
Abstract
Reinforcement corrosion is the risk most frequently cited to justify concrete durability research. The number of studies specifically devoted to corrosion propagation, once the object of most specialised papers, has declined substantially in recent years, whilst the number addressing initiation, particularly where induced by chlorides, has risen sharply. This article briefly describes the characteristics of steel corrosion in concrete that need to be stressed to dispel certain misconceptions, such as the belief that the corrosion zone is a pure anode. That is in fact seldom the case and as the zone is also affected by microcells, galvanic corrosion accounts for only a fraction of the corrosion rate. The role of oxygen in initiating corrosion, the scant amount required and why corrosion can progress in its absence are also discussed. Another feature addressed is the dependence of the chloride threshold on medium pH and the buffering capacity of the cement, since corrosion begins with acidification. Those general notions are followed by a review of the techniques for measuring corrosion, in particular polarisation resistance, which has proved to be imperative for establishing the processes involved. The inability to ascertain the area affected when an electrical signal is applied to large-scale elements is described, along with the concomitant need to use a guard ring to confine the current or deploy the potential attenuation method. The reason that measurement with contactless inductive techniques is not yet possible (because the area affected cannot be determined) is discussed. The method for integrating corrosion rate over time to find cumulative corrosion, Pcorr, is explained, together with its use to formulate the mathematical expressions for the propagation period. The article concludes with three examples of how to use corrosion rate to assess cathodic protection, new low-clinker cements or determine the chloride threshold with an integral accelerated service life method.
Similar content being viewed by others



Avoid common mistakes on your manuscript.
1 Introduction
Social appreciation for the built heritage is growing. The conservation and maintenance required to prevent its deterioration entails high costs, however, given the characteristics of all the technologies and processes involved in rehabilitation and repair. Beyond such economic considerations, deterioration diminishes functionality and may even pose the risk of accidents with the loss of human life due to unexpected collapse. Buildings and infrastructure form an essential part of developed societies, and as such their usability to the highest standards of safety is an indisputable priority. The material most widely used in the modern built heritage is concrete and corrosion of its steel reinforcement heads the list of its possible causes of decay.
Concrete underwent ceaseless development in the twentieth century for many reasons, one of which, directly related to this study, was that most design engineers deemed it to be eternally maintenance-free, in the understanding that cement alkalinity affords chemical protection for the steel and the cover constitutes a physical barrier much more effective than paint. It was not until the nineteen seventies and early eighties when Rilem began to explore reinforcement corrosion, a subject that aroused the interest of very few researchers. As new construction was booming and time-mediated damage had not yet been detected, only a handful of cases of corrosion had been identified, despite one very distressing instance of collapse [1]. By way of example of the scant attention lent to corrosion, when the author defended her Ph.D. thesis in 1973, she only found around 40 articles on the subject in all the journals on concrete published at the time. Pioneering the concern was Committee 60-CSC, Corrosion of Steel in Concrete, chaired by Peter Schiessl, who published a summary of the state of the art at the time [2].
Engineer Hans Arup, who also alerted to the problem early on, organised the first workshop devoted exclusively to corrosion in 1981. A photo of the participants is reproduced in Fig. 1. One of scientists in the picture, Kyosti Tuutti, published a much-cited and impactful thesis [3] in 1982, describing a two-period (initiation and propagation) model.

Participants in the workshop entitled ‘Corrosion of steel in concrete’ organised by Hans Arup at Copenhagen in 1981 In the first row from right to left: (1) ??, (2) K. Tuutti, (3) ??, 4) J.A. Gonzalez, (5) ??, 6) C. Locke, (7) C. Hansson, second row: (8) O. Vennesland, (9) O. Gjorv, (10) C. Page, (11) R. Bakker, (12) J. Dawson, (13) Byfors (14) P. Schiessl, (15) ??, (16) ??. (17) G. John (18) C. Andrade, third row: (19) ??, 20) ??, (21) Wilkins, (22) H. Arup, (23) ??, (24) ??, (25) ??, (26) ??, (27) ??
A number of Rilem committees were subsequently created, but only one on reinforcement corrosion, RILEM TC-154-EMC: “Electrochemical Techniques for Measuring Corrosion In Concrete” chaired by the author, published Recommendations on the techniques for measuring corrosion, which are still in effect [4,5,6,7]. Later committees went on to develop service life models, in particular in connection with chloride diffusion [8], or determine the steel depassivation limit or its governing parameters.
The many and very valuable scientific and technical papers that have been forthcoming in the interim attest to the economic implications of the subject and the scientific challenges posed. The author apologises for listing but a few of those papers here [9,10,11,12,13,14], chosen because they deal specifically with active steel corrosion.
This article summarises some of the principles associated with corrosion, its measurement and modelling in the propagation stage, as a tribute to Rilem in its 75th anniversary and as a token of my gratitude for the inspiration I have drawn from its working approach throughout my career. Rilem’s routine practice of creating short-lived technical committees, consistently mandated to explore innovative and topical concerns, expands the frontiers of knowledge, invariably challenging presumably established principles. This review calls some of those principles into question and identifies the many gaps still extant in our understanding of reinforcement corrosion.
2 Basic principles and mechanisms
Concrete corrosion is an electrochemical process, for it involves both chemical reactions and the circulation of an electrical current, in which the reagents are electrical charges (electrons and ions). In essence, corrosion is triggered when concrete pH at the interface with the steel drops to levels below around 8 [15], dissolving the passive layer that forms in the highly alkaline aqueous phase. Under those circumstances the iron atoms in the steel convert to positively charged ions (oxidation), generating an excess of electrons in the base metal:
To maintain its electrical neutrality, the metal in adjacent areas induces another reaction (reduction) which in neutral and basic media uses dissolved oxygen:
That oxidation–reduction (redox) mechanism supports the corrosion ‘cell’, an only apparently simple development, for neither oxidation nor reduction takes place in a single stage. Rather, both entail a series of intermediate species, whilst electron migration from the steel to the reagent depends on the so-called ‘tunnel effect’ to overcome the energy barrier involved. Therefore, although the basic mechanisms governing steel corrosion may be simplified, sight should not be lost of their complexity, a feature they share with nearly all chemical reactions taking place in successive or simultaneous stages. As a result, in the spontaneous reaction (corrosion-free potential), the total anode and the cathode current densities are equal and equal as well to the corrosion current density.
As some of the most significant characteristics of these basic processes and their governing parameters are often poorly understood, they are dealt with in the sections listed below.
- 1.
Corrosion cell and acidification of the corrosion zone
- 2.
Microcells and macrocells (corrosión rate and galvanic current)
- 3.
Effect of the oxygen
- 4.
The role of potential
- 5.
Composition of the rust formed
- 6.
Effect of temperature
Although these effects feed back into one another, they are addressed separately here for greater clarity.
- 1.
Corrosion cell and acidification of the corrosion zone
As Evans showed [16] nearly 100 years ago with his droplet experiment (Fig. 2), corrosion begins at the atomic level with the formation of microcells. When a drop of salt water is left on steel the cell forming in the centre of the drop is anodic because it contains less oxygen than the border of the droplet, which becomes cathodic. Evans proved that anodes and cathodes were formed (Fig. 1) by adding phenolphthalein (reduction generates hydroxyls, colouring the outer side of the drop pink) and sodium sulfocyanide (which when combined with iron yields a blue iron sulfocyanide complex). If left standing the colours mix in the drop due to the formation of many nano- and microcells. According to Pourbaix [15], the pH dips to around 3–5. Corrosion always entails local acidification in the zones affected [17].

a Evans droplet basic reactions (Note that water hydrolysis lowers the pH in the anodic zone and the formation of hydroxide ions raise it in the cathodic zone) and b some initial pitting in a rebar
Consequently, the buffering capacity of portlandite is of key importance not only in retarding carbonation but in chloride-induced depassivation in concrete. When the first iron atoms are released into the solution by the depassivating action of the chlorides, they hydrolyse the water, lowering the local pH. That effect is neutralised by the buffering capacity of the hydroxyls, sourced primarily from the portlandite, as well as from the alkalis, which repair the passive layer, mitigating the decrease in pH. The critical chloride content is reached when the amount of dissolved Fe++ ions, generating protons by inducing water hydrolysis, cannot be neutralised. Consequently, the Cl−/OH− ratio governs the chloride corrosion threshold. In chloride attack, acidification is local because the attack is localised and the rest of the pores retain their alkalinity, passivating the rest of the metal surface. Moreover, the polarisation and lower potential induced by cell formation beginning at the pit polarise the surrounds to the pit potential (Fig. 3). That creates a need for more chlorides to exceed the Cl− limit at the lower mixed potential induced by pit formation, ultimately protecting the surrounds. The attack therefore proceeds in depth instead of spreading laterally, which may nonetheless occur, depending on the degree of polarisation at the pit borders and pit growth.

