Pt Deposites on TiO2 for Photocatalytic H2 Evolution: Pt Is Not Only the Cocatalyst, but Also the Defect Repair Agent


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
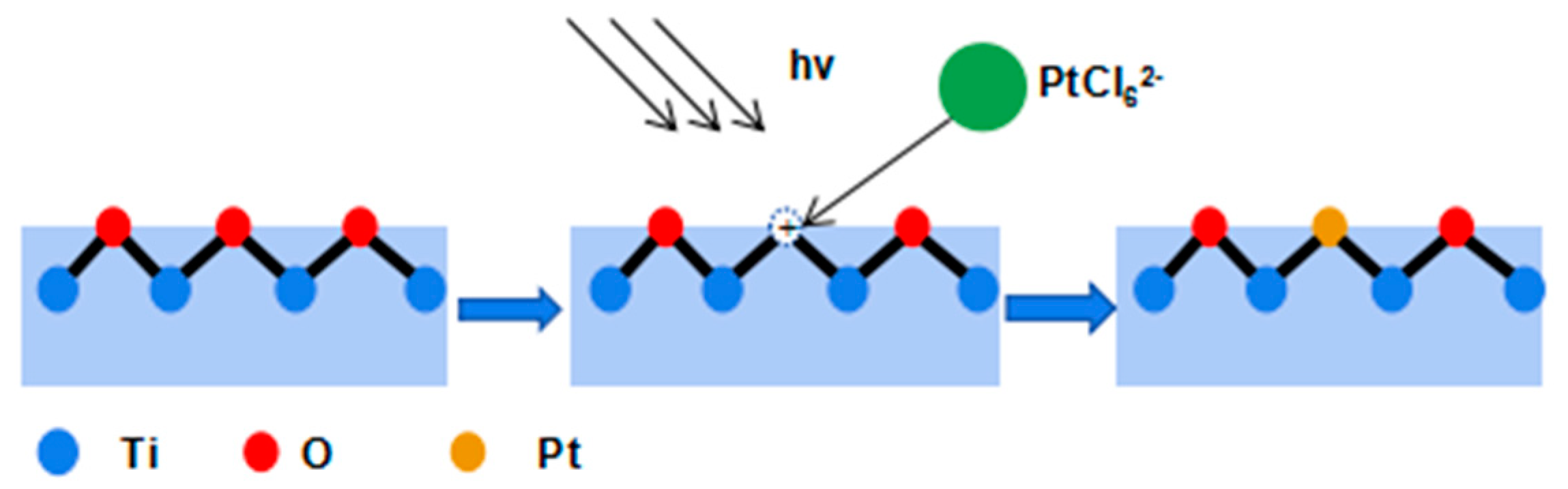{kind=link}
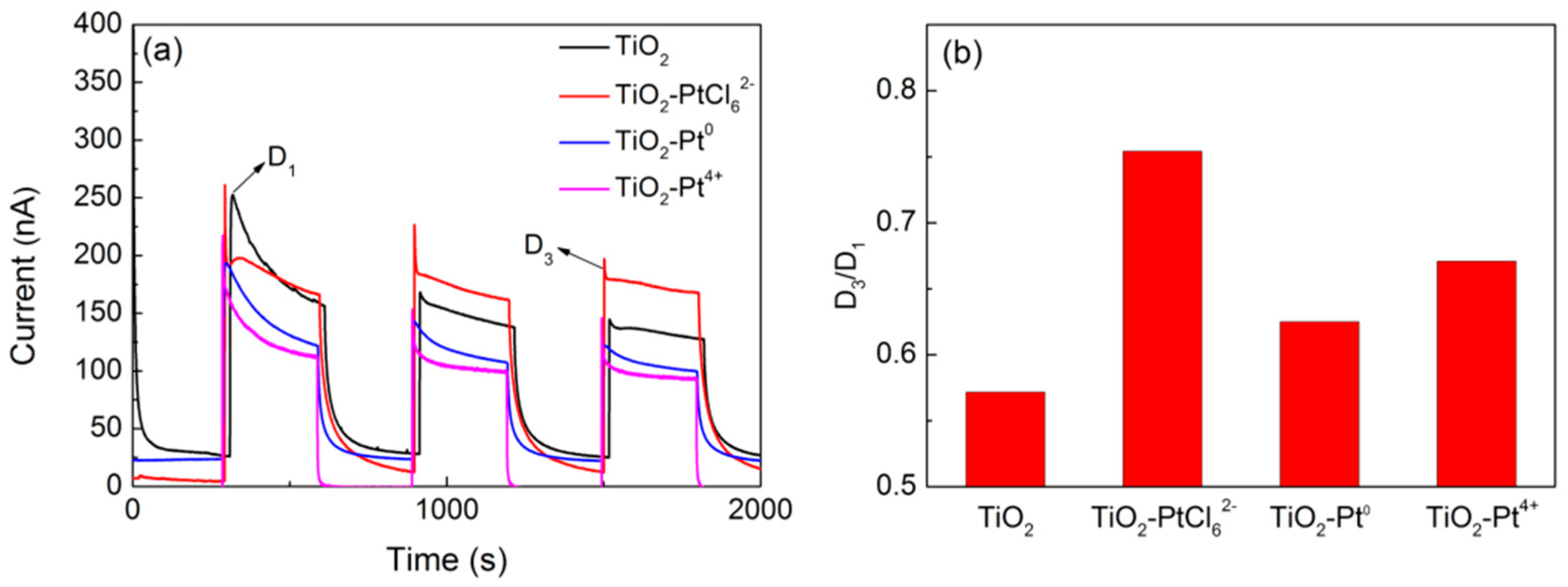{kind=link}
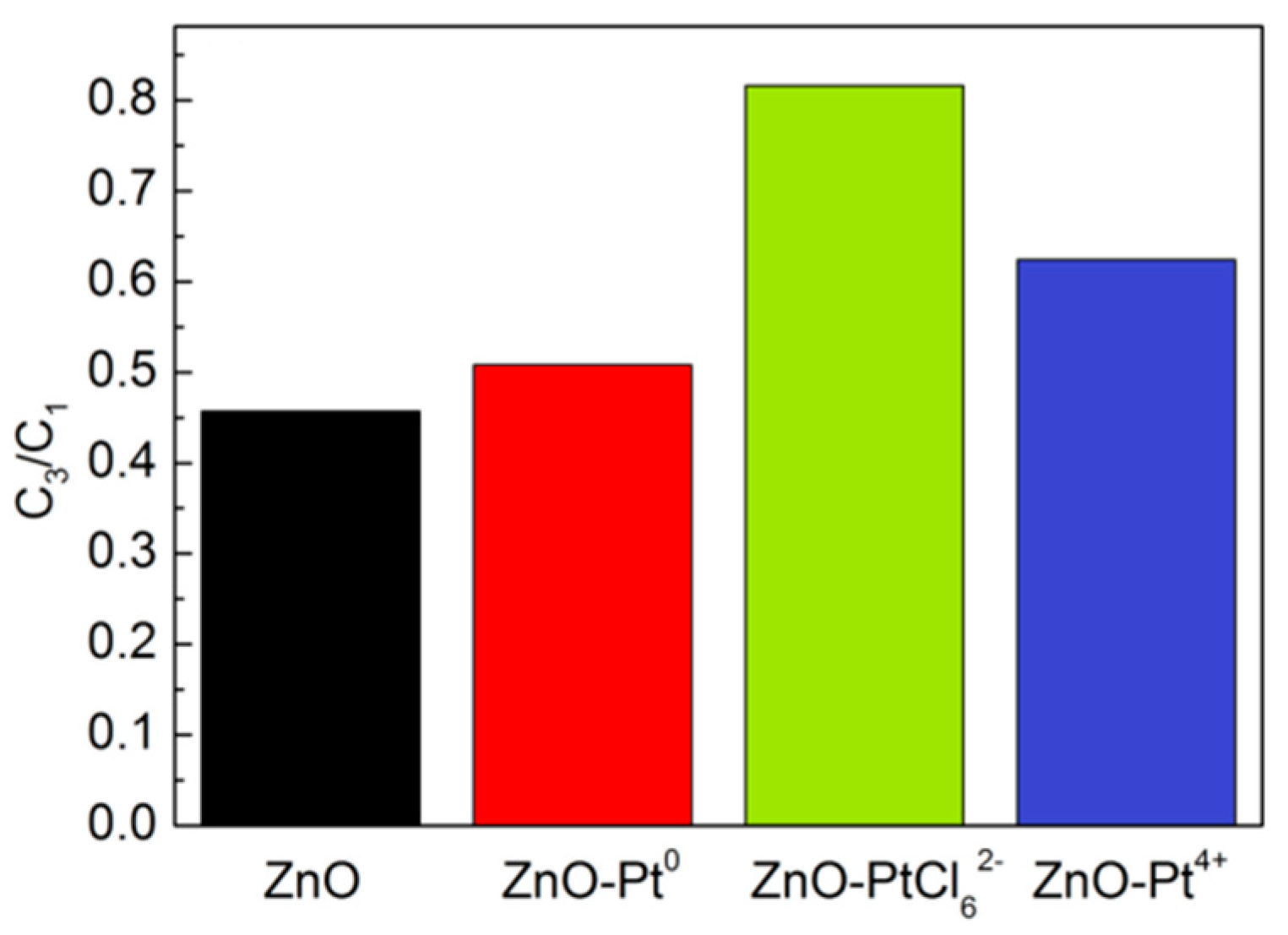{kind=link}
1. Introduction
2. Results and Discussions
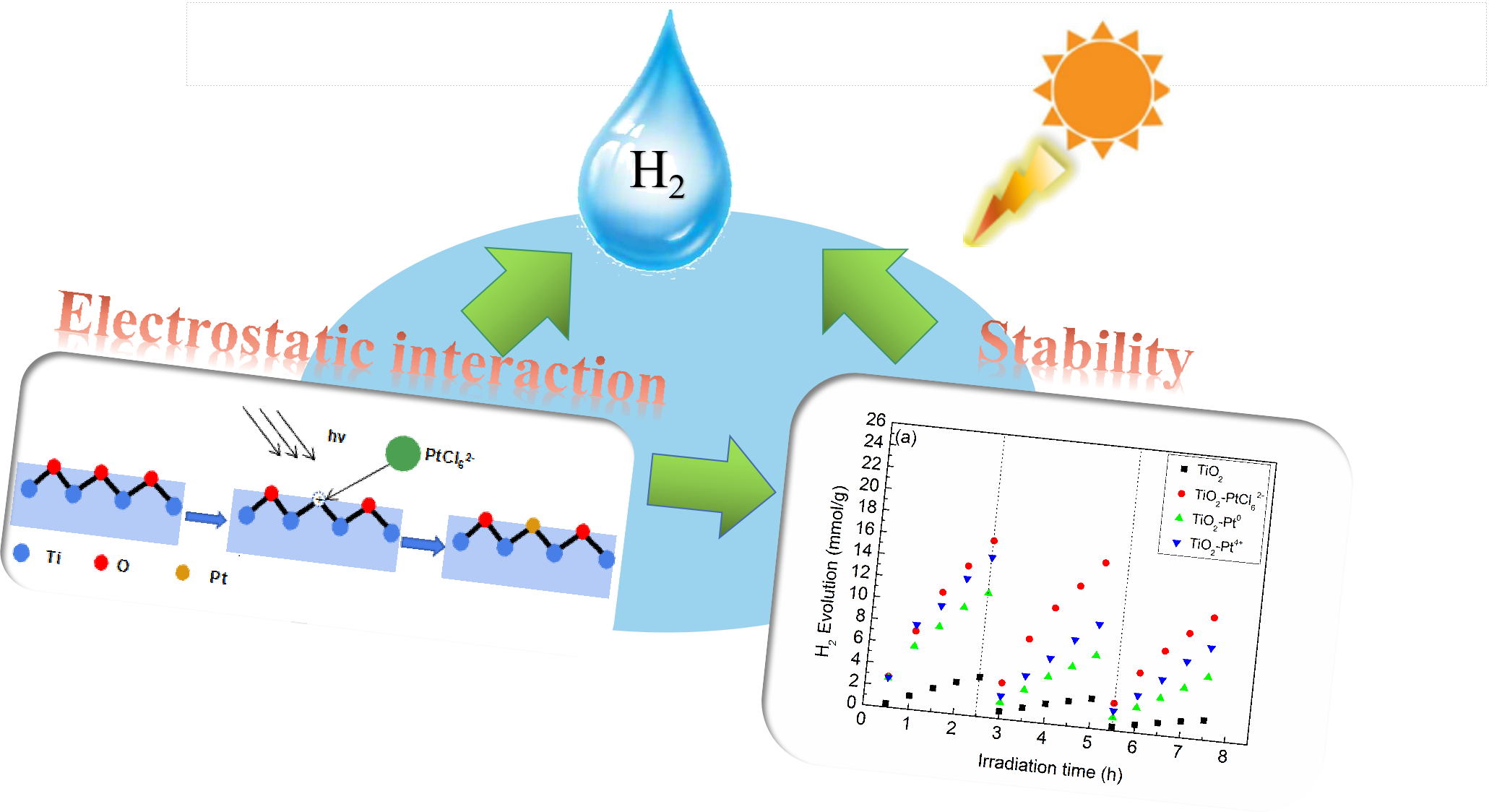
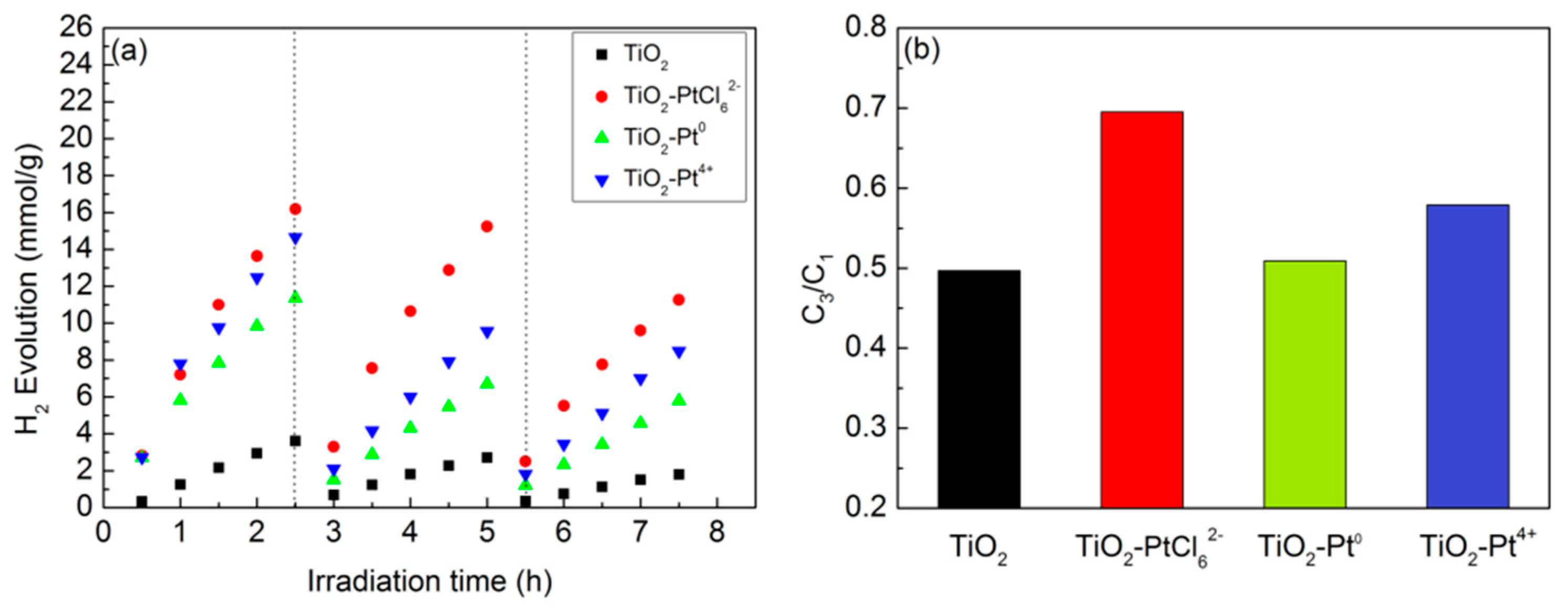
2.1. Photocatalytic H2 Evolution Activity
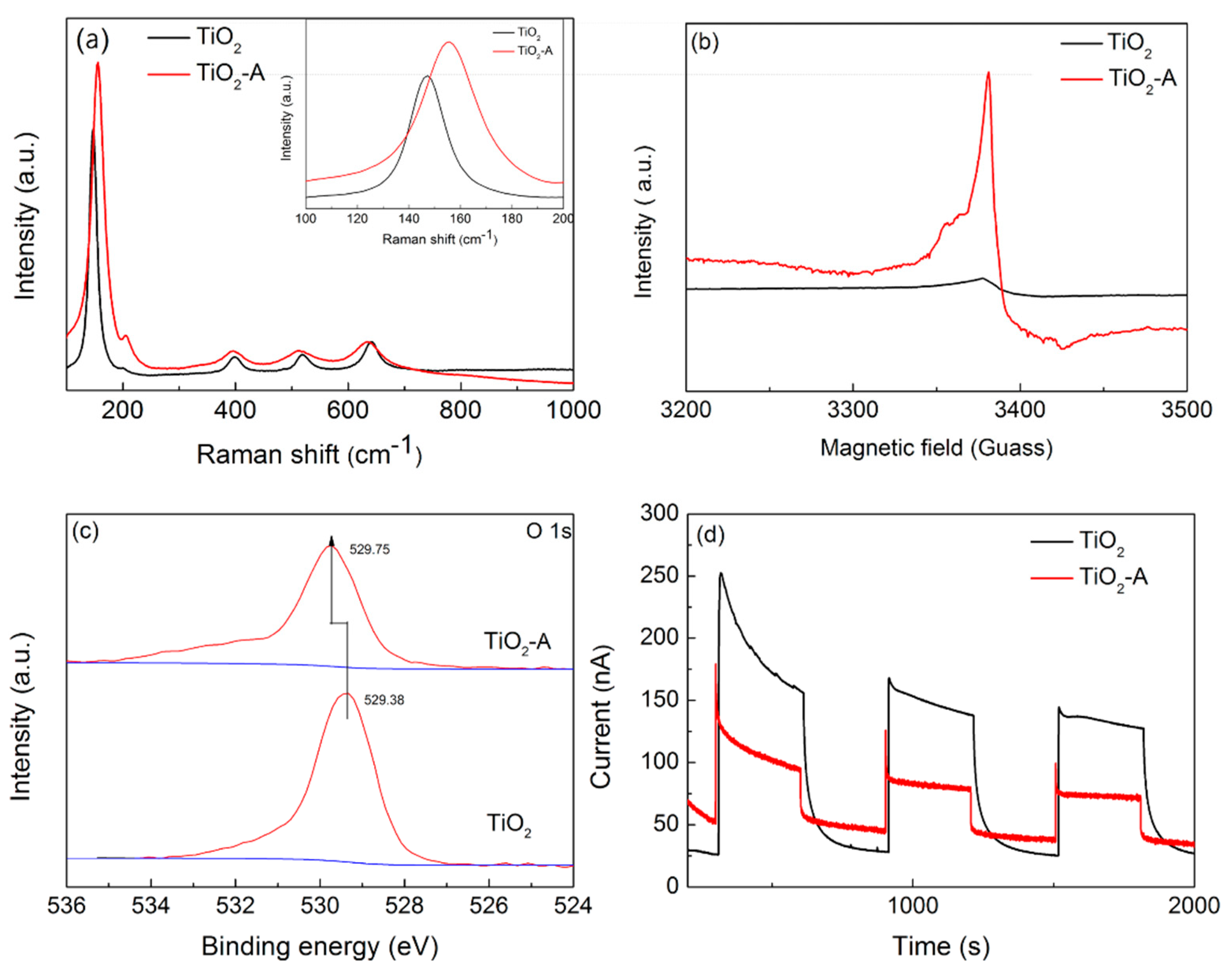
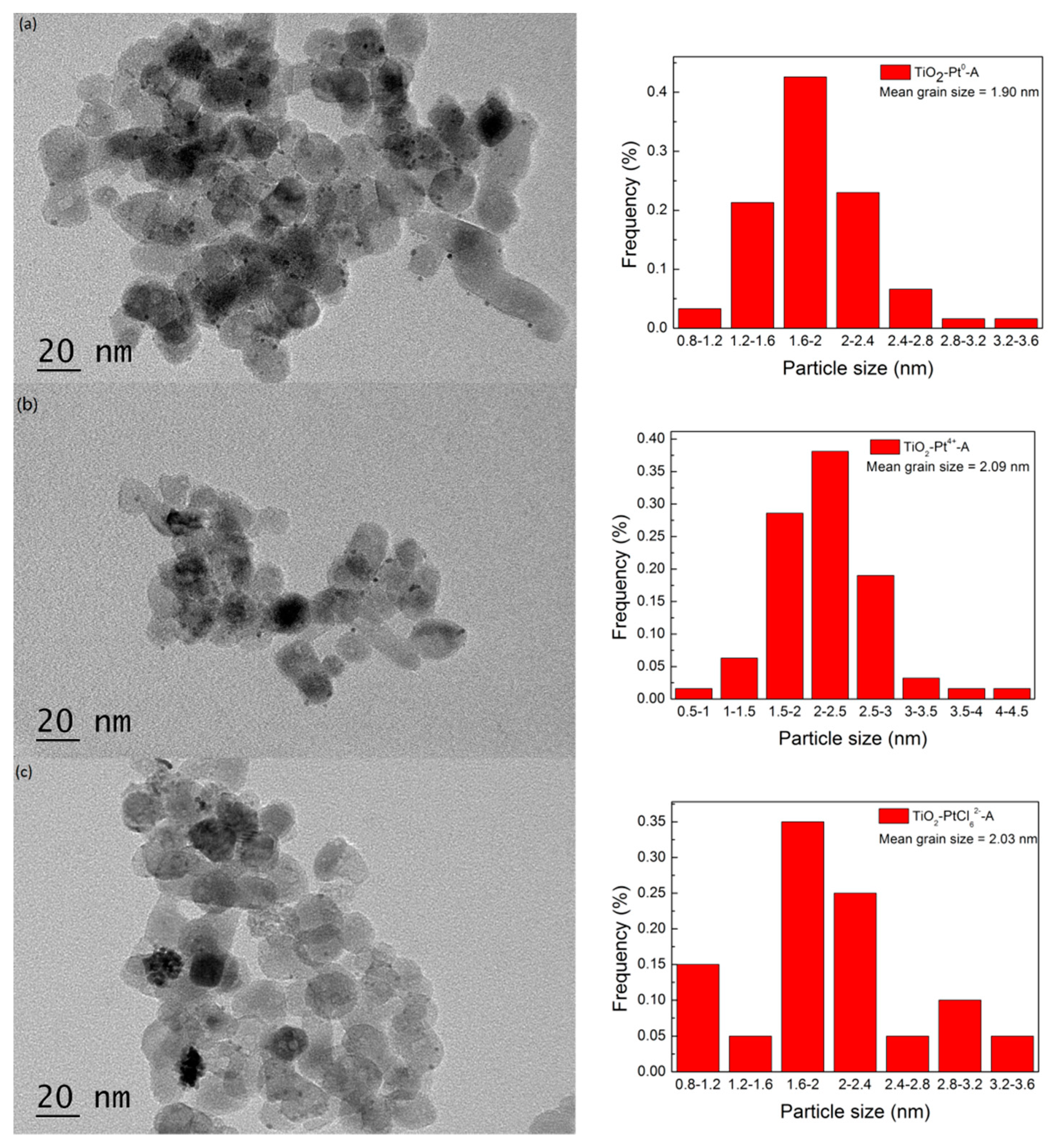
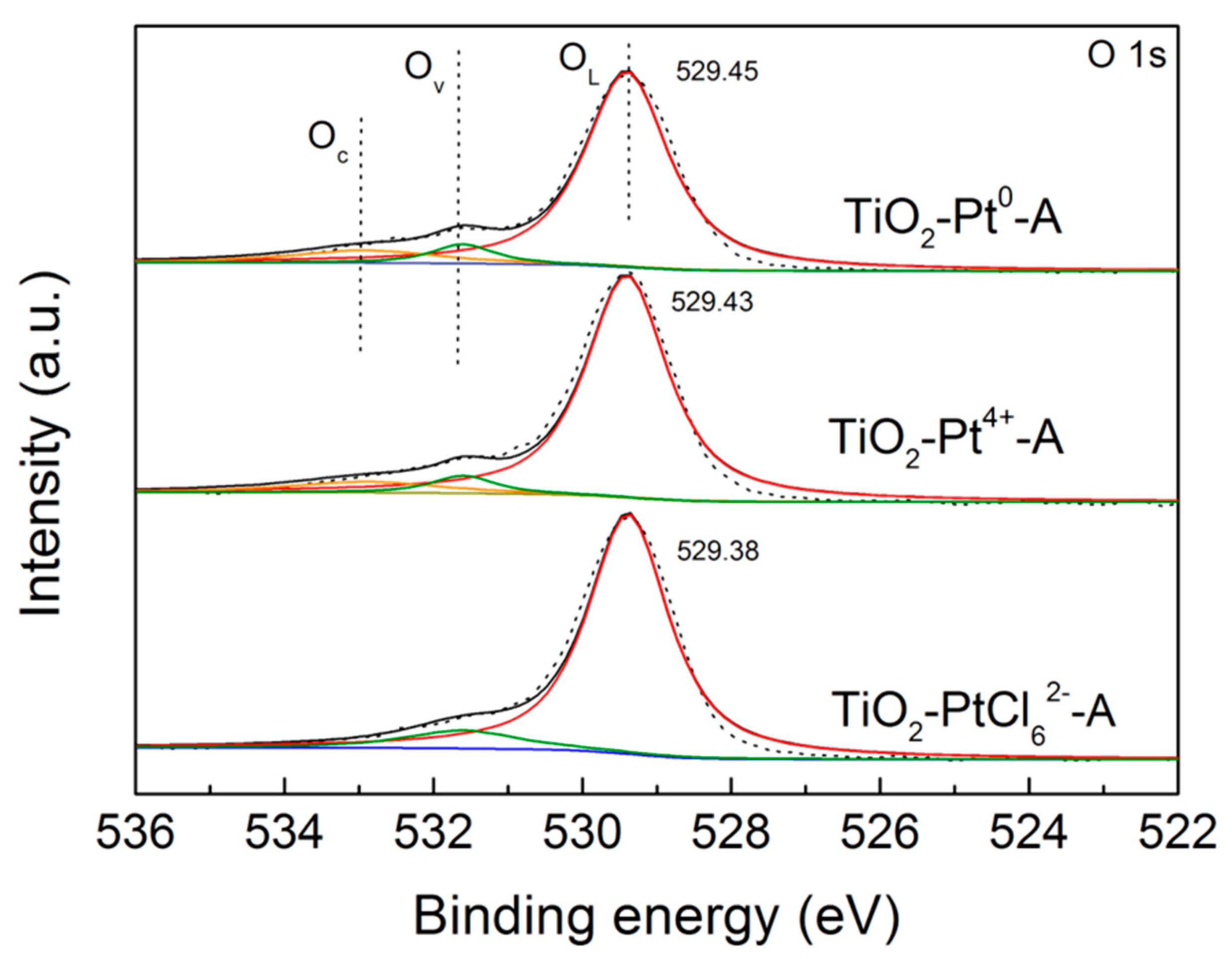
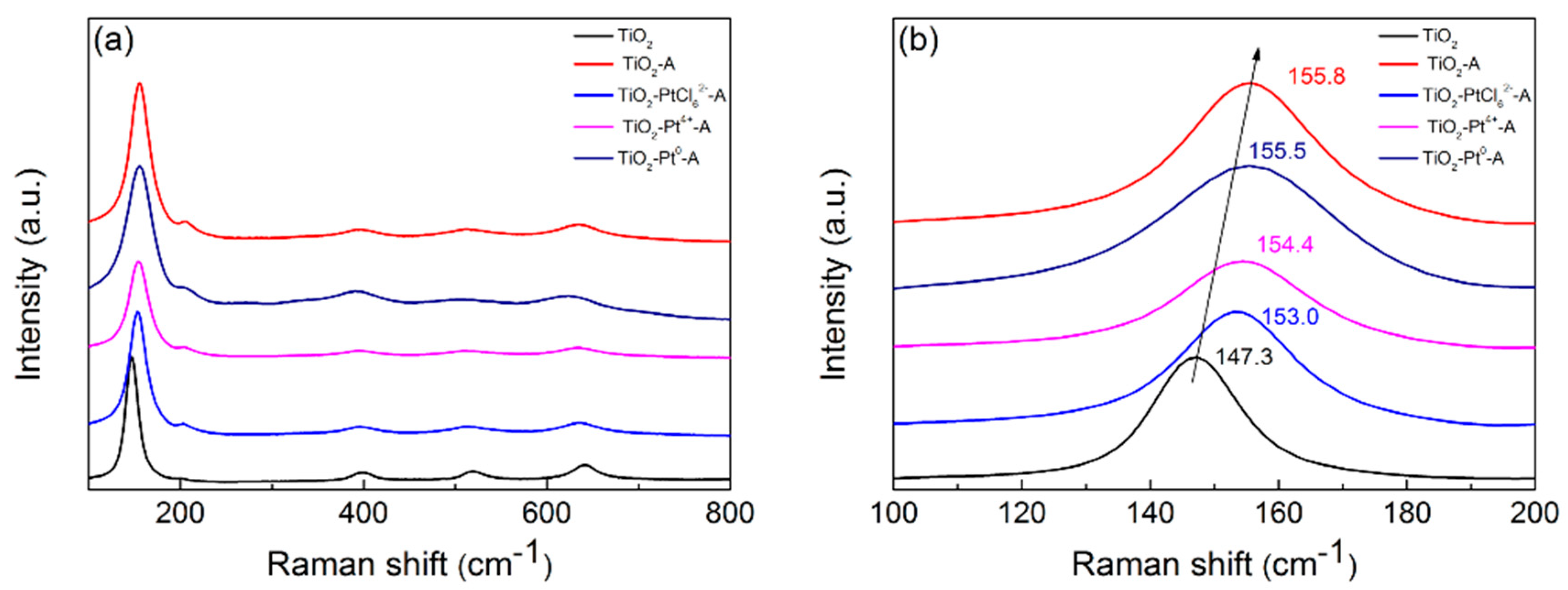
2.2. Changes of TiO2 Catalyst during Photocatalytic H2 Evolution
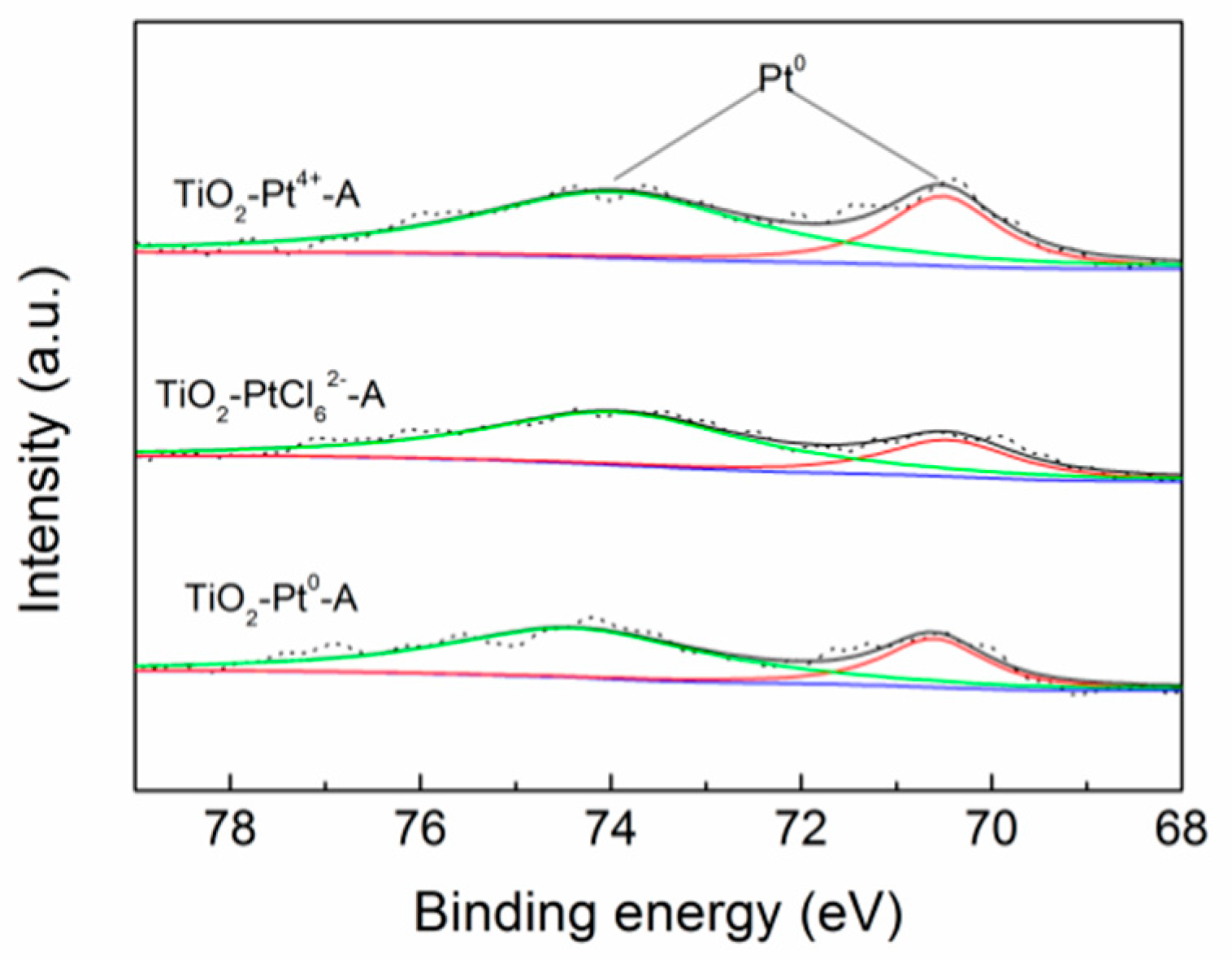
2.3. The Influences of Different Pt Sources on Photostability
3. Experimental Section
3.1. Materials
3.2. Preparation of Samples
3.2.1. TiO2-Pt0
3.2.2. TiO2-Pt4+ and TiO2-PtCl62−
3.3. Characterization
3.4. Photocurrent Tests
3.5. Catalytic Performances Measurement
3.5.1. TiO2 and TiO2-Pt0
3.5.2. TiO2-Pt4+ and TiO2-PtCl62
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nat. Lond. 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Wangac, D.; Ye, J.; Kako, T.; Kimura, T. Photophysical and photocatalytic properties of SrTiO3 doped with cr cations on different sites. J. Phys. Chem. B 2006, 110, 15824–15830. [Google Scholar] [CrossRef] [PubMed]
