
Abstract
Nanosized ZrO2 powders with near pure monoclinic, tetragonal, and cubic structures synthesized by various methods were used as catalysts for photocatalytic degradation of methyl orange. The structural and textural properties of the samples were analyzed by X-ray diffraction, Raman spectroscopy, TEM, UV-vis, X-ray photoelectron spectroscopy (XPS), and N2 adsorption measurements. The performance of synthesized ZrO2 nanoparticles in the photocatalytic degradation of methyl orange under UV light irradiation was evaluated. The photocatalytic activity of the pure monoclinic ZrO2 sample is higher than that of the tetragonal and cubic ZrO2 samples under optimum identical conditions. The characterization results revealed that monoclinic ZrO2 nanoparticles possessed high crystallinity and mesopores with diameter of 100 Å. The higher activity of the monoclinic ZrO2 sample for the photocatalytic degradation of methyl orange can be attributed to the combining effects of factors including the presence of small amount of oxygen-deficient zirconium oxide phase, high crystallinity, large pores, and high density of surface hydroxyl groups.
Similar content being viewed by others


Background
The rapid growth of the textile industry has led to the accumulation of various organic pollutants, with dyes accumulating in bodies of water as a particularly severe example. This type of aquatic pollution has indirect or direct adverse effects on the biosphere [1]. Photocatalysis is one promising approach to protect the aquatic environment based on its ability to oxidize low concentrations of organic pollutants in water [2,3]. In the past two decades, many oxide and sulfide semiconductors such as TiO2, ZnO, WO3, SrTiO3, ZnS, and CdS were applied as photocatalysts for environmental control technology and also a wide range of chemical reactions [4]. Recently, Kuriakose et al. [5,6], Cheng et al. [7], and Ren et al. [8] successfully employed ZnO- and TiO2-based nanomaterials for photocatalytic degradation of organic dyes. ZrO2 has been considered as a photocatalyst in different chemical reactions due to its relatively wide band gap value E g and the high negative value of the conduction band potential [9]. The reported band gap energy of ZrO2 range was between 3.25 and 5.1 eV, depending on the preparation technique of the sample [10].
It is reported that a good manipulation of ZrO2 morphological tuning, porous structure control, and crystallinity development is required in order to enhance the light harvesting capability, prolong the lifetime of photoinduced electron-hole pairs, and facilitate the reactant accessibility to surface active sites [11]. As ZrO2 is used in a wide variety of applications in addition to photocatalysis, the fabrication of identical ZrO2 nanoscale structures has been recently attracted a great deal of interest. Nanocrystalline ZrO2 with various attractive morphologies has been effectively prepared by different synthesis methods like hydrothermal synthesis, sol-gel synthesis, precipitation, and thermal decomposition [12].
It is well known that ZrO2 has three polymorphs [13]: monoclinic, tetragonal, and cubic. Preparation methods play an important role in determining the final crystal structure of ZrO2. Although the different surface properties on different ZrO2 polymorphs have been extensively studied [14,15], the effect of crystal structures on photocatalysis has rarely been investigated.
Nawale et al. [16] synthesized ZrO2 samples using thermal plasma reactor at different operating pressures. The sample which contained both tetragonal and monoclinic phases synthesized at 1.33 bar of operating pressure showed the highest photocatalytic activity. The presence of tetragonal phase along with monoclinic phase indicates the crystallographic rearrangement in ZrO2 due to the oxygen vacancies. The authors related the photocatalytic properties of ZrO2 with the trap levels present in it due to oxygen vacancies. It was observed that the photocatalytic response tracks the energy gap of the monoclinic phase which varies with the varying synthesis parameters.
Zhao et al. [17] used anodization method to synthesize ZrO2 nanotubes with a length of 25 μm, inner diameter of 80 nm, and wall thickness of 35 nm. The authors observed 97.6 decolorization percentage of methyl orange in 8 h at optimal pH value 2. Ismail et al. [18] synthesized 6-μm-thick anodic oxide film with nanotubular ZrO2 structure, and the authors tested the photocatalytic ability of the ZrO2 nanotubes. The authors reported 30% of methyl orange degradation under UV light in the presence of the cubic/tetragonal ZrO2 nanotubes after 120 min of reaction.
Jiang et al. [19] used zirconium foil to anodize in electrolyte containing 1 M (NH4)2SO4 and 0.25 wt.% NH4F to in situ construct the ZrO2 nanotubes on the surface. The authors reported that ZrO2 nanotubes showed excellent photocatalytic performance with methyl orange photodegradation rate of 94.4% after 240 min. They also claimed that photocatalysis performance was due to the hydroxyl group absorbing on the surface.
Shu et al. [20] synthesized tetragonal star-like ZrO2 nanostructures using hydrothermal synthesis method. The authors used ZrO2 nanostructures for the photodegradation of anionic dyes including methyl orange, in acidic, neutral, and weak basic aqueous solutions. They observed that the ZrO2 sample offered complete degradation of methyl orange within 60 min; however, authors have not studied the stability and reusability of the synthesized ZrO2 nanomaterial.
The objective of the present study is to synthesize nanocrystalline mesoporous monoclinic, tetragonal, and cubic ZrO2 samples with high surface area using fairly simple experimental procedures. In this work, nanosized pure monoclinic, tetragonal, and cubic ZrO2 samples were prepared and the physico-chemical properties of the samples were performed by different characterization techniques. The photocatalytic degradation of methyl orange over the three ZrO2 samples were studied and correlated to the phase structure, specific surface area, and electronic properties of the catalysts.
Methods
Materials
Zirconyl chloride, zirconium isopropoxide, sodium hydroxide solution, methyl orange, and hydrochloric acid were purchased from Aldrich, Dorset, England, UK. All chemicals used in this study were analytical grade and used directly without further purification. Deionzied water was used for the preparation of the methyl orange standard solution as well as the respective dilutions.
Synthesis of pure monoclinic, tetragonal, and cubic ZrO2 samples
Monoclinic ZrO2
A near pure monoclinic nanocrystalline ZrO2 was synthesized by following the method reported by Guo et al. [21]. The zirconyl chloride was dissolved in deionized water so that the final concentration of zirconium was 38.7 g per liter (0.42 M). Of the zirconyl chloride solution, 15 ml was added to 300 mL deionized water in a glass beaker, and then concentrated aqueous ammonia was added rapidly to the solution with constant stirring until pH 4.5. The resultant precipitate was aged in the mother liquor for 24 h. After filtration, it was washed several times with dilute ammonia and hot deionized water (80°C) until chloride ions were no longer detectable in the washing water (AgNO3 test) and then dried at 100°C for 12 h. The synthesized sample was calcined at 500°C for 3 h in air with a ramp rate of 1°C min−1 and kept isothermally for 3 h and was annotated as m-ZrO2.
