
Abstract
 (
( , Pr, Nd, Sm, and Gd) perovskite oxides have been synthesized and compared as cathode materials for intermediate temperature solid oxide fuel cells (SOFC). Both the electrical conductivity and the oxide ion vacancy concentration decrease from
, Pr, Nd, Sm, and Gd) perovskite oxides have been synthesized and compared as cathode materials for intermediate temperature solid oxide fuel cells (SOFC). Both the electrical conductivity and the oxide ion vacancy concentration decrease from  to Gd, which leads to a decrease in the electrocatalytic activity for oxygen reduction in SOFC. However, the thermal expansion coefficient (TEC) decreases favorably from
to Gd, which leads to a decrease in the electrocatalytic activity for oxygen reduction in SOFC. However, the thermal expansion coefficient (TEC) decreases favorably from  to Gd due to a decreasing ionicity of the Ln–O bond and a suppression of the tendency to lose oxygen from the lattice with increasing temperature. Therefore,
to Gd due to a decreasing ionicity of the Ln–O bond and a suppression of the tendency to lose oxygen from the lattice with increasing temperature. Therefore,  cathodes with an intermediate lanthanide ion such as
cathodes with an intermediate lanthanide ion such as  or
or  offer a trade-off between electrocatalytic activity and TEC. Also, the differences in dependence of the properties of the
offer a trade-off between electrocatalytic activity and TEC. Also, the differences in dependence of the properties of the  and
and  systems on the
systems on the  ions are discussed.
ions are discussed.
Export citation and abstract BibTeX RIS
The chemical reactivity and thermal expansion mismatch among the components as well as the limited choice of interconnect and cathode materials at the conventional operating temperatures  of solid oxide fuel cells (SOFC) have created enormous interest recently in intermediate temperature
of solid oxide fuel cells (SOFC) have created enormous interest recently in intermediate temperature  SOFC.1 However, the performance of the intermediate temperature SOFC is strongly dependent on the electrochemical properties of the cathode materials because the cathode overpotential for the oxygen reduction reaction (ORR) increases significantly at lower temperatures.2 Therefore, there is a need to develop alternate mixed ionic-electronic conductors that can replace the conventional
SOFC.1 However, the performance of the intermediate temperature SOFC is strongly dependent on the electrochemical properties of the cathode materials because the cathode overpotential for the oxygen reduction reaction (ORR) increases significantly at lower temperatures.2 Therefore, there is a need to develop alternate mixed ionic-electronic conductors that can replace the conventional  cathodes.
cathodes.
A desirable cathode material for the intermediate temperature SOFC should have high electronic and oxide ion conductivities, chemical and thermal expansion compatibility with the electrolyte, and high catalytic activity for the oxygen reduction reaction. In this regard, the Sr-doped lanthanum cobaltites  have been investigated intensively due to their high electronic and oxide ion conductivities.3, 4 However, the
have been investigated intensively due to their high electronic and oxide ion conductivities.3, 4 However, the  cathodes exhibit a large thermal expansion coefficient (TEC) and react readily with the yttria-stabilized zirconia (YSZ) electrolyte.5, 6 Accordingly, other Sr-doped lanthanide cobaltites
cathodes exhibit a large thermal expansion coefficient (TEC) and react readily with the yttria-stabilized zirconia (YSZ) electrolyte.5, 6 Accordingly, other Sr-doped lanthanide cobaltites 
 have also been investigated as cathode materials for intermediate temperature SOFC.7–12 Despite a scattered investigation of the
have also been investigated as cathode materials for intermediate temperature SOFC.7–12 Despite a scattered investigation of the  cathodes, a systematic investigation of the factors that control their electrocatalytic activity toward oxygen reduction reaction is scarcely available in the literature. With this perspective, we present here a comparison of the crystal chemistry, thermal expansion, electrical conductivity, chemical compatibility, and electrochemical performance of the
cathodes, a systematic investigation of the factors that control their electrocatalytic activity toward oxygen reduction reaction is scarcely available in the literature. With this perspective, we present here a comparison of the crystal chemistry, thermal expansion, electrical conductivity, chemical compatibility, and electrochemical performance of the  (
( , Pr, Nd, Sm, and Gd) cathodes and a correlation of the electrochemical performance to the lanthanide host cations. Additionally, the differences in the influence of the
, Pr, Nd, Sm, and Gd) cathodes and a correlation of the electrochemical performance to the lanthanide host cations. Additionally, the differences in the influence of the  ions on the properties of the
ions on the properties of the  and
and  systems are discussed.
systems are discussed.
Experimental
The  (
( , Pr, Nd, Sm, and Gd) cathode samples were synthesized by conventional solid-state reaction method. Required amounts of the lanthanide oxides (
, Pr, Nd, Sm, and Gd) cathode samples were synthesized by conventional solid-state reaction method. Required amounts of the lanthanide oxides ( ,
,  ,
,  ,
,  , or
, or  ),
),  , and
, and  were thoroughly mixed with ethanol using an agate mortar and pestle, and calcined in air first at
were thoroughly mixed with ethanol using an agate mortar and pestle, and calcined in air first at  for
for  . The calcined powders were then ground, pressed into pellets, and finally sintered at
. The calcined powders were then ground, pressed into pellets, and finally sintered at  for
for  . The sintering at
. The sintering at  was repeated again for
was repeated again for  after regrinding and repelletizing to improve the product homogeneity. The
after regrinding and repelletizing to improve the product homogeneity. The  (LSGM) electrolyte disks were prepared by firing required amounts of
(LSGM) electrolyte disks were prepared by firing required amounts of  ,
,  ,
,  , and MgO at
, and MgO at  for
for  , followed by pelletizing and sintering at
, followed by pelletizing and sintering at  for
for  .
