Materials for Pharmaceutical Dosage Forms: Molecular Pharmaceutics and Controlled Release Drug Delivery Aspects

Abstract
:1. Introduction
2. Controlled Drug Release Technology of Drugs
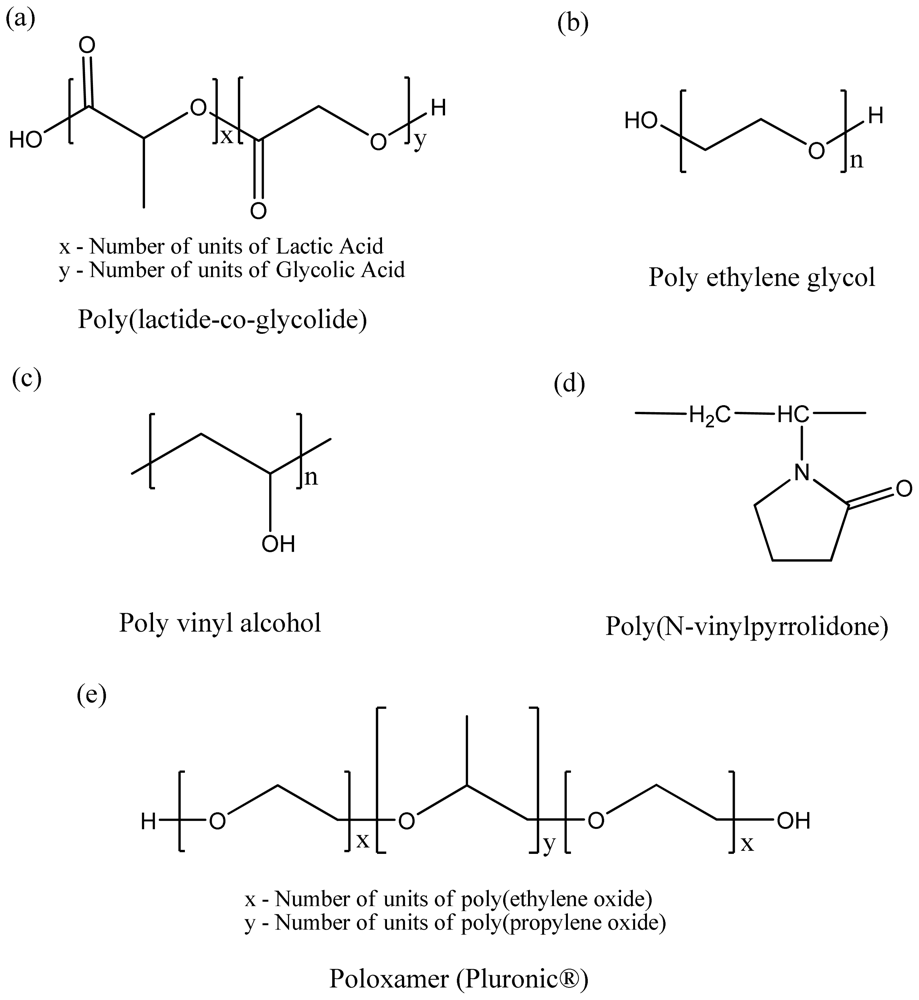
3. Types and Classes of Biodegradable and Biocompatible Pharmaceutical Polymers
3.1. Polyester-Based Synthetic Polymers
3.2. Natural Origin Polymers Used as Pharmaceutical Excipients
3.3. Homo vs. Diblock Copolymer vs. Triblock Copolymers
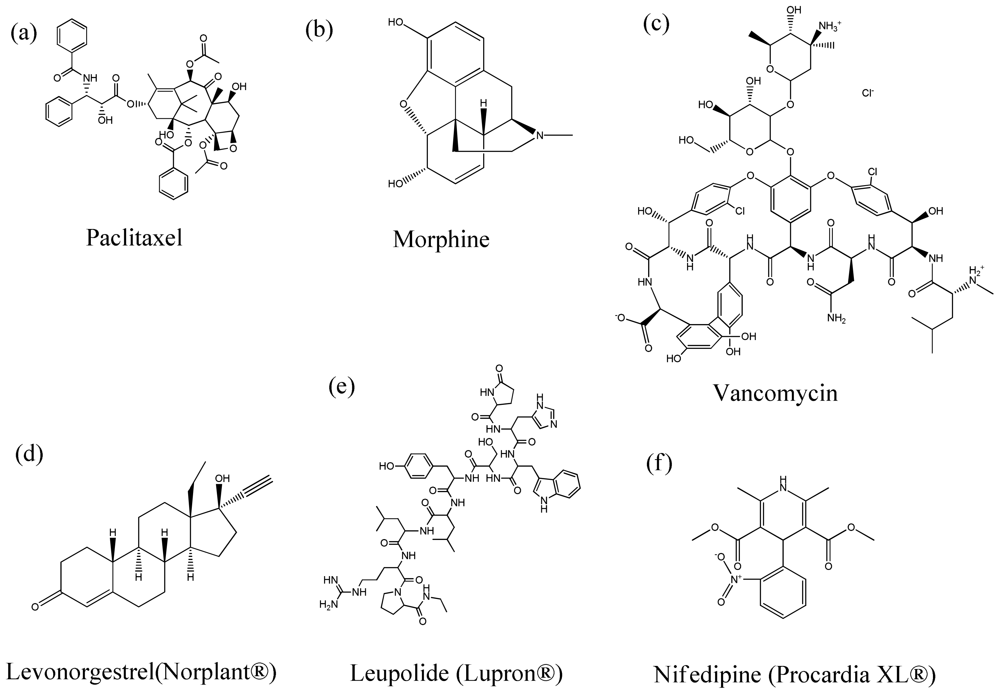
4. Therapeutic Agents Encapsulated in Polymeric Particles
5. Types of Polymeric Pharmaceutical/Drug Delivery Particles
5.1. Microparticles for Controlled Release Delivery
5.2. Nanoparticles for Controlled Release Delivery
6. Manufacturing/Particle Engineering Design of Polymeric Microparticles and Nanoparticles
6.1. Double-Emulsion Evaporation Methods
6.2. Supercritical Fluid (SCF) Technology
6.3. Supercritical Antisolvent Method
6.4. Spray Drying Particle Engineering Design
7. Marketed Controlled Release Polymeric Pharmaceutical Products and Clinical Trials
8. Conclusions
References
- Minko, T. Sinko, PJ, Ed.; Drug Delivery Systems-Controlled Drug Release. In Martns Physical Pharmacy and Pharmaceutical Sciences; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2006; pp. 667–672. [Google Scholar]
- Jantzen, GM; Robinson, JR. Banker, GS, Rhodes, CT, Eds.; Sustained- and Controlled-Release Drug Delivery Systems. In Modern Pharmaceutics; Marcel Dekker, Inc: New York, NY, USA, 2002; pp. 501–528. [Google Scholar]
- Longer, MA; Robinson, JR. Gennaro, AR, Ed.; Sustained-Release Drug Delivery Systems. In Remington’s Pharmaceutical Sciences; Mack Publishing Company: Easton, PA, USA, 1990; pp. 1676–1693. [Google Scholar]
- Ebube, NK; Hikal, AH; Wyandt, CM; Beer, DC; Miller, LG; Jones, AB. Sustained release of acetaminophen from heterogeneous matrix tablets: Influence of polymer ratio, polymer loading, and co-active on drug release. Pharm. Dev. Technol 1997, 2, 161–170. [Google Scholar]
- Miyazaki, Y; Yakou, S; Takayama, K. Study on jelly fig extract as a potential hydrophilic matrix for controlled drug delivery. Int. J. Pharm 2004, 287, 39–46. [Google Scholar]
- Panyam, J; Dali, MM; Sahoo, SK; Ma, W; Chakravarthi, SS; Amidon, GL; Levy, RJ; Labhasetwar, V. Polymer degradation and in vitro release of a model protein from poly (D,L-lactide-co-glycolide) nano- and microparticles. J. Control. Release 2003, 92, 173–187. [Google Scholar]
- Wischke, C; Schwendeman, SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm 2008, 364, 298–327. [Google Scholar]
- Fundueanu, G; Constantin, M; Dalpiaz, A; Bortolotti, F; Cortesi, R; Ascenzi, P; Menegatti, E. Preparation and characterization of starch/cyclodextrin bioadhesive microspheres as platform for nasal administration of Gabexate Mesylate (Foy) in allergic rhinitis treatment. Biomaterials 2004, 25, 159–170. [Google Scholar]
- Peng, H; Xiao, Y; Mao, X; Chen, L; Crawford, R; Whittaker, AK. Amphiphilic triblock copolymers of methoxy-poly(ethylene glycol)-b-poly(L-lactide)-b-poly(L-lysine) for enhancement of osteoblast attachment and growth. Biomacromolecules 2009, 10, 95–104. [Google Scholar]
- Na, DH; DeLuca, PP. PEGylation of octreotide: I. Separation of positional isomers and stability against acylation by poly (D,L-lactide-co-glycolide). Pharm. Res 2005, 22, 736–742. [Google Scholar]
- Na, DH; Lee, JE; Jang, SW; Lee, KC. Formation of acylated growth hormone-releasing peptide-6 by poly(lactide-co-glycolide) and its biological activity. AAPS Pharm. Sci. Tech 2007, 8, 43. [Google Scholar]
- Berkland, C; King, M; Cox, A; Kim, K; Pack, DW. Precise control of PLG microsphere size provides enhanced control of drug release rate. J. Control. Release 2002, 82, 137–147. [Google Scholar]
- Quaglia, F; Ostacolo, L; Nese, G; De Rosa, G; La Rotonda, MI; Palumbo, R; Maglio, G. Microspheres made of poly (epsilon-caprolactone)-based amphiphilic copolymers: potential in sustained delivery of proteins. Macromol. Biosci 2005, 5, 945–954. [Google Scholar]