Potential and pH values in a corrosion pit [15]
In carbonation, hydroxil ions from portlandite exert a buffering effect across the entire surface, attempting to offset the carbon dioxide-mediated reduction of pH: as the hydroxil ions are consumed calcium carbonate forms with the calcium ion of portlandite, the pH declines and corrosion is driven across the entire surface by many micro-anodes and micro-cathodes (Fig. 4). In that case acidity is governed by carbonate/bicarbonate ions, which tend to maintain a neutral pH at values of around 7.

Microcells generated by generalised carbonation-mediated depassivation
One important implication of acidification in the corroded areas is that the cathodic reaction may reduce not only the oxygen in the adjacent passive areas, but also the protons in the corrosion zone itself. Corrosion may consequently proceed independently via the interior microcells (self-catalysis) if the source of oxygen to the adjacent areas is interrupted (Fig. 3). In other words, protons may be reduced in a cathodic reaction, a development that has been detected in the form of H2 bubbles inside pits [18].
Acidification and the repassivation capacity of the passive layer are the factors that determine whether the attack is local (proceeding depth-wise) or laterally widespread. In stainless steels with a very high repassivation capacity, corrosion proceeds as very small pits that bore quickly inward. Therefore, whilst the use of stainless steel ensures very high chloride thresholds, once corrosion is triggered and the pits become active, the attack is very dangerous because the pit may grow depth-wise in a narrow area with a scant loss of material. In contrast, in black steel with a lower repassivation capacity, the pits are shallower and spread sideways.
- 2.
Microcells and macrocells (corrosión rate and galvanic current)
In microcell corrosion the anodes and cathodes are separated by just a few microns, whereas in macrocell corrosion they may be spaced at several centimetres or even metres. Metres-wide macrocell separation can only exist in concrete in marine media where electrical charges can circulate across long distances thanks to the low resistivity of seawater. Corrosion rate and galvanic current are different concepts. The reference area for expressing the galvanic current, which is the current circulating between the anodic and cathodic zones, is the anodic area only, whereas corrosion rate refers to the sum of the anodic and cathodic areas.
Corrosion, particularly in concrete, is invariably supported mostly by microcells, with macrocells contributing to the corrosion rate only marginally (see Figs. 3 and 4). If corrosion proceeds at the micron level, what role do passive zones much greater than the corrosion area play in local corrosion? Whilst along with galvanic current, Ig, they indisputably contribute to corrosion current density, but unless the anodic area is very small, they do not account for a majority proportion in the total corrosion current. In other words, the corrosion current measured is the sum of the microcell and the galvanic currents. The inference is that the galvanic current measurement alone cannot be directly likened, nor is it necessarily proportional, to the corrosion rate. This issue will be discussed more fully in the section on measuring techniques.
That corrosion is primarily supported by microcells in the corrosion zone was confirmed long ago, as the earlier reference to Evans’s droplet experiment shows. Atomic force microscopy (AFM) has recently corroborated those findings. In [19] a drop of saltwater lying on a steel plate was observed under an atomic force microscope (AFM) at different ages. Figure 5 shows, in a 5 micron window, that the ferrite acting as the anode dissolved whereas the cementite acting as the cathode remained intact (Fig. 5).

Three-dimensional AFM images of the sample surface after a 0 h 0 min; and b 2 h 15 min of exposure to a 0.05 M NaCl solution [19]
The behaviour clearly visible in Fig. 5 for the metal in a prestressing steel tendon in which the layers of cementite and ferrite alternate, may vary in other steel microstructures. Generic corrosion is summarised in Fig. 6, which shows that at the nano- and micro- levels some areas of the steel are anodic and others cathodic. The figure also describes the mechanism in which water is hydrolysed by Fe++, generating protons that acidify the anodic zones.

Representation of microcell activity
On a somewhat larger (millimetre) scale, the trial conducted by Mansfeld [20] with a Kelvin probe, likewise with a drop of saltwater, is also illustrative. As Fig. 7 shows, the potential is positive only on the outer side that acts as a cathode, whereas the potential on the inner side adopts very negative values.

Mansfeld experiment with a Kelvin probe [20]: potential profile for a drop of saltwater
The mixed or corrosion-free potential measured on a metal bar immersed in an electrolyte would lie in-between the anodic and cathodic semi-reaction corrosion potentials. The exact value would depend on how the process is conducted and whether the anode and cathode are coplanar and positioned face-to-face. As shown in Fig. 8, steel bars may be placed face-to-face in separate chambers connected by a salt bridge (Fig. 8, left) or in the same chamber separated by a concrete disc acting as a diffusion cell (Fig. 8, right). Electrical resistances are then introduced in the circuit between anolyte and catholyte to simulate different types of concrete and degrees of water saturation.

Setup for studying galvanic currents in concrete with face-to-face alignment
Figure 9 shows some of the results obtained from this experiment using galvanised steel – black steel couples. In the figure Sc = cathodic bar surface and SA = anodic (galvanised) bar surface [21].

Ecorr values for simulated macrocells versus the log of the resistance between anolyte and catholyte for an Fe/Zn galvanic couple [21]—[Legend: blank/solid squares: anodic corrosion potential before/after connecting anode and cathode; blank/solid circles: cathodic corrosion potential before/after connecting anode and cathode]. Ecorr values of simulated macrocells in function of the inert resistances for the couple Fe/Zn
Inasmuch as the salt bridge itself generates ohmic resistance, in the graph on the left in Fig. 9 the resistance drop prevents the potentials from concurring. The mixed potential is any value from the cathodic − 500 mV to the anodic − 1400 mV. In the middle graph in Fig. 9 the mixed potential continues to differ where the gap between anodic and cathodic potential is wide in high resistance values, but as that resistance lowers the mixed potential ultimately concurs with the anodic potential. Given that the anode is aeriated, it can support corrosion by itself with its microcells. The graph on the right depicts the situation when the anode is non-aeriated. Here also, if resistance in the medium is very high the potentials fail to concur. In contrast, when resistance declines both the anode and the cathode are polarised, yielding an intermediate mixed potential for the galvanic couple. The explanation is that due to non-aeriation, corrosion in the anode (right in the figure) is less supported by microcells, raising the proportion of galvanic current in the corrosion current.
That is, if the anodic zone is as aeriated as the cathode (middle of Fig. 9), the mixed potential is the anode potential, whereas the potential adopts an intermediate value when the corrosion zone has restricted access to oxygen. In addition to these factors, the value of the mixed potential is affected by the size of the cathodic relative to the anodic area. Another aspect that influences the contribution of the galvanic current to the corrosion rate is since in a given bar the anodic and cathodic zones are in the same plane (coplanar), the galvanic current is transmitted laterally, whereby the ‘transmission line model’ should be applied in its calculation [22] and may result of smaller proportion than face-to-face arrangement.
- 3.
Effect of the oxygen
The role of oxygen in corrosion generation and progress in concrete has been misconceived in some respects. Trying to control oxygen very seldom limits concrete corrosion. Oxygen is essential for depassivation but not for corrosion progression. The description of its role can be divided into two stages, as discussed below: under passive conditions and in the presence of active corrosion.
2.1 Passive conditions
For oxygen to feed the cathodic reaction it must be dissolved in the pore solution. In the absence of pollutants oxygen contributes to thickening the passive layer. The variation over time in the corrosion potential characteristic of a bar embedded in an undersea structure with a 7 cm cover is graphed in Fig. 10. The potential evolves more positive up to values of + 80 mVAg/AgCl, denoting steel passivity and the presence of a compact, late age passive layer due to an uninterrupted supply of oxygen. The figure also shows the fluctuation in corrosion potential in a bar immersed in a 0.5 M NaCl solution bubbled with nitrogen throughout the test to maintain the dissolved oxygen at 0.3 ppm or under. The potential under these conditions is much more cathodic because the low O2 concentration induces structural changes in the passive layer.

