2.1. HEMA/IPDI as Bis-GMA Substitute in Copolymers with TEGDMA
The substitution of Bis-GMA with HEMA/IPDI, in formulations with TEGDMA, provided the first type of copolymers. This was achieved by mixing HEMA/IPDI as well as Bis-GMA with TEGDMA, in 80:20 and 60:40 wt% ratios, and photopolymerization. For comparative purposes, HEMA/IPDI, Bis-GMAand TEGDMA homopolymers were produced.
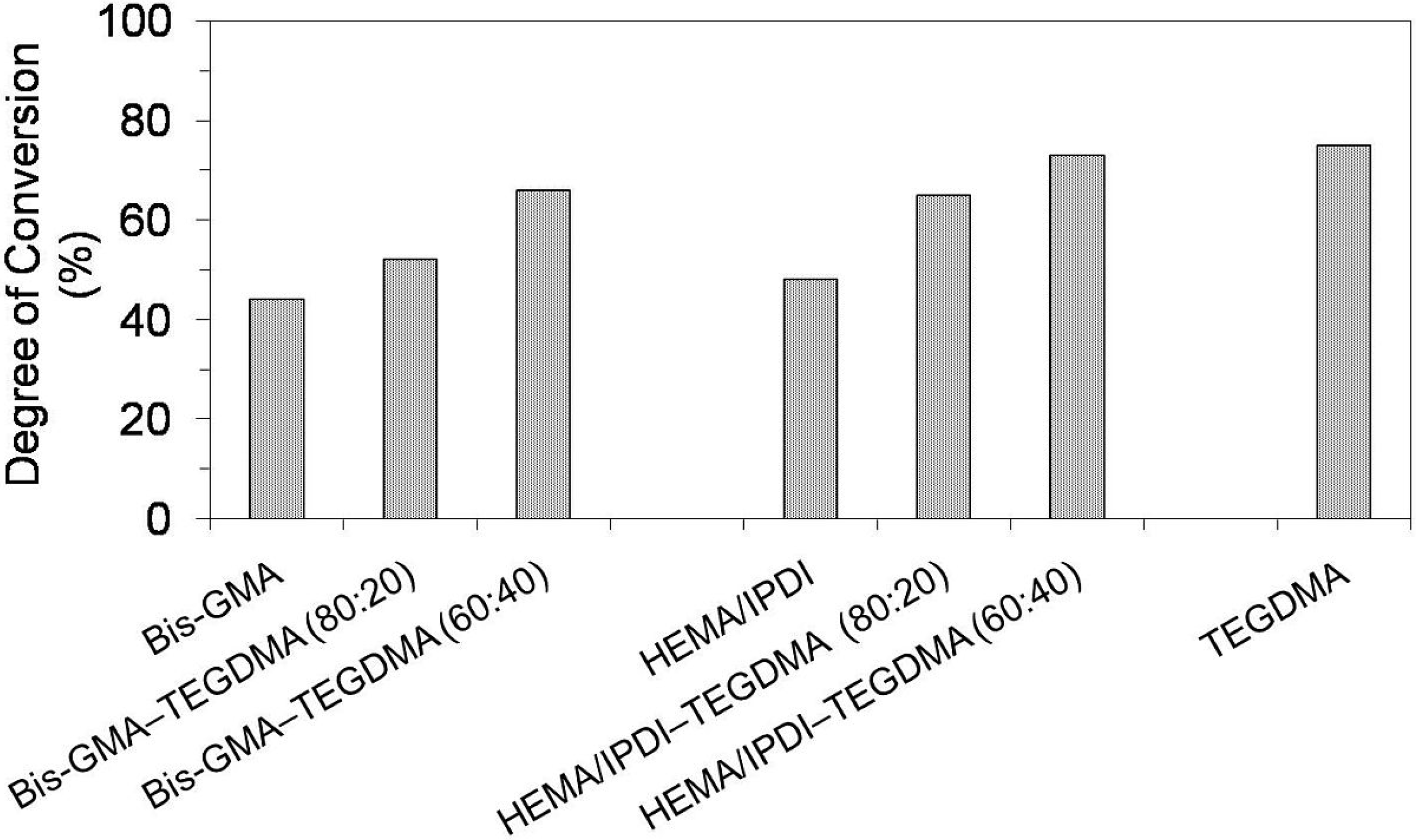
Figure 1 presents results for the degree of conversion (
DC) in homopolymer and copolymer networks. The
DC is crucial for understanding the physico-mechanical behavior of the polymer network [
8,
9]. When IR spectroscopy is applied to investigate the
DC in poly(dimethacrylate)s, the internal band ratio method with stretching vibrations of the aromatic ring, as a reference, is the most frequently used [
3,
8]. However, due to the lack of aromatic moieties in the majority of examined compositions, the carbonyl vibrations peak was chosen as an internal standard to calculate the
DC in polymers. This method is often used, when monomers have no aromatic rings [
8,
10]. However, it might produce a lower
DC than the method using an aromatic band as a standard, especially for monomers with a greater stiffness [
8].