- Konta, R.; Ishii, T.; Kato, H.; Kudo, A. Photocatalytic activities of noble metal ion doped SrTiO3 under visible light irradiation. J. Phys. Chem. B 2004, 35, 8992–8995. [Google Scholar] [CrossRef]
- Zhang, L.J.; Li, S.; Liu, B.K.; Wang, D.J.; Xie, T.F. Highly eEfficient CdS/WO3 photocatalysts: Z-scheme photocatalytic mechanism for their enhanced photocatalytic H2 evolution under visible light. ACS Catal. 2014, 4, 3724–3729. [Google Scholar] [CrossRef]
- Wang, X.; Liu, G.; Chen, Z.-G.; Li, F.; Wang, L.; Lu, G.; Cheng, H.-M. Enhanced photocatalytic hydrogen evolution by prolonging the lifetime of carriers in ZnO/CdS heterostructures. Chem. Commun. 2009, 3452. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yin, W.-J.; Peng, K.-L.; Wang, K.; Hu, Q.; Selloni, A.; Chen, F.-R.; Liu, L.-M.; Sui, M. Self-hydrogenated shell promoting photocatalytic H2 evolution on anatase TiO2. Nat. Commun. 2018, 9, 2752. [Google Scholar] [CrossRef]
- Wang, M.; Cai, L.; Wang, Y.; Zhou, F.; Xu, K.; Tao, X.; Chai, Y. Graphene-draped semiconductors for enhanced photocorrosion resistance and photocatalytic properties. J. Am. Chem. Soc. 2017, 139, 4144–4151. [Google Scholar] [CrossRef]