Corrosion potential in a bar embedded in undersea concrete and in another submerged in a non-aeriated solution
The experiment with the non-aeriated solution depicted in Fig. 10 confirms the near impossibility of lowering the oxygen content of a solution below 0.1 ppm by de-aerating with nitrogen [23], due to the oxygen impurities in this gas. The potential is consequently around − 500 mVAg/AgCl and not lower. If the oxygen level declines further, however, the potential would shift to more cathodic values (Fig. 11), entering the generalised corrosion region (Fig. 11) and, in the absence of depassivating ions, triggering passivation. Lower potential values would carry the current into the cathodic protection region, with the reduction of water as per the cathodic reaction:

Possible trends of polarization curves of steel embeded in concrete. If no breadown is found before around + 650 mVAg/AgCl the steel is passive. If the curve bends before such potential with a sudden increase in the current, pits were generated. In the case of generalized corrosion, no decrease of the current denoting passive film formation from around − 650 mVAg/AgCl is noticed and the current continuously increases
To put it briefly, under passive conditions the value of the corrosion potential is governed by oxygen. When the oxygen concentration declines, the potential shifts to more cathodic values, hampering depassivation. However, as Fig. 10 shows, oxygen has fairly ready access to the reinforcement not only in dry but also in saturated concrete, where the oxygen content in the pore solution suffices to maintain and raise steel passivity. The reason is that water contains around 8 ppm of oxygen, which is more than enough to heighten the passivation of concrete-embedded steel. Oxygen concentration in water depends on salt concentration and temperature and although oxygen solubility is lower in highly saline solutions, such situations are seldom present in concrete, even when it is underground or undersea.
The behaviour deducible from Fig. 10 likewise stands as evidence that cover thickness has no impact on oxygen availability around the reinforcement. That is, tantamount to saying that although corrosion at different depths logically begins at different times because it takes chloride ions longer to penetrate thicker covers, the corrosion rate is similar. Therefore, oxygen concentration is either the same at all depths or it does not control the process. Models that make corrosion rate dependent upon oxygen permeability are consequently erroneous.
2.2 The role of oxygen in depassivation and propagation of corrosion
As said, sufficient oxygen is normally present in concrete pore solutions to maintain passivity. That notwithstanding, in alkaline media it plays an essential role in the initiation of corrosion, but not in its propagation. In the presence of a high enough chloride content, oxygen at concentrations as small as a few tenths of ppm suffices to initiate corrosion (localised at the site where the oxygen is in contact with the metal surface). At low oxygen concentration, corrosion is much localised, as shown in Fig. 12 and Table 1. The figure plots the fluctuations in potential and corrosion rate in two bars immersed in saturated calcium hydroxide solutions, one with 0.5 M NaCl and the other with 2.5 M NaCl, bubbled with N2 gas throughout the test. (The bar in the 0.5 M solution is the same shown in Fig. 10) Despite an O2 content of < 1 ppm, corrosion was noticed, although even at the high corrosion rates measured, a post-test visual inspection of the bars immersed in the 2.5 M solution revealed only sporadic corrosion staining and very small pits. A bar tested for 5 d in 2.5 M NaCl (which exhibited a corrosion-free potential more anodic than that in the 0.5 M solution) is depicted in Table 1.

Corrosion potential (left) and corrosion rate (right) of steels immersed in 0.5 M and 2.5 M NaCl solutions
Rusting was more visible in the 30 day test at 0.5 M NaCl. The corrosion products formed along a line in the bar surface which may have been generated during manufacture (Table 1). Crevice corrosion was also observed at the edge of the tape used to delimit the working area. In both the 0.5 M and the 2.5 M NaCl solutions only the areas with surface imperfections such as seams or rolled laps, corroded. These imperfections seem to be the preferred sites for pit initiation. High Icorr values are indicative of active corrosion even when rust is not visible to the naked eye until it reaches certain dimensions (hence the readier visibility in the month-long than in the 5 day experiment).
When potential is not spontaneous but electrochemically induced (potentiostatic test), the results are as given in Table 2. At potential values more cathodic than − 650 mV (Ag/AgCl electrode), no pits are detected. Pits form at − 550 mV, while generalised corrosion appears at − 350 mV.
As commented, once the depassivation is produced and sufficient rust formed, oxygen enhances the corrosion rate but it is not necessary for the corrosion to progress, due to the protons and the Fe+++ ions may feed the cathodic reaction.
- 4.
The role of potential
The role of potential in depassivation is illustrated in Fig. 13, where breakdown potential is plotted against chloride concentration threshold. Further to the time-honoured fundamentals on breakdown potential in the presence of depassivating ions, for localised corrosion to take place, the potential must be greater than the potential at which passivity breaks down, i.e., when due to the presence of oxygen, the potential exceeds the breakdown potential threshold. Figure 13 [24], inspired by Pourbaix’s [15] findings, represents potentiostatic tests in mortars made with different types of cement [25].Different potentials are applied to bars embedded in specimens immersed in a NaCl solution and the current is recorded. When the amount of chlorides arriving to the bar surface exceeds the threshold, the current rises steeply, denoting breakdown of the passive layer. Potential, then, controls tolerance to one chloride threshold or another. The more positive the spontaneous potential, the more Fe+++ found in the passive layer (due to more oxygen present), the more vulnerable is passivity to breakdown and the lower is the chloride level required to initiate local corrosion. As the graph in Fig. 13 shows, when the potential applied is high, fewer chlorides are needed to induce corrosion, whereas when the potential shifts to negative values, the chloride threshold is higher. The graph mimics the natural pattern described earlier, in which a low enough potential prompts cathodic protection.

Briefly, potential is an indication of the level of oxygen present. Where it is not under − 650 mV, the risk of corrosion subsists and local or generalised corrosion may take place with scarce oxygen given sufficiently high chloride content [23]. In concrete semi-submerged in seawater, corrosion tends to begin in the more aeriated tidal zone, which constitutes a sacrificial anode for the rest of the structure, inasmuch as saltwater is a very good conductor. The differential aeriation between tidal and undersea zones gives rise to a galvanic macrocell. In the absence of such a difference in potential, corrosion may begin in the underwater zones. While not immune to corrosion, given the more cathodic potential present, such underwater zones fail to depassivate until a higher chloride concentration is reached.
- 5.
Composition of the rust formed
As the reaction products may vary widely and involve many intermediate species, establishing the basic mechanism calls for very specific studies and the application, for instance, of ring-disks [26] that are unusable in concrete. Although the description of the mechanism is normally simplified when intended for practical purposes, sight should not be lost of the complexity ensuing from its many stages and intermediate species [15, 26,27,28] with charge imbalances and solvated ions surrounded by different proportions of water molecules. The species involved include: [Fe(OH)+], [FeCl+], [Fe(OH)2], [Fe3O4], [γ-Fe2O3], [γ-FeOOH], or green rust gels with variable compositions [29], etc. The most stable rusts are mixes of hydroxides and oxides (called oxy-hydroxides).
The oxides involved in steel corrosion in concrete are the same as observed in its corrosion in the atmosphere, including nearly all the most common iron oxides [27], such as magnetite, goethite and lepidocrocite and, in the presence of chlorides, the akaganeite. Given such diversity, all attempts to relate oxide composition under real exposure conditions to corrosion mechanisms have proven futile, for composition fluctuates constantly, in keeping with the balance between acidity (in turn dependent upon chloride or bicarbonate ion concentration) and oxygen availability. When exposed to natural media, the composition and dynamics of the oxides present are neither predictable nor indicative of corrosion rate.
Although composition is variable, certain differences exist between the oxides detected in the early stages of corrosion, depending on whether it is carbonation- or chloride-induced (with the appearance in the latter of akaganeite, an oxide that takes up chlorides in its structure). Chlorides generally give rise to more soluble oxides that diffuse farther than those involved in carbonation.
Since the corrosion zone acidifies, the oxide ions form a suspension that progressively gels due to coagulation of the individual constituents (Figs. 14, 15). The figure shows that the OH−-solvated tetrahedron housing the Fe ion evolves into an octahedron that begins to coagulate with others, forming larger particles that ultimately constitute a network of crystalline oxides.