- Wei, H; Zhang, XZ; Zhou, Y; Cheng, SX; Zhuo, RX. Self-assembled thermoresponsive micelles of poly (N-isopropylacrylamide-b-methyl methacrylate). Biomaterials 2006, 27, 2028–2034. [Google Scholar]
- Kumar, N; Ravikumar, MN; Domb, AJ. Biodegradable block copolymers. Adv. Drug Deliv. Rev 2001, 53, 23–44. [Google Scholar]
- Choi, S; Baudys, M; Kim, SW. Control of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic rats. Pharm. Res 2004, 21, 827–831. [Google Scholar]
- Jaraswekin, S; Prakongpan, S; Bodmeier, R. Effect of poly(lactide-co-glycolide) molecular weight on the release of dexamethasone sodium phosphate from microparticles. J. Microencapsul 2007, 24, 117–128. [Google Scholar]
- Wei, G; Pettway, GJ; McCauley, LK; Ma, PX. The release profiles and bioactivity of parathyroid hormone from poly(lactic-co-glycolic acid) microspheres. Biomaterials 2004, 25, 345–352. [Google Scholar]
- Tracy, MA; Ward, KL; Firouzabadian, L; Wang, Y; Dong, N; Qian, R; Zhang, Y. Factors affecting the degradation rate of poly(lactide-co-glycolide) microspheres in vivo and in vitro. Biomaterials 1999, 20, 1057–1062. [Google Scholar]
- Tsukada, Y; Hara, K; Bando, Y; Huang, CC; Kousaka, Y; Kawashima, Y; Morishita, R; Tsujimoto, H. Particle size control of poly(dl-lactide-co-glycolide) nanospheres for sterile applications. Int. J. Pharm 2009, 370, 196–201. [Google Scholar]
- Sinha, VR; Bansal, K; Kaushik, R; Kumria, R; Trehan, A. Poly-epsilon-caprolactone microspheres and nanospheres: An overview. Int. J. Pharm 2004, 278, 1–23. [Google Scholar]
- Abuchowski, A; McCoy, JR; Palczuk, NC; van Es, T; Davis, FF. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem 1977, 252, 3582–3586. [Google Scholar]
- Esposito, P; Barbero, L; Caccia, P; Caliceti, P; D’Antonio, M; Piquet, G; Veronese, FM. PEGylation of growth hormone-releasing hormone (GRF) analogues. Adv. Drug Deliv. Rev 2003, 55, 1279–1291. [Google Scholar]
- Wu, Z; Chen, H; Liu, X; Zhang, Y; Li, D; Huang, H. Protein Adsorption on Poly(N-vinylpyrrolidone)-Modified Silicon Surfaces Prepared by Surface-Initiated Atom Transfer Radical Polymerization. Langmuir 2009, 25, 2900–2906. [Google Scholar]
- Mawad, D; Poole-Warren, LA; Martens, P; Koole, LH; Slots, TL; van Hooy-Corstjens, CS. Synthesis and characterization of radiopaque iodine-containing degradable PVA hydrogels. Biomacromolecules 2008, 9, 263–268. [Google Scholar]
- Fernandes, PA; Tzvetkov, G; Fink, RH; Paradossi, G; Fery, A. Quantitative analysis of scanning transmission X-ray microscopy images of gas-filled PVA-based microballoons. Langmuir 2008, 24, 13677–13682. [Google Scholar]
- D’Souza, SS; DeLuca, PP. Methods to assess in vitro drug release from injectable polymeric particulate systems. Pharm. Res 2006, 23, 460–474. [Google Scholar]
- Park, EJ; Na, DH; Lee, KC. In vitro release study of mono-PEGylated growth hormone-releasing peptide-6 from PLGA microspheres. Int. J. Pharm 2007, 343, 281–283. [Google Scholar]
- Sehra, S; Dhake, AS. Formulation and evaluation of sustained release microspheres of poly-lactide-co-glycolide containing tamoxifen citrate. J. Microencapsul 2005, 22, 521–528. [Google Scholar]
- Dailey, LA; Wittmar, M; Kissel, T. The role of branched polyesters and their modifications in the development of modern drug delivery vehicles. J. Control. Release 2005, 101, 137–149. [Google Scholar]
- Muschert, S; Siepmann, F; Leclercq, B; Carlin, B; Siepmann, J. Drug release mechanisms from ethylcellulose: PVA-PEG graft copolymer-coated pellets. Eur. J. Pharm. Biopharm 2009, 72, 130–137. [Google Scholar]
- Mumper, RJ; Duguid, JG; Anwer, K; Barron, MK; Nitta, H; Rolland, AP. Polyvinyl derivatives as novel interactive polymers for controlled gene delivery to muscle. Pharm. Res 1996, 13, 701–709. [Google Scholar]
- Hayama, M; Yamamoto, K; Kohori, F; Uesaka, T; Ueno, Y; Sugaya, H; Itagaki, I; Sakai, K. Nanoscopic behavior of polyvinylpyrrolidone particles on polysulfone/polyvinylpyrrolidone film. Biomaterials 2004, 25, 1019–1028. [Google Scholar]
- Buhler, V (Ed.) Kollidon® — Polyvinylpyrrolidone for the Pharmaceutical Industry; BASF: LUD, Germany, 1999.