- Hsieh, P.-Y.; Chiu, Y.-H.; Lai, T.-H.; Fang, M.-J.; Wang, Y.-T.; Hsu, Y.-J. TiO2 nanowire-supported sulfide hybrid photocatalysts for durable solar hydrogen production. ACS Appl. Mater. Interfaces 2018, 11, 3006–3015. [Google Scholar] [CrossRef]
- Yan, L.; Yang, F.; Tao, C.; Luo, X.; Zhang, L. Highly efficient and stable Cu2O–TiO2 intermediate photocatalytic water splitting. Ceram. Int. 2020, 46, 9455–9463. [Google Scholar] [CrossRef]
- Yang, Y.; Ling, Y.; Wang, G.; Liu, T.; Wang, F.; Zhai, T.; Tong, Y.; Li, Y. Photohole induced corrosion of titanium dioxide: Mechanism and solutions. Nano Lett. 2015, 15, 7051–7057. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Lin, J.; Wang, Y.; Xu, J.; Zhu, Y. Visible-light photocatalytic degradation of BiTaO4 photocatalyst and mechanism of photocorrosion suppression. J. Phys. Chem. C 2010, 114, 6472–6477. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, H.; Zong, R.; Zhu, Y. Photocorrosion suppression of ZnO nanoparticles via hybridization with graphite-like carbon and enhanced photocatalytic activity. J. Phys. Chem. C 2009, 113, 2368–2374. [Google Scholar] [CrossRef]
