

Abstract
Nicotinic acetylcholine receptors (nAChRs) are expressed widely in the CNS, and mediate both synaptic and perisynaptic activities of endogenous cholinergic inputs and pharmacological actions of exogenous compounds (e.g., nicotine and choline). Behavioral studies indicate that nicotine improves such cognitive functions as learning and memory. However, the mechanism of nicotine's action on cognitive function remains elusive. We performed patch-clamp recordings from hippocampal CA3 pyramidal neurons to determine the effect of nicotine on mossy fiber glutamatergic synaptic transmission. We found that nicotine in combination with NS1738, an α7 nAChR-positive allosteric modulator, strongly potentiated the amplitude of evoked EPSCs (eEPSCs), and reduced the EPSC paired-pulse ratio. The action of nicotine and NS1738 was mimicked by PNU-282987 (an α7 nAChR agonist), and was absent in α7 nAChR knock-out mice. These data indicate that activation of α7 nAChRs was both necessary and sufficient to enhance the amplitude of eEPSCs. BAPTA applied postsynaptically failed to block the action of nicotine and NS1738, suggesting again a presynaptic action of the α7 nAChRs. We also observed α7 nAChR-mediated calcium rises at mossy fiber giant terminals, indicating the presence of functional α7 nAChRs at presynaptic terminals. Furthermore, the addition of PNU-282987 enhanced action potential-dependent calcium transient at these terminals. Last, the potentiating effect of PNU-282987 on eEPSCs was abolished by inhibition of protein kinase A (PKA). Our findings indicate that activation of α7 nAChRs at presynaptic sites, via a mechanism involving PKA, plays a critical role in enhancing synaptic efficiency of hippocampal mossy fiber transmission.
Introduction
Nicotine, the main psychoactive compound in tobacco smoking, has been shown to enhance cognitive function in both humans and animals (Newhouse et al., 2004; Levin et al., 2006). Nicotine exerts its effects through the activation of nicotinic acetylcholine receptors (nAChRs), a family of cys-loop cationic receptor channels. In the mammalian brain, there are six α (2–7) and three β (2–4) nAChR subunits, which have been shown to be expressed in various cell types (Nashmi and Lester, 2006). In neurons, these subunits have been found at presynaptic and postsynaptic compartments (Fabian-Fine et al., 2001) as well as perisynaptic sites (Bürli et al., 2010). In the hippocampus, the most prevalent nAChRs are comprised of α7 and α4β2 subtypes (Jones et al., 1999; Albuquerque et al., 2009). Physiological studies have shown that α4β2 nAChRs have higher affinity for acetylcholine and slower desensitization rates, but less calcium permeability than α7 receptors (Albuquerque et al., 2009).
The granule cells in the dentate gyrus (DG) encode inputs from the entorhinal cortex, and relay the information to CA3 neurons by action potential (AP)-dependent synaptic transmission using their long mossy fiber axons (Nicoll and Schmitz, 2005). These mossy fiber axons form giant synapses (∼5 μm) onto the proximal dendrites of CA3 pyramidal neurons, and these synapses show low basal release probability and high-frequency-dependent plasticity (Nicoll and Schmitz, 2005; Bischofberger et al., 2006). Alterations in the synaptic efficacy of DG-CA3 mossy fiber synapses would either constrain or amplify this neuronal output. In fact, DG-CA3 mossy fiber synapses have been proposed to be the main gain control in the hippocampal circuit (Lee et al., 2013), and also reported to be the sites of neuromodulatory effects of GABA and kainate (Contractor et al., 2001; Schmitz et al., 2001; Ruiz et al., 2003). However, it is still unclear how nicotine and acetylcholine (through activation of nAChRs) regulate AP-dependent synaptic transmission at these synapses. Thus, the detailed mechanism of nicotine's action in this pathway remains elusive.
Indirect evidence suggests that nicotine acts, in part, at presynaptic terminals. Acute application of nicotine increased the frequency of miniature EPSCs in hippocampal neurons (Gray et al., 1996; Sharma and Vijayaraghavan, 2003; Sharma et al., 2008) and enhanced glutamate release from synaptosomes extracted from hippocampal mossy fibers (Bancila et al., 2009). However, the presence of nAChRs at mossy fiber terminals remains a matter of debate (Vogt and Regehr, 2001), and the effect of nicotine on mossy fiber glutamatergic transmission has not yet been studied directly. Therefore, we examined the modulation of nicotine on mossy fiber-evoked EPSCs (eEPSCs). We found that nicotine, via activation of the α7 nAChRs and protein kinase A (PKA), potentiated the amplitude of eEPSCs. In addition, we directly monitored α7 receptor-mediated calcium increases in mossy fiber terminals using GCaMP3 (Tian et al., 2009). We conclude that nicotine activates α7 nAChRs at mossy fiber terminals to increase glutamate release onto CA3 pyramidal neurons.
Materials and Methods
Hippocampal slice preparation.