Tetragonal ZrO2
Pure tetragonal ZrO2 was synthesized by the following reported procedure in the literature [22]. Zirconium oxychloride and ammonia solution (25% w/w) solutions were prepared using deionized water. First, 50 mL of 2.5 M ammonia solution was added to 50 mL of 0.1 M zirconium oxychloride solution drop by drop in a beaker and the mixture was stirred vigorously at room temperature for 4 h. The white zirconium hydroxide precipitates in time of addition of ammonia solution. The obtained precipitate was separated by centrifugation at 4,000 rpm, washed with water and ethanol for several times. Then, the precipitate was transferred into Teflon-lined autoclave, and the autoclave was kept at 100°C for 12 h. Finally, the white powder was calcined in furnace at 500°C for 3 h with a ramp rate of 1°C min−1 and kept at this temperature for 3 h and was annotated as t-ZrO2.
Cubic ZrO2
Cubic ZrO2 was synthesized by hydrothermal method reported my Tahir et al. [23]. In a Teflon vessel, 1 g of zirconium isopropoxide was dissolved in 6 mL of ethanol (99.8%) and then the Teflon vessel was kept in a desiccator containing a Petri dish filled with water at the bottom. The diffusion experiment was stopped after 12 h, followed by the addition of 25 mL of 10 M NaOH aqueous solution. Then, the reaction vessel was sealed into a stainless steel hydrothermal bomb, which was heated to 180°C for 18 h. After the autoclave was cooled down to room temperature, the products were filtered and repeatedly washed with 0.1 M HNO3, 1 N HCl, and deionized water. After drying under vacuum for 3 h, a white soft and fibrous powder was obtained. The obtained powder was calcined at 500°C for 3 h in air with a ramp rate of 1°C min−1 and kept isothermally for 3 h and was annotated as c-ZrO2.
Material characterization
X-ray powder diffraction (XRD) studies were performed for all of the prepared solid samples using a Bruker diffractometer (Bruker D8 advance target; Bruker AXS, GmbH, Karlsruhe, Germany). The patterns were run with copper Kα1 and a monochromator (λ = 1.5405 Å) at 40 kV and 40 mA. The crystallite size of the ZrO2 was calculated using Scherrer's equation;
where D is the average crystallite size of the phase under investigation, B is the Scherer constant (0.89), λ is the wavelength of the X-ray beam used (1.54056 A°), β 1/2 is the full width at half maximum (FWHM) of the diffraction peak, and θ is the diffraction angle. The identification of different crystalline phases in the samples was performed by comparing the data with the Joint Committee for Powder Diffraction Standards (JCPDS) files.
The Raman spectra of the samples were measured with a Bruker Equinox 55 FT-IR spectrometer equipped with an FRA106/S FT Raman module and a liquid N2-cooled Ge detector using the 1,064-nm line of a Nd:YAG laser with an output laser power of 200 mW.
A Philips CM200FEG microscope (Philips, Amsterdam, The Netherlands), 200 kV, equipped with a field emission gun was used for HRTEM analysis. The coefficient of spherical aberration was C s = 1.35 mm. The information limit was better than 0.18 nm. High-resolution images with a pixel size of 0.044 nm were taken with a CCD camera.
The textural properties of the prepared samples were determined from nitrogen adsorption/desorption isotherm measurements at −196°C using Autosorb automated gas sorption system (Quantachrome, Boynton Beach, FL, USA). Prior to measurement, each sample was degassed for 6 h at 150°C. The specific surface area, S BET, was calculated by applying the Brunauer-Emmett-Teller (BET) equation. The average pore radius was estimated from the relation 2V p/S BET, where V p is the total pore volume (at P/P0 = 0.975). Pore size distribution over the mesopore range was generated by the Barrett-Joyner-Halenda (BJH) analysis of the desorption branches, and the values for the average pore size were calculated.
The X-ray photoelectron spectroscopy (XPS) measurements were carried out by using a SPECS GmbH X-ray photoelectron spectrometer (SPECS, Berlin, Germany). Prior to analysis, the samples were degassed under vacuum inside the load lock for 16 h. The binding energy of the adventitious carbon (C 1 s) line at 284.6 eV was used for calibration, and the positions of other peaks were corrected according to the position of the C 1 s signal. For the measurements of high-resolution spectra, the analyzer was set to the large area lens mode with energy steps of 25 m eV and in fixed analyzer transmission (FAT) mode with pass energies of 34 eV and dwell times of 100 ms. The photoelectron spectra of the four samples were recorded with the acceptance area and angle of 5 mm in diameter and up to ±5°, respectively. The base pressure during all measurements was 5 × 10−9 mbar. A standard dual anode excitation source with Mg Kα (1,253.6 eV) radiation was used at 13 kV and 100 W.
The UV-vis absorption spectra in transmittance mode were recorded on a Thermo Scientific (Evolution 600 UV-vis; Thermo Fisher Scientific, Waltham, MA, USA) instrument. The optical bandgap of the samples is measured by plotting a (αhυ)2 versus hυ. The extrapolation of the straight line in the graph to (αhυ)2 = 0 gives the value of the energy band gap.
Photocatalytic degradation of methyl orange
Photocatalytic activity measurements were carried out in a homebuilt reactor. The reactor is a wooden box with dimensions of 100 cm height, 100 cm width, and 60 cm thickness, equipped with a 12-V transformer for an electric exhaust fan. Six 18 W UV lamps (60 cm × 2.5 cm) of approximately 350 to 400 nm (F20 T8 BLB) were used; the total power of the UV light at the surface of the test suspension measured with a Newport 918DUVOD3 detector (Newport Corporation, Irvine, CA, USA) and power meter was 13 W/m2. In a typical experiment, 100 mL of aqueous methyl orange solution (10 mg/L) was stirred (300 rpm) with 100 mg of the different photocatalysts. The resulting suspension was equilibrated by stirring for 1 h to stabilize the absorption of methyl orange dye over the surface of catalyst before exposing to the UV light. Samples were withdrawn at 10 min intervals, filtered through a 0.2-mm PTFE Millipore membrane filter (Millipore, Billerica, MA, USA) to remove suspended catalyst agglomerates, and finally analyzed using the UV-vis spectrometer (Thermo Fisher Scientific Evolution 160) in the range between 250 and 600 nm. The decolorization rate percentages of methyl orange were calculated by the following equation:
where C o is the concentration of methyl orange before illumination and C is the concentration after a certain irradiation time.