Clustering of Fe ion tetrahedra into octahedra and solvated particles

Rust in different physical conditons in concrete
These grains can migrate across the pores and spread, precipitating and clustering until they appear on the surface. When the resulting gels [29] find alkaline cathodic zones they coagulate further and precipitate, ceasing to spread as they convert into laminar rust or solid particles. They diffuse neither continuously nor at a continuous rate. Rather, diffusion adapts to the steady increase of concrete resistivity over time, rendering corrosion rate resistivity-dependent [30].
2.3 Cover cracking effect
If the reaction proceeds, the rust on the concrete migrates across its pores. Although the rust exerts pressure from the outset, due to the porosity of the steel/concrete interface the concrete is unaffected by the full thrust of the pressure. When the pores at the interface become saturated, the full radial pressure is transferred to the concrete, ultimately prompting cracking at the surface of the bar that extends to the surface of the concrete (Fig. 15). The more porous the concrete, then, the longer it takes for rust to exert effective pressure that could crack it. Very porous or moist concrete may not crack at all. The cantilevered roof over Maracaná Stadium, for instance, contained chlorides due to the haste with which it was built. Sixty years later, no cracks were detected even though the reinforcement had corroded completely and disappeared entirely in extensive areas [31].
As corrosion progresses the rust no longer migrates but remains at the bar/concrete interface where, depending on the steel microstructure or the presence of moisture in the concrete, it spreads by layers or cycles (Fig. 15). This rust detaches readily when accessed, but may afford misleading information, for the residual sound diameter may appear to be larger than the actual uncorroded diameter (Fig. 15g). Another fact worth mentioning is that section losses of 20% or higher may render the steel more brittle, for the hydrogen generated during corrosion may diffuse toward the steel.
If the pressure exerted by the lack of space to accommodate the rust forming suffices to crack the concrete, cracking begins at the reinforcement surface (Fig. 16) and proceeds toward the closest concrete surface, where it emerges and spreads practically linearly [32] with corrosion penetration, Pcorr between 10 and 50 microns. Corrosion-induced cracking has been a popular line of research in recent years, with any number of papers on analytical and numerical calculation methods. Many fewer experimental studies have been conducted, however [33, 34].

Left: Strain gauges on concrete in a prismatic specimen. Right: crack induced corrosion at initial stages
The general consensus is that as an attack of under 10 µm to 50 µm suffices to crack the cover, cracking may be a visual symptom of the presence of corrosion. The earlier corrosion is detected the better, whilst attempts to prevent it by adding fibres to the concrete or wrapping with carbon based textiles, may be counterproductive.
2.4 Alternative cathodic reactions
Another consideration to be borne in mind with respect to the oxides formed during corrosion is that the ferric oxides forming with Fe++ oxidation may in turn be reduced to ferrous ions, binding electrons and contributing to the cathodic reaction.
Whether corrosive action continues therefore depends not only on oxygen availability in the still passive zones. It may also be supported by the oxidation–reduction of the oxides forming in the corrosion zones [35] and by the proton reduction generated in water hydrolysis. That is what is meant when corrosion is said to be self-catalytic and explains that it may proceed with no or scant access to oxygen.
- 6.
Effects of temperature
Temperature is known to accelerate chemical reactions by reducing activation energy. The Arrhenius equation is normally used to calculate the effect of a change in temperature on the rate constant and therefore on the rate of reaction [36]. However, this equation can be applied (providing the result is considered an Apparent Activation Energy), if the concrete is saturated, but not if it can exchange moisture freely with the environment, because then the changes are due not only to the activation energy barrier. Thus, higher temperature entails evaporation, rendering Arrhenius inapplicable. In other words, temperature cycles involve variations in humidity inside concrete that preclude using Arrhenius equation.
Fluctuations in temperature also change the solubility of the components of the aqueous phase, which may not necessarily rise with temperature. Portlandite and sulfates, for instance, are less soluble at higher temperatures. Furthermore, temperatures of over 60 °C may decompose sulfoaluminate phases, releasing sulfates into the aqueous phase. Another significant effect of rising temperature is the decline in gas solubility in liquids. Oxygen is consequently expelled at higher temperatures, which translates into less rather than more corrosion. For all the foregoing, the Arrhenius equation is not recommended to be applied in corrosion in concrete.
By way of summary, all these developments make corrosion much more than a simplistic process involving anodes and cathodes in which iron oxidises and oxygen is reduced. A rigorous interpretation must take all the intermediate steps and the many stages governing them into consideration.
3 Corrosion rate and measuring techniques
Corrosion entails a loss of metal. Since in the case of reinforcement steel the aggressive substances penetrate the concrete from outside, the outcome is normally an asymmetrical loss of diameter (Fig. 17, left). The residual diameter, Φt, is therefore equal to the initial diameter, Φ0, less the corrosion penetration, Pcorr. When corrosion is localised (Fig. 17, right), the depth of the deepest pit (Ppit) determines the residual diameter.

Section and diameter loss due to generalized (left) and localized (right) corrosion
Loss can be measured gravimetrically, i.e., (a) by weighing the sample before and after testing it in specific environments, to find the cumulative loss, Pcorr, expressed in g/cm2 of area exposed to corrosion, or (b) by using electrochemical techniques able to measure the metal weight loss through Faraday’s law.
3.1 Electrochemical techniques
The oldest of such techniques, the corrosion potential measurement, is qualitative, indicating only the risk of corrosion, as specified in (4). A second technique, which measures concrete resistivity, which is an indication of the degree of material saturation (5), was recently proposed by the author to characterise overall concrete durability, as discussed in a subsequent section.
The technique that has been widely shown to quantify corrosion most accurately consists in measuring polarisation resistance, Rp, or linear polarisation (6). Over the years, this technique, first applied to concrete in the early nineteen seventies [37, 38] to study a series of corrosion inhibitors, has proven [39] to be the most appropriate, yet to be excelled by any other, for quantifying and monitoring material loss both in concrete and any other electrolyte/metal system. Inasmuch as corrosion is an electrochemical event, such techniques are indisputably the most suitable for quantification purposes.
Linear polarisation, Rp [6, 37], routinely used in all metal/electrolyte systems, consists in applying no more than 20–30 mV above the corrosion-free potential (ΔE) of the embedded steel to induce an electrical current (ΔI). The duration of the increase in potential should be at least 30–60 s to obtain the correct Rp value, for at shorter times the value is affected by the transitory electrical capacity induced by the double layer at the steel/electrolyte interface. Measurements lasting only a few seconds, such as in some devices, yield erroneous values due to the non-dissipation of that effect. The duration of testing to ensure optimal results is one of the features addressed in the earliest studies of this technique [37, 39].
The ratio between Rp and corrosion rate, Icorr, is known as the ‘Stern formula’.
Between 1970 and 1978 [37], further to an approach developed by the author, constant B in that expression when applied to concrete, was quantified by calibration with simultaneous gravimetric losses (Fig. 18) and application of Faraday’s law to convert µA/cm2 to corrosion penetration in mm/yr.