All animal procedures were conducted in accordance with National Institutes of Health animal welfare guidelines. Wild-type C57BL/6 male mice were purchased from Charles River, and α7 nAChR knock-out mice of either sex were purchased from The Jackson Laboratory. For acute slice preparations, mice (17–24 d old) were anesthetized with isoflurane and decapitated. Brains were quickly removed and placed into ice-cold cutting solution containing the following (in mm): 110 sucrose, 60 NaCl, 3 KCl, 7 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 28 NaHCO3, 1 ascorbate, 3 Na pyruvate, and 5 glucose, and saturated with oxygen (95%O2/5%CO2). Transverse hippocampal slices (300 μm) were cut on a VT1000 vibratome (Leica Biosystem). Slices were collected into oxygenated artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 3.5 KCl, 1.3 MgCl2, 2 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, and 10 glucose. After recovery at 35°C for 30 min, the slices were kept at room temperature before recording. The age range of animals was chosen based on mossy fiber synapse developmental profile (Amaral and Dent, 1981). For organotypic hippocampal slice cultures, brains were quickly removed from 8–10 d old mice into ice-cold cutting solution composed of minimum essential media supplemented with the following (in mm): 25 HEPES, 10 Tris base, 10 glucose, and 3 MgCl2, pH 7.2. Transverse hippocampal slices (350 μm) were placed onto the Transwell membrane inserts (Corning). The slice culture medium contained the following (in mm): a 2:1 mixture of basal medium Eagle and Earle's balanced salts solution, 20 NaCl, 5 NaHCO3, 0.2 CaCl2, 1.7 MgSO4, 48 glucose, 26.7 HEPES, 10 ml/L penicillin-streptomycin (Invitrogen), 1.32 mg/L insulin, and 5% horse serum (Invitrogen), pH 7.2. The slices were stored in a CO2 incubator at 34°C and fed twice a week with a half-change of media.
Electrophysiological recordings.
Data were collected using an Axopatch 200B amplifier and pClamp 10.2 software (Molecular Devices). Recording electrodes with resistances of 4–6 MΩ were filled with the following (in mm): 135 Cs gluconate, 2 EGTA, 0.2 MgCl2, 8 NaCl, 4 ATPMg, 0.3 GTPNa2, 10 HEPES, 5 phosphocreatine, and 5 QX314, pH 7.3. A bipolar tungsten-stimulating electrode was placed in the granule cell layer near the midpoint of the suprapyramidal blade of the DG. Synaptic events were evoked by a stimulus pulse (0.2 ms, 0.1 Hz) with an S88X stimulator (Grass Astro-Med). Whole-cell patch-clamp recordings were made at 30−32°C from CA3 pyramidal cells, which are identified by their morphology and position within the CA3b subfield via infra-red differential interference contrast microscopy. All cells were held at −70 mV, which is the experimentally tested reversal potential of GABAA current. The evoked synaptic currents were completely blocked by 10 μm CNQX, suggesting that they were predominantly mediated by AMPA receptors (Torborg et al., 2010). Mossy fiber EPSCs were selected based on the following criteria: paired-pulse ratio (PPR) >1.8 at a 50 ms interval and >70% EPSC inhibition with the Group II metabotropic glutamate receptor agonist 2-(2,3-dicarboxycyclopropy) glycine (DCG-IV; 2 μm; Kamiya et al., 1996). Miniature EPSCs (mEPSCs) were recorded in the presence of tetrodotoxin (TTX; 0.5 μm) and gabazine (10 μm). In some experiments, a picospritzer (General Valve) was used to apply nicotine rapidly and directly. Nicotine (10 μm) was pressure applied from the tip of a glass pipette (1–2 MΩ) at 10 psi for 10 min, which was placed 50 μm away from the proximal dendrites of recorded CA3 neurons. QX314, CNQX, DCG-IV, NS1738, KT5720, PKI 14–22 amide peptide, KN62, PNU-282987, and PNU-120596 were purchased from Tocris Bioscience. All other compounds were purchased from Sigma unless stated otherwise.
Data analysis.
Electrophysiological data were analyzed with Clampfit 10.2 (Molecular Devices), and OriginPro 8.5 (OriginLab). eEPSC peak amplitude was measured every 10 s, and three consecutive measurements were averaged to produce a mean response every 30 s. The mean baseline EPSC amplitude was obtained via averaging 5 min before the drug application, which was used to normalize the EPSC amplitude during nicotine application. Nicotine-induced changes were calculated by averaging the normalized amplitude of EPSCs during the 10 min drug application. The effect of different chemicals was assessed by comparing the mean normalized EPSC amplitudes; experiments were discarded if the baseline EPSC peak amplitude was unstable (>20% change during the first 15 min of baseline recording). The mEPSCs were analyzed with MiniAnalysis (Synaptosoft). Data are plotted as the mean ± SEM. Statistical tests were performed with unpaired or paired (for the same cell) Student's t tests.
Virus preparation and application.
Individual genes of interest were subcloned into an adeno-associated virus (AAV) vector under a synapsin promoter for neuronal expression. The following plasmids were obtained from Addgene: AAV vector with the synapsin promoter (Addgene plasmid no. 26972), GCaMP3 (Addgene plasmid no. 22692), and R-GECO1 (Addgene plasmid no. 32444). All viruses were packaged with the serotype 9 helper factors by the viral core facility at National Institute of Environmental Health Sciences. Viruses (5 nl) were applied by microinjection to either the DG or CA3 region of slices using a Drummond Nanoject (Drummond Scientific) 2 h after slicing. Experiments were performed 10 d after virus infection.
Calcium imaging.
Calcium imaging was performed with a Zeiss LSM 510 NLO META system coupled to an Axioskop 2FS microscope (Carl Zeiss). An Argon ion laser (488 nm) with long-pass (500 nm) and bandpass filters (500–550 nm) was used for GCaMP3 detection, and a Krypton laser (546 nm) with long-pass (560 nm) and bandpass filters (560–620 nm) was used for R-GECO1. Cultured hippocampal slices were imaged 10 d after viral infection. We placed a bipolar tungsten-stimulating electrode in the area of DG expressing GCaMP3 to evoke APs with an Isostim A320 stimulator (WPI). The mossy fiber terminals were imaged in the CA3 region. Zeiss Zen 2010 software was used for image acquisition and analysis, and terminals >3 μm in diameter were selected for our analysis. Mean intensity values of regions of interest were plotted after background subtraction. Only those experiments in which two 10 Hz train (5 s) stimulations separated by a 10 min interval yielded peak calcium rises within 10% of each others were used.