Results and discussion
X-ray powder diffraction
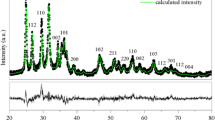
The XRD patterns of synthesized m-ZrO2, t-ZrO2, and c-ZrO2 samples and corresponding JCPDS reference patterns are shown in Figure 1. XRD pattern of the m-ZrO2 sample showed intensive diffraction patterns at 2θ = 24.2°, 28.2°, 31.4°, and 34.3° which are corresponding to monoclinic ZrO2 crystal phase [JCPDS 37-1484]. It is observed that there is one major peak at 2θ = 25.4° and another small peak at 22°, which are not indexed for monoclinic ZrO2 phase. These peaks can be indexed to the oxygen-deficient zirconium oxide, ZrO0.35 phase [JCPDS; 17-0385, hexagonal, space group P6322]. To determine the purity of monoclinic phase of the m-ZrO2 sample, volume percent of monoclinic and oxygen-deficient zirconium oxide phase present in the m-ZrO2 sample was determined from the integrated intensities of the diffraction peaks (−111) (111) of m-ZrO2 at 2θ = 28.5° and 31.5°, respectively, and the diffraction line (101) of oxygen deficient ZrO2 at 2θ = 25.4°. We used the expressions (3) and (4) reported in the literature [24].

XRD patterns of the different zirconia samples.
The percentages of monoclinic and oxygen deficient phases were found to be 97% and 3%, respectively, for the m-ZrO2 sample. There are no additional unindexed peaks in the m-ZrO2 sample.
XRD pattern of the t-ZrO2 sample showed peaks for pure tetragonal phase of ZrO2 [JCPDS 80-0965] at 2θ = 30.2°, 35.2°, 50.6°, and 60.2°. No additional peaks corresponding to any other phase was observed in XRD pattern of this sample. All of the diffraction peaks of the XRD pattern of the c-ZrO2 sample can be indexed to the standard pattern of the pure cubic phase of ZrO2. Peaks at 2θ = 30.3°, 35.14°, 50.48°, and 60.2° reveal the presence of (111), (200), (220), and (311) planes, respectively, of cubic ZrO2 according to JCPDS CAS number 27-0997. These observation indicates that the m-ZrO2 sample is near pure monoclinic; however, the t-ZrO2 and c-ZrO2 samples did not show presence of any additional phases or impurities indicating that these two phases are pure in composition. In addition, the intensities of diffraction peaks of the m-ZrO2 sample were much higher than those of the t-ZrO2 and c-ZrO2 samples indicating that the m-ZrO2 sample is highly crystalline than the t-ZrO2 and c-ZrO2 samples.
However, it is known that assignment of cubic and tetragonal structures, based solely on the X-ray diffraction analysis, can be misleading because the cubic and tetragonal structures (a 0 = 0.5124 nm for cubic and a 0 = 0.5094 nm and c 0 = 0.5177 nm for tetragonal structures) are very similar [25]. The authors also reported that the tetragonal structure can be distinguished from the cubic structure by the presence of the characteristic splitting of the diffraction peaks, whereas the cubic phase exhibits only single peaks. A significant line broadening obscured any clear distinction between the tetragonal and cubic polymorphs of ZrO2 (Figure 1). However, this detection is possible by measuring with high step counting times the (112) Bragg reflection of the tetragonal structure, which is forbidden in the cubic symmetry [26]. As it can be observed in Figure 1, a shift of the peak positions to higher 2θ values occurred. This shift may indicate a decrease in the lattice parameters.
The crystallite size was calculated using the Scherrer's equation (1). The average crystallite sizes of the monoclinic phase, calculated from the (111) diffraction peak was found to be 34 nm. Similarly, the average crystallite sizes, calculated from the (111) diffraction peak of the tetragonal and cubic phases, were found to be 17 and 20 nm for the t-ZrO2 and c-ZrO2 samples, respectively.
Raman spectroscopy
In order to confirm the crystalline structure of the samples, the Raman spectra of the samples were obtained and shown in Figure 2. From this figure, we can see that the m-ZrO2 sample showed several peaks centering at 183, 301, 335, 381, 476, 536, 559, 613, and 636 cm−1. The strong peaks are at 183, 335, and 476 cm−1. The exhibited bands are clearly indicating that the m-ZrO2 sample possessed dominant monoclinic phase of ZrO2 [27]. The t-ZrO2 sample showed peaks at 149, 224, 292, 324, 407, 456, and 636 cm−1, and the peak positions are in quite accordance with the reported values for tetragonal phase of ZrO2 [28].

Raman spectra of different zirconia samples.
The Raman spectrum for c-ZrO2 is characterized by a narrow band at 145 cm−1 and broad bands centered at 246, 301, 436, and 625 cm−1. Gazzoli et al. [29] reported that the Raman peak at 149 cm−1 is common for both of tetragonal and cubic phases, and cubic ZrO2 presents the strong band between 607 and 617 cm−1. The c-ZrO2 sample in this study clearly showed the broad peak centered at 625 cm−1, and this sample also shows poorly defined features related to the disordered oxygen sub-lattice whereas tetragonal ZrO2 exhibits several well-defined sharp bands because of the symmetry reduction [30]. In addition, the highly intense peaks at 292 and 636 cm−1 which are main characteristic bands of tetragonal ZrO2 cannot be found in the spectrum of the c-ZrO2 sample, which indicates the absence of tetragonal ZrO2 phase in this sample. Kontoyannis et al. [31] also reported that cubic ZrO2 shows amorphous-like Raman spectrum with one broad band at 530 to 670 cm−1. The features of Raman spectrum of c-ZrO2 as shown in Figure 2 is in accordance with the spectral results reported in the literature.
Transmission electron microscopy
The TEM images for the m-ZrO2, t-ZrO2, and c-ZrO2 samples are shown in Figure 3A,B,C, respectively. Tightly packed dumbbell-shaped particles can be observed in the low magnification TEM images of three samples. The average particle size for the m-ZrO2, t-ZrO2, and c-ZrO2 samples was found to be 24, 18, and 8 nm, respectively. There are conflicting reports in the literature regarding the phase structure of ZrO2 particles in smaller size (less than 10 nm). Some authors reported that cubic ZrO2 phase exists as fine nanoparticles [32], and few other researchers reported that tetragonal ZrO2 phase exists in smaller size than cubic phase [33]. However, in the present work, it is clear that in the c-ZrO2 sample, pure cubic ZrO2 phase possessed smaller particles size than the t-ZrO2 sample (pure tetragonal).