Figure 1.
The degree of conversion in Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
Figure 1.
The degree of conversion in Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
As can be seen in
Figure 1, the homopolymers had lower
DC values than copolymers. The lowest
DC in the Bis-GMA homopolymer, being of 40%, can be interpreted by low elasticity and mobility of the monomer. The Bis-GMA molecule is long and spacious; it has two
p-phenylene rings and two hydroxyl groups, involved in strong intermolecular hydrogen bonds. These features cause a dramatic increase in viscosity (1200 Pa·s,
Table 1) and a decrease in the
DC [
3]. HEMA/IPDI, when compared to Bis-GMA, is 99% less viscous (12 Pa·s,
Table 1) [
6]. Additionally, two elastic urethane linkages may offer an alternative polymerization path, causing chain transfer reactions, which increase the mobility of radical sites in the network [
3]. With regard to this aspect, HEMA/IPDI might be expected to polymerize to a more suitable
DC. However, the factor of high molecular stiffness, resulting from the cycloaliphatic IPDI core, seems to explain the relatively low
DC in the homopolymer, which is 48%.
As far as
Figure 1 is concerned, the degree of conversion in both groups of copolymers increased with increasing TEGDMA content. This behavior can be associated with two effects: the resin viscosity and the chemical structure allowing for molecular mobility [
11,
12]. The small size of the TEGDMA molecule and the high concentration of double bonds ensure a close proximity between radical sites, whereas its high flexibility and the absence of the hydrogen bond proton donor incur low resin viscosity (of 0.011 Pa·s [
7],
Table 1). The degree of conversion in HEMA/IPDI copolymers (65% and 73%) was higher than the
DC in corresponding Bis-GMA copolymers (52% and 66%). It could be generally concluded, that the copolymerization of dimethacrylates having hydrogen bond proton donors (UDMA and Bis-GMA) with TEGDMA, having only proton acceptors, improved the degree of conversion within the network. Additionally, it worked more effectively in systems composed of HEMA/IPDI, being the urethane-dimethacrylate, rather than in Bis-GMA systems. These results were in agreement with the findings from other studies, performed on similar dimethacrylate systems [
11,
12,
13].