- Sahoo, J; Murthy, PN; Biswal, SM. Formulation of sustained-release dosage form of verapamil hydrochloride by solid dispersion technique using Eudragit RLPO or Kollidon SR. AAPS Pharm. Sci. Technol 2009, 10, 27–33. [Google Scholar]
- Reis, RL; Neves, NM; Mano, JF; Gomes, ME; Marques, AP; Azevedo, HS (Eds.) Natural-Based Polymers for Biomedical Applications; CRC Press-Woodhead Publishing Limited: Boca Raton, FL, USA, 2008.
- Malafaya, PB; Silva, GA; Reis, RL. Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv. Drug Deliv. Rev 2007, 59, 207–233. [Google Scholar]
- Bonacucina, G; Di Martino, P; Piombetti, M; Colombo, A; Roversi, F; Palmieri, GF. Effect of plasticizers on properties of pregelatinised starch acetate (Amprac 01) free films. Int. J. Pharm 2006, 313, 72–77. [Google Scholar]
- Ma, X; Jian, R; Chang, PR; Yu, J. Fabrication and characterization of citric acid-modified starch nanoparticles/plasticized-starch composites. Biomacromolecules 2008, 9, 3314–3320. [Google Scholar]
- Coucke, D; Schotsaert, M; Libert, C; Pringels, E; Vervaet, C; Foreman, P; Saelens, X; Remon, JP. Spray-dried powders of starch and crosslinked poly(acrylic acid) as carriers for nasal delivery of inactivated influenza vaccine. Vaccine 2009, 27, 1279–1286. [Google Scholar]
- Wei, L; Cai, C; Lin, J; Chen, T. Dual-drug delivery system based on hydrogel/micelle composites. Biomaterials 2009, 30, 2606–2613. [Google Scholar]
- Ozbas-Turan, S; Akbuga, J; Aral, C. Controlled release of interleukin-2 from chitosan microspheres. J. Pharm. Sci 2002, 91, 1245–1251. [Google Scholar]
- d’Ayala, GG; Malinconico, M; Laurienzo, P. Marine derived polysaccharides for biomedical applications: chemical modification approaches. Molecules 2008, 13, 2069–2106. [Google Scholar]
- Pajic-Lijakovic, I; Plavsic, M; Nedovic, V; Bugarski, B. Investigation of Ca-alginate hydrogel rheological behaviour in conjunction with immobilized yeast cell growth dynamics. J. Microencapsul 2007, 24, 420–429. [Google Scholar]
- Liao, YH; Jones, SA; Forbes, B; Martin, GP; Brown, MB. Hyaluronan: pharmaceutical characterization and drug delivery. Drug Delivery 2005, 12, 327–342. [Google Scholar]
- Kang, JY; Chung, CW; Sung, JH; Park, BS; Choi, JY; Lee, SJ; Choi, BC; Shim, CK; Chung, SJ; Kim, DD. Novel porous matrix of hyaluronic acid for the three-dimensional culture of chondrocytes. Int. J. Pharm 2009, 369, 114–120. [Google Scholar]
- Lin, W; Coombes, AG; Davies, MC; Davis, SS; Illum, L. Preparation of sub-100 nm human serum albumin nanospheres using a pH-coacervation method. J. Drug. Target 1993, 1, 237–243. [Google Scholar]
- Weber, C; Coester, C; Kreuter, J; Langer, K. Desolvation process and surface characterisation of protein nanoparticles. Int. J. Pharm 2000, 194, 91–102. [Google Scholar]
- Wang, G; Siggers, K; Zhang, S; Jiang, H; Xu, Z; Zernicke, RF; Matyas, J; Uludag, H. Preparation of BMP-2 containing bovine serum albumin (BSA) nanoparticles stabilized by polymer coating. Pharm. Res 2008, 25, 2896–2909. [Google Scholar]
- Friess, W. Collagen-biomaterial for drug delivery. Eur. J. Pharm. Biopharm 1998, 45, 113–136. [Google Scholar]
- Schoof, H; Apel, J; Heschel, I; Rau, G. Control of pore structure and size in freeze-dried collagen sponges. J. Biomed. Mater. Res 2001, 58, 352–357. [Google Scholar]
- Sun, XD; Jeng, L; Bolliet, C; Olsen, BR; Spector, M. Non-viral endostatin plasmid transfection of mesenchymal stem cells via collagen scaffolds. Biomaterials 2009, 30, 1222–1231. [Google Scholar]
- Young, S; Wong, M; Tabata, Y; Mikos, AG. Gelatin as a delivery vehicle for the controlled release of bioactive molecules. J. Control. Release 2005, 109, 256–274. [Google Scholar]
- Adams, ML; Lavasanifar, A; Kwon, GS. Amphiphilic block copolymers for drug delivery. J. Pharm. Sci 2003, 92, 1343–1355. [Google Scholar]
- Kataoka, K; Harada, A; Nagasaki, Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv. Drug Deliv. Rev 2001, 47, 113–131. [Google Scholar]
- Otsuka, H; Nagasaki, Y; Kataoka, K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Deliv. Rev 2003, 55, 403–419. [Google Scholar]
- Lee, PY; Li, Z; Huang, L. Thermosensitive hydrogel as a Tgf-beta1 gene delivery vehicle enhances diabetic wound healing. Pharm. Res 2003, 20, 1995–2000. [Google Scholar]
- Frauke, PK; Breitenbach, A; Zange-Volland, R; Kissel, T. Brush-like branched biodegradable polyesters, part III. Protein release from microspheres of poly(vinyl alcohol)-graft-poly(D,L-lactic- co-glycolic acid). J. Control. Release 2001, 73, 7–20. [Google Scholar]
- Jung, T; Breitenbach, A; Kissel, T. Sulfobutylated poly(vinyl alcohol)-graft-poly(lactide-co-glycolide) s facilitate the preparation of small negatively charged biodegradable nanospheres. J. Control. Release 2000, 67, 157–169. [Google Scholar]
- Jung, T; Kamm, W; Breitenbach, A; Hungerer, KD; Hundt, E; Kissel, T. Tetanus toxoid loaded nanoparticles from sulfobutylated poly(vinyl alcohol)-graft-poly(lactide-co-glycolide): evaluation of antibody response after oral and nasal application in mice. Pharm. Res 2001, 18, 352–360. [Google Scholar]
