
Abstract
The kinetics of the hydrogen oxidation reaction (HOR) and hydrogen evolution reaction (HER) on polycrystalline platinum [Pt(pc)] and high surface area carbon-supported platinum nanoparticles (Pt/C) were studied in 0.1 M KOH using rotating disk electrode (RDE) measurements. After corrections of noncompensated solution resistance from ac impedance spectroscopy and of hydrogen mass transport in the HOR branch, the kinetic current densities were fitted to the Butler–Volmer equation using a transfer coefficient of 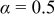 , from which HOR/HER exchange current densities on Pt(pc) and Pt/C were obtained, and the HOR/HER mechanisms in alkaline solution were discussed. Unlike the HOR/HER rates on Pt electrodes in alkaline solution, the HOR/HER rates on a Pt electrode in 0.1 M
, from which HOR/HER exchange current densities on Pt(pc) and Pt/C were obtained, and the HOR/HER mechanisms in alkaline solution were discussed. Unlike the HOR/HER rates on Pt electrodes in alkaline solution, the HOR/HER rates on a Pt electrode in 0.1 M 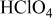 were limited entirely by hydrogen diffusion, which renders the quantification of the HOR/HER kinetics impossible by conventional RDE measurements. The simulation of the hydrogen anode performance based on the specific exchange current densities of the HOR/HER at
were limited entirely by hydrogen diffusion, which renders the quantification of the HOR/HER kinetics impossible by conventional RDE measurements. The simulation of the hydrogen anode performance based on the specific exchange current densities of the HOR/HER at 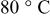 illustrates that in addition to the oxygen reduction reaction cell voltage loss on the cathode, the slow HOR kinetics are projected to cause significant anode potential losses in alkaline fuel cells for low platinum loadings (
illustrates that in addition to the oxygen reduction reaction cell voltage loss on the cathode, the slow HOR kinetics are projected to cause significant anode potential losses in alkaline fuel cells for low platinum loadings (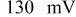 at
at 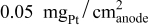 and
and 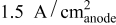 ), contrary to what is reported for proton exchange membrane fuel cells.
), contrary to what is reported for proton exchange membrane fuel cells.
Export citation and abstract BibTeX RIS
Fuel cells show promise to provide environmentally friendly energy conversion at high efficiency and power density. One of the major focus areas in proton exchange membrane fuel cell (PEMFC) research is the oxygen reduction reaction (ORR) on the cathode because the slow ORR kinetics are responsible for more than 50% of the overall cell voltage loss during PEMFC operation.1–3 Thus, even on platinum (Pt), the most active ORR catalyst in PEMFCs, the ORR overpotential is typically on the order of 300–400 mV,4 consistent with the very low ORR exchange current density of only  at
at  .5 The same applies to the ORR in alkaline electrolytes, where Pt has a similar ORR activity as in acid6, 7 and again is among the most active catalysts8 (including catalysts9, 10 with active sites composed of nitrogen-coordinated iron and/or cobalt displaying ORR activities comparable to Pt). The ORR overpotential loss in alkaline fuel cells (AFCs) or alkaline membrane fuel cells (AMFCs) is very similar to that in PEMFCs, i.e., the cathode overpotential loss remains the major factor limiting the overall energy conversion efficiency and performance of AFCs11 and AMFCs.10
.5 The same applies to the ORR in alkaline electrolytes, where Pt has a similar ORR activity as in acid6, 7 and again is among the most active catalysts8 (including catalysts9, 10 with active sites composed of nitrogen-coordinated iron and/or cobalt displaying ORR activities comparable to Pt). The ORR overpotential loss in alkaline fuel cells (AFCs) or alkaline membrane fuel cells (AMFCs) is very similar to that in PEMFCs, i.e., the cathode overpotential loss remains the major factor limiting the overall energy conversion efficiency and performance of AFCs11 and AMFCs.10
In contrast to the slow ORR kinetics, the kinetics of the hydrogen oxidation reaction (HOR) on Pt anode catalysts in a PEMFC are so fast that the cell voltage losses at the anode are negligible even for very low Pt loadings ( at anode Pt loadings of
at anode Pt loadings of  12). Due to the very high HOR exchange current density
12). Due to the very high HOR exchange current density  on Pt in acid electrolytes, its quantification is experimentally difficult, leading to a wide range of experimentally reported values in acid electrolytes at room temperature. For example, HOR exchange current densities measured by the rotating disk electrode (RDE) technique are reported to be on the order of
on Pt in acid electrolytes, its quantification is experimentally difficult, leading to a wide range of experimentally reported values in acid electrolytes at room temperature. For example, HOR exchange current densities measured by the rotating disk electrode (RDE) technique are reported to be on the order of  on polycrystalline Pt [Pt(pc)],13 Pt single-crystals,14 and carbon-supported Pt (Pt/C).15 However, much larger values of 24 to
on polycrystalline Pt [Pt(pc)],13 Pt single-crystals,14 and carbon-supported Pt (Pt/C).15 However, much larger values of 24 to  have been obtained by microelectrode studies on Pt/C16 and Pt(pc)17 or by Pt/C-based gas-diffusion electrodes.18 Moreover, the observed
have been obtained by microelectrode studies on Pt/C16 and Pt(pc)17 or by Pt/C-based gas-diffusion electrodes.18 Moreover, the observed  PEMFC performance independent of Pt anode loadings (varied between 0.4 and
PEMFC performance independent of Pt anode loadings (varied between 0.4 and  ) could only be rationalized by assuming very high HOR exchange current densities,19 and subsequent kinetic measurements in PEMFCs at
) could only be rationalized by assuming very high HOR exchange current densities,19 and subsequent kinetic measurements in PEMFCs at  indeed yielded
indeed yielded  values of
values of  .12 This ambiguity in the measurement of the HOR/hydrogen evolution reaction (HER) kinetics in acid can be related to the fact that it is experimentally very challenging to eliminate hydrogen mass-transport resistances in RDE measurements. For example, if the HOR/HER exchange current density were 10-fold larger than the diffusion limited HOR current density (
.12 This ambiguity in the measurement of the HOR/hydrogen evolution reaction (HER) kinetics in acid can be related to the fact that it is experimentally very challenging to eliminate hydrogen mass-transport resistances in RDE measurements. For example, if the HOR/HER exchange current density were 10-fold larger than the diffusion limited HOR current density ( at typical maximum rotation rates of 3600 rpm14), the quantification of kinetic constants for the HOR/HER would not be possible.
at typical maximum rotation rates of 3600 rpm14), the quantification of kinetic constants for the HOR/HER would not be possible.
As microelectrode studies on Pt(pc) actually suggest two orders of magnitude lower HOR/HER exchange current densities in alkaline compared to acidic electrolytes,17 RDE measurements may allow the quantification of HOR/HER kinetics in KOH. Previous RDE measurements of the HOR/HER kinetics in alkaline electrolytes on Pt single crystals have yielded exchange current densities on the order of  .20, 21 However, neither HOR/HER exchange current densities nor reaction mechanism details have been reported for industrially relevant carbon-supported Pt catalyst (Pt/C) catalysts in alkaline electrolyte. If they were on the order of only
.20, 21 However, neither HOR/HER exchange current densities nor reaction mechanism details have been reported for industrially relevant carbon-supported Pt catalyst (Pt/C) catalysts in alkaline electrolyte. If they were on the order of only  , it would imply that the anode overpotential losses in AFCs/AMFCs would be substantial, prohibiting the use of ultralow anode Pt loadings, which have been demonstrated successfully in PEMFCs (
, it would imply that the anode overpotential losses in AFCs/AMFCs would be substantial, prohibiting the use of ultralow anode Pt loadings, which have been demonstrated successfully in PEMFCs ( 12).
12).
In this study, we investigate the HOR/HER kinetics on Pt(pc) and Pt/C at different temperatures using RDE measurements combined with in situ uncompensated solution resistance measurements [i.e., iR-correction term via ac impedance]. In addition, the temperature-dependent HOR/HER exchange current densities can be well modeled using the Butler–Volmer equation, from which the mechanism of the HOR in an alkaline solution is discussed. Moreover, comparative measurements illustrate that the HOR/HER kinetics in acid electrolyte is orders of magnitude faster, which cannot be quantified by RDE measurements due to the Nernstian diffusion overpotential, as suggested previously.17, 22 Furthermore, we contrast the ORR activity of Pt(pc) and Pt/C in alkaline with acid, after which the performance of AFCs/AMFCs and PEMFCs using Pt/C at the anode and the cathode is compared.
Experimental
Electrode preparation
Thin-film electrodes of high surface area carbon-supported Pt catalyst were made with 46 wt % Pt (Pt/C) supplied by Tanaka Kikinzoku International, Inc. (TKK) following previous studies.23, 24 The average Pt nanoparticle (NP) diameter of the Pt/C determined by transmission electron microscopy was  based on at least 200 particle counts. Aqueous suspensions of Pt/C of
based on at least 200 particle counts. Aqueous suspensions of Pt/C of  were obtained by dispersing the catalyst in deionized (DI) water (
were obtained by dispersing the catalyst in deionized (DI) water ( , Millipore) using ultrasonication of the suspension in an ice bath.
, Millipore) using ultrasonication of the suspension in an ice bath.  of the Pt/C catalyst water suspension was deposited on a glassy carbon electrode (GCE) disk (5 mm diameter, Pine Instruments), which was polished with 0.05 μm alumina (Buehler), and dried in air at room temperature, resulting in a Pt loading of
of the Pt/C catalyst water suspension was deposited on a glassy carbon electrode (GCE) disk (5 mm diameter, Pine Instruments), which was polished with 0.05 μm alumina (Buehler), and dried in air at room temperature, resulting in a Pt loading of  on GCE. The Pt(pc) disk electrode (5 mm in diameter, Pine Instruments) was also polished with
on GCE. The Pt(pc) disk electrode (5 mm in diameter, Pine Instruments) was also polished with  alumina and was cleaned by ultrasonication in DI water.
alumina and was cleaned by ultrasonication in DI water.
Modification of the reference electrode
Glass corrodes in an alkaline solution and the arising contaminants (lead and silica) can significantly influence the ORR and the HOR/HER activity of Pt electrodes.25, 26 To minimize contamination from the gradual dissolution of glass, a fresh alkaline electrolyte was prepared for each electrochemical measurement at each temperature, and all electrochemical data were collected within 30 min. More importantly, we also modified the Luggin capillary, replacing the highly KOH-soluble Vycor glass tip of the Luggin capillary with a  thick Nafion film, analogous to what was shown previously.27 The short exposure time and modification of the Luggin capillary tip eliminated contamination; consequently, as shown later, the HOR/HER and ORR activities obtained in our study are equal to those reported for measurements in a Teflon cell.26
thick Nafion film, analogous to what was shown previously.27 The short exposure time and modification of the Luggin capillary tip eliminated contamination; consequently, as shown later, the HOR/HER and ORR activities obtained in our study are equal to those reported for measurements in a Teflon cell.26
Electrochemical measurements
The electrochemical measurements were carried out in a jacketed glass cell (Pine Instruments) connected to a circulating temperature-controlled water bath (Thermo Electron Corp.). As-prepared Pt/C thin-film electrodes or a Pt(pc) disk was mounted onto a rotator (Pine Instruments) and was immersed into 0.1 M potassium hydroxide (99.99% purity, Sigma-Aldrich) solution. A Pt wire served as the counter electrode and a saturated calomel electrode (Analytical Sensor, Inc.), immersed into the Nafion film-modified Luggin capillary filled with 0.1 M KOH, was used as the reference electrode. However, all potentials reported in this paper are referenced to the reversible hydrogen electrode (RHE) potential, calibrated in the same electrolyte by measuring HOR/HER currents on the Pt RDE, whereby the potential at zero current corresponds to 0 V vs RHE. After the electrolyte was saturated with argon, the cyclic voltammogram (CV) at 294 K was recorded between  and
and  vs RHE at a scan rate of 50 mV/s after it reached steady state.
vs RHE at a scan rate of 50 mV/s after it reached steady state.
The HOR/HER rates were measured using RDE measurements. After the electrolyte was saturated with pure hydrogen, polarization curves were recorded between  and
and  vs RHE at a sweep rate of 10 mV/s and rotation rates of 400, 900, 1600, and 2500 rpm. A freshly prepared 0.1 M KOH electrolyte was used for each measurement at each temperature, as described above. Exchange current densities
vs RHE at a sweep rate of 10 mV/s and rotation rates of 400, 900, 1600, and 2500 rpm. A freshly prepared 0.1 M KOH electrolyte was used for each measurement at each temperature, as described above. Exchange current densities  of the HOR/HER were obtained by fitting the experimental data to the Butler–Volmer equation. The HOR/HER was also measured on Pt(pc) in
of the HOR/HER were obtained by fitting the experimental data to the Butler–Volmer equation. The HOR/HER was also measured on Pt(pc) in  -saturated 0.1 M
-saturated 0.1 M  (70%, GFS Chemicals Inc.) using the same method and experimental conditions.
(70%, GFS Chemicals Inc.) using the same method and experimental conditions.
After measuring the HOR/HER at 294 K, the ORR kinetics were also measured on Pt(pc) and Pt/C in  -saturated 0.1 M KOH at 1600 rpm and 10 mV/s. The kinetic current at 0.9 V vs RHE was calculated based on the Koutecky–Levich equation
-saturated 0.1 M KOH at 1600 rpm and 10 mV/s. The kinetic current at 0.9 V vs RHE was calculated based on the Koutecky–Levich equation