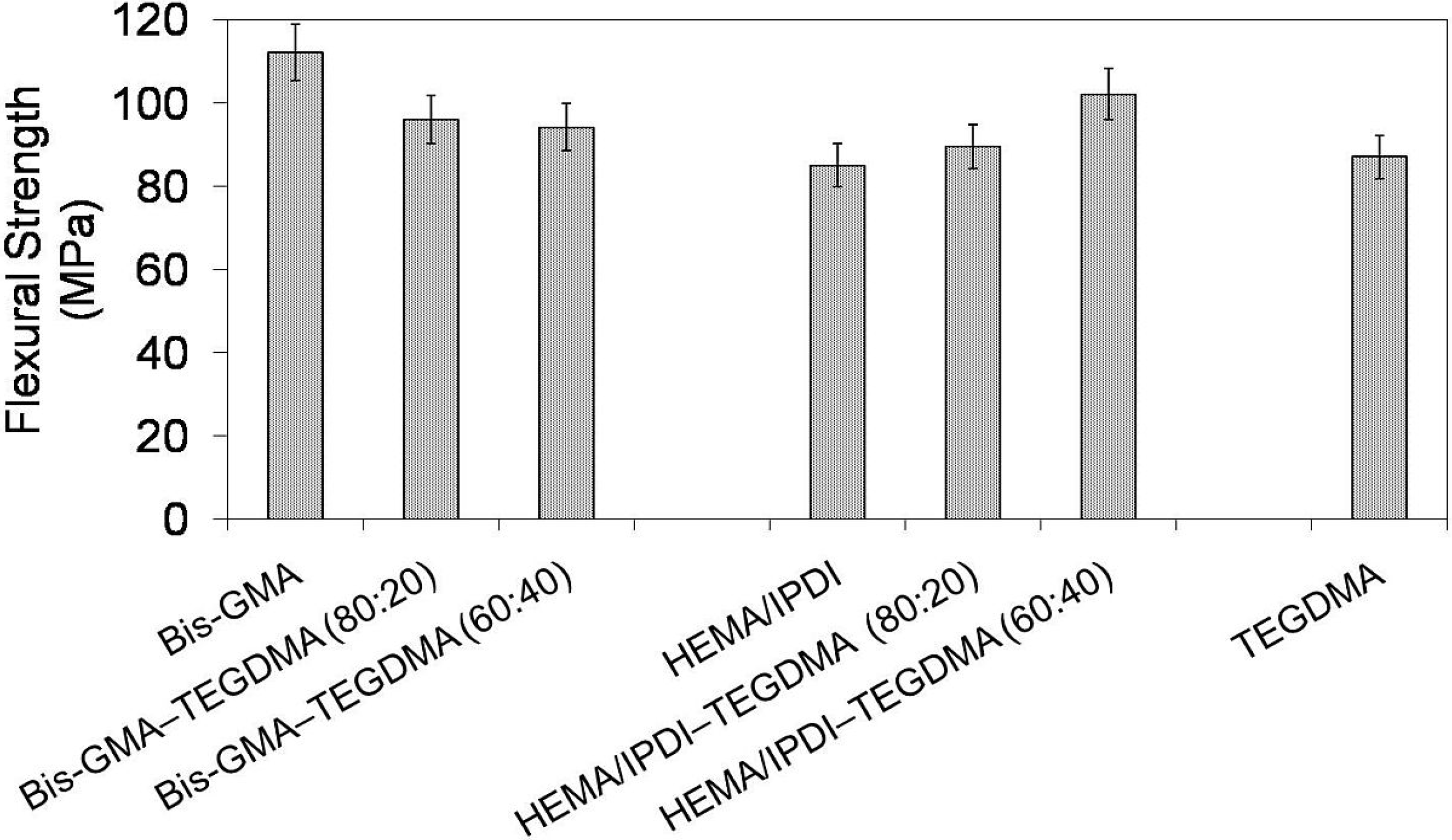
In
Figure 2 the relationship between flexural strength and polymer composition is depicted. The mechanical strength of investigated homopolymers ranges from 85 to 112 MPa and follows this order: σ
poly(HEMA/IPDI) < σ
poly(TEGDMA) < σ
poly(Bis-GMA). The flexural strength of the Bis-GMA–TEGDMA copolymers slightly decreased as the TEGDMA content increased. In contrast, the flexural strength of the HEMA/IPDI–TEGDMA copolymers increased as the TEGDMA content was raised. The HEMA/IPDI–TEGDMA (60:40) network had the highest flexural strength among the copolymers of this group, having a value of 102 MPa.