Results
Nicotine potentiates mossy fiber EPSCs via activation of the α7 nAChR
Although the application of nicotine has been shown to enhance AP-independent spontaneous synaptic transmission from CA3 pyramidal neurons (Gray et al., 1996; Sharma et al., 2008), the recordings could not discern among different glutamatergic inputs. CA3 pyramidal neurons receive at least three different glutamate inputs: mossy fiber axons to their proximal dendrites, extensive recurrent connections from axons of other CA3 neurons, and axons from the entorhinal cortex to their distal dendrites. To isolate the effect of nicotine in regulating hippocampal mossy fiber synaptic transmission, we examined its effect on eEPSCs in CA3 neurons using acute hippocampal slices. EPSCs were evoked at 0.1 Hz before, during, and after a 10 min bath application of nicotine (10 μm). The amplitudes of these eEPSCs were averaged over 10 min of the nicotine treatment for comparison with the mean amplitude before nicotine application. Nicotine alone failed to induce any significant change in eEPSC amplitude (−1.3 ± 6%, n = 5, p > 0.10; Fig. 1B, solid circle). We also tested the effect of nicotine applied rapidly and locally via pressure ejection. The puff pipette was placed at the proximal dendrite of CA3 neurons. We observed nicotine-induced inward currents ranging from 400 to 2600 pA from 10 of 15 recorded CA3 neurons, which often were accompanied by a burst of spontaneous EPSCs. These currents interfered with the accurate measurement of the eEPSC amplitude. For the other five cells that lacked a nicotine-induced response, we found that the pressure-applied nicotine showed a trend of increasing the mean normalized amplitude of the eEPSC (change of normalized EPSC 11.2 ± 18.5%, n = 5, p > 0.10), but which was not significant; this was similar to the effect of the bath application of nicotine alone. Because the ineffectiveness of nicotine could be due to the fast desensitization of the nAChR receptors (in particular the α7 subtype), we tested whether nicotine in combination with NS1738, a positive allosteric modulator (PAM) of the α7 nAChRs, may have any effect on eEPSCs. When applied alone, NS1738 did not change the amplitude of eEPSCs or evoke a detectable current (Timmermann et al., 2007). Interestingly, the coapplication of NS1738 (5 μm) and nicotine (10 μm) induced a significant increase in eEPSC amplitude of 83.6 ± 39% compared with baseline (n = 7, p < 0.05; Fig. 1B, open circle). The amplitude increase peaked after 2 min and remained elevated for the length of the nicotine application and after the washout of nicotine and NS1738. This result suggests that nicotine induces a pronounced effect on mossy fiber transmission through α7 nAChR activation.

Nicotine enhanced the amplitude of mossy fiber EPSCs with the addition of an α7 nAChR PAM. A, Representative traces of eEPSCs recorded from a CA3 pyramidal neuron before (left), during (middle), and after (right) application of nicotine (10 μm) and the α7 nAChR PAM NS1738 (5 μm). B, Normalized EPSC amplitude was plotted against the time of nicotine application (from 0 to 10 min) with (○) and without (●) NS1738 (5 μm). C, Normalized EPSC amplitude was plotted against the time of nicotine and NS1738 application (from 0 to 10 min) with (▴) and without (○) the presence of MLA (20 nm), an α7 nAChR-selective antagonist. D, The histogram shows nicotine-induced net change of normalized EPSC amplitude in listed conditions. Data shown are mean ± SEM; statistical significance was determined by Student's t test (*p < 0.05).
Next, to confirm that the effect was mediated by α7 nAChR, we tested whether the effect of nicotine could be blocked by antagonists of the two main subtypes of nAChRs that are predominantly expressed in the hippocampus, the α7 and α4β2 receptors. In the presence of the α7 nAChR antagonist MLA (20 nm), coapplication of nicotine and NS1738 failed to increase the peak amplitude of the eEPSCs (−8.2 ± 12%, n = 7, p < 0.05; Fig. 1C, filled triangle), demonstrating that the activation of the α7 nAChR is required for this enhancement. Interestingly in the presence of the non-α7 nAChR antagonist DHβE (1 μm), this enhancement was not significantly affected (data not shown). To further test for the involvement of the α7 nAChR in the enhancement of DG-CA3 transmission, we used the selective α7 nAChR agonist PNU-282987 (Bodnar et al., 2005). The application of PNU-282987 (0.5 μm) alone produced a similar enhancement of the eEPSC amplitude (101 ± 32%, n = 6, p > 0.10; Fig. 2A, filled squares; B) as nicotine and NS1738 coapplication, indicating that the activation of the α7 nAChR alone is sufficient to enhance this transmission. In slices from α7 nAChR knock-out mice, the application of PNU-282987 failed to enhance the eEPSC amplitude (−4.6 ± 9%, n = 5, p < 0.05; Fig. 2C, open squares; D), further confirming the role of the α7 receptor in this enhancement.