- Imanishi, A.; Okamura, T.; Ohashi, N.; Nakamura, R.; Nakato, Y. Mechanism of water photooxidation reaction at atomically flat TiO2 (Rutile) (110) and (100) surfaces: Dependence on solution pH. J. Am. Chem. Soc. 2007, 129, 11569–11578. [Google Scholar] [CrossRef] [PubMed]
- Weng, B.; Qi, M.-Y.; Han, C.; Tang, Z.-R.; Xu, Y.-J. Photocorrosion inhibition of semiconductor-based photocatalysts: Basic principle, current development, and future perspective. ACS Catal. 2019, 9, 4642–4687. [Google Scholar] [CrossRef]
- Sreethawong, T.; Yoshikawa, S. Comparative investigation on photocatalytic hydrogen evolution over Cu- Pd-, and Au-loaded mesoporous TiO2 photocatalysts. Catal. Commun. 2005, 6, 661–668. [Google Scholar] [CrossRef]
- Li, X.; Bi, W.; Zhang, Q.; Tao, S.; Chu, W.; Zhang, Q.; Luo, Y.; Wu, C.; Xie, Y. Single-atom Pt as co-catalyst for enhanced photocatalytic H2 evolution. Adv. Mater. 2016, 28, 2427–2431. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, D.; Lin, Y.; Liu, W.; Cao, L.; Liu, X.; Zhang, W.; Mou, X.; Fang, S.; Shen, X.; et al. Single Pt atom with highly vacant d-orbital for accelerating photocatalytic H2 evolution. ACS Appl. Energy Mater. 2018, 1, 6082–6088. [Google Scholar] [CrossRef]