where  is the measured current,
is the measured current,  is the kinetic current,
is the kinetic current,  is the diffusion limited current, ω is the rotation rate, and
is the diffusion limited current, ω is the rotation rate, and  is a constant related to the number of electrons transferred in a reaction, the gas concentration and diffusivity, as well as the electrolyte kinematic viscosity.28 The kinetic currents were then normalized to the electrochemical surface area (ESA) to obtain the specific activity and, in Pt/C, also to the Pt mass to obtain the mass activity.
is a constant related to the number of electrons transferred in a reaction, the gas concentration and diffusivity, as well as the electrolyte kinematic viscosity.28 The kinetic currents were then normalized to the electrochemical surface area (ESA) to obtain the specific activity and, in Pt/C, also to the Pt mass to obtain the mass activity.
Impedance measurements
The cell resistance was measured immediately after RDE measurements using VoltaMaster (Voltlab), taking the ac impedance spectra from 32 to 0.1 kHz and a voltage perturbation of 10 mV. The real part of the resistance at 1 kHz was taken as the cell resistance and was used to obtain the iR-free potential of the working electrode. The solution resistances measured at 275 and 314 K were 65 and  , respectively. These rather high noncompensated solution resistances in the relatively conductive 0.1 M KOH were due to the highly skewed potential distribution near the RDE surface,29 with half of the potential drop in the electrolyte phase occurring at a distance of only a disk radius from the disk surface.
, respectively. These rather high noncompensated solution resistances in the relatively conductive 0.1 M KOH were due to the highly skewed potential distribution near the RDE surface,29 with half of the potential drop in the electrolyte phase occurring at a distance of only a disk radius from the disk surface.
Results
Measurements of HOR/HER kinetics of Pt(pc)
The CV of a Pt(pc) disk in 0.1 M KOH (Fig. 1) shows the typical Pt–H underpotential deposition region (0.08–0.5 V), double-layer region (0.5–0.6 V), and Pt–oxide region (above 0.6 V), similar to that reported previously.30 The Pt–H peaks at  and
and  can be attributed to the Pt–H interaction on Pt(110) and Pt(100) planes, respectively, based on the CVs of Pt single-crystal surfaces in 0.1 M KOH.20, 21 To determine the Pt surface area, the positive potential window was restricted to 0.6 V to eliminate the interference from Pt oxide reduction in quantifying the double-layer contribution to the H adsorption/desorption region. The Pt surface area was then obtained by integrating the H adsorption/desorption region, subtracting the double-layer charging currents, and dividing the resulting coulombic charge by 2 and
can be attributed to the Pt–H interaction on Pt(110) and Pt(100) planes, respectively, based on the CVs of Pt single-crystal surfaces in 0.1 M KOH.20, 21 To determine the Pt surface area, the positive potential window was restricted to 0.6 V to eliminate the interference from Pt oxide reduction in quantifying the double-layer contribution to the H adsorption/desorption region. The Pt surface area was then obtained by integrating the H adsorption/desorption region, subtracting the double-layer charging currents, and dividing the resulting coulombic charge by 2 and  .23, 30 The thus obtained roughness factor of the Pt(pc) disk from four independent experiments, which was defined as
.23, 30 The thus obtained roughness factor of the Pt(pc) disk from four independent experiments, which was defined as  , was
, was  .
.