Figure 2.
The flexural strength of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
Figure 2.
The flexural strength of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
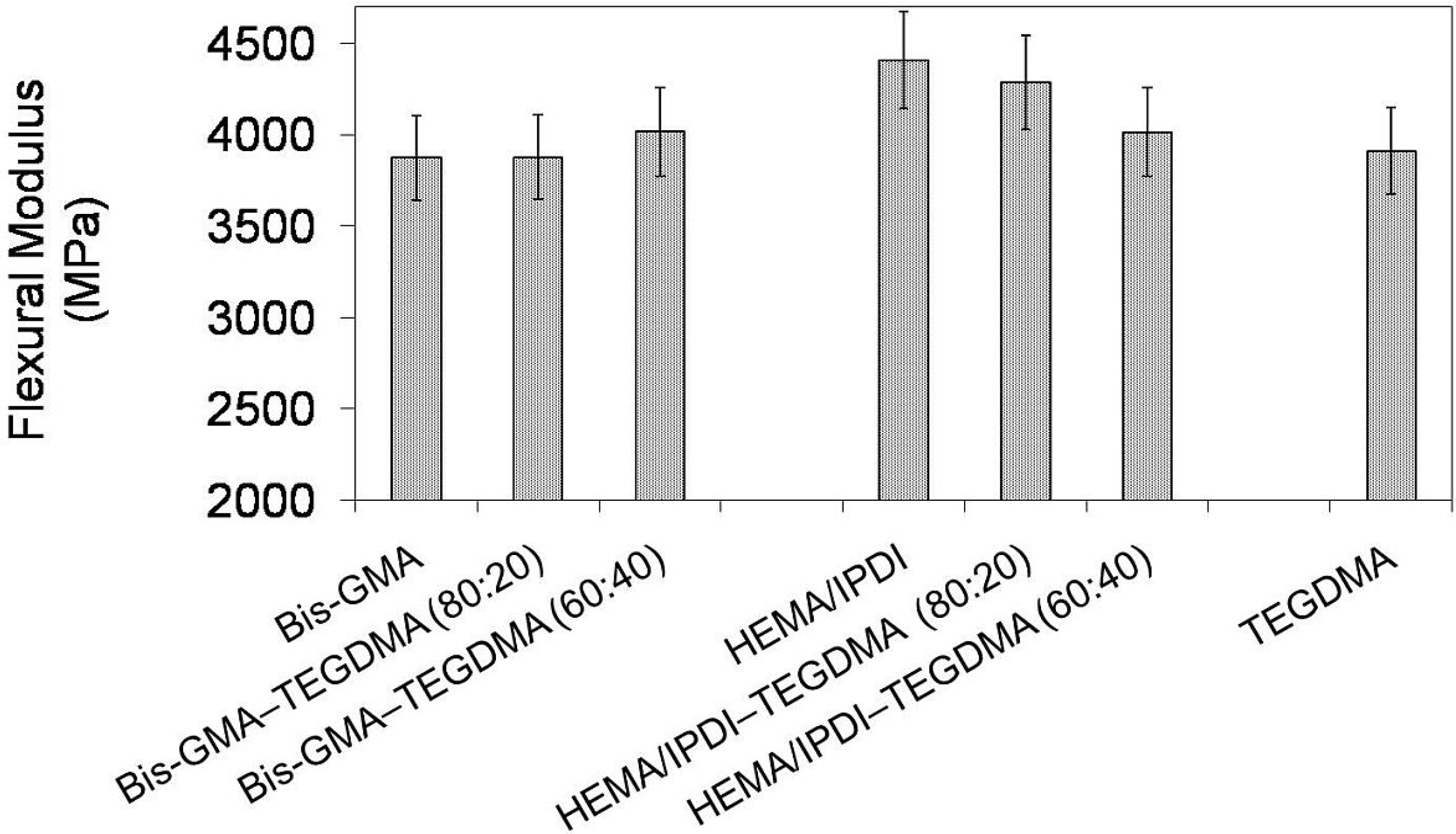
In
Figure 3 the relationship between flexural modulus and polymer compositions is depicted. One can observe the modulus of investigated homopolymers in the following order:
Epoly(Bis-GMA) <
Epoly(TEGDMA) <
Epoly(HEMA/IPDI) and in the range from 3872 to 4406 MPa. The modulus of the Bis-GMA–TEGDMA copolymers was raised with increasing TEGDMA fraction. In contrast, the modulus of the HEMA/IPDI–TEGDMA copolymers decreased with increasing TEGDMA content. The Bis-GMA–TEGDMA (80:20) copolymer had the lowest modulus in this group, of 3876 MPa, whereas the analogous HEMA/IPDI–TEGDMA (80:20) copolymer had the highest
E, of 4283 MPa.