TEM images of (A) m -ZrO 2 (B) t -ZrO 2 (C) c -ZrO 2 and HRTEM images of (D) m -ZrO 2 (E) t -ZrO 2 (F) c -ZrO 2 .
High-resolution transmission electron microscopy
In order to authenticate the ZrO2 phase existed in the samples, high-resolution transmission electron microscopy (HRTEM) was carried out on particles of the three samples. Figure 3D represents the HRTEM image of the m-ZrO2 sample. The image clearly showed well-resolved lattice fringes. The distance between the fringes was calculated to be 0.297 nm which can be attributed to the interplanar spacing corresponding to (111) plane of monoclinic ZrO2 [34]. The HRTEM image of t-ZrO2 was shown in Figure 3E. This image also showed well-resolved equidistant lattice fringes. The distance between the parallel fringes was calculated to be 0.296 nm which can be attributed to the well-recognized lattice d-spacing of (111) plane of tetragonal ZrO2 [35]. A typical HRTEM image of particles of the c-ZrO2 sample is shown in Figure 3F. The image shows equidistant parallel fringes which depict single crystalline nature of the particle. The distance between the parallel fringes was calculated to be 0.291 nm which is the well-recognized lattice d-spacing of (111) plane of cubic ZrO2 [36].
BET surface area
A typical nitrogen adsorption-desorption isotherms of the samples are shown in Figure 4. The adsorption-desorption patterns of the three ZrO2 samples belong to the typical IUPAC IV-type with the H2-type hysteresis loop, which is a characteristic of particles with uniform size and mesoporous structure [37]. From the figure, it is clear that all the three samples showed type IV isotherms with hysteresis loop at P/Po = 0.45 to 0.95. However, each sample exhibited a different type of hysteresis loop suggesting that pore size and shape were not same in these samples. The H2-type adsorption hysteresis can be explained as a consequence of the interconnectivity of pores. It was reported that in such systems, the distribution of pore sizes and pore shapes are not well defined or irregular. A sharp step on desorption isotherm is usually understood as a sign of interconnection of the pores. The shape of hysteresis loop of the m-ZrO2 sample suggesting that this sample possessed pores known as ‘ink-bottle’ type [38].

Nitrogen adsorption-desorption isotherms and pore size distribution (inset) of the samples.
The pore size distribution patterns of the synthesized ZrO2 samples were shown in the inset of Figure 4. Pore size distribution of the m-ZrO2 sample reveals a broad and monomodal distribution of the pore dimension in the mesoporous region. In addition, the c-ZrO2 sample showed a narrower pore size distribution than t-ZrO2. The porosity of the m-ZrO2 and t-ZrO2 samples appears to be arising from non-crystalline intra-aggregate voids and spaces formed as a result of inter-particle contact [39]. The narrow and broad bimodal distribution of pores can be observed in the case of the c-ZrO2 sample. The porosity of this sample appears to be framework porosity which corresponds to the porosity within the uniform channels of ZrO2 structure.
The textural properties of the synthesized ZrO2 samples from the adsorption-desorption data was tabulated in Table 1. Specific surface area of the m-ZrO2 sample is 65 m2g−1, an average pore radius of 50 Å, and a total pore volume of 0.626 cm3g−1. The t-ZrO2 and c-ZrO2 samples possessed the surface area of 74 and 204 m2g−1; the drastic increase of the surface area of the c-ZrO2 sample could be due to very small size of particles (TEM results). However, the c-ZrO2 sample possessed pores with small radius (19 Å) than the t-ZrO2 (28.3 Å) and m-ZrO2 (50 Å) samples.
X-ray photoelectron spectroscopy
It is known that XPS is a very sensitive tool in analyzing the chemical state of Zr cations in ZrO2 and its composites [40]. Figure 5A,B displays the XPS spectra of the Zr 3d and O 1 s core levels of the three samples, respectively. The peaks located at 181.3 and 183.8 eV are attributed to the spin-orbit splitting of the Zr 3d components, Zr 3d 5/2 and Zr 3d 3/2 . The binding energy of O 1 s in ZrO2 is located at 530.1 eV.

Deconvoluted XPS spectra of samples (A) Zr ( 3d and 5d ) (B) O ( 1 s ).
Deconvolution of the spectra produces peaks attributed to the existence of two kinds of zirconium species, referred as Zr2+ species with low binding energy in the range 180.7 to 181.4 eV and Zr4+ species with higher binding energy in the range of 182.1 to 182.3 eV. It should be noted that the fraction of Zr4+ species for all samples is larger compared to that of species Zr2+. It is reported that the binding energy of Zr4+ species in pure ZrO2 is around 182.6 eV [41]; however, slightly lower values compared to that of stoichiometric ZrO2 were observed especially for the sample m-ZrO2 (182.1 eV), probably, due to some oxygen deficiency. The position shift toward the lower binding energy might be associated with the holes created by oxygen vacancies in the ZrO2 lattice [42].
Kawasaki [43] reported that Zr 3d components, Zr 3d 5/2 and Zr 3d 3/2 for cubic ZrO2 can be observed at 182.0 and 184.4 eV, respectively. The same components for tetragonal ZrO2 and monoclinic ZrO2 samples appeared at 182.7 and 184.7 eV [44] and 182.2 eV and 184.6 eV [45], respectively. The binding energy values of Zr 3d components observed in this study are in accordance with values reported in the literature.
The O 1 s broad peaks can be deconvulated into three peaks at the corresponding position using XPS Casa Software, whose relative contents are shown in Table 2. Navio et al. [9] observed two types of oxygen species in the ZrO2 sample, oxygen species of ZrO2 and oxygen species of Zr-OH, whose binding energy is in the range of 529.8 to 530.3 and 530.9 to 532.2 eV, respectively. It was also reported that the oxygen species with binding energy 531.0 eV are attributed to Zr-OH groups [46]. All the three samples showed XPS peaks corresponding to Zr-OH, Zr4+-O, and Zr2+-O species in different proportions [43]. The m-ZrO2 sample showed highest 12.3 mass percentage Zr-OH groups bounded to Zr atom, while t-ZrO2 and c-ZrO2 have 8% and 6.1%, respectively. These features of the XPS spectra indicate that the c-ZrO2 and t-ZrO2 samples were regular surfaces and with no apparent defect relative to the Zr4+ species; this is rather significant. In fact, the m-ZrO2 sample showed surface defects with more surface hydroxyl groups.