The constant B was found to be 52 mV in the passive state and 26 mV in the active or corroded state, although 26 mV can be used in all cases inasmuch as an error of a factor of 2 in passive steel is negligible, in light of the gravimetric losses. Three important issues, have to be taken into account regarding this technique (6).
- 1.
As the area exposed to corrosion must be taken into account, Rp is expressed as Ωcm2 and hence corrosion rate in µA/cm2. Rp,ap is the value not normalised to the area of the polarised metal.
- 2.
In concrete the upper case ‘I’ rather than the lower case ‘i’ (=uniform corrosion current density) is used as an indication that corrosion may be localised (not visible in concrete). The use of icorr is only correct when corrosion is generalised and uniform.
- 3.
In the absence of coloured rust (=where the steel remains passive), Icorr is less than 0.1–0.2 µA/cm2 and it is greater than those values, when coloured rust is detected. Values of over 0.1 µA/cm2 may be temporarily recorded soon after the steel is immersed in the alkaline solution due to passive layer formation. Once the layer becomes uniform the Icorr values decline to under 0.1 µA/cm2, whether the steel is immersed in a solution or embedded in concrete. When the steel is actively corroding, Icorr values lower than 0.1 µA/cm2 may nonetheless be found when the concrete is dry.
The aforementioned technique is applicable to all systems, as shown by the mean corrosion rates for solutions with different pH used to simulate the values inside and outside a pit induced by chlorides (x) given in Fig. 19. In this figure, it is appreciable that only solutions with a pH of over 12.5 exhibit Icorr values of less than 0.1 µA/cm2. That does not mean, however, that corrosion begins at pH values of < 12.5, for the pH attendant upon carbonation-mediated corrosion has been shown to be around 8 (15). Depassivation as a result of a general decline in pH would, then, depend on the composition of the pore solution and call for values of around 8.

Icorr values (log scale) vs pH (aeriated and non-aeriated solutions)
3.2 Electrochemical impedance
Electrochemical impedance spectroscopy (EIS) delivers results very similar to those found with linear polarisation. As noted many years ago [39], the parameters are the same in the two, but expressed differently: as direct current in terms of time (linear polarisation) or as frequency (EIS). Since EIS is more work-intensive, its use is entirely unnecessary in most cases. Its application is only justified in specific studies where, for instance, the object is to determine capacity-related properties in the double layer of the concrete or the corrosion mechanisms involved. It is often used to establish concrete resistivity, which can be found quickly with a single pulse using only one frequency. It is not discussed in detail in this review paper in light of such specific applicability.
3.3 Contactless measurement
As accessing the reinforcement entails breaking the concrete cover, corrosion should ideally be measured on the surface, with no physical contact with the bar. That approach, which had never before been used to measure corrosion (even though the corrosive power of stray current had long been known), was shown by the author to be viable in small-scale specimens in a paper published in 2001 [42] and was subsequently patented in Spain (Patent No. ES 2 237 241 B). It was based on a simplification of the analogue electrical model (Fig. 20) [43].

Left: Arrangement of electrodes in the Inductive method, Middle: simplified analogue model, Right: polarization detected when applied external current
Contactless measurements in large specimens need to account for the reinforcement length (critical length, see later) reached by the electrical pulse, as is made with normal corrosion measurements. No publications were found by the authors on how to know this critical length in the contactless method. Then, the application to large structures of this new non-contact method with the same level of accuracy of the traditional one should be questioned, until the scientific basis and convincing results are published.
3.4 Measurements in small- and large-scale elements
Devices able to measure Rp are based on potentiostat-galvanostats, which can apply a fixed potential or current. The technique can be readily deployed in small specimens and all commercial devices include the option. The problem in large members in real structures is that as the counter-electrode is much smaller than the reinforcement area, the potential scatters across large distances, as shown in Fig. 21.

Scatter with distance of the steel bar of the current applied through the small counter
The problem of finding the critical length can be countered in one of two ways.
In the guard ring method for current confinement [44] the ring must be modulated [adapting the outer ring current to confine it to the circle pre-determined by the two inner Vss electrodes (Fig. 22)]. The current is not correctly confined without those electrodes (6). Only one commercial device correctly confines the current with this method; other market devices without such inner electrodes deliver erroneous corrosion rate values, under- or over-confining the central current.
Fig. 22 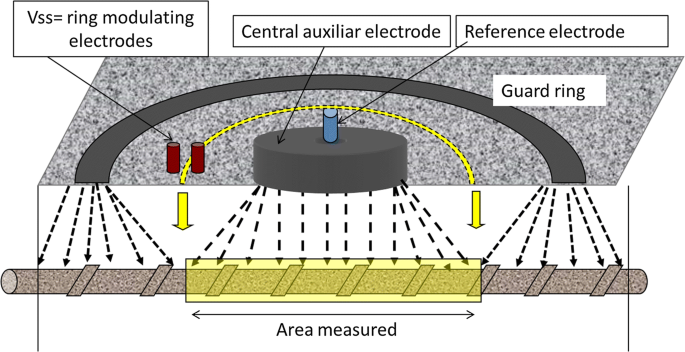
Modulated confinement of the current: uses a Guard ring which is controlled with the two (Vss) electrodes in between guard ring and central counter-electrode [44]
In potential attenuation method [45], electrodes aligned with the counter-electrode (Fig. 23) measure the distance reached by the potential applied and use the total area polarised (6). Only one commercial device (the one correctly confining the current) features this method, which should only be implemented when the structure is very moist and satisfactory confinement is not possible.
Fig. 23 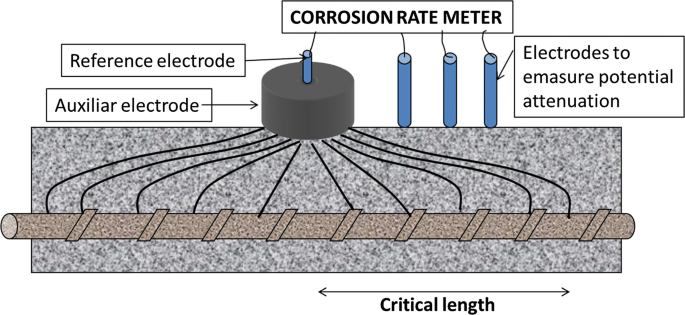
Attenuation of the potential method: the distance reached by the current is measured through the side electrodes at different distances [45]


All that need be done to verify whether a device delivers correct values is to measure samples containing passive steel. Devices that fail to confine the current satisfactorily cannot deliver values below 0.1 µA/cm2, the correct rate for passive steel. This is relevant, for instance, to determine whether corrosion inhibitors effectively reduce the corrosion rate [44].
3.5 Resistance control. Relationship between R p and R ohm
One issue with significant implications that has not been addressed sufficiently in the literature to date is that in concrete Icorr is subject to resistance control, i.e., is proportional to the resistance of the medium. Empirical verification of that fact [30] led to the so-called corrosion rate-resistivity (Icorr−ρ) diagram (Fig. 24), from which the following expression was derived:

Values of the relation Icorr-resistivity obtained in real structures and trend established
The relationship exhibits some scatter, depending on the temperature at which resistivity is measured and the porosities and resistivities of the concrete pore solution, although the general pattern is consistently observed. With that empirical relationship, Icorr can be related both to resistivity and hence to the effective diffusion coefficient [46], from which the apparent diffusion coefficient can be deduced. Thus,
which can be rearranged as:
This reasoning can be carried one step further, by again comparing that equation [12] to the basic equation for Rp [10], whereby Rp can be equated to resistivity:
In a concrete specimen with actively corroding steel and a resistivity of 20 kΩ·cm, for instance, the bar corrosion rate can be found with Eq. (15):
As the relationship only holds in the presence of active reinforcement corrosion, it is not applicable when the steel is still passive, for in such circumstances deducing the Rp from the ρ value would yield wholly erroneous results.
4 Propagation period model
Once the corrosion rate is quantified, the effect of any variable that alters it [47], such as inhibitors or the presence of moisture in carbonated or chloride-bearing concrete, can be studied, the condition of a real structure can be assessed, and the propagation period [48] quantified by integrating the value over time.
On those grounds and bearing in mind the structural consequences of corrosion [49] the Icorr values can be classified into four levels as shown in Fig. 25 and the difference between instantaneous corrosion rate, Icorr, and cumulative corrosion penetration, Pcorr (6), can be expressed.