Figure 3.
The flexural modulus of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
Figure 3.
The flexural modulus of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
In order to explain these contrary flexural property-composition relationships, for each group of copolymers, a different approach should be implemented. In the Bis-GMA–TEGDMA (80:20) system, Bis-GMA still seriously limited the system mobility and sterically isolated methacrylate groups against polymerization. The TEGDMA content was not high enough to significantly improve the degree of conversion and as a consequence, the modulus remained almost unchanged. On the other hand, TEGDMA influenced the flexural strength by decreasing its value. When the TEGDMA content was raised to 40 wt%, the degree of conversion increased from 52% to 66%. The improved copolymerization scale caused the decrease in the copolymer elasticity, thereby decreasing the strain at the break, from 96 to 94 MPa, and hence increasing the modulus, from 3876 to 4014 MPa. HEMA/IPDI, if compared to Bis-GMA, produced homopolymer networks of a higher degree of conversion and higher modulus. After copolymerization with TEGDMA, an increase in elasticity was observed, despite an increase in the DC. The reason for this could be due to the TEGDMA elasticity. The homopolymer of TEGDMA, characterized by the modulus of 3910 MPa, is more elastic than that of HEMA/IPDI. Consequently, the decrease in flexural modulus and the increase of flexural strength, proportional to the TEGDMA content, were observed.
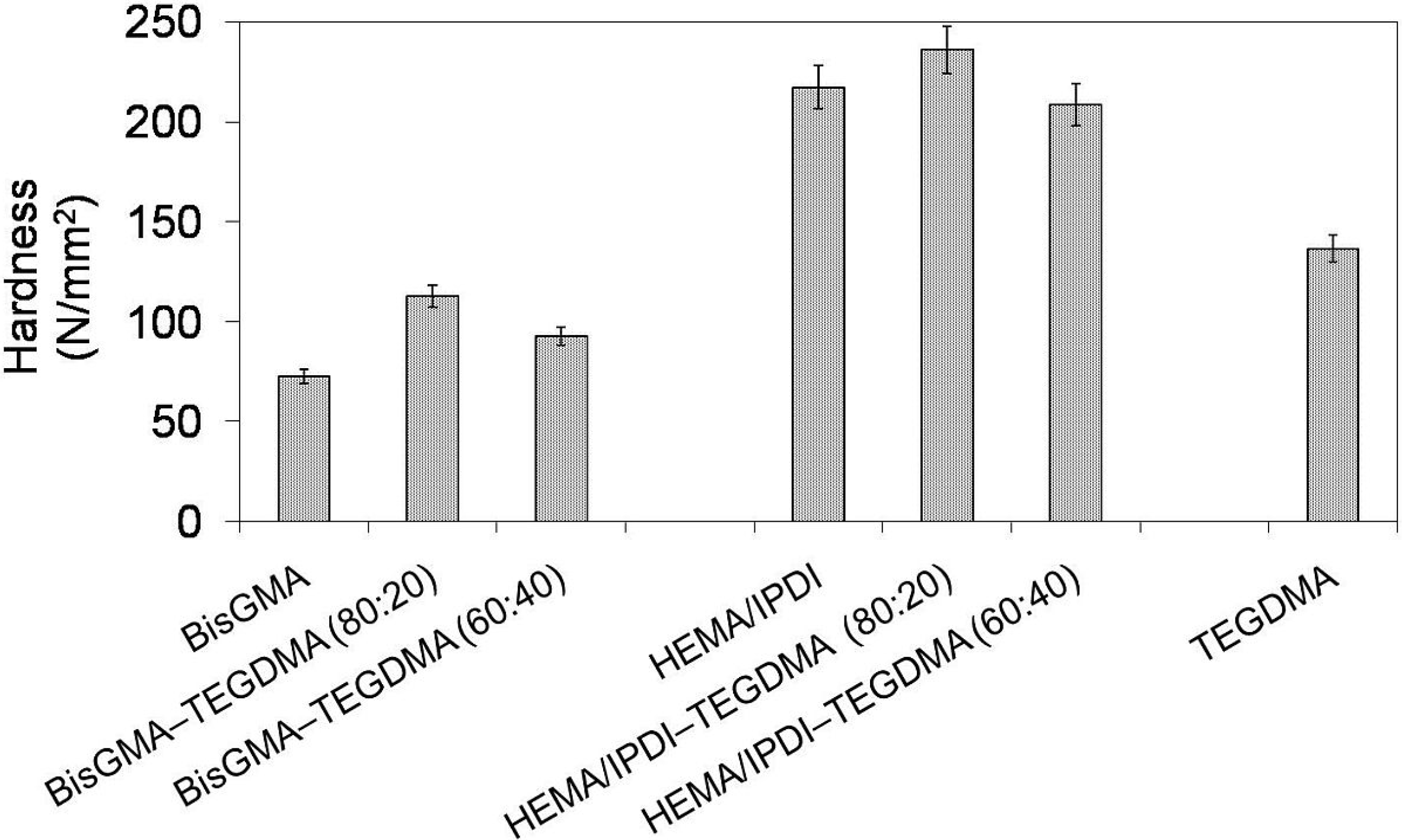
In
Figure 4 the relationship between Brinell hardness (
HB) and monomer composition is depicted. One can note that the hardness of investigated homopolymers follows this order:
HBpoly(Bis-GMA) <
HBpoly(TEGDMA) <
HBpoly(HEMA/IPDI) and ranges from 73 to 217 N/mm
2. The hardness of copolymers, after the initial growth, dropped with increase of TEGDMA content. Copolymers of HEMA/IPDI were characterized by significantly higher hardness (236 and 208 N/mm
2) than copolymers of Bis-GMA (113 and 93 N/mm
2). This behavior implies a strong influence of the degree of conversion on
HB. The initial increase in
HB resulted from the
DC increase. The consequent decrease in
HB, accompanied by an increase in the degree of conversion, was affected by the TEGDMA structure.
Figure 4.
The Brinell hardness of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
Figure 4.