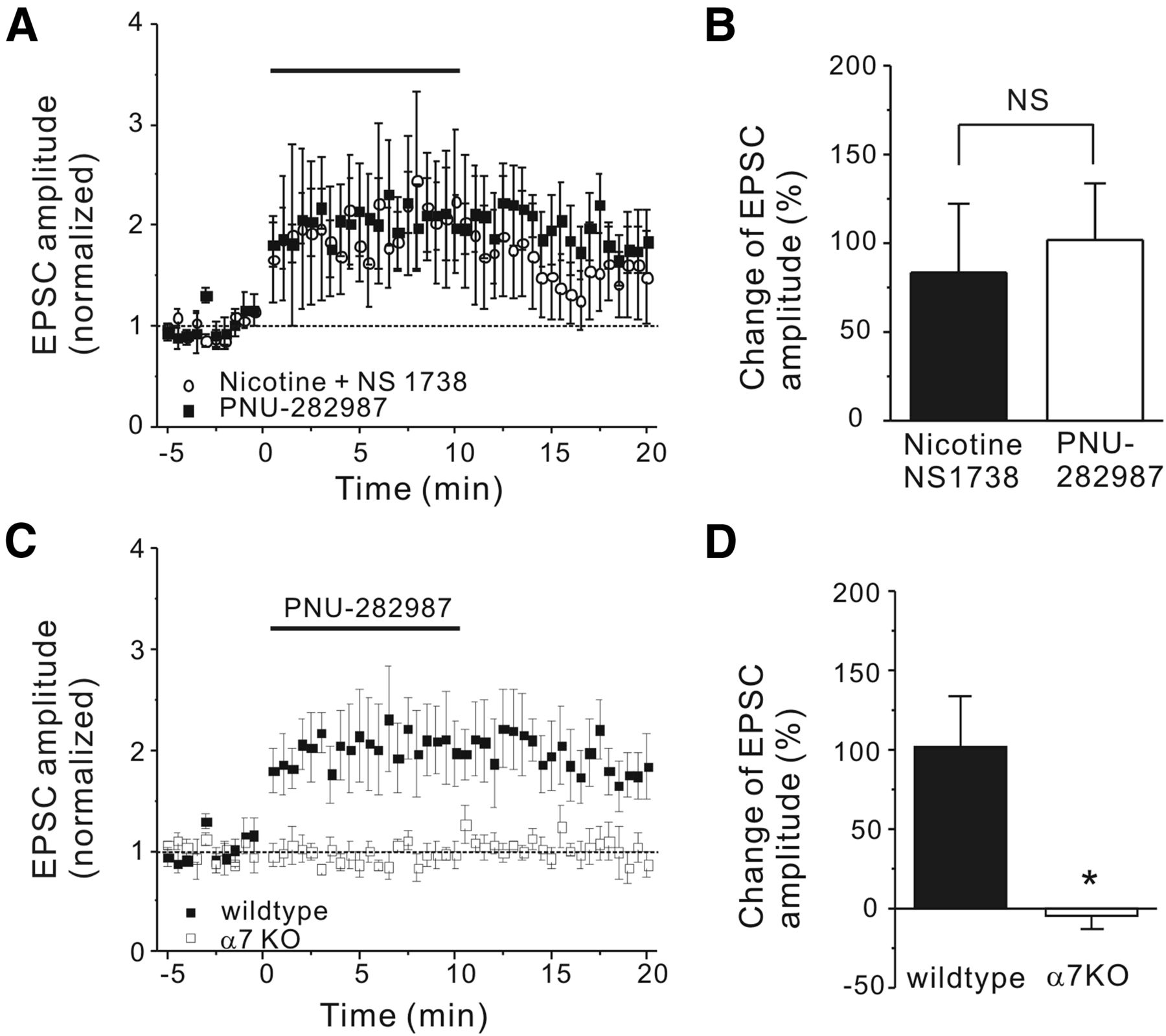
The α7-selective nAChR agonist PNU-282987 mimicked the action of nicotine and an α7 nAChR PAM in wild-type, but not α7 nAChR knock-out, neurons. A, Normalized EPSC amplitude was plotted against the application time (from 0 to 10 min) of PNU-282987 (0.5 μm, ■) and nicotine and NS1738 (○). B, The histogram shows the change of the normalized EPSC amplitude in listed conditions. C, Normalized EPSC amplitude was plotted against the application time of PNU-282987 (from 0 to 10 min) for wild-type (■) and α7 nAChR knock-out (□) neurons. D, The histogram shows that the PNU-282987-induced change of normalized EPSC amplitude is absent in α7 nAChR knock-out neurons. Data shown are mean ± SEM; statistical significance was determined by Student's t test (*p < 0.05; NS, p > 0.05).
Presynaptic locus of nicotine's action
Several previous reports in the hippocampus have shown that the α7 receptors are located both somatodendritically (postsynaptic) and presynaptically to regulate neurotransmitter release, particularly glutamate (Jones et al., 1999; Tu et al., 2009). In addition, immunohistochemistry has revealed a diffuse pattern of α7 nAChR-like immunoreactivity throughout cell bodies and processes of neurons in the DG, and CA3 and CA1 regions (Fabian-Fine et al., 2001). To investigate whether the activation of the α7 nAChR was enhancing the eEPSC amplitude through presynaptic mechanisms at the DG-CA3 synapses, we measured the PPR to probe the changes in presynaptic neurotransmitter release probability. PPR is calculated from the amplitude of EPSCs evoked by two stimuli applied at 50 ms intervals, 10 min before and after application of both nicotine and NS1738. The PPR was significantly reduced in the CA3 pyramidal neurons after application of nicotine and NS1738 (from 2.9 ± 0.3 to 1.5 ± 0.3, n = 8, paired Student's t test, p < 0.05; Fig. 3A–C), suggesting that nicotine and the α7 PAM increased glutamate release probability, and therefore most likely are acting through presynaptic α7 nAChRs. In addition, deletion of the α7 nAChRs abolished the effect of nicotine and NS1738 on PPR; the PPR remained unchanged in CA3 pyramidal neurons from α7 nAChR knock-out mice (3.9 ± 0.8 in ACSF and 4.0 ± 1.0 in nicotine and NS1738, n = 10, paired Student's t test, p = 0.68; Fig. 3D). In the absence of NS1738, while nicotine reduced the PPR ratio (from 2.1 ± 0.2 to 1.6 ± 0.2, n = 8, paired Student's t test, p = 0.15; data not shown), the effect was not significant. All of these data support the notion that activation of α7 nAChRs was critical for nicotine's action.