- Jung, T; Kamm, W; Breitenbach, A; Klebe, G; Kissel, T. Loading of tetanus toxoid to biodegradable nanoparticles from branched poly(sulfobutyl-polyvinyl alcohol)-g-(lactide-co-glycolide) nanoparticles by protein adsorption: a mechanistic study. Pharm. Res 2002, 19, 1105–1113. [Google Scholar]
- Batrakova, EV; Kabanov, AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J. Control. Release 2008, 130, 98–106. [Google Scholar]
- Kabanov, AV; Batrakova, EV; Alakhov, VY. Pluronic block copolymers for overcoming drug resistance in cancer. Adv. Drug Deliv. Rev 2002, 54, 759–779. [Google Scholar]
- Kabanov, AV; Batrakova, EV; Alakhov, VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J. Control. Release 2002, 82, 189–212. [Google Scholar]
- Dhanikula, AB; Panchagnula, R. Localized paclitaxel delivery. Int. J. Pharm 1999, 183, 85–100. [Google Scholar]
- Wang, Y; Yu, L; Han, L; Sha, X; Fang, X. Difunctional Pluronic copolymer micelles for paclitaxel delivery: synergistic effect of folate-mediated targeting and Pluronic-mediated overcoming multidrug resistance in tumor cell lines. Int. J. Pharm 2007, 337, 63–73. [Google Scholar]
- Huh, KM; Lee, SC; Cho, YW; Lee, J; Jeong, JH; Park, K. Hydrotropic polymer micelle system for delivery of paclitaxel. J. Control. Release 2005, 101, 59–68. [Google Scholar]
- Cheon, LS; Kim, C; Chan, KI; Chung, H; Young, JS. Polymeric micelles of poly(2-ethyl-2- oxazoline)-block-poly(epsilon-caprolactone) copolymer as a carrier for paclitaxel. J. Control. Release 2003, 89, 437–446. [Google Scholar]
- Le, GD; Gori, S; Luo, L; Lessard, D; Smith, DC; Yessine, MA; Ranger, M; Leroux, JC. Poly(N-vinylpyrrolidone)-block-poly(D,L-lactide) as a new polymeric solubilizer for hydrophobic anticancer drugs: in vitro and in vivo evaluation. J. Control. Release 2004, 99, 83–101. [Google Scholar]
- Xiong, XB; Uludag, H; Lavasanifar, A. Biodegradable amphiphilic poly(ethylene oxide)-block-polyesters with grafted polyamines as supramolecular nanocarriers for efficient siRNA delivery. Biomaterials 2009, 30, 242–253. [Google Scholar]
- Sun, TM; Du, JZ; Yan, LF; Mao, HQ; Wang, J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials 2008, 29, 4348–4355. [Google Scholar]
- Gupte, A; Ciftci, K. Formulation and characterization of Paclitaxel, 5-FU and Paclitaxel +5-FU microspheres. Int. J. Pharm 2004, 276, 93–106. [Google Scholar]
- Roullin, VG; Mege, M; Lemaire, L; Cueyssac, JP; Venier-Julienne, MC; Menei, P; Gamelin, E; Benoit, JP. Influence of 5-fluorouracil-loaded microsphere formulation on efficient rat glioma radiosensitization. Pharm. Res 2004, 21, 1558–1563. [Google Scholar]
- Shuai, X; Ai, H; Nasongkla, N; Kim, S; Gao, J. Micellar carriers based on block copolymers of poly(epsilon-caprolactone) and poly(ethylene glycol) for doxorubicin delivery. J. Control. Release 2004, 98, 415–426. [Google Scholar]
- Yadav, AK; Mishra, P; Mishra, AK; Jain, S; Agrawal, GP. Development and characterization of hyaluronic acid-anchored PLGA nanoparticulate carriers of doxorubicin. Nanomedicine 2007, 3, 246–257. [Google Scholar]
- Bae, Y; Nishiyama, N; Kataoka, K. In vivo antitumor activity of the folate-conjugated pH-sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartments. Bioconjug. Chem 2007, 18, 1131–1139. [Google Scholar]
- Maeda, M; Moriuchi, S; Sano, A; Yoshimine, T. New drug delivery system for water-soluble drugs using silicone and its usefulness for local treatment: application of GCV-silicone to GCV/HSV-tk gene therapy for brain tumor. J. Control. Release 2002, 84, 15–25. [Google Scholar]
- Park, K; Yang, JH; Choi, Y; Lee, C; Kim, SY; Byun, Y. Chemoprevention of 4-NQO-induced oral carcinogenesis by co-administration of all-trans retinoic acid loaded microspheres and celecoxib. J. Control. Release 2005, 104, 167–179. [Google Scholar]
- Sinha, VR; Bhinge, JR; Kumria, R; Kumar, M. Development of pulsatile systems for targeted drug delivery of celecoxib for prophylaxis of colorectal cancer. Drug Delivery 2006, 13, 221–225. [Google Scholar]
- D’Souza, R; Mutalik, S; Udupa, N. In Vitro and in Vivo preparation evaluations of bleomycin implants and microspheres Prepared with DL-poly (lactide-co-glycolide). Drug Dev. Ind. Pharm 2006, 32, 175–184. [Google Scholar]
- Shenoy, DB; D’Souza, RJ; Udupa, N. Poly(DL-lactide-co-glycolide) microporous microsphere-based depot formulation of a peptide-like antineoplastic agent. J. Microencapsul 2002, 19, 523–535. [Google Scholar]
- Chen, J; Cynkowski, T; Guo, H; Qin, K; Cabral-Lilly, D; Walters, K; Ashton, P. Morphine pharmacokinetics following intra-articular administration of a novel sustained release opioid (CDS-PM-101) for the relief of post-operative orthopaedic pain. J. Control. Release 2005, 101, 359–360. [Google Scholar]