Figure 1. CVs of Pt(pc) at  in Ar-saturated 0.1 M KOH at 50 mV/s with two different positive potential limits. The current density is referenced to the actual Pt surface area.
in Ar-saturated 0.1 M KOH at 50 mV/s with two different positive potential limits. The current density is referenced to the actual Pt surface area.
Figure 2a shows the HOR/HER polarization curves on Pt(pc) in 0.1 M KOH at 294 K as a function of rotation rate, with well-defined hydrogen mass transport controlled (limiting) current densities occurring at potentials above 0.2 V. They scale linearly with the inverse of the square root of the rotation rate, as shown by the Koutecky–Levich plot constructed at 0.5 V (see inset of Fig. 2a), yielding a zero  -axis intercept and a
-axis intercept and a  value (Eq. 1) of
value (Eq. 1) of  . The limiting current density of
. The limiting current density of  at 2500 rpm agrees excellently with the value obtained in a Teflon cell,26 where it was shown from KOH-induced glass corrosion would lead to substantially lower limiting current densities. In addition, our measured HOR half-wave potential of 0.065 V on Pt(pc) at 2500 rpm and 294 K (Fig. 2a) is actually slightly higher than that of 0.08 V obtained in a Teflon cell26 at essentially the same conditions. The identical diffusion limited current densities and the slightly higher HOR activity in our study compared to the data reported for measurements in a Teflon cell26 clearly prove that there is no contamination by glass corrosion products in our experiments, which was accomplished by the short exposure time and the elimination of the Vycor frit from the Luggin capillary.
at 2500 rpm agrees excellently with the value obtained in a Teflon cell,26 where it was shown from KOH-induced glass corrosion would lead to substantially lower limiting current densities. In addition, our measured HOR half-wave potential of 0.065 V on Pt(pc) at 2500 rpm and 294 K (Fig. 2a) is actually slightly higher than that of 0.08 V obtained in a Teflon cell26 at essentially the same conditions. The identical diffusion limited current densities and the slightly higher HOR activity in our study compared to the data reported for measurements in a Teflon cell26 clearly prove that there is no contamination by glass corrosion products in our experiments, which was accomplished by the short exposure time and the elimination of the Vycor frit from the Luggin capillary.

Figure 2. (a) HOR/HER polarization curves (positive-going scans) on Pt(pc) at different rotation rates at  . The inset shows a Koutecky–Levich plot at 0.5 V/RHE. (b) HOR/HER polarization curves on Pt(pc) at 1600 rpm at different temperatures (positive-going scans). The data were collected in
. The inset shows a Koutecky–Levich plot at 0.5 V/RHE. (b) HOR/HER polarization curves on Pt(pc) at 1600 rpm at different temperatures (positive-going scans). The data were collected in  -saturated 0.1 M KOH at 10 mV/s.
-saturated 0.1 M KOH at 10 mV/s.
The temperature dependence of the HOR limiting current densities is shown in Fig. 2b, illustrating the previously reported observation of decreasing HOR limiting current densities with decreasing temperatures. The limiting current of  at 275 K and 1600 rpm in Fig. 2b agrees well with that reported for
at 275 K and 1600 rpm in Fig. 2b agrees well with that reported for  single crystals at 275 K.20
single crystals at 275 K.20
The HOR/HER kinetic current densities  on Pt(pc) are shown as gray solid lines in Fig. 3a and 3b (at 275 and 314 K, respectively), which were obtained from correcting the polarization curves by the measured iR and the hydrogen mass transport in the HOR branch (Eq. 1). The voltage corrections associated with the iRs can be quite substantial at the higher end of the current density scale, amounting to, for example, 25 mV (275 K) and 12 mV (314 K) at
on Pt(pc) are shown as gray solid lines in Fig. 3a and 3b (at 275 and 314 K, respectively), which were obtained from correcting the polarization curves by the measured iR and the hydrogen mass transport in the HOR branch (Eq. 1). The voltage corrections associated with the iRs can be quite substantial at the higher end of the current density scale, amounting to, for example, 25 mV (275 K) and 12 mV (314 K) at  , as shown in the insets of Fig. 3a and 3b. Following the iR correction, the HOR/HER kinetic current densities were obtained by hydrogen mass transport resistance correction for the HOR branch using the Koutecky–Levich equation (Eq. 1). Only data below 80% of the HOR diffusion limited current density were used to minimize errors in the analysis, which can become substantial as the measured current approaches the mass transport limited current. Because hydrogen oversaturation and bubble formation can introduce mass transport resistances for the HER at high current densities, only data with HER current densities equal to or lower than
, as shown in the insets of Fig. 3a and 3b. Following the iR correction, the HOR/HER kinetic current densities were obtained by hydrogen mass transport resistance correction for the HOR branch using the Koutecky–Levich equation (Eq. 1). Only data below 80% of the HOR diffusion limited current density were used to minimize errors in the analysis, which can become substantial as the measured current approaches the mass transport limited current. Because hydrogen oversaturation and bubble formation can introduce mass transport resistances for the HER at high current densities, only data with HER current densities equal to or lower than  were used, where no bubble formation was observed and the measured current was rotation rate independent (between 900 and 2500 rpm). The thus obtained HOR/HER kinetic current densities
were used, where no bubble formation was observed and the measured current was rotation rate independent (between 900 and 2500 rpm). The thus obtained HOR/HER kinetic current densities  on Pt(pc) were then plotted in Fig. 3 (gray solid lines) at (a) 275 and (b) 314 K.
on Pt(pc) were then plotted in Fig. 3 (gray solid lines) at (a) 275 and (b) 314 K.