The Brinell hardness of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
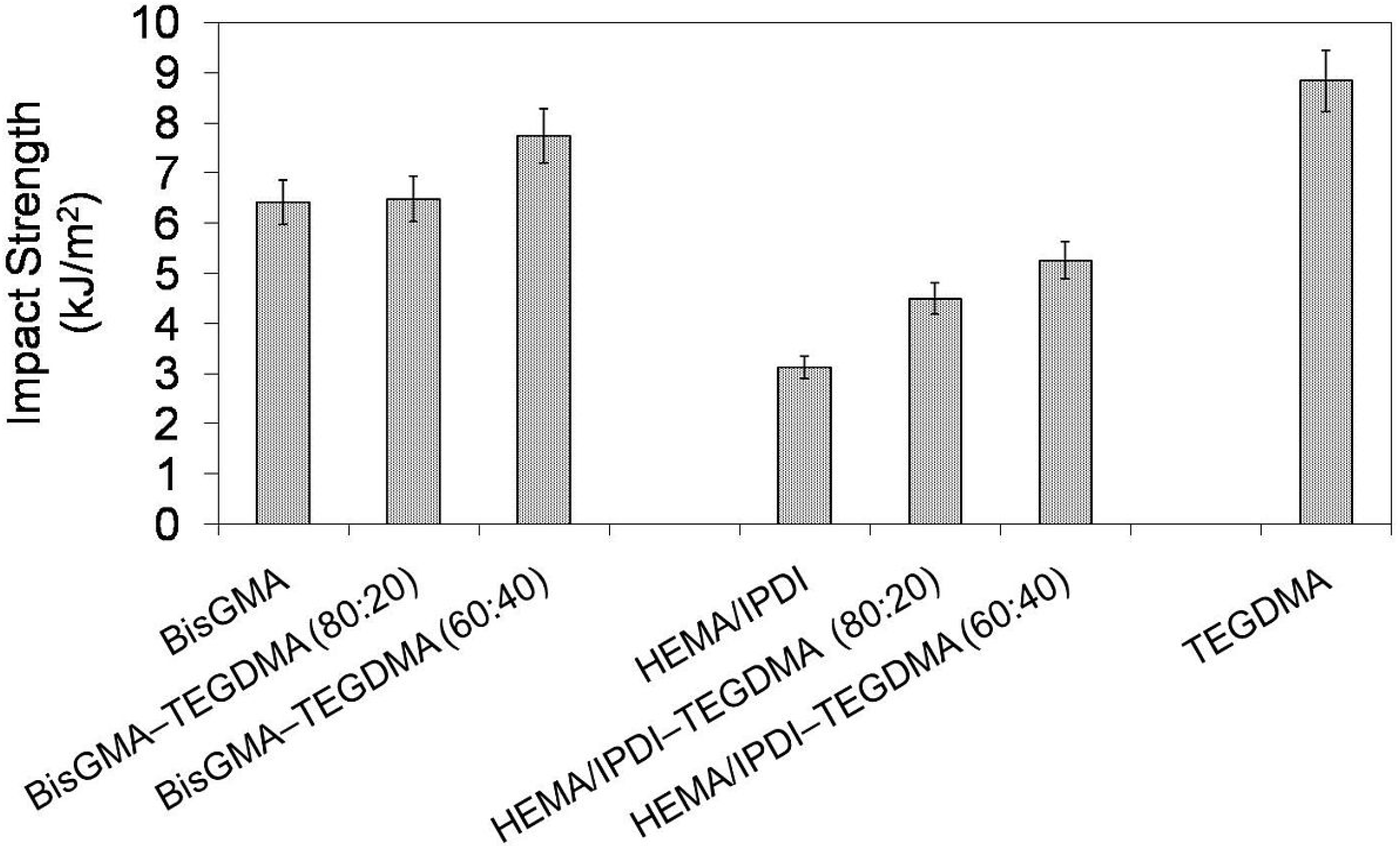
In
Figure 5 the relationship between impact strength and monomer composition is depicted. It may be seen that the HEMA/IPDI homopolymer had the lowest impact strength of 3.1 kJ/m
2, poly(Bis-GMA) had 6.4 kJ/m
2 and poly(TEGDMA) had the highest, of 8.8 kJ/m
2. Thereby, the order:
an poly(UDMA) <
an poly(Bis-GMA) <
an poly(TEGDMA), which was found in previous studies, was confirmed [
14]. The copolymers, in general, had better resistance to cracking than homopolymers. Copolymers of HEMA/IPDI were characterized by higher brittleness (
an = 4.5 and 5.3 kJ/m
2) than copolymers of Bis-GMA (
an = 6.5 and 7.7 kJ/m
2). The higher the TEGDMA content, the higher the impact resistance determined.
Figure 5.
The impact resistance of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
Figure 5.