- Morales, ME; Gallardo, LV; Calpena, AC; Domenech, J; Ruiz, MA. Comparative study of morphine diffusion from sustained release polymeric suspensions. J. Control. Release 2004, 95, 75–81. [Google Scholar]
- Liu, FI; Kuo, JH; Sung, KC; Hu, OY. Biodegradable polymeric microspheres for nalbuphine prodrug controlled delivery: in vitro characterization and in vivo pharmacokinetic studies. Int. J. Pharm 2003, 257, 23–31. [Google Scholar]
- Tiwari, SB; Murthy, TK; Pai, MR; Mehta, PR; Chowdary, PB. Controlled release formulation of tramadol hydrochloride using hydrophilic and hydrophobic matrix system. AAPS Pharm. Sci. Technol 2003, 4, E31. [Google Scholar]
- Dinarvand, R; Alimorad, MM; Amanlou, M; Akbari, H. In vitro release of clomipramine HCl and buprenorphine HCl from poly adipic anhydride (PAA) and poly trimethylene carbonate (PTMC) blends. J. Biomed. Mater. Res. Part A 2005, 75, 185–191. [Google Scholar]
- Kleppner, SR; Patel, R; McDonough, J; Costantini, LC. In vitro and in vivo characterization of a buprenorphine delivery system. J. Pharm. Pharmacol 2006, 58, 295–302. [Google Scholar]
- Seo, SA; Choi, HS; Khang, G; Rhee, JM; Lee, HB. A local delivery system for fentanyl based on biodegradable poly(L-lactide-co-glycolide) oligomer. Int. J. Pharm 2002, 239, 93–101. [Google Scholar]
- Seo, SA; Khang, G; Rhee, JM; Kim, J; Lee, HB. Study on in vitro release patterns of fentanyl-loaded PLGA microspheres. J. Microencapsulaytion 2003, 20, 569–579. [Google Scholar]
- Sendil, D; Bonney, IM; Carr, DB; Lipkowski, AW; Wise, DL; Hasirci, V. Antinociceptive effects of hydromorphone, bupivacaine and biphalin released from PLGA polymer after intrathecal implantation in rats. Biomaterials 2003, 24, 1969–1976. [Google Scholar]
- Lu, Y; Zhang, G; Sun, D; Zhong, Y. Preparation and evaluation of biodegradable flubiprofen gelatin micro-spheres for intra-articular administration. J. Microencapsul 2007, 24, 515–524. [Google Scholar]
- Fernandez-Carballido, A; Herrero-Vanrell, R; Molina-Martinez, IT; Pastoriza, P. Sterilized ibuprofen-loaded poly(D,L-lactide-co-glycolide) microspheres for intra-articular administration: effect of gamma-irradiation and storage. J. Microencapsul 2004, 21, 653–665. [Google Scholar]
- Thakkar, H; Sharma, RK; Mishra, AK; Chuttani, K; Murthy, RS. Celecoxib incorporated chitosan microspheres: in vitro and in vivo evaluation. J. Drug Targeting 2004, 12, 549–557. [Google Scholar]
- Thakkar, H; Sharma, RK; Mishra, AK; Chuttani, K; Murthy, RS. Efficacy of chitosan microspheres for controlled intra-articular delivery of celecoxib in inflamed joints. J. Pharm. Pharmacol 2004, 56, 1091–1099. [Google Scholar]
- Tuncay, M; Calis, S; Kas, HS; Ercan, MT; Peksoy, I; Hincal, AA. In vitro and in vivo evaluation of diclofenac sodium loaded albumin microspheres. J. Microencapsul 2000, 17, 145–155. [Google Scholar]
- Tuncay, M; Calis, S; Kas, HS; Ercan, MT; Peksoy, I; Hincal, AA. Diclofenac sodium incorporated PLGA (50:50) microspheres: formulation considerations and in vitro/in vivo evaluation. Int. J. Pharm 2000, 195, 179–188. [Google Scholar]
- La, SB; Okano, T; Kataoka, K. Preparation and characterization of the micelle-forming polymeric drug indomethacin-incorporated poly(ethylene oxide)-poly(beta-benzyl L-aspartate) block copolymer micelles. J. Pharm. Sci 1996, 85, 85–90. [Google Scholar]
- Shin, IG; Kim, SY; Lee, YM; Cho, CS; Sung, YK. Methoxy poly(ethylene glycol)/epsilon-caprolactone amphiphilic block copolymeric micelle containing indomethacin. I. Preparation and characterization. J. Control. Release 1998, 51, 1–11. [Google Scholar]
- Sendil-Keskin, D; Altunay, H; Wise, DL; Hasirci, V. In vivo pain relief effectiveness of an analgesic-anesthetic carrying biodegradable controlled release rod systems. J. Biomater. Sci., Polym. Ed 2003, 14, 497–514. [Google Scholar]
- Loughlin, RG; Tunney, MM; Donnelly, RF; Murphy, DJ; Jenkins, M; McCarron, PA. Modulation of gel formation and drug-release characteristics of lidocaine-loaded poly(vinyl alcohol)-tetraborate hydrogel systems using scavenger polyol sugars. Eur. J. Pharm. Biopharm 2008, 69, 1135–1146. [Google Scholar]
- Moss, GP; Gullick, DR; Woolfson, AD; McCafferty, DF. Mechanical characterization and drug permeation properties of tetracaine-loaded bioadhesive films for percutaneous local anesthesia. Drug Dev. Ind. Pharm 2006, 32, 163–174. [Google Scholar]
- Ratajczak-Enselme, M; Estebe, JP; Dollo, G; Chevanne, F; Bec, D; Malinovsky, JM; Ecoffey, C; Le Corre, P. Epidural, intrathecal and plasma pharmacokinetic study of epidural ropivacaine in PLGA-microspheres in sheep model. Eur. J. Pharm. Biopharm 2009, 72, 54–61. [Google Scholar]
- Kiremitci, AS; Ciftci, A; Ozalp, M; Gumusderelioglu, M. Novel chlorhexidine releasing system developed from thermosensitive vinyl ether-based hydrogels. J. Biomed. Mater. Res. Part B 2007, 83, 609–614. [Google Scholar]
- Bigucci, F; Luppi, B; Musenga, A; Zecchi, V; Cerchiara, T. Chitosan salts coated with stearic acid as colon-specific delivery systems for vancomycin. Drug Delivery 2008, 15, 289–293. [Google Scholar]