Diffuse-reflectance UV-vis
Figure 6 represents the UV-vis absorption spectra of the three ZrO2 samples. It is known that all ZrO2 polymorphs are very similar in vibrational structure, and a minor variation in their band frequencies or intensities infers small differences in the Zr4+ distribution in Zr-O sites and the oxygen vacancies and other structural defects [47]. Herrera et al. [48] reported that UV-vis spectra of monoclinic Fe-doped ZrO2 display two bands at around 245 and 320 nm, which are associated with charge transfer transitions.

UV-vis absorption spectra of the samples.
Li et al. [49] indicated that the ZrO2 sample with pure monoclinic ZrO2 nanoparticles showed a pronounced absorption peak at 270 nm, and tetragonal and cubic ZrO2 nanoparticles show an absorption peak at 314 nm. The absorptions in the range 250 to 350 nm were assigned to O2 → Zr4+ charge transfer transitions with Zr in low coordination states (possibly six) either isolated or present in small ZrxOy clusters [10].
Band gap of all the ZrO2 samples was determined by establishing the relation between hυ and (αhυ)2. The obtained data indicated that the band gap energy for m-ZrO2 (3.25 eV) is lower compared with t-ZrO2 (3.58 eV) and c-ZrO2 (4.33 eV). Crystal structure plays an important role in the electronic structure of ZrO2. This effect is most significant in the d-electron-derived conduction bands (CBs). It is reported that the reduction of the CB gap between the Zr 4d (x2−y2, z2) and the Zr 4d (xy, yz, zx) CBs is present in cubic ZrO2 and disappears in tetragonal ZrO2, and also, substantial volume expansion was observed in the case of monoclinic ZrO2 due to the hybridization of Zr 4d CBs into a new single Zr 4d CB [50].
It was also reported that the pure tetragonal and monoclinic ZrO2 nanoparticles showed energy band gaps of 4.0 and 3.5 eV, respectively [47]. These values are very similar to the values reported in the literature reports. Emeline et al. [51] determined an energy band gap of 5.0 eV for monoclinic ZrO2 thin films calcined at 550°C, and Chang and Doong [52] determined an energy gap of 5.7 eV for the same sample at the same temperature. However, Navio et al. [9] reported an energy band gap of 3.7 eV for monoclinic ZrO2 powders prepared by sol-gel method. These authors claim that the decrease in the band gap energy could be attributed to a highly disordered structure, as a result of the conditions used in the preparation technique. As a consequence of structural defects, some energy levels are introduced into the semiconductor band gap that allow transitions of lower energy and therefore lead to a decrease of the band gap energy.
Photocatalytic degradation of methyl orange
The photocatalytic activity of the three ZrO2 samples was determined by monitoring the degradation of the methyl orange dye. A blank experiment was carried out to confirm that the photo-degradation reaction did not proceed without the presence of either catalyst or the UV radiation. Figure 7 shows the change in the UV-vis absorbance spectra of methyl orange solution (10 ppm) with different irradiation intervals over the ZrO2 samples.

UV-visible absorption changes of methyl orange aqueous solution at 25°C in the presence of the ZrO 2 samples.
As mentioned in the experimental section, the catalyst was equilibrated with the methyl orange solution to check for adsorption of the dye on the solid photocatalyst. The spectra depicted in Figure 7 were recorded after the equilibration of the photocatalyst. The UV-vis absorption spectra of methyl orange have a strong characteristic peak at 465 nm and a weak absorption peak at 274 nm.
These absorption peaks become weak and disappear along with the extension of reaction time. The UV-vis results indicate that methyl orange was degraded during the reaction. The decrease in the absorbance of the solution was due to the destruction of the homo- and hetero-polyaromatic rings present in the dye molecules. The m-ZrO2 sample was found to be the most effective catalyst in comparison with the t-ZrO2 and c-ZrO2 samples under identical optimized conditions (Figure 7). Complete degradation of adsorbed dye molecules was observed within 110 min for the m-ZrO2 sample, whereas 180 and 240 min were required for the complete the degradation of adsorbed dye molecules for the t-ZrO2 and c-ZrO2 samples under the similar conditions, respectively. The decolorization efficiency of the ZrO2 samples was calculated using Equation 2. Figure 8 showed degradation efficiency of methyl orange aqueous solution at 25°C in the presence of the three ZrO2 samples. The m-ZrO2 photocatalyst showed 99% degradation of methyl orange in 110 min of reaction; however, the t-ZrO2 and c-ZrO2 catalysts showed 90% and 80% degradation in the same reaction time, respectively.

Decolorization efficiency of methyl orange aqueous solution at 25°C in presence of ZrO 2 samples.
It was reported that the structures of nanoscale favor the movement or transfer of electrons and holes generated inside the crystals to the surface [53], which also enhances the photocatalytic activity. The photocatalytic activity of ZrO2 appears to be strongly dependent on the surface composition. Bachiller-Baeza et al. [54] reported that Lewis acid sites were more abundant on monoclinic ZrO2 than on tetragonal ZrO2, and the former brought about stronger surface adsorption sites concerning CO2 adsorption than the latter. Ma et al. [55] determined and compared the surface properties of ZrO2 polymorphs. It was found that ZrO2 polymorphs exhibited different surface hydroxyl and acid-base properties. These differences had great influence on the behavior of CO adsorption and reaction. They also showed that monoclinic ZrO2 possessed better adsorption properties than the other ZrO2 structures.
The adsorption capacity of methyl orange per gram of each catalyst was determined under identical conditions. It was observed that m-ZrO2 (1.95 mg g−1) possessed better adsorption capacity than the t-ZrO2 (1.05 mg g−1) and c-ZrO2 (0.65 mg g−1) samples. The photocatalytic activities observed in this study show similar trend that the m-ZrO2 sample showed high photocatalytic activity even though it possessed less surface area. ZrO2 nanomaterials have been investigated previously in the photocatalytic degradation of methyl orange; the time period required for the degradation of methyl orange tends to be 120 min or greater. Here, the synthesized pure monoclinic ZrO2 nanoparticles offered 99% of efficiency in 110 min of reaction time. The observed photocatalytic degradation activity is substantially higher than the activity reported for the ZrO2 samples in the literature.