.  (GDC) cermet
(GDC) cermet  anode was synthesized by the glycine-nitrate combustion method.13
anode was synthesized by the glycine-nitrate combustion method.13
The products thus obtained were characterized by X-ray diffraction (XRD) and the lattice parameters were obtained by analyzing the XRD data with the Rietveld method. Brunauer, Emmett, and Teller (BET) surface area was measured with a Quantachrom Autosorb-1 surface area and pore size analyzer. The average oxidation state of Co and the oxygen content at room temperature were determined by iodometric titration14 with fine powders obtained by ballmilling the oxides and sieving them a few times; the fine powder was necessary to have a quantitative dissolution of the oxides in a mixture of 10% KI and  HCl solution during iodometric titration. Thermogravimetric analysis (TGA) and thermal expansion data were collected with a Perkin-Elmer series 7 thermal analysis system. The TGA experiments were carried out from room temperature to
HCl solution during iodometric titration. Thermogravimetric analysis (TGA) and thermal expansion data were collected with a Perkin-Elmer series 7 thermal analysis system. The TGA experiments were carried out from room temperature to  with a heating rate of
with a heating rate of  and a cooling rate of
and a cooling rate of  in air. The TECs of sintered samples were measured from room temperature to
in air. The TECs of sintered samples were measured from room temperature to  with a heating/cooling rate of
with a heating/cooling rate of  . Electrical conductivity of the samples was measured by a four-probe dc method in the temperature range of
. Electrical conductivity of the samples was measured by a four-probe dc method in the temperature range of  . The bulk densities of the sintered samples were measured by the Archimedes method. All the samples used for thermal expansion and conductivity measurements had densities of
. The bulk densities of the sintered samples were measured by the Archimedes method. All the samples used for thermal expansion and conductivity measurements had densities of  of theoretical values. The specimens for the reactivity tests were obtained by mixing the cathode powders with the LSGM electrolyte in a 1:1 weight ratio. The reactivity tests were then carried out at
of theoretical values. The specimens for the reactivity tests were obtained by mixing the cathode powders with the LSGM electrolyte in a 1:1 weight ratio. The reactivity tests were then carried out at  for
for  in air. The microstructures of the cathodes were studied with a JEOL LSM-5610 scanning electron microscope.
in air. The microstructures of the cathodes were studied with a JEOL LSM-5610 scanning electron microscope.  measurements of single cells were carried out using a three-electrode configuration which allowed for separating and monitoring the cathode overpotentials during operation. Pt paste was used as the reference electrode. The
measurements of single cells were carried out using a three-electrode configuration which allowed for separating and monitoring the cathode overpotentials during operation. Pt paste was used as the reference electrode. The  cathodes and NiO-GDC cermet anode were prepared by screen printing onto
cathodes and NiO-GDC cermet anode were prepared by screen printing onto  thick LSGM electrolyte pellet, followed by firing for
thick LSGM electrolyte pellet, followed by firing for  at
at  for the cathode and
for the cathode and  for the anode. The geometrical area of the electrode was
for the anode. The geometrical area of the electrode was  . Humidified
. Humidified  (
(
 at
at  ) and air were supplied as fuel and oxidant, respectively, at a rate of
) and air were supplied as fuel and oxidant, respectively, at a rate of  .
.
Results and Discussion
X-ray diffraction showed all the  samples to be single-phase perovskite solid solutions without any detectable impurity phases, which is consistent with the report of Ohbayashi et al.12 While the
samples to be single-phase perovskite solid solutions without any detectable impurity phases, which is consistent with the report of Ohbayashi et al.12 While the  sample was found to be rhombohedral, all others with
sample was found to be rhombohedral, all others with  , Nd, Sm, and Gd were found to be orthorhombic. The lattice parameters, lattice volume, pseudo-cubic lattice parameter, and tolerance factor
, Nd, Sm, and Gd were found to be orthorhombic. The lattice parameters, lattice volume, pseudo-cubic lattice parameter, and tolerance factor  for all the
for all the  samples are summarized in Table I. The pseudo-cubic lattice parameter decreases from
samples are summarized in Table I. The pseudo-cubic lattice parameter decreases from  to Gd due to a decrease in the ionic radius of the
to Gd due to a decrease in the ionic radius of the  ion. The Goldschmidt tolerance factor
ion. The Goldschmidt tolerance factor  can be used as a measure of the deviation of the
can be used as a measure of the deviation of the  perovskite structure from the ideal cubic symmetry,
perovskite structure from the ideal cubic symmetry,
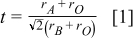
where  ,
,  , and
, and  are, respectively, the radii of
are, respectively, the radii of  ,
,  , and
, and  ions. A decreasing
ions. A decreasing  due to a decreasing ionic radius from
due to a decreasing ionic radius from  to
to  causes a lowering of the crystal symmetry and an increasing bending of the O–Co–O bond angle from the ideal value of 180°.
causes a lowering of the crystal symmetry and an increasing bending of the O–Co–O bond angle from the ideal value of 180°.
Table I. Lattice parameters, lattice volume, pseudocubic lattice parameter  , tolerance factor
, tolerance factor  , and chemical analysis data of
, and chemical analysis data of  .