Presynaptic nAChRs mediated nicotine's action on mossy fiber EPSC amplitude. A, Representative traces of paired EPSCs evoked with 50 ms interval were shown before and during application of nicotine and the α7 nAChR PAM NS1738. B, PPRs of EPSCs were plotted for each individual experiment. Summary of data showed a significant reduction in PPR for wild-type mice (C), but not α7 nAChR knock-out mice (D) after 10 min application of nicotine and the α7 nAChR PAM. E, Normalized EPSC amplitude was plotted against the application time of nicotine and α7 nAChR PAM (from 0 to 10 min) with standard (○) and BAPTA (10 mm)-containing internal solution (♦). F, The histogram indicates that the nicotine and the α7 nAChR PAM-induced net change of normalized EPSC amplitude remains the same after postsynaptic dialysis of BAPTA (10 mm). Data shown are mean ± SEM; statistical significance was determined by Student's t test (*p < 0.05; NS, p > 0.05).
To further differentiate the synaptic location of nicotine's action, we measured the change of frequency and amplitude of spontaneous mEPSCs. In the presence of nicotine and NS1738, the frequency of mEPSCs recorded from CA3 pyramidal neurons increased from 1.2 ± 0.2 to 2.1 ± 0.8 Hz (n = 10, paired Student's t test, p < 0.05; data not shown), while the amplitude of these mEPSCs remained unchanged (from 29.6 ± 3.2 to 29.2 ± 3.3 pA, n = 10, paired Student's t test, p = 0.75; data not shown). This suggests that postsynaptic modification does not contribute much to the nicotine and α7 PAM increased glutamate release.
Because α7 nAChRs are significantly more permeable to calcium than α4β2 receptors (Bertrand et al., 1993; Séguéla et al., 1993; Fayuk and Yakel, 2004; Fucile, 2004), it is possible that intracellular or intraterminal calcium increases due to α7 nAChR activation may be responsible for the increased eEPSC amplitude. We evaluated the potential role of postsynaptic calcium by dialyzing CA3 pyramidal neurons with the calcium chelator BAPTA (10 mm) through the recording electrodes; this did not block the enhancement of the eEPSCs induced by nicotine and NS1738 (131.5 ± 46.8%, n = 6, p > 0.10; Fig. 3E, filled diamonds; F).
Functional α7 nAChR at mossy fiber terminals
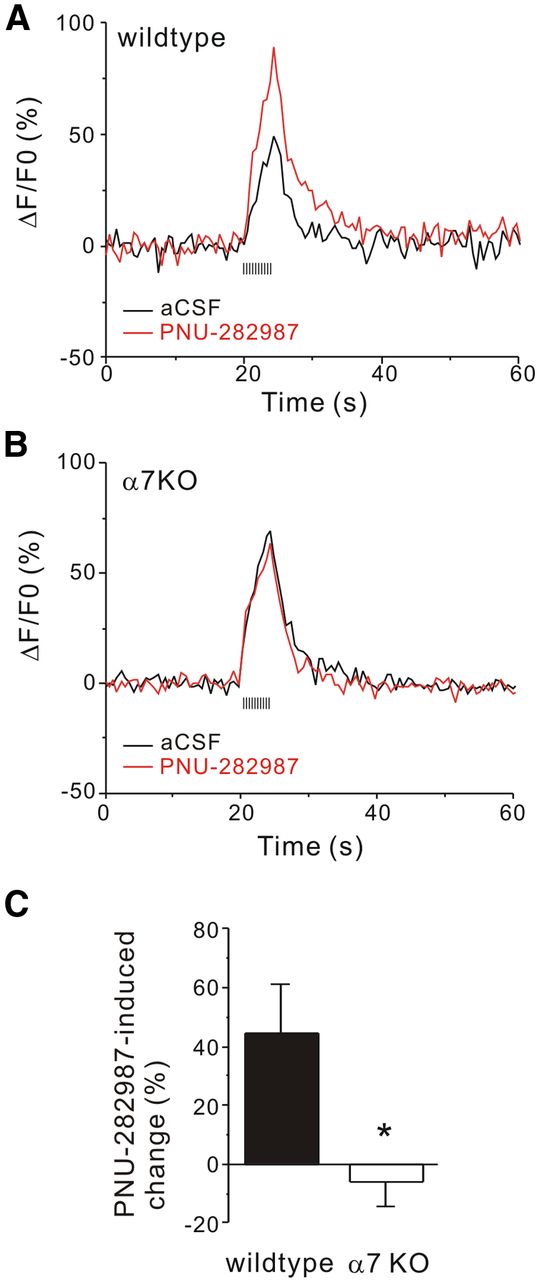
Our data have shown that the effect of nicotine is dependent on α7 nAChR activation, likely at presynaptic sites. However, the presence of α7 nAChRs on mossy fiber terminals is still controversial (Gray et al., 1996; Vogt and Regehr, 2001). To test this hypothesis, we used optical recordings to determine the location of the α7 nAChR-mediated responses in the hippocampal mossy fiber pathway via a genetically encoded calcium indicator, GCaMP3, in cultured hippocampal slices. The GCaMP3 gene was expressed under a synapsin promoter for neuronal specificity, and the GCaMP3-AAV virus was injected locally into the DG granule cell layer to limit the expression of GCaMP3 to DG granule cells and their mossy fiber terminals. We also infected CA3 neurons (by direct injection into the CA3 region) with the red genetically encoded calcium indicator R-GECO1 (also under a synapsin promoter in the AAV virus; Zhao et al., 2011; Fig. 4A). GCaMP3 expression was observed mainly in DG granule cells, which send axons out of the hilar region to the CA3 stratum lucidum, also visible in our preparation. Under higher magnification (Fig. 4A, right), we could identify the green mossy fiber terminals based on their size (>3 μm in diameter) and location (near the proximal dendrites of CA3 pyramidal neurons). The calcium imaging experiments were performed in the presence of CNQX and APV (to block glutamate channels), and TTX (to block voltage-gated sodium channels). To reduce desensitization of α7 nAChRs and to maximize the calcium signal, choline (an α7-selective agonist; 10 mm) was applied with PNU-120596, an α7 nAChR PAM (10 μm; Hurst et al., 2005). GCaMP3 fluorescence signals were sampled every 6 s for 10 min before and after the application of choline and PNU-120596. As shown in Figure 4B, after the bath application of choline and PNU-120596, there was a prominent increase in GCaMP3 fluorescence (indicative of an intracellular calcium rise) in the mossy fiber terminals; this calcium rise peaked quickly and returned to baseline levels within 2 min, most likely due to the desensitization of the α7 nAChRs. Furthermore, this calcium rise was absent in slices from α7 knock-out mice (30.7 ± 4.3% for wild-type terminals, n = 10; 4.0 ± 1.9% for α7 knock-out terminals, n = 8; Fig. 4B,C), indicating that the calcium rise was dependent on the activation of the α7 nAChRs. These data indicate that mossy fiber terminals contain functional α7 nAChRs that when activated, induce a rise in terminal calcium concentration.