Figure 3. HOR/HER measured kinetic current densities (gray solid lines) on Pt(pc) in 0.1 M KOH at 10 mV/s and their fit to the Butler–Volmer equation (Eq. 2) with  (dashed black lines): (a) 275 and (b) 314 K. The HOR/HER kinetic current densities were obtained from iR-corrected polarization curves and are corrected for hydrogen mass transport in the HOR branch using Eq. 1. The corresponding insets show the HOR/HER polarization curves before (solid black lines) and after (dashed red lines) iR correction.
(dashed black lines): (a) 275 and (b) 314 K. The HOR/HER kinetic current densities were obtained from iR-corrected polarization curves and are corrected for hydrogen mass transport in the HOR branch using Eq. 1. The corresponding insets show the HOR/HER polarization curves before (solid black lines) and after (dashed red lines) iR correction.
The HOR/HER exchange current densities  on Pt(pc) were extracted subsequently by fitting the HOR/HER kinetic current densities to the Butler–Volmer equation (dashed black lines in Fig. 3a and 3b) as a function of the HOR/HER overpotential
on Pt(pc) were extracted subsequently by fitting the HOR/HER kinetic current densities to the Butler–Volmer equation (dashed black lines in Fig. 3a and 3b) as a function of the HOR/HER overpotential 

where α represents the transfer coefficient,  is Faraday's constant (96,485 As/mol),
is Faraday's constant (96,485 As/mol),  is the universal gas constant (8.314 J/mol/K), and
is the universal gas constant (8.314 J/mol/K), and  is the temperature in Kelvin. In fitting all the HOR/HER data in this study, α and
is the temperature in Kelvin. In fitting all the HOR/HER data in this study, α and  were first set as variables, where the best fit was obtained for
were first set as variables, where the best fit was obtained for  . Therefore, the reported
. Therefore, the reported  values herein were obtained by setting
values herein were obtained by setting  . Fig. 3 shows that the simple Butler–Volmer equation with
. Fig. 3 shows that the simple Butler–Volmer equation with  perfectly fits the experimental HOR/HER kinetics on Pt(pc) at both 275 and 314 K, but equally good fits were obtained at intermediate temperatures, corresponding to Tafel slopes (TS) defined as
perfectly fits the experimental HOR/HER kinetics on Pt(pc) at both 275 and 314 K, but equally good fits were obtained at intermediate temperatures, corresponding to Tafel slopes (TS) defined as  with values of 109 mV/dec at 275 K and 125 mV/dec at 314 K. The symmetry of the anodic (HOR) and cathodic (HER) current densities with respect to the reversible potential suggests that the HOR and HER have the same reaction mechanism and intermediates, which is discussed in more detail later. The above determined potential-independent TSs of
with values of 109 mV/dec at 275 K and 125 mV/dec at 314 K. The symmetry of the anodic (HOR) and cathodic (HER) current densities with respect to the reversible potential suggests that the HOR and HER have the same reaction mechanism and intermediates, which is discussed in more detail later. The above determined potential-independent TSs of  disagree with the variable TSs ranging from
disagree with the variable TSs ranging from  to
to  at low and high overpotentials, respectively, reported for the HOR/HER on
at low and high overpotentials, respectively, reported for the HOR/HER on  in 0.1 M KOH.20, 21 However, the origin of this discrepancy is simply a misinterpretation of the definition of the TS because the latter can only be obtained from a plot of
in 0.1 M KOH.20, 21 However, the origin of this discrepancy is simply a misinterpretation of the definition of the TS because the latter can only be obtained from a plot of  vs η only if η is significantly larger in value than roughly one-half of the TS,31 so that the fitting of a TS to the low overpotential region in these studies20, 21 is incorrect. Consequently, only their reported value of
vs η only if η is significantly larger in value than roughly one-half of the TS,31 so that the fitting of a TS to the low overpotential region in these studies20, 21 is incorrect. Consequently, only their reported value of  at high overpotentials is determined correctly and is reasonably close to the TS obtained in our work.
at high overpotentials is determined correctly and is reasonably close to the TS obtained in our work.
Based on fitting at least three independent data sets at each temperature, specific HOR/HER exchange current densities of Pt(pc), normalized to the Pt ESA, are  ,
,  , and
, and  at 275, 294, and 314 K, respectively. At 294 K, our value of
at 275, 294, and 314 K, respectively. At 294 K, our value of  (listed in Table I) is larger than the
(listed in Table I) is larger than the  reported for Pt(pc) in 0.5 M NaOH 17 and for Pt(110), which was the most active surface for the HOR/HER among the three low index Pt single-crystal faces reported in an early study.21 The origin of this discrepancy is likely caused by both the lack of iR correction in these studies and electrolyte contamination effects, which is supported by the fact that the more recently measured HOR/HER specific exchange current density on Pt(110) at 293 K of
reported for Pt(pc) in 0.5 M NaOH 17 and for Pt(110), which was the most active surface for the HOR/HER among the three low index Pt single-crystal faces reported in an early study.21 The origin of this discrepancy is likely caused by both the lack of iR correction in these studies and electrolyte contamination effects, which is supported by the fact that the more recently measured HOR/HER specific exchange current density on Pt(110) at 293 K of  20 is much larger and comparable to our value on Pt(pc) (the exchange current density given in Ref. 20 must be multiplied by a factor of 2 to be consistent with the definition used in Eq. 2 here). In addition, the activation energy of the HOR/HER on Pt(pc) was obtained by plotting
20 is much larger and comparable to our value on Pt(pc) (the exchange current density given in Ref. 20 must be multiplied by a factor of 2 to be consistent with the definition used in Eq. 2 here). In addition, the activation energy of the HOR/HER on Pt(pc) was obtained by plotting  vs
vs  , where a linear relationship was found between 275 and 314 K. A representative data set is shown in Fig. 4. Based on four independent sets of repeat measurements, the HOR/HER activation energy
, where a linear relationship was found between 275 and 314 K. A representative data set is shown in Fig. 4. Based on four independent sets of repeat measurements, the HOR/HER activation energy  on Pt(pc) was determined to be
on Pt(pc) was determined to be  , which is listed in Table I. This value agrees reasonably with 23 kJ/mol obtained for Pt(110) in the same electrolyte.20
, which is listed in Table I. This value agrees reasonably with 23 kJ/mol obtained for Pt(110) in the same electrolyte.20
Table I. Summary of average HOR/HER exchange current densities  , mass activity
, mass activity  at
at  , and activation energy for Pt(pc) and Pt/C in 0.1 M KOH.
, and activation energy for Pt(pc) and Pt/C in 0.1 M KOH.
 
|
 
|
 (kJ/mol) (kJ/mol) | |
|---|---|---|---|
| Pt(pc) |
 
| – |
 
|
| Pt/C |
 
|
 
|
 
|
The asterisk indicates the number of independent repeat experiments. Activation energies  were obtained by fitting the data between
were obtained by fitting the data between  and
and  (Fig. 4). Data were iR- and mass-transport corrected as described in the text.
(Fig. 4). Data were iR- and mass-transport corrected as described in the text.

Figure 4. Representative Arrhenius plots of the HOR/HER exchange current densities on Pt(pc) (black squares) and Pt/C (red circles) in 0.1 M KOH. The calculated HOR/HER activation energies  are
are  for Pt(pc) and
for Pt(pc) and  for Pt/C based on four and two sets of repeat measurements, respectively.
for Pt/C based on four and two sets of repeat measurements, respectively.
Measurements of HOR/HER kinetics of Pt/C
The same HOR/HER electrochemical measurements as on Pt(pc) were made on Pt/C. Fig. 5 depicts the CV of Pt/C in 0.1 M KOH at 294 K and 50 mV/s, where the current was normalized to the true Pt surface area. The observed features resemble those of Pt(pc) except that it has higher currents in the double-layer region due to the capacitive contribution from the high surface area carbon support. Using the double-layer-corrected Pt–H underpotential deposition region, the specific surface area of Pt/C was determined to be  , corresponding to a roughness factor of
, corresponding to a roughness factor of  for thin-film RDE electrodes of Pt/C.
for thin-film RDE electrodes of Pt/C.