.
| Ln |
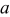 (Å) (Å) |
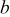 (Å) (Å) |
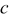 (Å) (Å) | Lattice volume 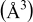
|
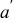 (Å) (Å) |
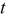
| Oxidation state of Co | Oxygen content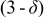
|
|---|---|---|---|---|---|---|---|---|
| La | 5.4079(4) | - | - | 112.5 | 4.8274 | 0.965 | 3.38 | 2.99 |
| Pr | 5.3733(5) | 5.4240(5) | 7.5962(8) | 221.4 | 3.8110 | 0.957 | 3.39 | 3.00 |
| Nd | 5.3656(5) | 5.4148(4) | 7.5917(7) | 220.6 | 3.8064 | 0.953 | 3.38 | 2.99 |
| Sm | 5.3564(6) | 5.3814(5) | 7.5792(8) | 218.5 | 3.7943 | 0.946 | 3.39 | 3.00 |
| Gd | 5.3578(9) | 5.3654(4) | 7.5718(8) | 217.7 | 3.7897 | 0.941 | 3.40 | 3.00 |
The average oxidation state of Co and the oxygen content values determined at room temperature by the iodometric titration are given in Table I for the  oxides. The oxygen contents remains at 3.0 irrespective of the lanthanide ion. The TGA plots comparing the variations of oxygen contents in
oxides. The oxygen contents remains at 3.0 irrespective of the lanthanide ion. The TGA plots comparing the variations of oxygen contents in  with temperature are shown in Fig. 1. The degree of oxygen loss decreases from
with temperature are shown in Fig. 1. The degree of oxygen loss decreases from  to Gd, suggesting a stronger binding of the oxide ions to the lattice. This is consistent with a report that the enthalpies of formation of solid oxides
to Gd, suggesting a stronger binding of the oxide ions to the lattice. This is consistent with a report that the enthalpies of formation of solid oxides  and their lattice energies increase from
and their lattice energies increase from  to Gd.15 The TGA data imply that the tendency for oxygen nonstoichiometry (vacancy concentration) in
to Gd.15 The TGA data imply that the tendency for oxygen nonstoichiometry (vacancy concentration) in  (
( , Pr, Nd, Sm, and Gd) decreases in the order
, Pr, Nd, Sm, and Gd) decreases in the order  .
.
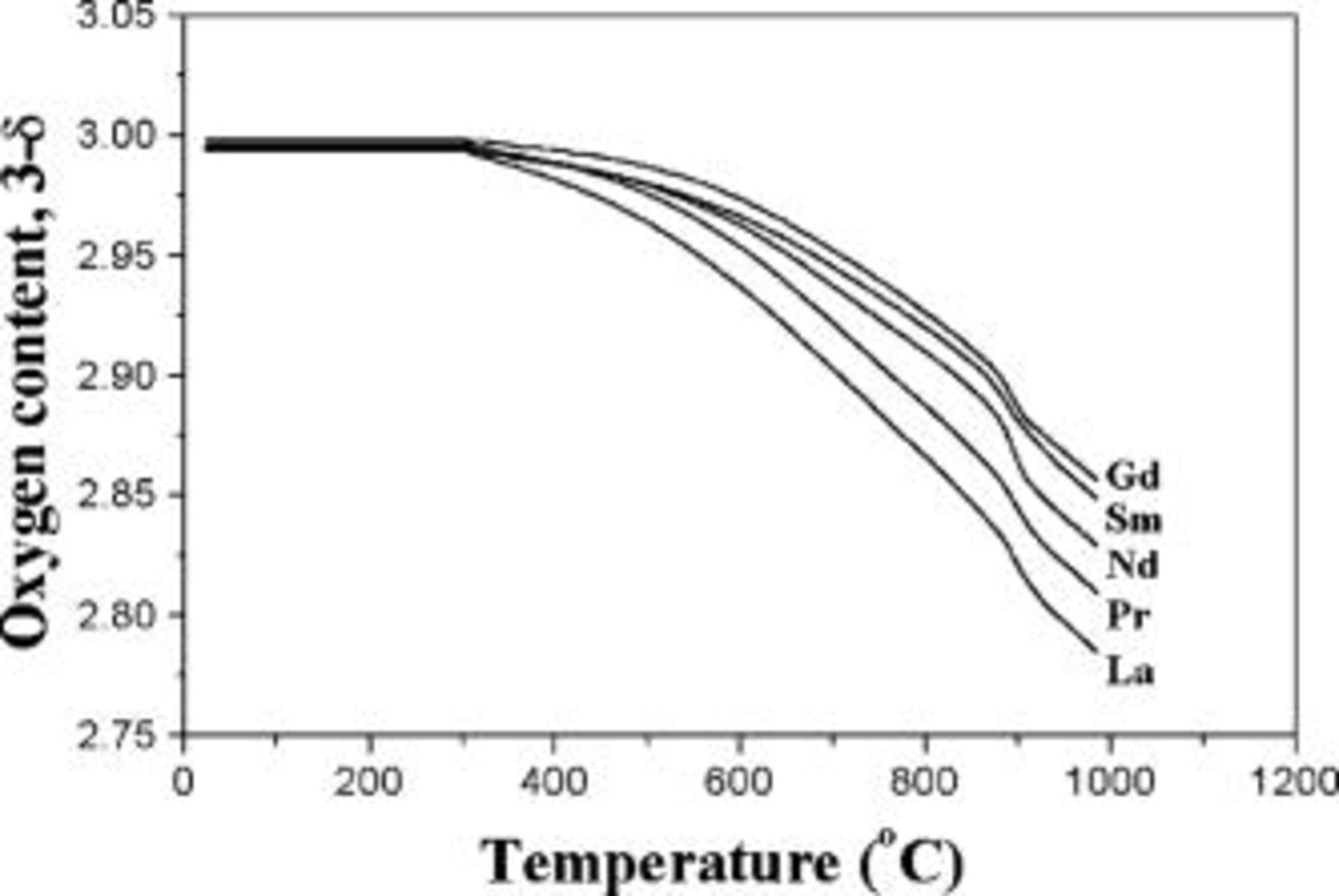
Figure 1. Comparison of the TGA plots of  (
( , Pr, Nd, Sm, and Gd) recorded in air with a heating rate of
, Pr, Nd, Sm, and Gd) recorded in air with a heating rate of  .
.