It is known that usually high surface area of photocatalyst enhances dye adsorption and subsequent photocatalytic activity. However, it was also reported that the amount of dye adsorption on a catalyst also depends on the adsorption coefficient. Thus, a high adsorption coefficient on a low surface area material could lead to the same amount of adsorbed material per gram of catalyst as low adsorption coefficient on a high surface area material. This could be the reason why the rate (per gram basis) of methyl orange degradation is high for m-ZrO2 (low surface area) and c-ZrO2 (high surface area).
The m-ZrO2 sample possesses the smallest surface area (65 m2g−1) and largest pore size, while the c-ZrO2 has the largest surface area of 204 m2g−1 and smallest pore size. It is interesting that m-ZrO2 exhibited higher photcatalytic activity than c-ZrO2. Guo et al. [56] studied the influence of the pore structure of TiO2 on its photocatalytic performance. They observed that the photocatalytic activity of nanometer TiO2 is less than that of mesoporous TiO2.
Shao et al. [57] synthesized ZrO2-TiO2 composites which possessed different surface areas and textural properties. Composite samples showed surface area in the range of 270 to 80 m2g−1; however, the sample which has low surface area showed high photocatalytic activity due to it optimum Ti-Zr composition. It is clear from the literature reports that it is possible for catalyst, which possessed low surface area, to offer better photocatalytic activity than the catalyst which possessed high surface area. This is due to the fact that photocatalytic activity can be influenced by several other factors such as crytallinity, composition, particle size distribution, porosity, band gap, and surface hydroxyl density [58].
The process of photocatalytic degradation of methyl orange over ZrO2 catalysts can be described as follows. The first step involves adsorption of the dye onto the surface of ZrO2 nanostructure sample. Exposure of dye adsorbed ZrO2 nanostructures with UV light leads to generation of electron-hole (e−-h+) pairs in ZrO2 as indicated in Equation 5. The photogenerated electrons in the conduction band of ZrO2 interact with the oxygen molecules adsorbed on ZrO2 to form superoxide anion radicals (*O2 −) (Equation 6). The holes generated in the valence band of ZrO2 react with surface hydroxyl groups to produce highly reactive hydroxyl radicals (*OH) (Equation 7). These photogenerated holes can lead to dissociation of water molecules in the aqueous solution, producing radicals (Equation 8). The highly reactive hydroxyl radicals (*OH) and superoxide radicals (*O2 −) can react with methyl orange dye adsorbed on ZrO2 nanostructures and lead to its degradation as represented in Equations 9 and 10.
The processes leading to photocatalytic degradation of methyl orange and the mechanism over the mesoporous ZrO2 nanostructures were represented in Figure 9. It was reported that the enhanced photocatalytic activity of mesoporous structure to crystalline ZrO2 nanomaterial was due to the light harvesting capability, prolong the life of the photoinduced electron-hole pairs, and facilitate the reactant accessibility to surface active sites [11]. In the photocatalytic degradation process, the increase in photocatalytic activity is associated with efficient separation of photogenerated electrons and holes. If a surface defect state is able to trap electrons or holes, recombination can be restricted. The presence of oxygen-deficient ZrO0.35 impurity in the m-ZrO2 sample could also be responsible for the oxygen vacancies and they are acting as electron accepters to trap electrons and interstitial oxygen act as shallow trappers for holes, both of which prevent the recombination of photogenerated electrons and holes, thereby increasing the efficiency in the m-ZrO2 sample. In addition, the m-ZrO2 sample clearly possess the large pores, which can effectively facilitate both higher reactant accessibility to the surface active sites and more efficient multiple light scattering inside the pore channels [59].

Schematic representation of the processes leading to photocatalytic degradation.
It is also known that the photocatalytic redox reaction mainly takes place on the surface of the photocatalysts and so the surface properties significantly influence the efficiency of catalyst [60]. Additionally, the surface hydroxyl groups of ZrO2 are acidic to a certain degree, and the proportion of transformed azo structure increases into quinoid structure under acidic conditions (Figure 10). It is also reported that quinoid structure is more likely to be degraded than azo structure [61]. From the XRD patterns, it is clear that the m-ZrO2 sample has the highest crystallinity (sharpest peaks, largest crystals). This is also reflected in the Raman spectra. A higher crystallinity is usually believed to be beneficial for photocatalysis because of the amount of defect sites in the structure (which usually act as recombination centers). In comparison, the m-ZrO2 sample synthesized in this work possessed relatively high pore volume, pore size, and high density of hydroxyl groups.

Structural forms of methyl orange under acidic and basic conditions.
We also tested the reusability of the m-ZrO2 catalyst for subsequent cycles of methyl orange degradation under optimized reaction conditions. Many of the reported photocatalysts have not been used for further degradation studies due to the fact that they undergo photocorrosion, by the direct illumination with light, and hence their photostability is diminished for further runs. For the reusability study, we collected the white-colored catalyst remained after the reaction, washed, dried at 100°C for 30 min, and used it for further reactions. The catalyst was found to be active for 5 cycles without any major deactivation, and more than 95% degradation was achieved in all experiments within 110 min using the m-ZrO2 catalyst. The reusability of the m-ZrO2 nanoparticles was ascribed to the low photocorrosive effect and high catalytic stability of the synthesized m-ZrO2 sample.
Conclusions
Nanosize crystalline porous ZrO2 nanoparticles with pure monoclinic, tetragonal, and cubic phases were synthesized by different preparation methods. The photocatalytic performance of the three ZrO2 samples for the degradation of methyl orange was evaluated. Under the optimized reaction conditions, the m-ZrO2 sample comparatively showed a higher methyl orange degradation activity than the t-ZrO2 and c-ZrO2 samples. The pronounced photocatalytic activity for m-ZrO2 catalyst was mainly attributed to combining effects of factors including the presence of small amount of oxygen-deficient zirconium oxide phase, high crystallinity, broad pore size distribution, and high density of surface hydroxyl groups.
References
Roselin LS, Selvin R. Photocatalytic treatment and reusability of textile dyeing effluents from cotton dyeing industries. Sci Adv Mater. 2011;3:113–9.
Ameen S, Akhtar MS, Kim YS, Yang OB, Shin HS. Synthesis and characterization of novel poly(1-naphthylamine)/zinc oxide nanocomposites: application in catalytic degradation of methylene blue dye. Colloid Polym Sci. 2010;288:1633–8.
Neelakandeswari N, Sangami G, Dharmaraj N. Taek N K, Kim H Y: Spectroscopic investigations on the photodegradation of toluidine blue dye using cadmium sulphide nanoparticles prepared by a novel method. Spectrochim Acta, Part A. 2011;78:1592–8.