Quantification of the propagation period from several corrosion rates
Icorr values of over 100 µA/cm2, equivalent to a diameter loss of over 1 mm/year, are extremely high, for the very highest values measured in concrete [48] are on the order of 100–200 µA/cm2. Those should consequently be the maxima used to artificially accelerate corrosion with an external current [33]. Tests with Icorr > 200 µA/cm2 create unrealistic conditions that yield types of rust differing from those actually found due to the high acidity generated by such high currents.
Integrating Icorr over time has also provided insight into the effect of climate, leading to the establishment of patterns and the definition of the yearly representative corrosion rate, Icorr,REP. Figure 26 reproduces an example of instantaneous corrosion rate in carbonated concrete over several years [50] and Fig. 25b the respective cumulative corrosion penetration values, Pcorr. The graphs in Fig. 25c, d show the same parameters for concrete with chlorides and the Fig. 25e, f are of concretes submerged in seawater.

a Evolution of the instantaneous corrosion rate of a carbonated specimen, b Accumulated corrosion penetration (mm) of the carbonated specimen, c evolution of the instantaneous corrosion rate of a specimen mixed with 3% CaCl2 and d accumulated corrosion of the specimen in c, e evolution of the instantaneous corrosion rate of specimen immersed in sea water, f accumulated corrosion of the specimen in sea water
The inference drawn from these behaviours is that corrosion can evolve over time presenting different trends. Either the corrosion evolves linearly (Fig. 25 line A) or accelerately (line C) or it may attenuate (line B). The difference can be impacted by cracking. In carbonated concrete, once formed, the cracked region on the steel surface remains dry longer than the uncracked zones (Fig. 24a, b). When cracks form in underwater structures, the opposite occurs, for the ingress of seawater across the cracks accelerates corrosion (Fig. 24e, f). In summary, the propagation period can be expressed as a linear relationship between time and Pcorr when Icorr is constant, or as two bilinear periods that may also be expressed mathematically as exponential or potential, (Fig. 27).

Bilinear trend of the propagation period [51]
A Representative yearly corrosion rate, Icorr, REP can be obtained from the slope of the evolution of Pcorr with time. In the linear trend the value will be unique when in the bilear trend, there would be two representative corrosion rate.
4.1 Diameter loss and structural implications
Although the relationship between corrosion and structural decay is not addressed in this paper, diameter loss in reinforcement is acknowledged as the basic parameter for quantifying the loss of bearing capacity (Fig. 28) [43]. In future, this link should be used for developing better models of corroded structures in order to optimize the calculation of the residual safety.

Structural decay curve in Manual Contecvet [43]
5 Examples of use of corrosion rate measurements
Three examples are presented for the practical application of the principles described before.
- 1.
Verification of the efficacy of cathodic protection
- 2.
Accelerated service life testing: diffusion coefficient, chloride threshold and corrosion rate in concrete; inhibitor efficacy
- 3.
Corrosion resistance of new binders.
5.1 Verification of the efficacy of cathodic protection
The uncertainties associated with the verification of cathodic protection efficacy are well known, for none of the methods tested to date ensures that the steel has repassivated or corrosion has been reduced to negligible levels. The methods most commonly used are:
‘instant-off’ potential measurement
potential change after 4 h of depolarisation
24 h depolarisation potential.
Efficacy can be accurately measured, however, if in addition to any of these methods, Rp is measured on site. Icorr should be measured by suitably confining the current (modulated confinemenet). The condition can be measured when the steel is disconnected from the power source. Depolarising after 4–24 h, its Icorr value indicates whether or not the steel is passive. Figure 29 illustrates the case of a bridge. In the left of the figure it is represented the decay in potential registered when depolarizing after 6 months with cathodic protection. The decay is smaller than the required (100 mV) while in the right of the figure is represented the Icorr before and after the 6 months. The values of Icorr have however shown a clear decrease in the corrosion rate.

Left: Depolarizing decay after 6 months of application of cathodic protection. Right: the values of corrosion rate before and after 6 months of application of the PC
5.2 Accelerated service life testing: diffusion coefficient, chloride threshold and corrosion rate in concrete; inhibitor efficacy
Accelerated testing to determine the service life of a concrete mix is one of the challenges facing specialists today. The procedure normally involves testing the mix for chloride penetration, either under accelerated or natural diffusion, to find the diffusion coefficient (Dap). That, in conjunction with the respective age factor value, the surface concentration and an assumed chloride threshold, can be used to predict steel depassivation time.
Present tests [52] deliver only the value of, coefficient Dap and of surface concentration: all the others are assumed. An accelerated ‘integral’ method has been developed [53] to find the values of the service life prediction parameters determining:
surface concentration (likewise derivable from other tests)
the chloride threshold
corrosion rate after depassivation.
The method, now standardised in Spain [54], consists in embedding one bar each in two cubic concrete specimens and conducting an accelerated chloride migration test by applying 12 V between two electrodes until the steel depassivates, as shown in Fig. 30). The subsequent steps are as follows.

Set up for the accelerated Integral method for chloride ingress testing
The drop in exterior voltage is interrupted in one of the specimens, allowing corrosion to proceed for at least 15 days to measure the Icorr values that appear spontaneously.
The other specimen is split open to:
Extract a sample of the concrete in contact with the chloride solution from which to find the chloride surface concentration prevailing during the test
Observe the corrosion induced in the steel by the chloride front visualised by spraying the material with silver chloride
Extract a sample of the concrete/steel interface to determine the (threshold) chloride content that induced such corrosion.
The time to depassivation is then used to calculate the apparent diffusion coefficient (Fig. 31) through the application of the equation of Table 3. Figure 31 also shows the chloride threshold measured.
Fig. 31 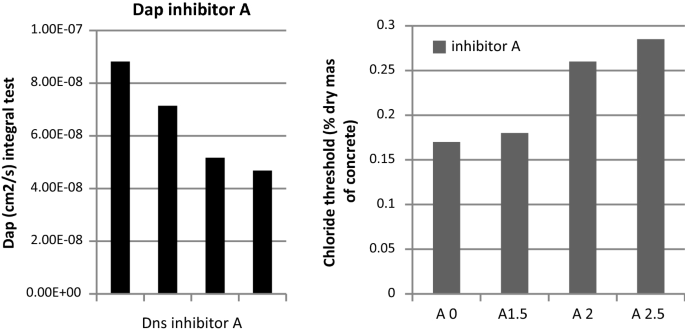
Values of (left) Apparent Diffusion coefficient and chloride threshold (right) for different concentration of inhibitor. A0 is the reference without inhibitor
Table 3 Equations to calculate the Dap from the integral test [53]

Figure 32 shows the registration of the corrosion rate Icorr with time from the initiation of the test. In the figure several concentrations of the same inhibitor are compared. It is evident that A0 with no inhibitor presents the earliest depassivation while the rest present later depassivation times as the inhibitor concentration increases. The findings were reasonable and while the long-term results regarding inhibition power are unknown, comparisons that help classify the starting materials in the concrete may be drawn from the short-term data. Until now it was found that the values obtained with this accelerated test are consistent with those found with the natural diffusion test [55].