Figure 5. CVs of Pt/C at  in Ar-saturated 0.1 M KOH at 50 mV/s with two different positive potential limits. The current density is referenced to the actual Pt surface area.
in Ar-saturated 0.1 M KOH at 50 mV/s with two different positive potential limits. The current density is referenced to the actual Pt surface area.
HOR/HER kinetic current densities of Pt/C RDE electrodes are shown in Fig. 6 and were extracted from HOR/HER polarization curves following processes analogous to what was used for Pt(pc) in Fig. 3. The hydrogen transport limited current densities on Pt/C RDE electrodes (not shown) were within 10% of those shown for Pt(pc) in Fig. 2. The fitting of the HOR/HER kinetic current densities using the Butler–Volmer equation with  was excellent, from which the exchange current density
was excellent, from which the exchange current density  was obtained. The experimentally extracted and fitted kinetic current densities at 275 and 304 K are shown in Fig. 6 as examples, whereas fits at intermediate temperatures (data not shown) are equally good. The specific exchange current density of the HOR/HER on Pt/C at 294 K is
was obtained. The experimentally extracted and fitted kinetic current densities at 275 and 304 K are shown in Fig. 6 as examples, whereas fits at intermediate temperatures (data not shown) are equally good. The specific exchange current density of the HOR/HER on Pt/C at 294 K is  , which is comparable to that obtained on Pt(pc) (Table I). Normalized to Pt mass, the mass exchange current density of Pt/C is
, which is comparable to that obtained on Pt(pc) (Table I). Normalized to Pt mass, the mass exchange current density of Pt/C is  . A representative Arrhenius plot of
. A representative Arrhenius plot of  on Pt/C vs
on Pt/C vs  ranging from 275 to 304 K is shown in Fig. 4. Based on two independent repeat measurements, Pt/C has an HOR/HER activation energy of
ranging from 275 to 304 K is shown in Fig. 4. Based on two independent repeat measurements, Pt/C has an HOR/HER activation energy of  , which is essentially identical to that on Pt(pc) (Table I), considering experimental uncertainty.
, which is essentially identical to that on Pt(pc) (Table I), considering experimental uncertainty.

Figure 6. HOR/HER measured kinetic current densities (gray solid lines) on Pt/C in 0.1 M KOH at 10 mV/s and their fit to the Butler–Volmer equation (Eq. 2) with  (dashed black lines): (a) 275 and (b) 314 K. The HOR/HER kinetic current densities were obtained from iR-corrected polarization curves and were corrected for hydrogen mass transport in the HOR branch using Eq. 1. The corresponding insets show the HOR/HER polarization curves before (solid black lines) and after (dashed red lines) iR correction.
(dashed black lines): (a) 275 and (b) 314 K. The HOR/HER kinetic current densities were obtained from iR-corrected polarization curves and were corrected for hydrogen mass transport in the HOR branch using Eq. 1. The corresponding insets show the HOR/HER polarization curves before (solid black lines) and after (dashed red lines) iR correction.
There are essentially identical specific exchange current densities, activation energies, and transfer coefficients for the HOR/HER on Pt(pc) thus on Pt/C, and it is hypothesized that there is no significant Pt particle-size effect for the HOR/HER in 0.1 M KOH. Considering the roughly 10-fold lower specific exchange current densities reported for Pt(111) compared to Pt(110),20 future studies on Pt NPs of different shapes are needed to examine this hypothesis further. To our knowledge, the HOR/HER kinetics on Pt/C in alkaline electrolytes have never been reported before and this is the first comparison of the HOR/HER kinetics of Pt(pc) and Pt NPs.
Discussion
Comparing HOR/HER kinetic data with proposed mechanisms in alkaline electrolyte
Due to the experimental difficulties of measuring the kinetic reaction rates of very fast reactions, the HOR/HER mechanism on Pt remains inconclusive and under active investigation. This is particularly true for HOR/HER in acid as some authors argue that the rate of the HOR/HER is so high that conventional RDE measurements cannot provide sufficiently high hydrogen mass-transport rates, which would be required to unambiguously quantify kinetic rates of the HOR/HER12, 17 (this is discussed in more detail below).
Nevertheless, the HOR/HER proceeds through a combination of several of the following elementary steps: the dissociative adsorption of  without electron transfer (Tafel reaction, Eq. 3) or with simultaneous electron transfer (Heyrovsky reaction, Eq. 4) and the discharge of adsorbed hydrogen
without electron transfer (Tafel reaction, Eq. 3) or with simultaneous electron transfer (Heyrovsky reaction, Eq. 4) and the discharge of adsorbed hydrogen  (Volmer reaction, Eq. 5), whereby the asterisk represents an adsorption site on Pt32–34
(Volmer reaction, Eq. 5), whereby the asterisk represents an adsorption site on Pt32–34



Although the Heyrovsky and Volmer reactions as written above apply for the HOR/HER in acidic electrolytes, they can also be written for the reactions occurring in an alkaline solution32


The possible HOR/HER mechanisms based on these three elementary reactions are thus the Tafel–Volmer (Eq. 3, 5 or 3, 7) or the Heyrovsky–Volmer (Eq. 4, 5 or 6, 7) reaction sequence. The simplest kinetic HOR/HER models assume that one of the reactions in each of the two reaction sequences are rate-determining steps (rds),32, 34 which leads to four limiting cases: Tafel–Volmer (rds), Heyrovsky–Volmer (rds), Heyrovsky (rds)–Volmer, and Tafel (rds)–Volmer.
In the following, we review and discuss the kinetic HOR/HER expressions for the first two cases (Volmer reaction being rds) in an alkaline solution, which was first derived by Vetter.32 The anodic reaction rate of Eq. 7 is proportional to the surface coverage of  ,
,  , and the activity of
, and the activity of  , whereas the cathodic reaction rate is proportional to the coverage of unoccupied surface sites
, whereas the cathodic reaction rate is proportional to the coverage of unoccupied surface sites  as well as the activity of
as well as the activity of  . Using the Butler–Volmer equation (Eq. 2) to describe this elementary reaction step, the kinetic expression can be written as
. Using the Butler–Volmer equation (Eq. 2) to describe this elementary reaction step, the kinetic expression can be written as

where  represents the electrode potential across the solid/liquid interface measured against an arbitrary reference electrode. At the reversible potential of the Volmer reaction
represents the electrode potential across the solid/liquid interface measured against an arbitrary reference electrode. At the reversible potential of the Volmer reaction  , Eq. 8 can be rewritten as
, Eq. 8 can be rewritten as

where  is the exchange current density of the Volmer reaction. Dividing Eq. 8 by Eq. 9 yields
is the exchange current density of the Volmer reaction. Dividing Eq. 8 by Eq. 9 yields

in the assumed case that the Volmer reaction is the rds and  corresponds to the overpotential
corresponds to the overpotential  of the overall HOR/HER. Furthermore, if the HOR/HER kinetics are measured in a reasonably narrow potential range (e.g., from −0.075 to
of the overall HOR/HER. Furthermore, if the HOR/HER kinetics are measured in a reasonably narrow potential range (e.g., from −0.075 to  in our study), the variation in
in our study), the variation in  is small (i.e.,
is small (i.e.,  ). To a first approximation, the terms
). To a first approximation, the terms  and
and  can be approximated to be one, as the potential dependence of the exponential terms influences kinetic current densities much more strongly. Thus, Eq. 10 can be approximated as
can be approximated to be one, as the potential dependence of the exponential terms influences kinetic current densities much more strongly. Thus, Eq. 10 can be approximated as