- Nahar, M; Mishra, D; Dubey, V; Jain, NK. Development, characterization, and toxicity evaluation of amphotericin B-loaded gelatin nanoparticles. Nanomedicine 2008, 4, 252–261. [Google Scholar]
- Jia, Y; Joly, H; Omri, A. Liposomes as a carrier for gentamicin delivery: development and evaluation of the physicochemical properties. Int. J. Pharm 2008, 359, 254–263. [Google Scholar]
- Lecaroz, C; Gamazo, C; Renedo, MJ; Blanco-Prieto, MJ. Biodegradable micro- and nanoparticles as long-term delivery vehicles for gentamicin. J. Microencapsul 2006, 23, 782–792. [Google Scholar]
- Gillissen, M; Steendam, R; van der Laan, A; Tijsma, E. Development of doxycycline-eluting delivery systems based on SynBiosys biodegradable multi-block copolymers. J. Control. Release 2006, 116, 90–92. [Google Scholar]
- Mundargi, RC; Srirangarajan, S; Agnihotri, SA; Patil, SA; Ravindra, S; Setty, SB; Aminabhavi, TM. Development and evaluation of novel biodegradable microspheres based on poly(d,l-lactide-co-glycolide) and poly(epsilon-caprolactone) for controlled delivery of doxycycline in the treatment of human periodontal pocket: in vitro and in vivo studies. J. Control. Release 2007, 119, 59–68. [Google Scholar]
- Giovagnoli, S; Tsai, T; DeLuca, PP. Formulation and release behavior of doxycycline-alginate hydrogel microparticles embedded into pluronic F127 thermogels as a potential new vehicle for doxycycline intradermal sustained delivery. AAPS Pharm-Sci-Tech 2010, 11, 212–220. [Google Scholar]
- Capan, Y; Jiang, G; Giovagnoli, S; Na, KH; DeLuca, PP. Preparation and characterization of poly(D,L-lactide-co-glycolide) microspheres for controlled release of human growth hormone. AAPS Pharm. Sci. Technol 2003, 4, 28. [Google Scholar]
- Chen, S; Singh, J. Controlled release of growth hormone from thermosensitive triblock copolymer systems: In vitro and in vivo evaluation. Int. J. Pharm 2008, 352, 58–65. [Google Scholar]
- Na, DH; Lee, KC; DeLuca, PP. PEGylation of octreotide: II. Effect of N-terminal mono-PEGylation on biological activity and pharmacokinetics. Pharm. Res 2005, 22, 743–749. [Google Scholar]
- Brache, V; Faundes, A; Alvarez, F; Garcia, AG. Transition from Norplant to Jadelle in a clinic with extensive experience providing contraceptive implants. Contraception 2006, 73, 364–367. [Google Scholar]
- Dasaratha, DM; Vema, K; Jayakumar, R; Vamsadhara, C. Preparation and characterization of injectable microspheres of contraceptive hormones. Int. J. Pharm 2003, 268, 23–29. [Google Scholar]
- Dhanaraju, MD; Rajkannan, R; Selvaraj, D; Jayakumar, R; Vamsadhara, C. Biodegradation and biocompatibility of contraceptive-steroid-loaded poly (DL-lactide-co-glycolide) injectable microspheres: in vitro and in vivo study. Contraception 2006, 74, 148–156. [Google Scholar]
- Pai, SS; Tilton, RD; Przybycien, TM. Poly(ethylene glycol)-modified proteins: implications for poly(lactide-co-glycolide)-based microsphere delivery. AAPS J 2009, 11, 88–98. [Google Scholar]
- Wang, SH; Zhang, LC; Lin, F; Sa, XY; Zuo, JB; Shao, QX; Chen, GS; Zeng, S. Controlled release of levonorgestrel from biodegradable poly(D,L-lactide-co-glycolide) microspheres: in vitro and in vivo studies. Int. J. Pharm 2005, 301, 217–225. [Google Scholar]
- Cheng, YH; Illum, L; Davis, SS. Schizophrenia and drug delivery systems. J. Drug. Targeting 2000, 8, 107–117. [Google Scholar]
- Hans, ML; Maxwell, C; Ehrlichman, RS; Metzger, K; Liang, Y; Siegel, SJ; Lowman, AM. Evaluation of in vitro release and in vivo efficacy of mPEG-PLA-haloperidol conjugate micelle-like structures. J. Biomed. Mater. Res. Part B 2007, 83, 422–430. [Google Scholar]
- Lu, Y; Tang, X; Cui, Y; Zhang, Y; Qin, F; Lu, X. In vivo evaluation of risperidone-SAIB in situ system as a sustained release delivery system in rats. Eur. J. Pharm. Biopharm 2008, 68, 422–429. [Google Scholar]
- Lu, Y; Yu, Y; Tang, X. Sucrose acetate isobutyrate as an in situ forming system for sustained risperidone release. J. Pharm. Sci 2007, 96, 3252–3262. [Google Scholar]
- Agnihotri, SA; Aminabhavi, TM. Controlled release of clozapine through chitosan microparticles prepared by a novel method. J. Control. Release 2004, 96, 245–259. [Google Scholar]
- Nahata, T; Saini, TR. Optimization of formulation variables for the development of long acting microsphere based depot injection of olanzapine. J. Microencapsul 2008, 25, 426–433. [Google Scholar]
- Guthmann, C; Lipp, R; Wagner, T; Kranz, H. Development of a novel osmotically driven drug delivery system for weakly basic drugs. Eur. J. Pharm. Biopharm 2008, 69, 667–674. [Google Scholar]
- McClelland, GA; Sutton, SC; Engle, K; Zentner, GM. The solubility-modulated osmotic pump: in vitro/in vivo release of diltiazem hydrochloride. Pharm. Res 1991, 8, 88–92. [Google Scholar]
- Mohammadi-Samani, S; Adrangui, M; Siahi-Shadbad, MR; Nokhodchi, A. An approach to controlled-release dosage form of propranolol hydrochloride. Drug Dev. Ind. Pharm 2000, 26, 91–94. [Google Scholar]