Hoffmann MR, Martin ST, Choi W, Bahnemann DW. Environmental applications of semiconductor photocatalysis. Chem Rev. 1995;95:69–96.
Kuriakose S, Satpati B, Mohapatra S. Enhanced photocatalytic activity of Co doped ZnO nanodisks and nanorods prepared by a facile wet chemical method. Phys Chem Chem Phys. 2014;16:12741–9.
Kuriakose S, Choudhary V, Satpati B, Mohapatra S. Facile synthesis of Ag-ZnO hybrid nanospindles for highly efficient photocatalytic degradation of methyl orange. Phys Chem Chem Phys. 2014;16:17560–8.
Cheng C, Amini A, Zhu C, Xu Z, Song H, Wang N. Enhanced photocatalytic performance of TiO2-ZnO hybrid nanostructures. Sci Reports. 2014;4:1481–6.
Ren L, Li Y, Hou J, Zhao X, Pan C. Preparation and enhanced photocatalytic activity of TiO2 nanocrystals with internal pores. ACS Appl Mater Interfaces. 2014;6:1608–15.
Navio JA, Hidalgo MC, Colon G, Botta SG, Litter MI. Preparation and physicochemical properties of ZrO2 and Fe/ZrO2 prepared by a sol-gel technique. Langmuir. 2001;17:202–10.
Botta SG, Navio JA, Hidalgo MC, Restrepo GM, Litter MI. Photocatalytic properties of ZrO2 and Fe/ZrO2 semiconductors prepared by a sol-gel technique. J Photochem Photobiol A Chem. 1999;129:89–99.
Sreethawong T, Ngamsinlapasathian S, Yoshikawa S. Synthesis of crystalline mesoporous-assembled ZrO2 nanoparticles via a facile surfactant-aided sol-gel process and their photocatalytic dye degradation activity. Chem Eng J. 2013;228:256–62.
Sohn JR, Ryu SG. Surface characterization of chromium oxide-zirconia catalyst. Langmuir. 1993;9:126–31.
Gao PT, Meng LJ, dos Santos MP, Teixeira V, Andritschky M. Study of ZrO2-Y2O3 films prepared by RF magnetron reactive sputtering. Thin Solid Films. 2000;377:32–6.
Ma ZY, Yang C, Wei W, Li WH, Su YH. Surface properties and CO adsorption on zirconia polymorphs. J Mol Catal A Chem. 2005;227:119–24.
Pokrovski K, Jung KT, Bell AT. Investigation of CO and CO2 adsorption on tetragonal and monoclinic zirconia. Langmuir. 2001;17:4297–303.
Nawale AB, Kanhe NS, Bhoraskar SV, Mathe VL, Das AK. Influence of crystalline phase and defects in the ZrO2 nanoparticles synthesized by thermal plasma route on its photocatalytic properties. Mater Res Bull. 2012;47:3432–9.
Zhao J, Wang X, Zhang L, Hou X, Li Y, Tang C. Degradation of methyl orange through synergistic effect of zirconia nanotubes and ultrasonic wave. J Hazar Mater. 2011;188:231–4.
Ismail S, Ahmad ZA, Berenov A, Lockman Z. Effect of applied voltage and fluoride ion content on the formation of zirconia nanotube arrays by anodic oxidation of zirconium. Corros Sci. 2011;53:1156–64.
Jiang W, He J, Zhong J, Lu J, Yuan S, Liang B. Preparation and photocatalytic performance of ZrO2 nanotubes fabricated with anodization process. Appl Sur Sci. 2014;307:407–13.
Shu Z, Jiao X, Chen D. Hydrothermal synthesis and selective photocatalytic properties of tetragonal star-like ZrO2 nanostructures. Cryst Eng Comm. 2013;15:4288–94.
Guo GY, Chen YL. A nearly pure monoclinic nanocrystalline zirconia. J Solid State Chem. 2005;178:1675–82.
Rezaei M, Alavi SM, Sahebdelfar S, Xinmei L, Yan ZF. Synthesis of mesoporous nanocrystalline zirconia with tetragonal crystallite phase by using ethylene diamine as precipitation agent. J Mater Sci. 2007;42:7086–92.
Tahir MN, Gorgishvili L, Li J. Facile synthesis and characterization of monocrystalline cubic ZrO2 nanoparticles. Solid State Sci. 2007;9:1105–9.
Calafat A. The influence of preparation conditions on the surface area and phase formation of zirconia. Stud Surf Sci Catal. 1998;118:837–43.
Srinivasan R, De Angelis RJ, Ice G, Davis BH. Identification of tetragonal and cubic structures of zirconia using synchrotron x-radiation source. J Mater Res. 1991;6:1287–92.
Abdala PM, Fantini MC, Craievich AF, Lamas DG. Crystallite size-dependent phases in nanocrystalline ZrO2-Sc2O3. Phys Chem Chem Phys. 2010;12:2822–9.
Mokhtar M, Basahel SN, Ali TT. Effect of synthesis methods for mesoporous zirconia on its structural and textural properties. J Mater Sci. 2013;48:2705–13.
Bersani D, Lottici PP, Rangel G, Ramos E, Pecchi G, Gomez R, et al. Micro-Raman study of indium doped zirconia obtained by sol-gel. J Non-Crystalline Solids. 2004;345–346:116–9.
Gazzoli D, Mattei G, Valigi M. Raman and X-ray investigations of the incorporation of Ca2+ and Cd2+ in the ZrO2 structure. J Raman Spectrosc. 2007;38:824–31.
Chervin CN, Clapsaddle BJ, Chiu HW, Gash AE, Satcher JH, Kauzlarich SM. Aerogel synthesis of yttria-stabilized zirconia by a non-alkoxide sol-gel route. Chem Mater. 2005;17:3345–51.
Kontoyannis CG, Orkoula M. Quantitative determination of the cubic, tetragonal and monoclinic phases in partially stabilized zirconias by Raman spectroscopy. J Mater Sci. 1994;29:5316–20.
Ray JC, Patil RK, Pramanik P. Chemical synthesis and structural characterization of nanocrystalline powders of pure zirconia and yttria stabilized zirconia (YSZ). J Eur Ceram Soc. 2000;20:1289–95.
Garvie RC. The occurrence of metastable tetragonal zirconia as a crystallite size effect. J Phys Chem. 1965;69:1238–43.
Zhao N, Pan D, Nie W, Ji X. Two-phase synthesis of shape-controlled colloidal zirconia nanocrystals and their characterization. J Am Chem Soc. 2006;128:10118–24.