Formally, Eq. 11 is equivalent to the Butler–Volmer equation (Eq. 2), which was used to fit the HOR/HER kinetic current densities in our study, providing an excellent fit with all the experimental data (Fig. 3 and 6).
An analogous derivation assuming the Heyrovsky reaction to be the rds is given in Ref. 32, yielding the same expression as Eq. 11 in the limit that  . Therefore, the Butler–Volmer equation given by Eq. 11 describes the HOR/HER kinetics in the vicinity of the HOR/HER equilibrium potential for three of the four limiting cases, i.e., for the Tafel–Volmer(rds), Heyrovsky–Volmer(rds), and Heyrovsky(rds)–Volmer mechanisms.32, 34 A very different kinetic expression is obtained only for the Tafel(rds)–Volmer mechanism,32, 34 so that it can be excluded as a possible HOR/HER mechanism in alkaline electrolytes on the basis of our measurements.
. Therefore, the Butler–Volmer equation given by Eq. 11 describes the HOR/HER kinetics in the vicinity of the HOR/HER equilibrium potential for three of the four limiting cases, i.e., for the Tafel–Volmer(rds), Heyrovsky–Volmer(rds), and Heyrovsky(rds)–Volmer mechanisms.32, 34 A very different kinetic expression is obtained only for the Tafel(rds)–Volmer mechanism,32, 34 so that it can be excluded as a possible HOR/HER mechanism in alkaline electrolytes on the basis of our measurements.
In recent density functional theory (DFT) calculations, a Tafel–Volmer mechanism was assumed and successfully described the HER activity trends of a wide range of metal catalysts in an acid electrolyte.35 This and later DFT33 calculations, nominally evaluated for pH 0, should apply equally to alkaline pH, as long as the potential scale is referenced to RHE. In the later DFT study by the same group on the HER mechanism on Pt(111),33 however, it was concluded that the activation barrier at zero overpotential of the Volmer reaction  is substantially lower than that of the Tafel
is substantially lower than that of the Tafel  and Heyrovsky
and Heyrovsky  reactions, suggesting that either one or both of the latter reactions would be rds. The calculated transfer coefficients for the Tafel and Heyrovsky reactions were
reactions, suggesting that either one or both of the latter reactions would be rds. The calculated transfer coefficients for the Tafel and Heyrovsky reactions were  and
and  , respectively.33 One major discrepancy between experimental data and their DFT calculations noted by the authors is the fact that the calculated activation barriers (0.6–0.8 eV) are so much larger than the experimentally reported activation energies on
, respectively.33 One major discrepancy between experimental data and their DFT calculations noted by the authors is the fact that the calculated activation barriers (0.6–0.8 eV) are so much larger than the experimentally reported activation energies on  in acid electrolytes (
in acid electrolytes ( 14) measured via RDE. As argued below, meaningful HOR/HER kinetics in an acid electrolyte cannot be obtained by RDE measurements, so that these experimental activation energies are not meaningful. However, experimental RDE-based activation energies in 0.1 M KOH, where the HOR/HER kinetics are slow, should be reliable and, indeed, the value of
14) measured via RDE. As argued below, meaningful HOR/HER kinetics in an acid electrolyte cannot be obtained by RDE measurements, so that these experimental activation energies are not meaningful. However, experimental RDE-based activation energies in 0.1 M KOH, where the HOR/HER kinetics are slow, should be reliable and, indeed, the value of  on Pt(111) in 0.1 M KOH reported by Schmidt et al.20 is quite comparable with the DFT-based values, considering the expected uncertainties of the latter. If one were to assume that the basic learnings from the DFT model33 are correct, i.e., that the Volmer reaction would have the lowest activation barrier and that the Heyrovsky reaction would have a transfer coefficient of
on Pt(111) in 0.1 M KOH reported by Schmidt et al.20 is quite comparable with the DFT-based values, considering the expected uncertainties of the latter. If one were to assume that the basic learnings from the DFT model33 are correct, i.e., that the Volmer reaction would have the lowest activation barrier and that the Heyrovsky reaction would have a transfer coefficient of  , our experimental observation that the HOR/HER on Pt in 0.1 M KOH can be described by a simple Butler–Volmer equation with
, our experimental observation that the HOR/HER on Pt in 0.1 M KOH can be described by a simple Butler–Volmer equation with  would favor the Heyrovsky(rds)–Volmer mechanism. However, this hypothesis is very tentative. Further experimental studies on the effect of pH and the partial pressure of
would favor the Heyrovsky(rds)–Volmer mechanism. However, this hypothesis is very tentative. Further experimental studies on the effect of pH and the partial pressure of  on the HOR/HER kinetics should be carried out to explore its mechanism in more detail.
on the HOR/HER kinetics should be carried out to explore its mechanism in more detail.
Comparing HOR/HER kinetics in acid with alkaline electro lyte
Although HOR/HER kinetics on Pt in alkaline can be measured accurately from conventional RDE measurements with iR correction, we compare here the HOR/HER kinetics in acid and alkaline and show that the HOR/HER kinetics on Pt in acid cannot be separated from the hydrogen diffusion overpotential in RDE measurements due to the very fast reaction rate. To illustrate this point, we conducted RDE measurements of the HOR/HER on Pt(pc) in 0.1 M  , including appropriate correction of the noncompensated solution resistance measured by ac impedance. Fig. 7a shows the polarization curve of the HOR/HER on the Pt(pc) electrode at 1600 rpm and 294 K before (solid black lines) and after (dotted red lines) iR correction and the Koutecky–Levich plot demonstrates that hydrogen mass transport limited current densities are obtained at already 0.1 V. The dashed black lines represent the calculated Nernstian diffusion overpotential
, including appropriate correction of the noncompensated solution resistance measured by ac impedance. Fig. 7a shows the polarization curve of the HOR/HER on the Pt(pc) electrode at 1600 rpm and 294 K before (solid black lines) and after (dotted red lines) iR correction and the Koutecky–Levich plot demonstrates that hydrogen mass transport limited current densities are obtained at already 0.1 V. The dashed black lines represent the calculated Nernstian diffusion overpotential  , assuming ideal reversibility of the HOR/HER, i.e., infinitely fast reaction kinetics22, 32
, assuming ideal reversibility of the HOR/HER, i.e., infinitely fast reaction kinetics22, 32