- Nokhodchi, A; Momin, MN; Shokri, J; Shahsavari, M; Rashidi, PA. Factors affecting the release of nifedipine from a swellable elementary osmotic pump. Drug Delivery 2008, 15, 43–48. [Google Scholar]
- Wang, X; Nie, SF; Li, W; Luan, L; Pan, W. Studies on bi-layer osmotic pump tablets of water-insoluble allopurinol with large dose: in vitro and in vivo. Drug Dev. Ind. Pharm 2007, 33, 1024–1029. [Google Scholar]
- He, L; Gong, T; Zhao, D; Zhang, ZR; Li, L. A novel controlled porosity osmotic pump system for sodium ferulate. Pharmazie 2006, 61, 1022–1027. [Google Scholar]
- Rani, M; Mishra, B. Comparative in vitro and in vivo evaluation of matrix, osmotic matrix, and osmotic pump tablets for controlled delivery of diclofenac sodium. AAPS Pharm. Sci. Technol 2004, 5, 71. [Google Scholar]
- Makhija, SN; Vavia, PR. Controlled porosity osmotic pump-based controlled release systems of pseudoephedrine. I. Cellulose acetate as a semipermeable membrane. J. Control. Release 2003, 89, 5–18. [Google Scholar]
- Kang, F; Singh, J. Effect of additives on the release of a model protein from PLGA microspheres. AAPS Pharm-Sci-Tech 2001, 2, 30. [Google Scholar]
- Blanco, D; Alonso, MJ. Protein encapsulation and release from poly(lactide-co-glycolide) microspheres: effect of the protein and polymer properties and of the co-encapsulation of surfactants. Eur. J. Pharm. Biopharm 1998, 45, 285–294. [Google Scholar]
- Sandor, M; Enscore, D; Weston, P; Mathiowitz, E. Effect of protein molecular weight on release from micron-sized PLGA microspheres. J. Control. Release 2001, 76, 297–311. [Google Scholar]
- Misra, SK; Ansari, TI; Valappil, SP; Mohn, D; Philip, SE; Stark, WJ; Roy, I; Knowles, JC; Salih, V; Boccaccini, AR. Poly(3-hydroxybutyrate) multifunctional composite scaffolds for tissue engineering applications. Biomaterials 2010, 31, 2806–2815. [Google Scholar]
- Francis, L; Meng, D; Knowles, JC; Roy, I; Boccaccini, AR. Multi-functional P(3HB) microsphere/45S5 Bioglass-based composite scaffolds for bone tissue engineering. Acta Biomater 2010, 6, 2773–2786. [Google Scholar]
- Mourino, V; Boccaccini, AR. Bone tissue engineering therapeutics: Controlled drug delivery in three-dimensional scaffolds. J. R. Soc. Interface 2009, 7, 209–227. [Google Scholar]
- Dani, BA; Raiche, AT; Puleo, DA; DeLuca, PP. A study of the antiresorptive activity of salmon calcitonin microspheres using cultured osteoclastic cells. AAPS Pharm. Sci. Technol 2002, 3, E21. [Google Scholar]
- Raiche, AT; Puleo, DA. Association polymers for modulated release of bioactive proteins. IEEE Eng. Med. Biol. Mag 2003, 22, 35–41. [Google Scholar]
- Jeon, JH; Thomas, MV; Puleo, DA. Bioerodible devices for intermittent release of simvastatin acid. Int. J. Pharm 2007, 340, 6–12. [Google Scholar]
- Jeon, JH; Piepgrass, WT; Lin, YL; Thomas, MV; Puleo, DA. Localized intermittent delivery of simvastatin hydroxyacid stimulates bone formation in rats. J. Periodontol 2008, 79, 1457–1464. [Google Scholar]
- Jeon, JH; Puleo, DA. Alternating release of different bioactive molecules from a complexation polymer system. Biomaterials 2008, 29, 3591–3598. [Google Scholar]
- Mansour, HM; Rhee, YS; Wu, X. Nanomedicine in Pulmonary Delivery. Int. J. Nanomed 2009, 4, 299–319. [Google Scholar]
- Barichello, JM; Morishita, M; Takayama, K; Nagai, T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev. Ind. Pharm 1999, 25, 471–476. [Google Scholar]
- Kumar, PS; Ramakrishna, S; Saini, TR; Diwan, PV. Influence of microencapsulation method and peptide loading on formulation of poly(lactide-co-glycolide) insulin nanoparticles. Pharmazie 2006, 61, 613–617. [Google Scholar]
- Hafeli, UO. Magnetically modulated therapeutic systems. Int. J. Pharm 2004, 277, 19–24. [Google Scholar]
- Cheng, J; Teply, BA; Jeong, SY; Yim, CH; Ho, D; Sherifi, I; Jon, S; Farokhzad, OC; Khademhosseini, A; Langer, RS. Magnetically responsive polymeric microparticles for oral delivery of protein drugs. Pharm. Res 2006, 23, 557–564. [Google Scholar]
- Mundargi, RC; Babu, VR; Rangaswamy, V; Patel, P; Aminabhavi, TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J. Control. Release 2008, 125, 193–209. [Google Scholar]
- Florence, AT. Florence, AT, Siepmann, J, Eds.; Pharmaceutical Aspects of Nanotechnology. In Modern Pharmaceutics: Volume 2-Applications and Advances; Informa Healthcare: New York, NY, USA, 2009; pp. 453–492. [Google Scholar]
- Hickey, AJ; Mansour, HM. Rathbone, MJ, Hadgraft, J, Roberts, MS, Lane, ME, Eds.; Formulation Challenges of Powders for the Delivery of Small Molecular Weight Molecules as Aerosols. In Modified-Release Drug Delivery Technology; Informa Healthcare: New York, NY, USA, 2008; pp. 573–602. [Google Scholar]
- York, P; Kompella, UB; Shekunov, BY (Eds.) Supercritical Fluid Technology for Drug Product Development; Informa Healthcare: New York, NY, USA, 2004.