Values of Icorr with time recorded during the integral test which show that the A0, without inhibitor, depassivates first and the increasing concentrations of inhibitor give progressive later depassivation times
This accelerated method delivers more experimental parameters than the diffusion coefficient test alone. More specifically, it constitutes an accelerated method for determining critical chloride content.
5.3 Corrosion resistance in new binders
The intense efforts presently being made to lower the clinker content in cement as a way of reducing its carbon footprint entail the use of additions of different types. The smaller proportion of portlandite attendant upon the reduction in clinker content translates into less effective buffering of the decline in pH induced by corrosion. As noted earlier, the buffering capacity of the so-called ‘alkaline reserve’ in cement plays a pivotal role in long-term concrete durability.
Steel passivation capacity or corrosion resistance in any type of cement, including new binders with much less clinker, has traditionally been studied on the grounds of electrochemical techniques. The methodology was exemplified in [55].
A thorough study of corrosion resistance calls for specific passivation tests and plotting polarisation curves for different amounts of chlorides, a discussion of which lies outside the object of this article. Be it said, however, the comparison with a type I reference cement can be made by testing the chloride and carbonation resistance.
Chloride resistance can be determined with the ‘integral’ method described above. Mortars or concretes can be prepared with the cements to be tested, comparing with a cement type I the Dap’s, the chloride thresholds, the surface concentrations and the corrosion rates.
Carbonation resistance calls for two types of specimens.
Cubic non-reinforced specimens are used to observe carbonation penetration in natural or accelerated (≤ 3% CO2) tests.
The second test type comprises reinforced specimens with a thin (around 0.5 cm) concrete cover to shorten the time required for carbonation to reach the steel/concrete interface. CO2 concentrations of up to 20% may be applied for accelerated carbonation, as the aim is full carbonation rather than rate comparison. After the specimen is fully carbonated, it is exposed to high humidity (submerged conditions) to induce corrosion. The Icorr values observed for the new binders are then compared to those found for a type I reference cement.
Experiments of this nature are summarised in [55] and Fig. 33. Reports of resistance to depassivation must specify whether chloride or carbonation testing was involved. Concrete resistivity and cement buffering capacity are both instrumental in chloride-mediated depassivation. In contrast, buffering capacity is the predominant mechanism for resisting the effects of carbonation.