The temperature dependence of the electrical conductivity of  (
( , Pr, Nd, Sm, and Gd) is shown in Fig. 2. All the samples exhibit metallic conduction and have conductivity values of
, Pr, Nd, Sm, and Gd) is shown in Fig. 2. All the samples exhibit metallic conduction and have conductivity values of  at
at  . The faster decrease in conductivity at higher temperatures could be due to the formation of significant amount of oxide ion vacancies as indicated by the TGA data (Fig. 1). The formation of oxide ion vacancies is accompanied by a reduction of
. The faster decrease in conductivity at higher temperatures could be due to the formation of significant amount of oxide ion vacancies as indicated by the TGA data (Fig. 1). The formation of oxide ion vacancies is accompanied by a reduction of  to
to  , resulting in a decrease in the charge carrier concentration and Co–O covalency. Also, the oxide ion vacancies will perturb the O–Co–O periodic potential and covalent interaction.16 These factors lead to a decrease in electrical conductivity at elevated temperatures.
, resulting in a decrease in the charge carrier concentration and Co–O covalency. Also, the oxide ion vacancies will perturb the O–Co–O periodic potential and covalent interaction.16 These factors lead to a decrease in electrical conductivity at elevated temperatures.
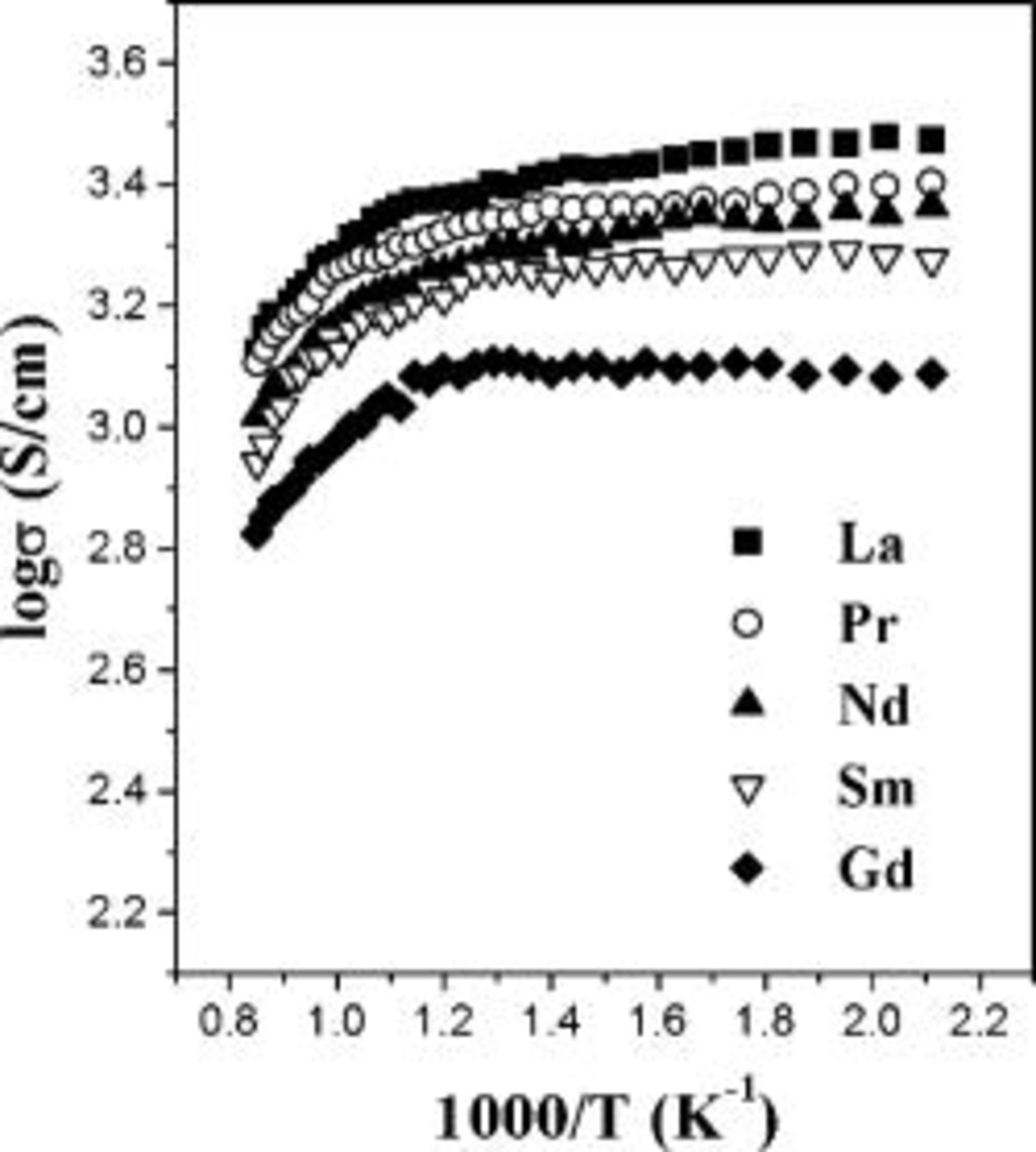
Figure 2. Comparison of the temperature dependence of the electrical conductivity of  (
( , Pr, Nd, Sm, and Gd) in air.
, Pr, Nd, Sm, and Gd) in air.
