work by Menetrier et al. ,14 an estimate for the Li loss from the cathode of approximately 25% can be determined. As a result, the cathode composition at the time of disassembly is approximately 

Figure 2.
 MAS NMR spectra for a cycled
MAS NMR spectra for a cycled  cathode. Isotropic resonances are seen at 60 ppm and 0.3 ppm; all the other peaks are spinning sidebands from 25kHz spinning speed. The
cathode. Isotropic resonances are seen at 60 ppm and 0.3 ppm; all the other peaks are spinning sidebands from 25kHz spinning speed. The  MAS NMR spectra were acquired by using a π/4 pulse length of 0.5 μs with 10 s delays between scans at 272.1 MHz for lithium. 1 M LiCl in distilled water was used as an external chemical shift reference at 0.0 ppm.
MAS NMR spectra were acquired by using a π/4 pulse length of 0.5 μs with 10 s delays between scans at 272.1 MHz for lithium. 1 M LiCl in distilled water was used as an external chemical shift reference at 0.0 ppm.
Figure 3 shows the experimentally determined losses of Li from the  cathode (determined by using the same approach as described above for cell A) as a function of cycle number for five other similar cells (B through F) cycled at room temperature. A positive correlation exists between the percent of lithium not reintercalated into the cathode at full discharge and the cycle number. The Li loss at higher temperatures is clearly higher: the Li loss for cell A, cycled at 60°C for only 3000 cycles, is intermediate between that of cell E and F, which have been cycled 6500 and 9000 times.
cathode (determined by using the same approach as described above for cell A) as a function of cycle number for five other similar cells (B through F) cycled at room temperature. A positive correlation exists between the percent of lithium not reintercalated into the cathode at full discharge and the cycle number. The Li loss at higher temperatures is clearly higher: the Li loss for cell A, cycled at 60°C for only 3000 cycles, is intermediate between that of cell E and F, which have been cycled 6500 and 9000 times.

Figure 3. Experimentally determined Li loss from  by
by  MAS NMR spectra for cycled
MAS NMR spectra for cycled  cathodes.
cathodes.
The Li that remains intercalated in the anode can also be directly monitored by acquiring the  NMR spectra of the anode. Figure 4 shows the
NMR spectra of the anode. Figure 4 shows the  MAS NMR spectra of a cycled carbon anode before and after rinsing three times with dimethylcarbonate (DMC). Washing was performed to remove residual electrolyte and salt. The weaker peak at 45 ppm, whose intensity remains unchanged on rinsing is assigned to the
MAS NMR spectra of a cycled carbon anode before and after rinsing three times with dimethylcarbonate (DMC). Washing was performed to remove residual electrolyte and salt. The weaker peak at 45 ppm, whose intensity remains unchanged on rinsing is assigned to the  and/or
and/or  phase,16
17 which shows that even though the cell was fully discharged, some intercalated lithium remains. A broader resonance, centered around 10 ppm is also observed, whose intensity is noticeably changed by rinsing. This resonance can be deconvoluted into at least 3 resonances at 0, 8, and 16 ppm. The peak near 0 ppm is assigned to the SEI and dried electrolyte, consistent with its change of intensity on washing. The shifts of the peaks at 8 and 16 ppm appear to be slightly too large to be attributed to any diamagnetic components and on this basis are tentatively assigned to
phase,16
17 which shows that even though the cell was fully discharged, some intercalated lithium remains. A broader resonance, centered around 10 ppm is also observed, whose intensity is noticeably changed by rinsing. This resonance can be deconvoluted into at least 3 resonances at 0, 8, and 16 ppm. The peak near 0 ppm is assigned to the SEI and dried electrolyte, consistent with its change of intensity on washing. The shifts of the peaks at 8 and 16 ppm appear to be slightly too large to be attributed to any diamagnetic components and on this basis are tentatively assigned to  16
23 However, it is somewhat surprising that the intensities would decrease with rinsing, suggesting that these peaks may be associated with
16
23 However, it is somewhat surprising that the intensities would decrease with rinsing, suggesting that these peaks may be associated with  either on or near the surface of the carbon particles.
either on or near the surface of the carbon particles.