- Xing, J.; Jiang, H.B.; Chena, J.; Li, Y.H.; Wu, L.; Yang, H.G.; Zheng, L.R.; Wang, H.; Hu, P.; Zhao, H.; et al. Active sites on hydrogen evolution photocatalyst. J. Mater. Chem. A 2013, 1, 15258–15264. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, Y.; Jiang, J.; Rong, Y.; Wang, Y.; Wu, Y.; Pan, C. Characterization of oxygen vacancy associates within hydrogenated TiO2: A positron annihilation study. J. Phys. Chem. C 2012, 116, 22619–22624. [Google Scholar] [CrossRef]
- Ünlü, M.S. Digital detection of nanoparticles: Viral diagnostics and multiplexed protein and nucleic acid assays. MRS Proc. 2015, 1720, 1720–1725. [Google Scholar] [CrossRef]
- Kuvarega, A.T.; Mamba, B.B. Double walled carbon Nanotube/TiO2 nanocomposites for photocatalytic dye degradation. J. Nanomater. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.; Arora, A.K.; Sridharan, V. Raman line shapes of optical phonons of different symmetries in anatase TiO2 nanocrystals. J. Phys. Chem. C 2009, 113, 16927–16933. [Google Scholar] [CrossRef]
- Choi, H.C.; Jung, Y.M.; Bin Kim, S. Size effects in the Raman spectra of TiO2 nanoparticles. Vib. Spectrosc. 2005, 37, 33–38. [Google Scholar] [CrossRef]
- Du, M.; Chen, Q.; Wang, Y.; Hu, J.; Meng, X. Synchronous construction of oxygen vacancies and phase junction in TiO2 hierarchical structure for enhancement of visible light photocatalytic activity. J. Alloys Compd. 2020, 830, 154649. [Google Scholar] [CrossRef]