At a given temperature, the electrical conductivity of the  (
( , Pr, Nd, Sm, and Gd) samples decreases from
, Pr, Nd, Sm, and Gd) samples decreases from  to Gd. This can be understood by considering the changes in the structural parameters. As seen in Table I, the decreasing tolerance factor
to Gd. This can be understood by considering the changes in the structural parameters. As seen in Table I, the decreasing tolerance factor  with decreasing ionic radius from
with decreasing ionic radius from  to
to  increases the bending of the O–Co–O bonds (lowers the O–Co–O bond angle from 180°), which results in a decrease in the overlap between the
increases the bending of the O–Co–O bonds (lowers the O–Co–O bond angle from 180°), which results in a decrease in the overlap between the  and
and  orbitals and bandwidth. Thus, the decreasing covalency of the Co–O bonds and the increasing electron localization from
orbitals and bandwidth. Thus, the decreasing covalency of the Co–O bonds and the increasing electron localization from  to
to  causes a decrease in electrical conductivity.
causes a decrease in electrical conductivity.
The average thermal expansion coefficients (TECs) of the  samples measured in the range of
samples measured in the range of  in air are given in Table II. The TEC value decreases from
in air are given in Table II. The TEC value decreases from  to Gd. A similar trend has been observed in the analogous
to Gd. A similar trend has been observed in the analogous  17 and
17 and  18 systems also. In general, ionic bonds have a larger thermal expansion than the covalent bonds. Therefore, the variations in TEC can be understood by considering the ionic character of the Ln–O bonds. Mori et al.19 have also discussed the variations of TEC in the analogous lanthanum manganites
18 systems also. In general, ionic bonds have a larger thermal expansion than the covalent bonds. Therefore, the variations in TEC can be understood by considering the ionic character of the Ln–O bonds. Mori et al.19 have also discussed the variations of TEC in the analogous lanthanum manganites  (
( -earth) in terms of the ionic character of the
-earth) in terms of the ionic character of the  bond. The percent ionic character of a bond is related to the electronegativity difference between the bonded atoms
bond. The percent ionic character of a bond is related to the electronegativity difference between the bonded atoms  and
and  in the
in the  bond by the following empirical relationship:20
bond by the following empirical relationship:20
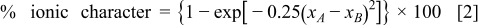
where  and
and  are the electronegativities of the
are the electronegativities of the  and
and  atoms, respectively. Based on the Pauling electronegativity values,21 one can, therefore, understand that the decreasing TEC from
atoms, respectively. Based on the Pauling electronegativity values,21 one can, therefore, understand that the decreasing TEC from  to Gd is due to the increasing electronegativity of Ln and the decreasing ionic character of the Ln–O bond from
to Gd is due to the increasing electronegativity of Ln and the decreasing ionic character of the Ln–O bond from  to Gd.
to Gd.
Table II. BET surface area, average crystallite size, and average thermal expansion coefficient (TEC) of  .
.
| Ln | BET surface area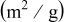
| Average crystallite size |
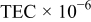 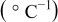
|
|---|---|---|---|
| La | 4.6 | 510 | 21.3 |
| Pr | 5.2 | 480 | 19.5 |
| Nd | 4.0 | 550 | 18.7 |
| Sm | 3.7 | 580 | 18.0 |
| Gd | 4.8 | 500 | 17.1 |
bAverage crystallite size was estimated with the line broadening of XRD peaks, and the error bar is  .
.
Additionally, the variations in TEC could also be related to the tendency to form oxide ion vacancies as the sample is heated. The formation of oxide ion vacancies can cause an increase in TEC due to the reduction of the smaller  ions to the larger
ions to the larger  ions.3, 22 Furthermore, TEC is inversely proportional to the binding energy between the ions in the lattice.23 Hayashi et al.24 have reported that the TEC of GDC increases with the amount of oxide ion vacancies due to a reduction in the binding energy of the metal-oxygen bonds. Therefore, the increasing Ln–O bond strength15 and the decreasing degree of oxygen loss, as indicated by the TGA data in Fig. 1, from
ions.3, 22 Furthermore, TEC is inversely proportional to the binding energy between the ions in the lattice.23 Hayashi et al.24 have reported that the TEC of GDC increases with the amount of oxide ion vacancies due to a reduction in the binding energy of the metal-oxygen bonds. Therefore, the increasing Ln–O bond strength15 and the decreasing degree of oxygen loss, as indicated by the TGA data in Fig. 1, from  to Gd can be considered to cause a decrease in TEC. In addition, the larger TEC of the cobaltites is partly related to the transition of the
to Gd can be considered to cause a decrease in TEC. In addition, the larger TEC of the cobaltites is partly related to the transition of the  ions from a low spin (
ions from a low spin ( ,
,  ) to high spin (
) to high spin ( ,
,  ) state.25 A suppression of such spin-state transitions by the decreasing amount of
) state.25 A suppression of such spin-state transitions by the decreasing amount of  ions (or the decreasing amount of oxide ion vacancies) at elevated temperatures on going from
ions (or the decreasing amount of oxide ion vacancies) at elevated temperatures on going from  to Gd could also contribute to the decreasing TEC.
to Gd could also contribute to the decreasing TEC.
The influence of the formation of oxide ion vacancies on TEC is also supported by the fact that the decrease in the TEC value on going from  to Gd in
to Gd in  (
( , measured at
, measured at  ) is larger than that found with the analogous
) is larger than that found with the analogous  system (
system ( , measured at
, measured at  ).17 The increasing tendency to form oxide ion vacancies on going from
).17 The increasing tendency to form oxide ion vacancies on going from  to
to  causes a faster increase (20%) in TEC in the cobaltite system, while the absence of formation of significant amount of oxygen vacancies in the manganite system for
causes a faster increase (20%) in TEC in the cobaltite system, while the absence of formation of significant amount of oxygen vacancies in the manganite system for  results in a slower increase (14%) in TEC. Thus, the change in TEC on going from
results in a slower increase (14%) in TEC. Thus, the change in TEC on going from  to Gd in the manganite system is mainly due to the decreasing ionicity of the Ln–O bond while that in the cobaltite system is due to both the decreasing ionicity of the Ln–O bond and the differences in the formation of oxide ion vacancies. Furthermore, the larger TEC of the cobaltites
to Gd in the manganite system is mainly due to the decreasing ionicity of the Ln–O bond while that in the cobaltite system is due to both the decreasing ionicity of the Ln–O bond and the differences in the formation of oxide ion vacancies. Furthermore, the larger TEC of the cobaltites  compared to that of the manganites
compared to that of the manganites  is due to the formation of oxide vacancies, spin-state transitions associated with
is due to the formation of oxide vacancies, spin-state transitions associated with  , and the relatively weaker Co–O bond compared to the Mn–O bond.