Figure 4.
 MAS NMR spectra for the anode of cell A showing the presence of intercalated lithium at 45 ppm.
MAS NMR spectra for the anode of cell A showing the presence of intercalated lithium at 45 ppm.
Because we were not able to further resolve the  resonances corresponding to LiF in the anode or cathode SEI,
resonances corresponding to LiF in the anode or cathode SEI,  NMR was used to identify all the fluorine-containing components.
NMR was used to identify all the fluorine-containing components.  NMR is more sensitive to small changes in the local environment, because the chemical shift range is larger compared to
NMR is more sensitive to small changes in the local environment, because the chemical shift range is larger compared to  and thus components such as LiF are more easily resolved.
and thus components such as LiF are more easily resolved.
Figure 5 shows the  MAS NMR spectra for both the cathode and anode taken from cell A. The LiF resonance appears at
MAS NMR spectra for both the cathode and anode taken from cell A. The LiF resonance appears at  The resonance at
The resonance at  is from the
is from the  in the dried electrolyte. The remaining resonances at
in the dried electrolyte. The remaining resonances at  and
and  are assigned to polyvinylidene fluoride (PVDF), which is used as an electrode binder. Rinsing of the anode with DMC results in a decrease in the integrated intensity of both the dried electrolyte and LiF peaks. However, while the dried electrolyte peak is eliminated, a significant portion of the LiF peak remains. Spectral simulation and integration yield values for the percentage of total
are assigned to polyvinylidene fluoride (PVDF), which is used as an electrode binder. Rinsing of the anode with DMC results in a decrease in the integrated intensity of both the dried electrolyte and LiF peaks. However, while the dried electrolyte peak is eliminated, a significant portion of the LiF peak remains. Spectral simulation and integration yield values for the percentage of total  signal (excluding dried electrolyte) from LiF content in the cathode, anode, and rinsed anode, respectively for cell A. Normalizing this with the PVDF content (Eq. 1) in each electrode (1:3 cathode to anode) allows for a direct comparison of the amount of LiF formed at each electrode (0.25 : 3.44 : 2.16 for cathode : unrinsed anode: rinsed anode, respectively). As a result, 8.5 and 13 times as much LiF content is present at the rinsed and unrinsed anode surface compared to the unrinsed cathode, respectively.
signal (excluding dried electrolyte) from LiF content in the cathode, anode, and rinsed anode, respectively for cell A. Normalizing this with the PVDF content (Eq. 1) in each electrode (1:3 cathode to anode) allows for a direct comparison of the amount of LiF formed at each electrode (0.25 : 3.44 : 2.16 for cathode : unrinsed anode: rinsed anode, respectively). As a result, 8.5 and 13 times as much LiF content is present at the rinsed and unrinsed anode surface compared to the unrinsed cathode, respectively.

Figure 5.
 MAS NMR spectra of
MAS NMR spectra of  cathode and unrinsed and rinsed MCMB anode. The
cathode and unrinsed and rinsed MCMB anode. The  MAS NMR spectra were acquired by using a π/2 pulse length of 2.3 μs with 10 s delays between scans at 564.6 MHz for Fluorine.
MAS NMR spectra were acquired by using a π/2 pulse length of 2.3 μs with 10 s delays between scans at 564.6 MHz for Fluorine.  was used as the
was used as the  reference set to 0 ppm.
reference set to 0 ppm.
Figure 6 shows the quantitative results from the series of cells cycled at room temperature from 3.0 to 4.2 V 4500, 5500, 6000, 6500, and 9000 times (cells B-F). These were also fully discharged prior to disassembly. The graph shows a positive correlation for the amount of LiF, determined by  NMR, at the cathode surface and the percent of Li loss from the cathode (determined by
NMR, at the cathode surface and the percent of Li loss from the cathode (determined by  NMR). Cell F, which was cycled for 9000 times and showed a 30% Li loss from the cathode, as determined by the 89 ppm
NMR). Cell F, which was cycled for 9000 times and showed a 30% Li loss from the cathode, as determined by the 89 ppm  NMR shift of the
NMR shift of the  resonance, contained twice as much LiF as cell C (cycled 5500 times; 15% Li loss from the cathode). In all the cells much more LiF was found to be present on the anode compared to the cathode.
resonance, contained twice as much LiF as cell C (cycled 5500 times; 15% Li loss from the cathode). In all the cells much more LiF was found to be present on the anode compared to the cathode.