Activation of α7 nAChR-evoked intracellular calcium rise at mossy fiber terminals of wild-type neurons. A, Representative images show a cultured hippocampal slice expressing GCaMP3 (green) at dentate and R-GECO1 (red) at CA3 region. Scale bars: Left, 100 μm; right, 5 μm. The white arrowheads (right) point to the mossy fiber terminals (green). B, The averaged measurement of fluorescence changes of mossy fiber terminals was plotted against time of the bath application of choline (10 mm) and PNU-120596 (5 μm; from 0 to 10 min). Measurements were done for both wild-type (green) and α7 nAChR knock-out (black) slices. The dashed line indicates the duration of choline and PNU-120596 perfusion. To prevent movement during imaging, we perfused the bath at a slow rate, which resulted in the delay of the response initiation. C, The summary data showed that coapplication of choline and PNU-120596-evoked calcium rises in wild-type, but not α7 nAChR knock-out, mossy fiber terminals (*p < 0.05, Student's t test).
AP-dependent calcium rise is enhanced by α7 nAChR activation
Based on our findings of paired-pulse experiments, we hypothesize that activation of the α7 nAChR on glutamatergic mossy fiber terminals enhanced release probability, which is in close relationship with intracellular calcium levels (Neher and Sakaba, 2008). Next, we asked whether activation of these α7 nAChRs modulated the AP-dependent calcium rise in these terminals. To do this, we examined the calcium rise evoked by APs at these terminals before and after 10 min application of the α7 nAChR agonist PNU-282987 in the presence of CNQX and APV. The stimulation electrode was placed at the granule cell layer of the DG, and mossy fiber terminals were imaged in the stratum lucidum of the CA3 region. To maximize the calcium signals, 50 APs were evoked at 10 Hz frequency, and we monitored the fluorescence change of the GCaMP3 signals every 0.5 s before, during, and after stimulation. The train of APs evoked a robust calcium rise, which peaked at the end of the stimulation train and returned to baseline within 20 s. Bath application of PNU-282987 (0.5 μm) did not change the basal calcium level. However, in the presence of PNU-282987, the peak of the AP-induced calcium rise was significantly increased (by 44.7 ± 17%, n = 10 experiments, each experiment contains 8–12 terminals) without a change in the kinetics (Fig. 5A). In slices from α7 nAChR knock-out mice, PNU-282987 had no effect on the AP-induced calcium transients (−5.8 ± 9%, n = 8 experiments, each experiment contains 8–12 terminals; Fig. 5B,C). This suggests that the enhancement of the AP-induced calcium rise by PNU-282987 requires the activation of the α7 nAChR, and provides a mechanism for the enhanced glutamate release by activation of α7 nAChRs at the mossy fiber terminals.

Application of α7 nAChR agonist potentiated AP-evoked intracellular calcium rise at mossy fiber terminals. The representative recordings illustrate intracellular calcium transients evoked by 10 Hz stimulation trains before (black line) and 10 min after the bath application of PNU-282987 (red line) in wild-type (A) and α7 nAChR knock-out (B) terminals. The dotted line denoted the timing of 10 Hz train stimulation for 5 s. C, Summary data shows the averaged net change induced by PNU-282987 on AP-dependent calcium peak between wild-type and α7 nAChR knock-out terminals (*p < 0.05, Student's t test).
PKA activity is required for α7 nAChR-mediated modulation
The increase in calcium levels at the mossy fiber terminals could activate a variety of calcium-dependent kinases such as CaMKII or PKA through calmodulin and calcium-activated adenylyl cyclases (Xia and Storm, 2005). Our data showed that the α7 nAChR-mediated potentiation of eEPSC amplitude persisted for 5–10 min after the removal of the agonist, suggesting that calcium-dependent signaling molecules might contribute to this prolonged action. Because PKA has frequently been shown to regulate both short- and long-term plasticity at mossy fiber terminals (Weisskopf et al., 1994; Weisskopf and Nicoll, 1995), we tested whether PKA activity is required for the α7 nAChR-mediated modulation by either bath application of its cell-permeable blocker KT5720 (200 nm), or by dialyzing neurons with the PKA inhibitory peptide (PKI; 100 nm) through the recording electrode. We found that the α7 nAChR agonist PNU-282987 failed to enhance the amplitude of eEPSCs in the presence of KT5720 (−3.03 ± 5.6%, n = 6, p < 0.05; Fig. 6A,B). Interestingly, while the inhibition of PKA postsynaptically with PKI dialysis appeared to reduce the α7 nAChR-mediated eEPSC enhancement, the difference was not statistically significant (38.4 ± 20.%, n = 5, p > 0.10; Fig. 6C,D), suggesting that the α7 nAChR-mediated eEPSC enhancement requires PKA activity predominantly at presynaptic mossy fiber terminals.