where  is the hydrogen diffusion limited current density. Clearly, after iR correction, the experimental current densities of the HOR/HER are indistinguishable from the Nernstian diffusion overpotential and because the latter assumes infinitely fast reaction kinetics, it is not reasonable to attempt to extract kinetic rates for the HOR/HER on Pt in acid using the RDE method, consistent with previous conclusions.17 Consequently, the several orders of magnitude lower HOR/HER exchange current density values from RDE measurements (0.1 to
is the hydrogen diffusion limited current density. Clearly, after iR correction, the experimental current densities of the HOR/HER are indistinguishable from the Nernstian diffusion overpotential and because the latter assumes infinitely fast reaction kinetics, it is not reasonable to attempt to extract kinetic rates for the HOR/HER on Pt in acid using the RDE method, consistent with previous conclusions.17 Consequently, the several orders of magnitude lower HOR/HER exchange current density values from RDE measurements (0.1 to  13, 14, 36) compared to those obtained from microelectrode16, 17 or gas-diffusion electrode12, 18 measurements are caused by both the rather low hydrogen diffusion limited current densities in the RDE configuration (on the order of several
13, 14, 36) compared to those obtained from microelectrode16, 17 or gas-diffusion electrode12, 18 measurements are caused by both the rather low hydrogen diffusion limited current densities in the RDE configuration (on the order of several  ) as well as the lack of iR correction. Kinetic currents cannot be measured with reasonable precision if the exchange current density is one or two orders of magnitude larger than the diffusion limited current density. Consequently, many RDE-based studies on the HOR/HER on Pt in an acid electrolyte report TS values in the vicinity of
) as well as the lack of iR correction. Kinetic currents cannot be measured with reasonable precision if the exchange current density is one or two orders of magnitude larger than the diffusion limited current density. Consequently, many RDE-based studies on the HOR/HER on Pt in an acid electrolyte report TS values in the vicinity of  ,14, 36 which simply reflects the apparent TS of the Nernstian diffusion overpotential (Eq. 12). Finally, based on RDE and microelectrode data for the HOR on Pt in acid electrolytes, Wang et al.37 recently developed a complex kinetic model, which suggests that the HOR proceeds through parallel Heyrovsky–Volmer and Tafel–Volmer mechanisms. However, considering the difficulty of extracting HOR kinetics on Pt in an acid electrolyte from RDE measurements, the complexity of the model is perhaps not warranted by the uncertainties associated with available electrochemical data.
,14, 36 which simply reflects the apparent TS of the Nernstian diffusion overpotential (Eq. 12). Finally, based on RDE and microelectrode data for the HOR on Pt in acid electrolytes, Wang et al.37 recently developed a complex kinetic model, which suggests that the HOR proceeds through parallel Heyrovsky–Volmer and Tafel–Volmer mechanisms. However, considering the difficulty of extracting HOR kinetics on Pt in an acid electrolyte from RDE measurements, the complexity of the model is perhaps not warranted by the uncertainties associated with available electrochemical data.

Figure 7. (a) HOR/HER polarization curve on Pt(pc) in 0.1 M  at 1600 rpm before (
at 1600 rpm before ( , solid black lines) and after (
, solid black lines) and after ( , dotted red lines) iR correction. The inset is the Koutecky–Levich plot obtained at
, dotted red lines) iR correction. The inset is the Koutecky–Levich plot obtained at  for the HOR at different rotation rates. (b) HOR/HER polarization curve on Pt(pc) in 0.1 M KOH at 1600 rpm before (
for the HOR at different rotation rates. (b) HOR/HER polarization curve on Pt(pc) in 0.1 M KOH at 1600 rpm before ( , solid black lines) and after (
, solid black lines) and after ( , dotted red lines) iR correction; the dashed black lines are the Nernstian diffusion overpotential calculated according to Eq. 12. The data are shown for the positive-going scans at 10 mV/s at
, dotted red lines) iR correction; the dashed black lines are the Nernstian diffusion overpotential calculated according to Eq. 12. The data are shown for the positive-going scans at 10 mV/s at  . The
. The  -axis potential range in (b) is double of that in (a).
-axis potential range in (b) is double of that in (a).
However, the iR-corrected polarization curve of the HOR/HER on Pt(pc) in 0.1 M KOH under the same experimental conditions deviates substantially from the Nernstian diffusion overpotential, as demonstrated in Fig. 7b. This is caused by the much slower HOR/HER kinetics on Pt in KOH, so that the diffusion limited current density is only reached at  (the diffusion limited current densities are essentially the same in both 0.1 M KOH and 0.1 M
(the diffusion limited current densities are essentially the same in both 0.1 M KOH and 0.1 M  ). Because the difference between the Nernstian diffusion overpotential relationship and the experimental data in KOH is very large, kinetic parameters for the HOR/HER can be extracted easily without interference from mass transport resistances. As was shown, this is not the case in an acidic electrolyte (see Fig. 7a), which is the reason why reliable HOR/HER kinetics on Pt with the RDE method can be obtained in alkaline but not in acid electrolytes. The fact that the HOR/HER kinetics on Pt are slower in alkaline compared to an acid electrolyte was noted before,17 but its cause remains unclear and, as discussed, is not captured in recent DFT models of the HER.6 Recent theoretical calculations38–40 about the HOR in acid and alkaline solutions suggest that the bond distance between Pt surface atoms and
). Because the difference between the Nernstian diffusion overpotential relationship and the experimental data in KOH is very large, kinetic parameters for the HOR/HER can be extracted easily without interference from mass transport resistances. As was shown, this is not the case in an acidic electrolyte (see Fig. 7a), which is the reason why reliable HOR/HER kinetics on Pt with the RDE method can be obtained in alkaline but not in acid electrolytes. The fact that the HOR/HER kinetics on Pt are slower in alkaline compared to an acid electrolyte was noted before,17 but its cause remains unclear and, as discussed, is not captured in recent DFT models of the HER.6 Recent theoretical calculations38–40 about the HOR in acid and alkaline solutions suggest that the bond distance between Pt surface atoms and  intermediates is shorter in alkaline than in an acid electrolyte, which would imply a stronger Pt–H binding energy in an alkaline electrolyte, which in principle could result in lower HOR/HER kinetics.35
intermediates is shorter in alkaline than in an acid electrolyte, which would imply a stronger Pt–H binding energy in an alkaline electrolyte, which in principle could result in lower HOR/HER kinetics.35
Predicted performance of AFCs/AMFCs vs PEMFCs using Pt/C
In this section, we estimate and compare the performance of AFCs and PEMFCs using Pt/C on the anode and the cathode. We first examine the implications of the slow HOR/HER kinetics on Pt in an alkaline electrolyte on the projected HOR overpotential in an AFC/AMFC using a Pt/C anode catalyst. To obtain the HOR/HER exchange current density at the typical fuel cell temperature of  , we extrapolated our experimental
, we extrapolated our experimental  values to
values to  using the Arrhenius plot, as shown in Fig. 8a. Quite clearly, the estimated specific exchange current density in an alkaline electrolyte on Pt/C is at least two orders of magnitude lower than that reported for PEMFCs.12 To predict the fuel cell relevant Pt mass activity in KOH as a function of overpotential, we then used the Butler–Volmer equation (Eq. 2) combined with the specific surface area of either our 46 wt % Pt/C catalyst
using the Arrhenius plot, as shown in Fig. 8a. Quite clearly, the estimated specific exchange current density in an alkaline electrolyte on Pt/C is at least two orders of magnitude lower than that reported for PEMFCs.12 To predict the fuel cell relevant Pt mass activity in KOH as a function of overpotential, we then used the Butler–Volmer equation (Eq. 2) combined with the specific surface area of either our 46 wt % Pt/C catalyst  or the 5 wt % Pt/C catalyst tested in PEMFCs
or the 5 wt % Pt/C catalyst tested in PEMFCs  .12 The resulting polarization curves in KOH are compared to that measured in PEMFCs,12 which is shown in Fig. 8b. Considering an envisaged anode catalyst loading of
.12 The resulting polarization curves in KOH are compared to that measured in PEMFCs,12 which is shown in Fig. 8b. Considering an envisaged anode catalyst loading of  and an operating current density of
and an operating current density of  , corresponding to a required mass activity of
, corresponding to a required mass activity of  , Fig. 8b illustrates that one would expect a hydrogen anode overpotential in an AFC/AMFC of 130–150 mV depending on the specific Pt surface area. This is in stark contrast to the negligibly small anode overpotential under the same conditions measured in a PEMFC. This projection clearly emphasizes that ultralow Pt anode loadings are not feasible in AFCs/AMFCs (in contrast to PEMFCs) and that novel anode catalysts must be developed for these applications, which either have a much higher mass activity for platinum group metals (PGM)-based anodes or are PGM-free.
, Fig. 8b illustrates that one would expect a hydrogen anode overpotential in an AFC/AMFC of 130–150 mV depending on the specific Pt surface area. This is in stark contrast to the negligibly small anode overpotential under the same conditions measured in a PEMFC. This projection clearly emphasizes that ultralow Pt anode loadings are not feasible in AFCs/AMFCs (in contrast to PEMFCs) and that novel anode catalysts must be developed for these applications, which either have a much higher mass activity for platinum group metals (PGM)-based anodes or are PGM-free.