Figure 6. Comparison of the relative LiF content found on (a) the cathodes and (b) the anodes from cells B, C, D, E, and F, using PVDF as a calibration standard vs. Li loss from the cathode.
At the moment there is no clear correlation between LiF content on the anode and the percent of Li loss from the cathode. The anode of Cell B contains more LiF than the other anodes which performed to a larger number of cycles. If irreversible loss of Lithium was the only mechanism to contribute to cell failure, it would be expected that the resultant composition of the cathode of cell B to be approximately  However; because the cathode for cell B shows a very low Lithium loss of
However; because the cathode for cell B shows a very low Lithium loss of 
 as determined by NMR, this would imply that the cell may have failed due to a different mechanism than the one responsible for the other cell failures. Cell B did contain more Super P carbon added to the MCMB carbon in comparison to the amount used in the other cells. This may suggest that the carbon Super P may also play a role in SEI formation, i.e., its higher concentration may result in more LiF in the SEI on the anode. More experiments are in progress to test this hypothesis. The anode for cell F (9000 cycles and 30% lithium loss from cathode) shows a lower concentration of LiF than the other less cycled anodes. Again, this is indicating that the dominant route for failure in this cell is different.
as determined by NMR, this would imply that the cell may have failed due to a different mechanism than the one responsible for the other cell failures. Cell B did contain more Super P carbon added to the MCMB carbon in comparison to the amount used in the other cells. This may suggest that the carbon Super P may also play a role in SEI formation, i.e., its higher concentration may result in more LiF in the SEI on the anode. More experiments are in progress to test this hypothesis. The anode for cell F (9000 cycles and 30% lithium loss from cathode) shows a lower concentration of LiF than the other less cycled anodes. Again, this is indicating that the dominant route for failure in this cell is different.
Temperature clearly affects SEI formation. Cell A cycled at a higher temperature (60°C: Fig. 2 and 4) showed more LiF on both the anode and cathode when compared to the other cells (B-F) cycled at room temperature for a similar extrapolated number of cycles. Interestingly, the results from cell A do not fall on the correlation shown in Fig. 6 for cells B, C, D, E, and F, indicating that the high Li loss seen at higher temperatures (60°C) does not directly correlate with LiF formation. This suggests the following reaction for decomposition of the electrolyte
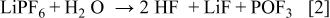
Conclusions
The results presented in this paper show that  NMR spectroscopy of fully discharged
NMR spectroscopy of fully discharged  cathodes can be used to estimate the percentage of Li not reintercalated into the cathode, for cells cycled under a variety of conditions. The lost Li is incorporated into the SEI at both the anode and cathode and also remains intercalated in the graphitic carbon at the anode. The large
cathodes can be used to estimate the percentage of Li not reintercalated into the cathode, for cells cycled under a variety of conditions. The lost Li is incorporated into the SEI at both the anode and cathode and also remains intercalated in the graphitic carbon at the anode. The large  chemical shift range allows LiF in the SEI at both the cathode and anode to be identified. Using PVDF as an internal calibration standard enables a direct quantitative comparison of LiF content between the electrodes. In all cells presented, more LiF was formed on the surface of the anode than on the cathode. A positive correlation was established for the amount of LiF formed on the cathode with both the number of cycles and the percent of Li loss from the cathode. No apparent correlation was found for LiFvs. cycle number for the anodes which may result from different failure mechanisms within the cells.
chemical shift range allows LiF in the SEI at both the cathode and anode to be identified. Using PVDF as an internal calibration standard enables a direct quantitative comparison of LiF content between the electrodes. In all cells presented, more LiF was formed on the surface of the anode than on the cathode. A positive correlation was established for the amount of LiF formed on the cathode with both the number of cycles and the percent of Li loss from the cathode. No apparent correlation was found for LiFvs. cycle number for the anodes which may result from different failure mechanisms within the cells.
Acknowledgment
We thank the Director of Central Intelligence Postdoctoral Research Fellowship Program for funding. Special thanks go to Dr. Martine Ziliox at the State University of New York at Stony Brook for her invaluable help with the high field spectrometers.
Hunter College of City University of New York assisted in meeting the publication costs of this article.