- Williams, JR; Clifford, AA; al-Saidi, SH. Supercritical fluids and their applications in biotechnology and related areas. Mol. Biotechnol 2002, 22, 263–286. [Google Scholar]
- Mishima, K. Biodegradable particle formation for drug and gene delivery using supercritical fluid and dense gas. Adv. Drug Deliv. Rev 2008, 60, 411–432. [Google Scholar]
- Lee, LY; Wang, CH; Smith, KA. Supercritical antisolvent production of biodegradable micro-and nanoparticles for controlled delivery of paclitaxel. J. Control. Release 2008, 125, 96–106. [Google Scholar]
- Li, W; Zhang, J; Zhang, C; Feng, X; Han, B; Yang, G. Synthesis of alpha-chymotrypsin/polymer composites by a reverse micelle/gas antisolvent method. Colloids Surf. B. Biointerfaces 2007, 59, 11–15. [Google Scholar]
- Steckel, H; Pichert, L; Muller, BW. Influence of process parameters in the ASES process on particle properties of budesonide for pulmonary delivery. Eur. J. Pharm. Biopharm 2004, 57, 507–512. [Google Scholar]
- Bleich, J; Muller, BW. Production of drug loaded microparticles by the use of supercritical gases with the aerosol solvent extraction system (ASES) process. J. Microencapsul 1996, 13, 131–139. [Google Scholar]
- Palakodaty, S; York, P; Pritchard, J. Supercritical fluid processing of materials from aqueous solutions: the application of SEDS to lactose as a model substance. Pharm. Res 1998, 15, 1835–1843. [Google Scholar]
- Chow, AH; Tong, HH; Chattopadhyay, P; Shekunov, BY. Particle engineering for pulmonary drug delivery. Pharm. Res 2007, 24, 411–437. [Google Scholar]
- Dunbar, CA; Concessio, NM; Hickey, AJ. Evaluation of atomizer performance in production of respirable spray-dried particles. Pharm. Dev. Technol 1998, 3, 433–441. [Google Scholar]
- Elversson, J; Millqvist-Fureby, A; Alderborn, G; Elofsson, U. Droplet and particle size relationship and shell thickness of inhalable lactose particles during spray drying. J. Pharm. Sci 2003, 92, 900–910. [Google Scholar]
- Elversson, J; Millqvist-Fureby, A. Particle size and density in spray drying-effects of carbohydrate properties. J. Pharm. Sci 2005, 94, 2049–2060. [Google Scholar]
- Gilani, K; Najafabadi, AR; Barghi, M; Rafiee-Tehrani, M. The effect of water to ethanol feed ratio on physical properties and aerosolization behavior of spray dried cromolyn sodium particles. J. Pharm. Sci 2005, 94, 1048–1059. [Google Scholar]
- Maa, YF; Costantino, HR; Nguyen, PA; Hsu, CC. The effect of operating and formulation variables on the morphology of spray-dried protein particles. Pharm. Dev. Technol 1997, 2, 213–223. [Google Scholar]
- Rogers, TL; Johnston, KP; Williams, RO. Solution-based particle formation of pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray-freezing technologies. Drug Dev. Ind. Pharm 2001, 27, 1003–1015. [Google Scholar]
- Maa, YF; Prestrelski, SJ. Biopharmaceutical powders: particle formation and formulation considerations. Curr. Pharm. Biotechnol 2000, 1, 283–302. [Google Scholar]
- Costantino, HR; Firouzabadian, L; Hogeland, K; Wu, C; Beganski, C; Carrasquillo, KG; Cordova, M; Griebenow, K; Zale, SE; Tracy, MA. Protein spray-freeze drying. Effect of atomization conditions on particle size and stability. Pharm. Res 2000, 17, 1374–1383. [Google Scholar]
- Yu, Z; Rogers, TL; Hu, J; Johnston, KP; Williams, RO. Preparation and characterization of microparticles containing peptide produced by a novel process: spray freezing into liquid. Eur. J. Pharm. Biopharm 2002, 54, 221–228. [Google Scholar]
- Hu, J; Johnston, KP; Williams, RO. Stable amorphous danazol nanostructured powders with rapid dissolution rates produced by spray freezing into liquid. Drug Dev. Ind. Pharm 2004, 30, 695–704. [Google Scholar]
- D’Souza, SS; Faraj, JA; DeLuca, PP. A model-dependent approach to correlate accelerated with real-time release from biodegradable microspheres. AAPS Pharm-Sci-Tech 2005, 6, E553–564. [Google Scholar]
- D’Souza, SS; Selmin, F; Murty, SB; Qiu, W; Thanoo, BC; DeLuca, PP. Assessment of fertility in male rats after extended chemical castration with a GnRH antagonist. AAPS J 2004, 6, 94–99. [Google Scholar]
- Kane, JM; Eerdekens, M; Lindenmayer, JP; Keith, SJ; Lesem, M; Karcher, K. Long-acting injectable risperidone: efficacy and safety of the first long-acting atypical antipsychotic. Am. J. Psychiatry 2003, 160, 1125–1132. [Google Scholar]
- Kostanski, JW; Thanoo, BC; DeLuca, PP. Preparation, characterization, and in vitro evaluation of 1- and 4-month controlled release orntide PLA and PLGA microspheres. Pharm. Dev. Technol 2000, 5, 585–596. [Google Scholar]
- Okada, H; Doken, Y; Ogawa, Y; Toguchi, H. Preparation of three-month depot injectable microspheres of leuprorelin acetate using biodegradable polymers. Pharm. Res 1994, 11, 1143–1147. [Google Scholar]
- Woo, BH; Na, KH; Dani, BA; Jiang, G; Thanoo, BC; DeLuca, PP. In vitro characterization and in vivo testosterone suppression of 6-month release poly (D,L-lactide) leuprolide microspheres. Pharm. Res 2002, 19, 546–550. [Google Scholar]
- Matsumura, Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Adv. Drug Deliv. Rev 2008, 60, 899–914. [Google Scholar]
- Kanagale, P; Patel, V; Venkatesan, N; Jain, M; Patel, P; Misra, A. Pharmaceutical development of solid dispersion based osmotic drug delivery system for nifedipine. Curr. Drug Deliv 2008, 5, 306–311. [Google Scholar]



{kind=link}
{kind=link}
| Polymeric inactive ingredients for FDA-approved drug products | |
|---|---|
| Polyester-based synthetic polymers | PLGA (for IM, SC uses) Poloxamer (for oral, topical, IV opthalmic, SC uses) Polyvinylpyrrolidone ethylcellulose (for oral use) Sodium pyrrolidone carboxylate (for topical use) Povidone (for oral, intra-articular, IM, Intrauterine, topical, SC, respiratory, opthalmic uses) PLA (for IM use) PEG (for oral, respiratory, topical, IM, IV, opthalmic uses) PVA (for auricular, IM, intraocular, topical uses) KOLLIDON VA 64 (for oral use) |
| Natural-origin polymers | Starch (for oral, IV, IM, topical) Hyaluronate (for intra-articular, IM, intravitreal, topical uses), Human albumin (for IV, SC, Oral uses) Gelatin (for IM, SC, IV, oral topical uses) Alginic acid (for opthalmic and oral uses) Collagen (for topical use) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mansour, H.M.; Sohn, M.; Al-Ghananeem, A.; DeLuca, P.P. Materials for Pharmaceutical Dosage Forms: Molecular Pharmaceutics and Controlled Release Drug Delivery Aspects. Int. J. Mol. Sci. 2010, 11, 3298-3322. https://doi.org/10.3390/ijms11093298
Mansour HM, Sohn M, Al-Ghananeem A, DeLuca PP. Materials for Pharmaceutical Dosage Forms: Molecular Pharmaceutics and Controlled Release Drug Delivery Aspects. International Journal of Molecular Sciences. 2010; 11(9):3298-3322. https://doi.org/10.3390/ijms11093298
Chicago/Turabian StyleMansour, Heidi M., MinJi Sohn, Abeer Al-Ghananeem, and Patrick P. DeLuca. 2010. "Materials for Pharmaceutical Dosage Forms: Molecular Pharmaceutics and Controlled Release Drug Delivery Aspects" International Journal of Molecular Sciences 11, no. 9: 3298-3322. https://doi.org/10.3390/ijms11093298