The action of α7 nAChR agonist was abolished by inhibition of PKA activity, but not CaMKII activity. A, Normalized EPSC amplitude was plotted against the application time (from 0 to 10 min) of PNU-282987 in the absence (■) and presence of KT5720 (○). B, The histogram shows that KT5720 abolished the PNU-282987-induced change of normalized EPSC amplitude. C, Normalized EPSC amplitude was plotted against the application time of PNU-282987 (from 0 to 10 min) recorded with internal solution without (■) and with PKI (○). D, The histogram shows PKI failed to abolish PNU-282987-induced net change of normalized EPSC amplitude. E, Normalized EPSC amplitude was plotted against the application time of PNU-282987 (from 0 to 10 min) in the absence (■) and presence of KN62 (○). F, The histogram shows that KN62 did not affect the PNU-282987-induced net change of normalized EPSC amplitude (*p < 0.05; NS, p > 0.05, Student's t test).
A recent study showed that CaMKII activity was required for the AP-independent and nicotine (through the α7 receptor)-mediated increase in spontaneous mEPSCs in CA3 neurons (Sharma et al., 2008). We tested whether CaMKII activity was required for the α7 nAChR-mediated modulation on the AP-dependent increase in eEPSC as well. While bath application of the CaMKII inhibitor KN62 (10 μm) appeared to reduce the PNU-282987-induced enhancement of eEPSCs (68.3 ± 22%, n = 6, p > 0.10; Fig. 6E,F), the effect was not significant, suggesting that CaMKII plays a less crucial role in the evoked glutamate release.
Discussion
We have shown that activation of α7 nAChRs enhances AP-dependent mossy fiber glutamate transmission. Furthermore our data suggest that these α7 receptors are located on the presynaptic mossy fiber terminals, and their activation increased intraterminal calcium levels during the AP that enhanced mossy fiber glutamatergic transmission that was dependent on the activity of PKA.
We report here several findings suggesting that activation of the α7 nAChR itself is sufficient to enhance mossy fiber transmission: (1) the potentiating effect of nicotine on mossy fiber EPSC amplitude was observed only in the presence of an α7 nAChR PAM; (2) the effect was prominent at a relatively high concentration of nicotine (10 μm); (3) the effect and was blocked by a low concentration of MLA (20 nm), an α7 nAChR-selective antagonist; (4) PNU-282987, the α7 nAChR-selective agonist, produced similar effects as the combination of nicotine and the α7 nAChR PAM; and (5) genetic deletion of the α7 nAChR prevented the PNU-282987-induced potentiation. Therefore, our findings indicate that α7 nAChRs mediate this effect.
Many studies have shown widespread distribution of mRNA for α7 nAChRs, as well as α-bungarotoxin binding throughout the DG, CA3, and CA1 regions of the hippocampal formation (Fabian-Fine et al., 2001; Adams et al., 2002). Detailed immunogold studies in the CA1 region revealed immunogold particles labeling α7 nAChRs at both presynaptic and postsynaptic compartments (Fabian-Fine et al., 2001). Functional α7 nAChR-mediated currents have also been reported in CA3 and CA1 pyramidal neurons (Grybko et al., 2011; Gu et al., 2012), and therefore it is possible that α7 nAChRs are located at both mossy fiber boutons and postsynaptic crescents. Therefore nicotine could be activating either presynaptic or postsynaptic α7 nAChRs (or both) to exert its action. Our results show that nicotine, in combination with an α7 PAM, increased the amplitude of eEPSCs with a decreased PPR, an indication of a presynaptic locus of action (Zucker and Regehr, 2002). In addition, we found that nicotine and the α7 PAM did not change the amplitude of the mEPSCs recorded from CA3 pyramidal neurons, suggesting that a postsynaptic component does not play an important role in this action. Our findings are also consistent with previous findings showing that nicotine enhances the frequency of mEPSCs recorded in CA3 pyramidal neurons (Gray et al., 1996; Sharma et al., 2008). The potentiating effect of nicotine on glutamate release has been frequently attributed to the activation α7 nAChRs in a variety of brain regions (McGehee et al., 1995; Gray et al., 1996; Maggi et al., 2004), although a few studies have also reported that activation of the α4β2 nAChRs could modulate glutamate release as well (Lambe et al., 2003; Garduño et al., 2012).
The presence of functional α7 nAChRs at the mossy fiber terminals has been a matter of debate due to conflicting results using the calcium dye Fura-2 (Gray et al., 1996; Vogt and Regehr, 2001). We selectively expressed the genetically encoded calcium indicator GCaMP3 in DG granule cells to enhance specificity of expression, and used an α7 nAChR PAM to abolish the desensitization of the α7 nAChR and enhance the calcium signal. By comparing data obtained from wild-type and α7 nAChR knock-out mice, our results clearly demonstrate the presence of α7 nAChR-dependent calcium increases at the mossy fiber terminals. Furthermore, since we used various blockers (TTX, CNQX, and APV) to eliminate any contribution from synaptic inputs, the calcium responses should be due directly to activation of the α7 nAChR. Thus, we have provided new direct evidence of functional α7 nAChRs at mossy fiber terminals that enhance glutamate release. The source of the calcium increases would be mainly comprised of the calcium influx directly through the α7 nAChR itself (Fayuk and Yakel, 2007; Gilbert et al., 2009), and any calcium-induced calcium release from intracellular calcium stores (Sharma and Vijayaraghavan, 2003; Sharma et al., 2008). Although calcium influx through voltage-gated calcium channels (VGCC) previously was shown not to contribute significantly to the α7 nAChR-mediated presynaptic effect (Dickinson et al., 2008), other studies reported that calcium influx through VGCCs could contribute to some degree (Barrantes et al., 1995; del Barrio et al., 2011).