, and the relatively weaker Co–O bond compared to the Mn–O bond.
The BET surface area and the average crystallite size of the  cathode powders do not vary significantly with the
cathode powders do not vary significantly with the  ions as seen in Table II. Therefore, the influence of the geometrical morphology of the prepared powders on the electrochemical performance could be neglected in this study. Figure 3 shows the scanning electron microscopy (SEM) micrographs of the
ions as seen in Table II. Therefore, the influence of the geometrical morphology of the prepared powders on the electrochemical performance could be neglected in this study. Figure 3 shows the scanning electron microscopy (SEM) micrographs of the  cathodes after screen printing them onto the LSGM electrolyte followed by firing at
cathodes after screen printing them onto the LSGM electrolyte followed by firing at  for
for  . Porous cathode layers of about
. Porous cathode layers of about  thick with homogeneous microstructure that are well adhered to the dense LSGM electrolyte are seen. In addition, X-ray powder diffraction patterns of the
thick with homogeneous microstructure that are well adhered to the dense LSGM electrolyte are seen. In addition, X-ray powder diffraction patterns of the  and LSGM mixtures after heating at
and LSGM mixtures after heating at  for
for  show that there is no interfacial reaction between the cathode compositions and the LSGM electrolyte (Fig. 4).
show that there is no interfacial reaction between the cathode compositions and the LSGM electrolyte (Fig. 4).

Figure 3. SEM micrographs of the  cathode-LSGM electrolyte assemblies after firing at
cathode-LSGM electrolyte assemblies after firing at  for
for  : (a)
: (a)  ; (b)
; (b)  ; (c)
; (c)  ; (d)
; (d)  ; and (e)
; and (e)  .
.
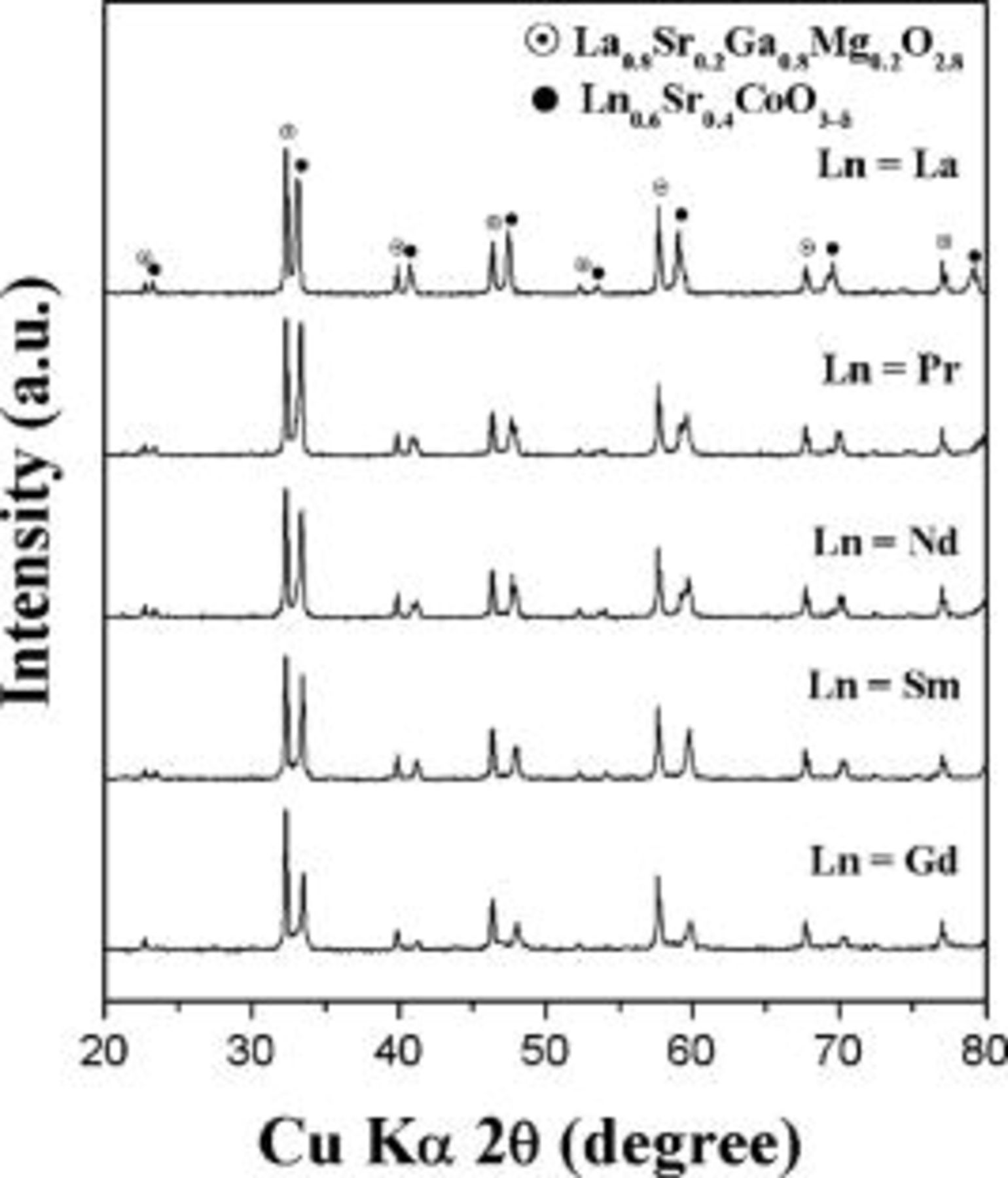
Figure 4. X-ray powder diffraction patterns recorded after heating the  cathode and the LSGM electrolyte powders at
cathode and the LSGM electrolyte powders at  for
for  .