Kasatkin I, Girgsdies F, Ressler T, Caruso RA, Schattka JH, Urban J, et al. HRTEM observation of the monoclinic-to-tetragonal (m-t) phase transition in nanocrystalline ZrO2. J Mater Sci. 2004;39:2151–7.
Inorganic Crystal Structure Database, FIZ Karlsruhe and the National Institute of Standards and Technology, Karlsruhe.2014, http://icsd.fiz-karlsruhe.de/search/index.xhtml. Accessed 10 Sept 2014.
Rouquerol F, Rouquerol J, Sing K. Adsorption by powders and porous solid: principle, methodology, and applications. San Diego: Academic; 1999.
McBain JW. An explanation of hysteresis in the hydration and dehydration of gels. J Am Chem Soc. 1935;57:699–700.
Basahel SN, Ali TT, Narasimharao K, Bagabas AA, Mokhtar M. Effect of iron oxide loading on the phase transformation and physicochemical properties of nanosized mesoporous ZrO2. Mater Res Bull. 2012;47:3463–72.
Wang W, Guo HT, Gao JP, Dong XH, Qin QX. XPS, UPS and ESR studies on the interfacial interaction in Ni-ZrO2 composite plating. J Mater Sci. 2000;35:1495–9.
Ardizzone S, Bianchi CL. XPS characterization of sulphated zirconia catalysts: the role of iron. Surf Inter Anal. 2000;30:77–80.
Dongare MK, Dongare AM, Tare VB, Kemniz E. Synthesis and characterization of copper-stabilized zirconia as an anode material for SOFC. Solid State Ionics. 2002;455:152–6.
Kawasaki KA. Positions of photoelectron and auger lines on the binding energy scale. Japan: XPS International; 1997. p. 7.
Ram S, Mondal A. X-ray photoelectron spectroscopic studies of Al3+ stabilized t-ZrO2 of nanoparticles. Appl Sur Sci. 2004;221:237–47.
Gredelj S, Gerson AR, Kumar S, Cavallaro GP. Characterization of aluminium surfaces with and without plasma nitriding by X-ray photoelectron spectroscopy. Appl Surf Sci. 2001;174:240–50.
Ardizzone S, Cattania MG, Lazzari P, Sarti M. Bulk, surface and double layer properties of zirconia polymorphs subjected to mechanical treatments. Mater Chem Phys. 1991;28:399–412.
Rashad MM, Baioumy HM. Effect of thermal treatment on the crystal structure and morphology of zirconia nanopowders produced by three different routes. J Mater Process Tech. 2008;195:178–85.
Herrera G, Montoya N, Domenech-Carbo A, Alarcon J. Synthesis, characterization and electrochemical properties of iron-zirconia solid solution nanoparticles prepared using a sol-gel technique. Phys Chem Chem Phys. 2013;15:19312–21.
Li N, Dong B, Yuan W, Gao Y, Zheng L, Huang Y, et al. ZrO2 nanoparticles synthesized using ionic liquid microemulsion. J Dispersion Sci Technol. 2007;28:1030–3.
French RH, Glass SJ, Ohuchi FS, Xu YN, Ching WY. Experimental and theoretical determination of the electronic structure and optical properties of three phases of ZrO2. Phys Rev B Condens Matter. 1994;49:5133–42.
Emeline V, Kuzmin GN, Purevdorj D, Ryabchuk VK, Serpone N. Spectral dependencies of the quantum yield of photochemical processes on the surface of wide band gap solids. 3. Gas/solid systems. J Phys Chem B. 2000;104:2989–99.
Chang SM, Doong RA. Inter band transitions in sol-gel-derived ZrO2 films under different calcination conditions. Chem Mater. 2007;19:4804–10.
Wang WW, Zhu YJ, Yang LX. ZnO-SnO2 hollow spheres and hierarchical nanosheets: hydrothermal preparation, formation mechanism, and photocatalytic properties. Adv Funct Mater. 2007;17:59–64.
Bachiller-Baeza B, Rodriguez-Ramos I, Guerrero-Ruiz A. Interaction of carbon dioxide with the surface of zirconia polymorphs. Langmuir. 1998;14:3556–64.
Ma Z-Y, Yang C, Wei W, Li W-H, Sun Y-H. Surface properties and CO adsorption on zirconia polymorphs. J Mol Catal A Chem. 2005;227:119–24.
Guo B, Shen H, Shu K, Zeng Y, Ning W. The study of the relationship between pore structure and photocatalysis of mesoporous TiO2. J Chem Sci. 2009;121:317–21.
Shao GN, Imran SM, Jeon SJ, Engole M, Abbas N, Haider MS, et al. Sol-gel synthesis of photoactive zirconia-titania from metal salts and investigation of their photocatalytic properties in the photodegradation of methylene blue. Powder Technol. 2014;258:99–109.
Ahmed S, Rasul MG, Brown R, Hashi MA. Influence of parameters on the heterogeneous photocatalytic degradation of pesticides and phenolic contaminants in wastewater: a short review. J Environ Manage. 2011;92:311–30.
Jantawasu P, Sreethawong T, Chavadej S. Photocatalytic activity of nanocrystalline mesoporous-assembled TiO2 photocatalyst for degradation of methyl orange monoazo dye in aqueous wastewater. Chem Eng J. 2009;155:223–33.
Xu C, Mei L. Synthesis and enhanced photocatalytic activity of hierarchical ZnO nanostructures. J Nanosci Nanotech. 2013;13:513–6.
Rhodes MD, Bell AT. The effects of zirconia morphology on methanol synthesis from CO and H2 over Cu/ZrO2 catalysts Part I. Steady-state studies J Catal. 2005;233:198–209.
Acknowledgements
This project was funded by Saudi Basic Industries Corporation (SABIC) and the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant number MS/14-325-1433. The authors therefore acknowledge with thanks SABIC and DSR technical and financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors have contributed to the final manuscript of the present investigation. SB and KN have defined the research topic. TA, KN, and MM involved in the preparation, the characterization, and photocatalytic experiments. KN, TA, and MM wrote the manuscript. SB provided important suggestions on the draft of the manuscript. All authors examined and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Basahel, S.N., Ali, T.T., Mokhtar, M. et al. Influence of crystal structure of nanosized ZrO2 on photocatalytic degradation of methyl orange. Nanoscale Res Lett 10, 73 (2015). https://doi.org/10.1186/s11671-015-0780-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11671-015-0780-z