Based on our data from paired-pulse experiments, activation of the α7 nAChR enhanced release probability, which is in close relationship with intracellular calcium levels (Neher and Sakaba, 2008). Taking advantage of GCaMP3's low calcium binding affinity, we found that the peak of the AP-evoked calcium transient was enhanced by the α7 nAChR agonist PNU-282987, although PNU-282987 had no direct effect on the basal calcium level. Similar action of presynaptic nAChRs on AP-evoked calcium transients was also reported in CA1 pyramidal neurons (Szabo et al., 2008). This effect also resembles the short-term synaptic potentiation due to the residual calcium of repetitive electrical stimulation (Magleby and Zengel, 1976; Zucker and Regehr, 2002). Changes in the AP-induced calcium transients would lead to changes in the amount of neurotransmitter released at the presynaptic terminals (Jackson et al., 1991). In addition, an α7 nAChR-mediated pathway was also suggested to tightly control the refilling of the readily releasable pool in dopamine terminals (Turner, 2004). Last, α7 nAChRs were reported to be associated with the plasma membrane calcium-ATPase pump isoform 2 (PMCA2), which clears away calcium and primes calcium storage in the endoplasmic reticulum upon α7 nAChR activation (Gómez-Varela et al., 2012).
Our results show that the α7 nAChR-mediated potentiation of eEPSC amplitude persisted for 5–10 min after the removal of the agonist, and similar long-lasting effects of nAChR agonists on neurotransmitter release were observed by others as well (Léna and Changeux, 1997; Zhong et al., 2008). These data suggest that calcium-dependent signaling molecules could regulate the glutamate release machinery to sustain nicotinic enhancement of glutamate release. Our data demonstrate that the activity of PKA is necessary for the α7 nAChR-mediated potentiation of eEPSC amplitude. PKA has been shown to regulate synaptic plasticity throughout the hippocampus (Pelkey et al., 2008; Wozny et al., 2008; Caiati et al., 2013), and particularly in mossy fiber terminals (Mellor et al., 2002; Rodríguez-Moreno and Sihra, 2004, 2011; Nicoll and Schmitz, 2005). Moreover, inhibition of PKA activity blocked the nAChR-mediated effect on synaptic plasticity in the hippocampus (Welsby et al., 2009). Recently, the α7 nAChR and adenylyl cyclase 1 (AC1) were shown to be associated physically and functionally in epithelium (Maouche et al., 2013). Stimulated by both calcium and calmodulin, AC1 is a suitable candidate to couple the α7 nAChR to PKA. Moreover, AC1 is concentrated in the mossy fiber tract in mature hippocampus (Conti et al., 2007). However, to date, direct evidence of nAChR-mediated PKA activation in neurons remains elusive. PKA has been shown to phosphorylate rab3A, RIMα, synaptotagmin 12, and other presynaptic proteins to enhance neurotransmitter release (Lonart and Sudhof, 1998; Lonart et al., 1998; Castillo et al., 2002; Menegon et al., 2006; Kaeser-Woo et al., 2013). The release machinery associated with α7 nAChR activation remains to be identified in future studies. Although CaMKII was shown to play an important role in nicotine's effect on the AP-independent glutamate release (Sharma et al., 2008), the α7 nAChR-dependent effect on the AP-dependent glutamate release that we have shown here employs a different signaling pathway. The different signaling pathways could be due to calcium dynamics at the terminals, animal age, or experimental temperature. In the prefrontal cortex, nicotine induced an increase in frequency and amplitude of spontaneous EPSCs, but did not affect the amplitude of evoked glutamatergic transmission from layer II/III to layer V pyramidal neurons (Couey et al., 2007). Moreover, mossy fiber-CA3 synapses showed different homeostatic adaptations between 11 and 21 d in vitro (Lee et al., 2013). Finally, the response of nAChRs to their agonists also depends on experimental temperature (Jindrichova et al., 2012).
Either activation of the α7 nAChR with agonists, or potentiation with PAMs (e.g., NS1738), has been shown to improve hippocampal-dependent learning and memory (Bitner et al., 2007; Timmermann et al., 2007; Yaguchi et al., 2009). Conversely, genetic deletion and pharmacological inhibition of the α7 nAChR in the hippocampus results in significant learning and memory impairments (Levin et al., 2002; Fernandes et al., 2006; Nott and Levin, 2006; Young et al., 2007; Levin et al., 2009), and worsens the learning and memory deficits in a mouse model of Alzheimer's disease (Hernandez et al., 2010). Our findings shed light on the potential cellular mechanisms of these positive cognitive actions of α7 nAChR agonists and PAMs, as well as providing further insight in the development of therapeutic treatments for cognition impairments. For example, schizophrenic patients are often heavy smokers (de Leon and Diaz, 2005), which has been suggested to be a form of self-medication to mitigate their cognitive dysfunctions (Levin et al., 1996). Our study provides a basis for the hypothesis that nicotine enhances the transmission from DG to CA3 to compensate for a loss of synaptic connection in schizophrenic patients (Kolomeets et al., 2005, 2007). Previous electron micrograph studies showed that cholinergic afferents formed direct synaptic contacts on mossy fiber terminals (Léránth and Frotscher, 1987, 1989). It will be important to determine the effect of α7 nAChR activation through these (and potentially other) cholinergic inputs at mossy fiber terminals in the future.
Footnotes
This research was funded by the National Institute of Environmental Health Sciences Intramural Research Program/National Institutes of Health. We thank Pattie Lamb for genotyping and mouse colony management, and Jeff Tucker for technical support of confocal microscopy. We appreciate Drs. Georgia Alexander, Serena Dudek, and Christian Erxleben for the helpful reading and suggestions on this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Jerrel L. Yakel, Laboratory of Neurobiology, NIEHS/NIH, 111 T. W. Alexander Drive, Durham, NC 27709. yakel{at}niehs.nih.gov





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}