.
The variations of the cell voltage, power density, and overpotential with current density at  are compared in Fig. 5 for the various
are compared in Fig. 5 for the various  cathodes. The power density decreases and the overpotential increases from
cathodes. The power density decreases and the overpotential increases from  to Gd. The cathode overpotential for oxygen reduction reaction is closely related to both the electronic and oxide ion conductivities of the cathode materials because the electrocatalytic reaction at a porous cathode is limited by the kinetics of oxygen exchange and diffusion as well as the charge transfer. Both the electrical conductivity (Fig. 2) and the concentration of oxide ion vacancies (Fig. 1) decrease from
to Gd. The cathode overpotential for oxygen reduction reaction is closely related to both the electronic and oxide ion conductivities of the cathode materials because the electrocatalytic reaction at a porous cathode is limited by the kinetics of oxygen exchange and diffusion as well as the charge transfer. Both the electrical conductivity (Fig. 2) and the concentration of oxide ion vacancies (Fig. 1) decrease from  to Gd in
to Gd in  ; oxide ion conductivity is proportional to the amount of oxide ion vacancies. Additionally, Kharton et al.26 have reported that a decrease in the radius of the
; oxide ion conductivity is proportional to the amount of oxide ion vacancies. Additionally, Kharton et al.26 have reported that a decrease in the radius of the  ions in
ions in  causes a decrease in the size of the anion transfer channel and an increase in the (Ln,Sr)–O bond energy, as evident from the decreasing cell volume in Table I. This suggests that the oxide ion conductivity of
causes a decrease in the size of the anion transfer channel and an increase in the (Ln,Sr)–O bond energy, as evident from the decreasing cell volume in Table I. This suggests that the oxide ion conductivity of  would decrease in the order
would decrease in the order  , which is consistent with that found by Kharton et al.26 for
, which is consistent with that found by Kharton et al.26 for  , Pr, and Nd from oxide ion conductivity measurements. Gao et al.27 have also reported that high oxide ion vacancy concentration in the surface of cathode could improve the dissociation of oxygen molecule
, Pr, and Nd from oxide ion conductivity measurements. Gao et al.27 have also reported that high oxide ion vacancy concentration in the surface of cathode could improve the dissociation of oxygen molecule  (ad) into atomic oxygen
(ad) into atomic oxygen  . Thus, the decreasing electronic and oxide ion conductivities from
. Thus, the decreasing electronic and oxide ion conductivities from  to Gd in
to Gd in  leads to a decrease in the oxygen exchange, transport speed of oxide ions, and charge transfer kinetics, which in turn results in a decrease in the electrochemical performance.
leads to a decrease in the oxygen exchange, transport speed of oxide ions, and charge transfer kinetics, which in turn results in a decrease in the electrochemical performance.
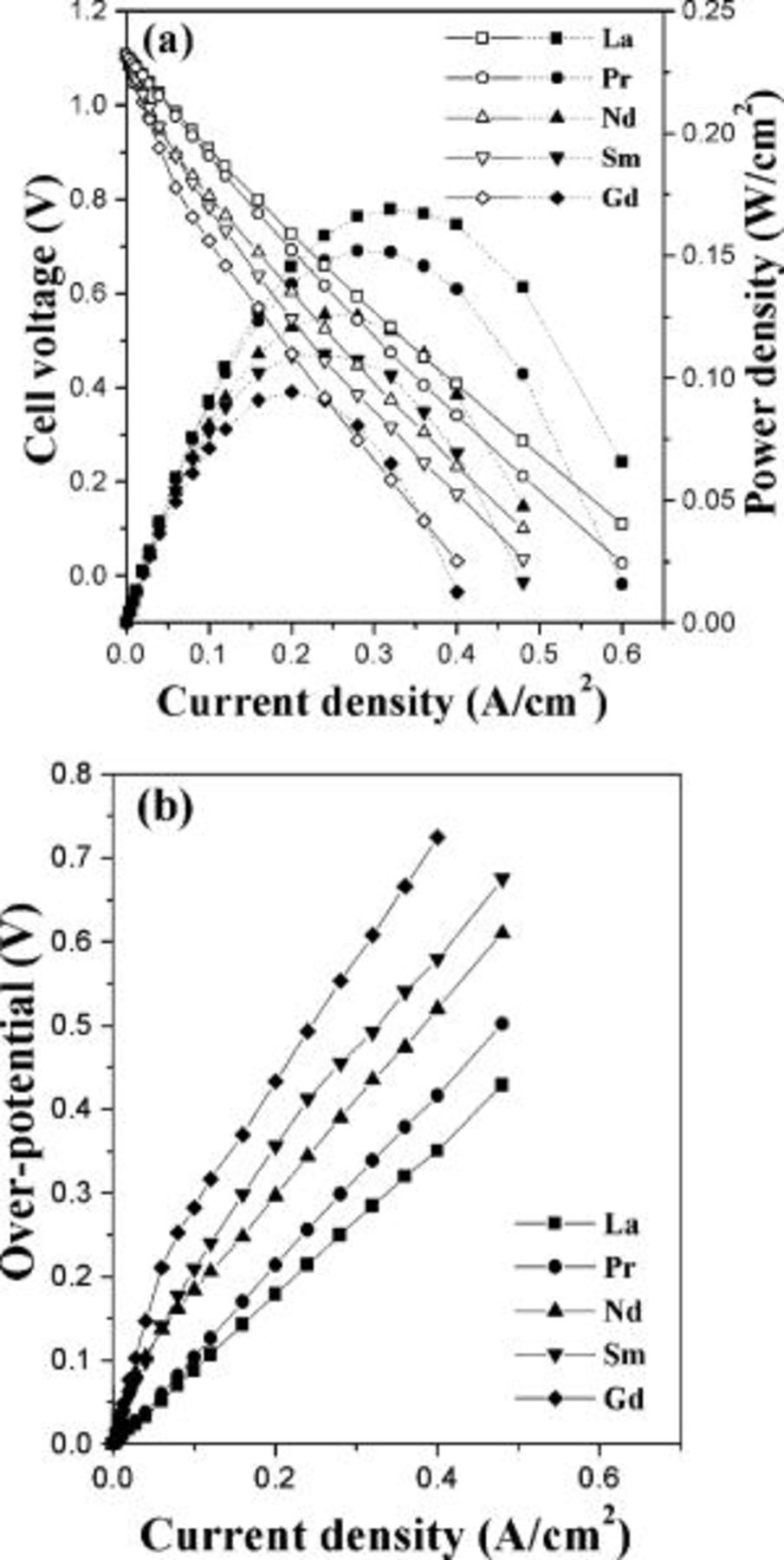
Figure 5. Electrochemical performance data of the  single cells at
single cells at  : variations of the (a)
: variations of the (a)  curves (open symbols) and power densities (closed symbols) and (b) cathode overpotential.