- Mao, C.; Zuo, F.; Hou, Y.; Bu, X.; Feng, P. In Situ preparation of a Ti3+self-doped TiO2 film with enhanced activity as photoanode by N2H4 reduction. Angew. Chem. Int. Ed. 2014, 53, 10485–10489. [Google Scholar] [CrossRef]
- Wang, G.; Wang, H.; Ling, Y.; Tang, Y.; Yang, X.; Fitzmorris, R.C.; Wang, C.; Zhang, J.Z.; Li, Y. Hydrogen-treated TiO2 nanowire arrays for photoelectrochemical water splitting. Nano Lett. 2011, 11, 3026–3033. [Google Scholar] [CrossRef]
- Lin, L.; Ren, W.; Wang, C.; Asiri, A.M.; Zhang, J.; Wang, X. Crystalline carbon nitride semiconductors prepared at different temperatures for photocatalytic hydrogen production. Appl. Catal. B Environ. 2018, 231, 234–241. [Google Scholar] [CrossRef]
- Saad, A.; Odrinski, A.; Tivanov, M.S.; Drozdov, N.; Fedotov, A.; Gremenok, V.; Mazanik, A.; Patryn, A.; Zalesski, V.; Zaretskaya, E. Investigation of defects in Cu(In, Ga)(S, Se)2 films using the photocurrent decay technique. J. Mater. Sci. Mater. Electron. 2008, 19, 371–374. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, Y.; Yang, X.; Gui, J.; Li, Y.; Peng, H.; Liu, D.; Qiu, J. Pt/ZnO@C nanocable with dual-enhanced photocatalytic performance and superior photostability. Langmuir 2017, 33, 4452–4460. [Google Scholar] [CrossRef]