Scheme of testing new binders
6 Final comments
Steel corrosion takes place in concrete in much the same way as in the atmosphere or the soil, although cement generates a unique alkaline medium. Corrosion is triggered by local carbonation or chloride attack-mediated acidification and proceeds primarily via microcells. Corrosion zones in concrete, even where involving a single pit, are not pure anodes connected to a cathode. Galvanic currents between corrosion and passive zones are not equivalent to corrosion rate and in fact account in general for a very small percentage of that rate. Their measurement may therefore detect only part of the process.
Oxygen is needed to trigger corrosion in concrete, but as tenths of ppm suffice for localised corrosion where chloride content is high, the process may occur even underwater. Less aeriated zones cannot therefore be said to be immune to corrosion. Oxygen is not required for corrosion to proceed (due to water hydrolysis and the reduction of the oxides formed), although its presence intensifies the process, which is enhanced by the sum of all the cathodic processes.
Concrete was one of the first systems in which the instantaneous corrosion rate, also known as ‘polarisation resistance’ and ‘linear polarisation’ (alluding to the fact that a small alteration in potential is used to induce a linear increase in current), was measured. The technique has been widely used ever since it was described in the ASTM G59-97 (2014) standard for all metal/electrolyte systems. Attempts to discredit it have been unable to disprove its good correlation with weight losses simultaneously measured by gravimetry and converted using Faraday’s law. At this writing its direct current version (more readily interpreted than as electrochemical impedance) continues to be the easiest and most convenient technique for quantifying measuring corrosion in any metal/electrolyte system.
The application of Rp to real structures is influenced by the quasi-infinite area of the reinforcement spraying longer than the auxiliary electrode border: inasmuch as the actual area of the metal polarised by the signal must be known, the area of the auxiliary electrode is not the correct reference. That circumstance necessitates specific devices in which measurement is based on modulated current confinement or in measuring the attenuation of potential with the distance from the auxiliary electrode border. Although a method for contactless corrosion measurement was published in 2001, to date no market device has proven able to take accurate measurements in large-scale structures (despite claims to the contrary), for the area affected has not yet been determined nor have studies on its calibration been forthcoming.
Periodic measurement of corrosion rate has provided the grounds for calculating cumulative corrosion and proposing corrosion propagation period models. In outdoor structures, seasonal cycles can be applied to show that corrosion may be constant if expressed as a yearly mean (Icorr,REP). In ‘resistance control’ of corrosion, also shown to exist in concrete, the rate is proportional to resistivity. This provides grounds for certain analogies, such as deducing corrosion rate from resistivity (under conditions of active corrosion) or relating it to the effective diffusion coefficient.
An incorrect diagnosis of the causes of corrosion and how it proceeds is known to be one of the major reasons for the limited durability of concrete repairs. Corrosion rate measurement is essential to studying and diagnosing progression of this process in a given structure. Activity in each zone of the structure can only be reliably determined by measuring corrosion rate. Engineers engaging in the repair of deteriorated structures must acquire more specialised knowledge to ensure effective intervention. Structural behaviour is in itself complex, but the basic processes triggering corrosion are much more complex than the mere presence of a corrosion cell with an anode and cathode. Another challenge to be confronted is to understand the structural behaviour of concrete with microcracks around the reinforcement, for with the concomitant impact on its bond with the steel it ceases to behave as a composite material.
Intense basic research on reinforcement corrosion will continue to be needed in the years to come to fill the existing gaps in the knowledge of its effect on structural behaviour and, using the findings, to develop calculation tools to accurately assess the residual strength and hence the safety of the structures concerned.
Change history
16 December 2019
The article Propagation of reinforcement corrosion: principles, testing and modelling, written by Carmen Andrade, was originally published electronically on the publisher’s internet portal (currently SpringerLink) on 26 December 2018 without open access.
References
Isecke B (1983) Failure analysis of the collapse of Berlin Congress Hall. In: Crane AP (ed) Corrosion of reinforcement in concrete, chapter 5. Elseiver, Amsterdam, p 79
Schiessl P (1989) Corrosion of Steel in Concrete. Report of RILEM TC 60-CSC RILEM, Chapman & Hall, New York
Tuutti K (1982) Corrosion of steel in concrete. Swedish Cement and Concrete Institute (CBI), Stockholm. pp 4–82
Elsener B, Andrade C, Gulikers J, Polder R, Raupach M (2003) RILEM TC 154-EMC: electrochemical techniques for measuring metallic corrosion half-cell potential measurements—potential mapping on reinforced concrete structures. Mater Struct 36:461–471
Polder R, Andrade C, Elsener B, Vennesland O, Gulikers J, Weidert R, Raupach M (2000) Test methods for on-site measurement of resistivity of concrete”, RILEM TC 154-EMC: electrochemical techniques for measuring metallic corrosion. Mater Struct 33:603–611
Andrade C, Alonso C, Gulikers J, Polder R, Cigna R, Vennesland Ø, Salta M, Raharinaivo A, Elsener B (2004) RILEM TC 154-EMC: electrochemical Techniques for Measuring Metallic Corrosion. Recommendations Test methods for on-site corrosion rate measurement of steel reinforcement in concrete by means of the polarization resistance method. Mater Struct 37(273):623–643
Vennesland Ø, Raupach M, Andrade C (2007) Recommendation of Rilem TC 154-EMC: ‘‘electrochemical techniques for measuring corrosion in concrete- measurements with embedded probes”. Mater Struct 40:745–758
Castellote M, Andrade C (2006) Round-Robin test on methods for determining chloride transport in concrete. RILEM recommendation of TC-178-” testing and modeling chloride penetration in concrete”. Mater Struct 39:990–995
Page CL, Treadaway KWJ (1982) Aspects of the electrochemistry of steel in concrete. Narute 297(5862):109–115
Gouda VK (1966) Anodic polarization measurements of corrosion and corrosion inhibition of steel in concrete. Br Corros J 2:13
Kaesche H (1959) Testing corrosion danger of steel reinforcement due to admixtures in concrete. Zement-Kalk-Gips 7:289
Pedeferri P (1989) Corrosione e protezione delle strutture metalliche e in cemento armato negli ambienti naturali-, Clup-Milano. Piazza Leonardo da Vinci, 32, Milano
Cigna R (1966) Studio sula corrosione dei ferri affogarti in malte cementicie effettuato mediante curve i polarizzacione. L’Industria Italiana del cementpo, p 740
Whiting D (1981) Rapid determination of the chloride permeability of concrete-, Report no. FHWA-RD-81-119, NTIS DB no. 82140724, Federal Highway Administration, Washington, DC, p 174
Pourbaix M (1973) Lectures in electrochemical corrosion. Plenum Press, New York
Evans UR (1948) Metallic corrosion, passivity and protection. Longmans Green and Co, New York
Galvele J (1983) Pitting Corrosion-Treatise on Materials Science and Technology 23:1–57
Pickering HW, Frankenthal RP (1972) On the mechanism of localized corrosion of iron and stainless steel: I. Electrochem Stud J Electrochem Soc 119(10):1297–1304
Sanchez J, Fullea J, Anmdrade C, Gaitero JJ, Porro A (2008) A study of the early corrosion of a high strength steel in a diluted sodium chloride solution. Corros Sci 50:1820–1824
Mansfeld F (1997) Potential distribution in the Evans drop experiment. Corros Sci 39(2):409–413
Andrade C, Rodriguez-Maribona I (1992) Macrocell versus microcell corrosion of reinforcement placed in parallel. Corrosion- NACE, paper no 194
Andrade C, Rodriguez-Maribona I, Feliu S, Gonzalez JA, Feliu S Jr (1992) The effect of macrocells between active and passive areas of steel reinforcements. Corros Sci 33(2):237–249
Andrade C, Fullea J, Toro L, Martinez I, Rebolledo N (2012) Reinforcement corrosion in chloride media in absence of oxygen. RILEN event TC -226 CNM- Long term oerformance of cementitous barriers and reinforced concrete in Nuclear Power Plant and Radioactive Waste Storage and Disposal- NUCPERF - Cadarache- France
Alonso C, Castellote M, Andrade C (2002) Chloride threshold dependence of pitting potential of reinforcements. Electr Acta 47:3469–3481
Izquierdo D, Alonso C, Andrade C, Castellote M (2004) Potentiostatic determination of chloride threshold values for rebar depassivation. Experimental and statistical study. Electrochim Acta 49:2731–2739
Joiret S, Keddam M, Nóvoa XR, Pérez MC, Rangel C (2002) Use of EIS, ring-disk electrode, EQCM and Raman spectroscopy to study the film of oxides formed on iron in 1 M NaOH. Cem Concr Compos 24(1):7–15
Stratmann M, Bohnenkamp K (1983) Engell, H-J- Electrochemical study of phase-transitions in rust layers. Corros Sci 23(9):969–985
Albani A, Gassa LM, Zerbino JO, Vilche JR, Arvia AJ (1990) Comparative study of the passivity and the breakdown of passivity of polycrystalline iron in different alkaline solutions. Elecfrochimica Acta 35(9):1437–1444
Thierry D, Persson D, Leygraf C, Boucherit N, Hugot-le Goff A (1991) Raman spectroscopy and XPS investigations of anodic corrosion films formed on Fe-Mo alloys in alkaline solutions. Corros Sci 32(3):273–284
Andrade C, Fullea J, Alonso C (1999) The use of the graph corrosion rate-resistivity in the measurement of the corrosion current. In: Rilem Proceedings no. 18: Measurement and Interpretation of the on-Site Corrosion Rate. pp 157–165
Andrade C, Pazini E (2012) Opciones teóricas de actuación en la marquesina del estadio de Maracana- PATORREB-2012- 4º Congreso de Patologia y Rehabilitacion de edificios-Santiago de Compostela
Alonso C, Andrade C, Rodriguez J, Díez JM (1998) Factors controlling cracking of concrete affected by reinforcement corrosion. Mater Struct 31:435–441
Andrade C, Alonso C, Molina FJ (1993) Cover cracking as a function of rebar corrosion: Part I - Experimental test. Mater Struct 26:453–464
Torres-Acosta AA, Sagüés AA (2004) Concrete Cracking by Localized steel corrosion. Geometric effects. ACI Mater J 101:501–507
Andrade C, Keddam M, Novoa XR, Perez MC, Rangel CM, Takenouti H (2001) Electrochemical behavious of steel rebars in concrete: influence of environmental factors and cement chemistry. Electrochim Acta 46:3905–3912
Andrade C, García S, Toro L, Alonso C, Castellote M (2006) Reinforcement corrosion in chloride environment of different concentrations. Concrete Durability and Service Life Planning (ConcreteLife’06)- Ein Bokek, Israel
Andrade C, Gónzalez JA (1978) Quantitative measurements of corrosion rate of reinforcing steels embedded in concrete using polarization resistance measurements-. Werkst Korros 29:515
Stern M, Geary AJ (1957) Electrochemical polarization: I. A. theoretical analysis of the shape of polarization curves. Electrochem Soc 104(1):56–63
González JA, Algaba S, Andrade C (1980) Corrosion of reinforcing bars in carbonated concrete. Br Corros J 3:135–139
Andrade C, Rebolledo N (2009) Corrosion of reinforced concrete made with different binders and exposed for 20 years in natural sea water. In: 2nd international RILRM workshop on concrete durability and service life planning- ConcreteLife’09- Haifa, Israel
Garces P, Andrade C, Saez A, Alonso MC (2005) Corrosion of reinforcing steel in neutral and acid solutions simulating the electrolytic environments in the micropores of concrete in the propagation period. Corros Sci 47:289–306
Andrade C, Martinez I, Alonso C, Fullea J (2001) New advanced electrochemical techniques for on site measurements of reinforcement corrosion. Mater Constr 51(263–264):97–107
Andrade C, Sanchez J, Martinez I, Rebolledo N (2011) Analogue circuit of the inductive polarization resistance. Electrochim Acta 56:1874–1880
Feliú S, González JA, Feliú S Jr, Andrade C (1990) Confinement of the electrical signal or in situ measurement of polarization resistance in reinforced concrete. ACI Mater J 87:457
Feliú S, González JA, Andrade C (1996) Multiple-electrode method for estimating the polarization resistance in large structures. J Appl Electrochem 26:305–309
Andrade C (1993) “Calculation of chloride diffusion coefficients in concrete from ionic migration measurements”. Cement Concr Res 23:724–742
Andrade C, Sanchez J, Fullea J, Rebolledo N, Tavares F (2012) On-site corrosion rate measurements:3D simulation and representative values. Mater Corros 63(12):1154–1164
Andrade C (2017) Reliability analysis of corrosion onset: initiation limit state. J Struct Integr Maint 2(4):200–208
CONTECVET -A validated user’s manual for assessing the residual life of concrete structures, DG Enterprise, CEC, (2001). (The manual can be downloaded from the web site of http://www.ietcc.csic.es/index.php/es/publicaciones-2/manual-contecvet
Andrade C, Castillo A (2010) Water content of concrete in natural atmospheres and its impact in the corrosion parameters. In: International RILEM conference on material science- MATSCI, Aachen-Germany. Springer, Berlin. vol II, pp 43–51
Andrade C, Rebolledo N (2017) Generic modelling of propagation of reinforced concrete damage. In: Proceedingsfib Symposium -Maastricht-The Netherland
CEN/TS 12390-11: 2013: Testing hardened concrete. Determination of the chloride resistance of concrete. Unidirectional diffusion
Andrade C, Rebolledo N (2014) Accelerated evaluation of chloride corrosion by means of the integral test. Structural 186:09
UNE 83992-2-Concrete durability. Test methods. Chloride penetration tests on concrete. Part 2: Integral accelerated method. Structural 186, p 09 (2014)
Andrade C, Bujak R (2013) Effects of some mineral additions to Portland cement on reinforcement corrosion. Cem Concr Res 53:59–67
Acknowledgements
The author acknowledges the collaboration of all those contributed to the work presented here. The original research reported in this review paper was supported by the following grants: public Agencies belonging to the several Ministries of Research of Spain, the European Commission (BRITE-EURAM and Marie-Curie -Nanocem grant) and the private firm GEOCISA (corrosion-rate meter). Also would like to mention Prof. José Calleja who had the vision to start the study of reinforcement corrosion from the end of 1960’s in the Institute of Construction Sciences of Spain.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that she has no conflict of interest.
Additional information
The original version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Andrade, C. Propagation of reinforcement corrosion: principles, testing and modelling. Mater Struct 52, 2 (2019). https://doi.org/10.1617/s11527-018-1301-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-018-1301-1