curves (open symbols) and power densities (closed symbols) and (b) cathode overpotential.
However, considering the long-term performance,  may encounter a faster degradation in electrochemical performance compared to
may encounter a faster degradation in electrochemical performance compared to  due to the larger difference in TEC (Table II) between the
due to the larger difference in TEC (Table II) between the  cathode
cathode  and the LSGM electrolyte
and the LSGM electrolyte  ,28 which could cause microcracks or delamination. Additionally, Qiu et al.29 have reported that
,28 which could cause microcracks or delamination. Additionally, Qiu et al.29 have reported that  with smaller lanthanide ions is less reactive with the YSZ electrolyte than
with smaller lanthanide ions is less reactive with the YSZ electrolyte than  . Therefore, the
. Therefore, the  cathodes with an intermediate lanthanide ion such as
cathodes with an intermediate lanthanide ion such as  may be preferred, considering the trade-off between electrochemical performance and other parameters like TEC and reactivity.
may be preferred, considering the trade-off between electrochemical performance and other parameters like TEC and reactivity.
Unlike in the  system, the cathode overpotential in the
system, the cathode overpotential in the  system, however, does not seem to depend strongly on the lanthanide host cation.17 This could be related to the differences in the formation of oxide ion vacancies and oxide ion conductivity. Wen et al.30 have reported that the
system, however, does not seem to depend strongly on the lanthanide host cation.17 This could be related to the differences in the formation of oxide ion vacancies and oxide ion conductivity. Wen et al.30 have reported that the  concentration increases proportionately with the
concentration increases proportionately with the  concentration in the
concentration in the  system and oxide ion vacancies do not exist even at high temperatures. Because the
system and oxide ion vacancies do not exist even at high temperatures. Because the  system17 does not contain oxide ion vacancies, the change in lanthanide host cations may not significantly affect the oxygen exchange kinetics, and the oxygen reduction reaction could be rather limited by the three-phase boundary or surface diffusion, resulting in no strong correlation between the lanthanide host cations and the electrochemical performance.
system17 does not contain oxide ion vacancies, the change in lanthanide host cations may not significantly affect the oxygen exchange kinetics, and the oxygen reduction reaction could be rather limited by the three-phase boundary or surface diffusion, resulting in no strong correlation between the lanthanide host cations and the electrochemical performance.
Conclusions
The electrocatalytic activity of  (
( , Pr, Nd, Sm, and Gd) for oxygen reduction reaction in SOFC decreases from
, Pr, Nd, Sm, and Gd) for oxygen reduction reaction in SOFC decreases from  to Gd. This trend could be understood to be due to the decreasing electrical and oxide ion conductivities caused, respectively, by an increasing bending of the O–Co–O bonds and a decreasing oxide ion vacancy concentration. However, going from
to Gd. This trend could be understood to be due to the decreasing electrical and oxide ion conductivities caused, respectively, by an increasing bending of the O–Co–O bonds and a decreasing oxide ion vacancy concentration. However, going from  to Gd provides an important advantage of a decrease in TEC, which could be understood to be due to the decreasing ionicity of the Ln–O bonds, the suppression of the reduction of the
to Gd provides an important advantage of a decrease in TEC, which could be understood to be due to the decreasing ionicity of the Ln–O bonds, the suppression of the reduction of the  ions to
ions to  ions, and the decreasing amount of oxide ion vacancies at elevated temperatures. These two opposing factors with decreasing
ions, and the decreasing amount of oxide ion vacancies at elevated temperatures. These two opposing factors with decreasing  size may make the
size may make the  cathodes with an intermediate lanthanide ion more attractive for practical cells. Additionally, the lack of a strong dependence of the catalytic activity of the analogous
cathodes with an intermediate lanthanide ion more attractive for practical cells. Additionally, the lack of a strong dependence of the catalytic activity of the analogous  system on the
system on the  ions unlike in the cobaltite system could be understood by considering the absence of oxide ion vacancies for
ions unlike in the cobaltite system could be understood by considering the absence of oxide ion vacancies for  .
.
Acknowledgment
This work was supported by the Welch Foundation grant F-1254 .
University of Texas at Austin assisted in meeting the publication costs of this article.