- Hu, Y.; Song, X.; Jiang, S.; Wei, C. Enhanced photocatalytic activity of Pt-doped TiO2 for NOx oxidation both under UV and visible light irradiation: A synergistic effect of lattice Pt4+ and surface PtO. Chem. Eng. J. 2015, 274, 102–112. [Google Scholar] [CrossRef]
- Samsudin, M.F.R.; Jayabalan, P.J.; Ong, W.J.; Ng, Y.H.; Sufian, S. Photocatalytic degradation of real industrial poultry wastewater via platinum decorated BiVO4/g-C3N4 photocatalyst under solar light irradiation. J. Photochem. Photobiol. A Chem. 2019, 378, 46–56. [Google Scholar] [CrossRef]
- Li, J.; Niu, L.; He, X. Enhanced visible-light activity of Ti3+ self-doped TiO2 with co-exposed {001} and {101} facets. Micro Nano Lett. 2018, 13, 514–517. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, Y.; Zhang, J.; Pan, C. Raman spectroscopy: A new approach to measure the percentage of anatase TiO2 exposed (001) facets. J. Phys. Chem. C 2012, 116, 7515–7519. [Google Scholar] [CrossRef]
- Kim, H.; Oh, M.-H.; Yang, B.L. Photocorrosion of polyaniline-ZnS–ZnO photoelectrode for water splitting. Thin Solid Films 2020, 693, 137678. [Google Scholar] [CrossRef]