Figure 8. (a) HOR/HER specific exchange current density of Pt/C in a PEMFC at 353 K and 
 (solid circles, from Ref. 12) compared to that obtained in 0.1 M KOH in this study (solid squares) and extrapolated to 353 K (open squares). (b) Projected HOR overpotential vs Pt mass activity in KOH based either on
(solid circles, from Ref. 12) compared to that obtained in 0.1 M KOH in this study (solid squares) and extrapolated to 353 K (open squares). (b) Projected HOR overpotential vs Pt mass activity in KOH based either on  for the 46 wt % Pt/C catalyst used in this study (black solid lines) or on
for the 46 wt % Pt/C catalyst used in this study (black solid lines) or on  for the 5 wt % Pt/C catalyst used in Ref. 12 (gray dashed lines); this is compared with mass activities measured on 5 wt % Pt/C in a PEMFC at 353 K (red lines, from Ref. 12).
for the 5 wt % Pt/C catalyst used in Ref. 12 (gray dashed lines); this is compared with mass activities measured on 5 wt % Pt/C in a PEMFC at 353 K (red lines, from Ref. 12).
Lastly, we compare the ORR kinetics of Pt/C in alkaline with that in acid and discuss the implication in the performance of AFCs/AMFCs relative to PEMFCs. As Pt NPs are shown to exhibit a much lower ORR activity than Pt bulk surfaces in acid,2, 41 we examined here the ORR specific activity of both Pt(pc) and Pt/C in 0.1 M KOH at 294 K and 
 . The activities at 0.9 V are summarized in Table II. The ORR activity of Pt(pc) is
. The activities at 0.9 V are summarized in Table II. The ORR activity of Pt(pc) is  , which is consistent with the value of
, which is consistent with the value of  reported in a Teflon cell26 (confirming our measurements of ORR activity are free of glass-cell related contamination effects). Unlike HOR/HER kinetics on Pt, the ORR specific activity of Pt/C is roughly two times lower than that of Pt(pc) (Table II), indicating a Pt particle-size effect on the ORR kinetics in alkaline. The ORR specific activities of Pt/C and Pt(pc), and the mass activities of Pt/C in 0.1 M KOH are identical to those in 0.1 M
reported in a Teflon cell26 (confirming our measurements of ORR activity are free of glass-cell related contamination effects). Unlike HOR/HER kinetics on Pt, the ORR specific activity of Pt/C is roughly two times lower than that of Pt(pc) (Table II), indicating a Pt particle-size effect on the ORR kinetics in alkaline. The ORR specific activities of Pt/C and Pt(pc), and the mass activities of Pt/C in 0.1 M KOH are identical to those in 0.1 M  within the experimental error42 (Table II). This is perhaps not surprising because the ORR rate-controlling formation of adsorbed oxygen-containing species
within the experimental error42 (Table II). This is perhaps not surprising because the ORR rate-controlling formation of adsorbed oxygen-containing species  occurs at essentially the same potential vs RHE in both electrolytes.43 For practical applications, the identical ORR activities in acid and base predict that the ORR overpotential on a Pt cathode in AFCs/AMFCs would be the same as that of PEMFCs under the same conditions [i.e., under the same mass specific current density
occurs at essentially the same potential vs RHE in both electrolytes.43 For practical applications, the identical ORR activities in acid and base predict that the ORR overpotential on a Pt cathode in AFCs/AMFCs would be the same as that of PEMFCs under the same conditions [i.e., under the same mass specific current density  , temperature, and oxygen partial pressure].
, temperature, and oxygen partial pressure].
Table II. Summary of ORR specific activity  and mass activity
and mass activity  at 0.9 V vs RHE,
at 0.9 V vs RHE, 
 , and
, and  in 0.1 M KOH and 0.1 M
in 0.1 M KOH and 0.1 M  for Pt(pc) and Pt/C.
for Pt(pc) and Pt/C.
  in 0.1 M KOH in 0.1 M KOH |
  in 0.1 M KOH in 0.1 M KOH |
  in 0.1 M in 0.1 M 
|
  in 0.1 M in 0.1 M 
| |
|---|---|---|---|---|
| Pt(pc) |
 
| — |
  42 42
| — |
| Pt/C |
 
|
 
|
  42 42
|
  42 42
|
The asterisk indicates the number of independent repeat experiments. Data were obtained from the positive-going scans at 10 mV/s and were iR- and mass-transport corrected as described in the text.
Conclusion
Our RDE measurements show that the HOR/HER on Pt(pc) and Pt/C in an alkaline electrolyte have very comparable specific exchange current densities (showing no size effect of HOR/HER on Pt) of  at 294 K, and activation energies of
at 294 K, and activation energies of  . In addition, based on the fact that the kinetics of the HOR/HER in our study are very well described by the Butler–Volmer equation and considering recent DFT findings,33 it is hypothesized that the mechanism of the HOR/HER in KOH may go though the Heyrovsky–Volmer mechanism with the Heyrovsky step as the rds.
. In addition, based on the fact that the kinetics of the HOR/HER in our study are very well described by the Butler–Volmer equation and considering recent DFT findings,33 it is hypothesized that the mechanism of the HOR/HER in KOH may go though the Heyrovsky–Volmer mechanism with the Heyrovsky step as the rds.
Analogous RDE measurements of the HOR/HER on Pt(pc) in 0.1 M  clearly demonstrate that the HOR/HER current densities in an acid solution simply follow the Nerstian hydrogen diffusion overpotential relationship, which is based on assuming reversible, i.e., infinitely fast HOR/HER kinetics. This implies that the HOR/HER exchange current densities in an acid electrolyte are one or more orders of magnitude larger than the typical hydrogen diffusion limited current density and can therefore not be quantified in RDE measurements. This finding is consistent with the much higher HOR/HER exchange current densities in measurements based on microelectrode or gas-diffusion electrodes.
clearly demonstrate that the HOR/HER current densities in an acid solution simply follow the Nerstian hydrogen diffusion overpotential relationship, which is based on assuming reversible, i.e., infinitely fast HOR/HER kinetics. This implies that the HOR/HER exchange current densities in an acid electrolyte are one or more orders of magnitude larger than the typical hydrogen diffusion limited current density and can therefore not be quantified in RDE measurements. This finding is consistent with the much higher HOR/HER exchange current densities in measurements based on microelectrode or gas-diffusion electrodes.
Our measurements and analysis suggest that the HOR/HER kinetics on Pt(pc) and Pt/C are several orders of magnitude slower in alkaline compared to acid electrolyte. Therefore, the use of Pt/C anode catalysts in AFCs/AMFCs would require high loadings and thus become a significant cost factor unlike in PEMFCs, where very low Pt anode loadings are sufficient. However, the use of Pt/C cathode catalysts in AFCs/AMFCs would result in comparable ORR potential losses as in PEMFCs, but other non-noble metal-based cathode catalysts are available for AFCs/AMFCs with ORR activity comparable to Pt/C. Therefore, the development of highly efficient catalysts toward HOR in alkaline electrolyte is one of the critical challenges to make AFCs/AMFCs more practical.
Acknowledgment
This work was supported in part by the DOE Hydrogen Initiative program under award no. DE-FG02-05ER15728 , an Air Products Faculty Excellence grant, and the Toyota Motor Co. This research made use of the Shared Experimental Facilities supported by the MRSEC Program of the National Science Foundation under award no. DMR 08-19762 .
Massachusetts Institute of Technology assisted in meeting the publication costs of this article.