- Jaramillo-Páez, C.; Navio, J.; Hidalgo, M.C.; Macías, M. ZnO and Pt-ZnO photocatalysts: Characterization and photocatalytic activity assessing by means of three substrates. Catal. Today 2018, 313, 12–19. [Google Scholar] [CrossRef]
- Fu, H.; Xu, T.; Zhu, S.; Zhu, Y. Photocorrosion inhibition and enhancement of photocatalytic activity for ZnO via hybridization with C60. Environ. Sci. Technol. 2008, 42, 8064–8069. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, Z.; Cai, Y.; Ji, J.; Tang, C.; Yu, S.; Zou, W.; Dong, L. Pt Deposites on TiO2 for Photocatalytic H2 Evolution: Pt Is Not Only the Cocatalyst, but Also the Defect Repair Agent. Catalysts 2020, 10, 1047. https://doi.org/10.3390/catal10091047
Shu Z, Cai Y, Ji J, Tang C, Yu S, Zou W, Dong L. Pt Deposites on TiO2 for Photocatalytic H2 Evolution: Pt Is Not Only the Cocatalyst, but Also the Defect Repair Agent. Catalysts. 2020; 10(9):1047. https://doi.org/10.3390/catal10091047
Chicago/Turabian StyleShu, Zhan, Yandi Cai, Jiawei Ji, Changjin Tang, Shuohan Yu, Weixin Zou, and Lin Dong. 2020. "Pt Deposites on TiO2 for Photocatalytic H2 Evolution: Pt Is Not Only the Cocatalyst, but Also the Defect Repair Agent" Catalysts 10, no. 9: 1047. https://doi.org/10.3390/catal10091047