The impact resistance of Bis-GMA, HEMA/IPDI and TEGDMA homopolymers as well as Bis-GMA–TEGDMA and HEMA/IPDI–TEGDMA copolymers.
The impact resistance of poly(urethane-dimethacrylate)s, due to its lower than expected values, remains a subject of great concern [
9,
14,
15]. As shown in earlier works, the fracture behavior of dimethacrylate polymer networks is a complex function of the monomer size and stiffness, the degree of conversion and the polymer morphology [
9,
14,
15]. Homo- and copolymerizations of UDMA, Bis-GMA and TEGDMA lead to the formation of strongly heterogeneous morphologies, composed of microgel agglomerates of different sizes and crosslink densities [
9,
14,
16,
17,
18,
19]. Previous XRPD studies on these homopolymers [
14] as well as on their acrylate analogues [
9] have shown that monomers having groups involved in hydrogen bonds with both the proton donor as well as with the acceptor, form more massive clusters than TEGDMA, having only the proton acceptor. Consequently, the homopolymers of UDMA and Bis-GMA were more brittle than the TEGDMA homopolymer.
Impact strength of the Bis-GMA homopolymer was shown to be twice as high as the impact strength of the HEMA/IPDI homopolymer. This behavior does not seem to be caused by the degree of conversion or microgel agglomerate dimensions. The latter network had higher crosslink density, resulting from a higher concentration of double bonds (
Table 1) as well as a higher degree of conversion (
Figure 1). Both monomers form strong hydrogen bonds and they organize themselves into microgel agglomerates of similar sizes through polymerization. The reason for the weak poly(UDMA) impact resistance might be the overall crosslink density in a less crosslinked matrix, in which highly crosslinked microgels are embedded. In the HEMA/IPDI as well as the Bis-GMA polymer networks, two kinds of crosslinks can be distinguished: permanent, covalent crosslinks and physical crosslinks, resulting from hydrogen bonds. The NH···N and NH···O hydrogen bonds, present in the UDMA system, are weaker than the O–H···O hydrogen bonds, present in the Bis-GMA (
Table 3) [
11,
20]. Hydrogen bonds between urethane groups are most probably not strong enough to bond microgel agglomerates in poly(UDMA) and to withstand high impact energies. The poly(Bis-GMA), where microgel agglomerates are even bigger, was shown to be tougher, since physical crosslinks are stronger. Finally, it could be concluded that impact resistance of poly(dimethacrylate)s depends on the following factors. When the polymer network morphology is less heterogeneous and consists of small clusters the impact resistance is good. When the network morphology is more heterogeneous, with massive clusters, the strength of hydrogen bonds determines the crack resistance. This situation is observed for polymer networks produced from stiff monomers, having groups involved in hydrogen bonding. The higher the strength of physical bonding, the higher the impact strength observed.
Table 3.
Types of hydrogen bonds in studied dimethacrylates and corresponding energies [
20].
Table 3.
Types of hydrogen bonds in studied dimethacrylates and corresponding energies [20].
| Type of Hydrogen Bond | Energy (kJ/mol) |
|---|
| O–H···N | 29 |
| O–H···O | 21 |
| N–H···N | 13 |
| N–H···O | 8 |
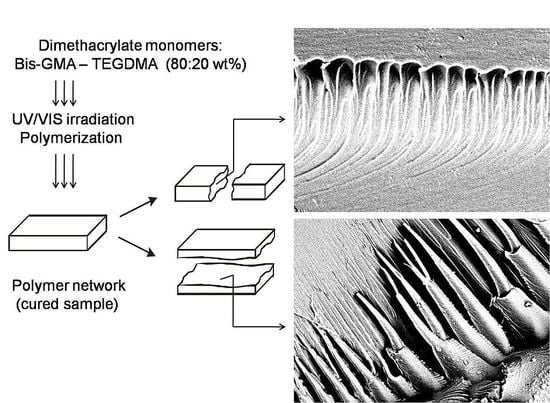
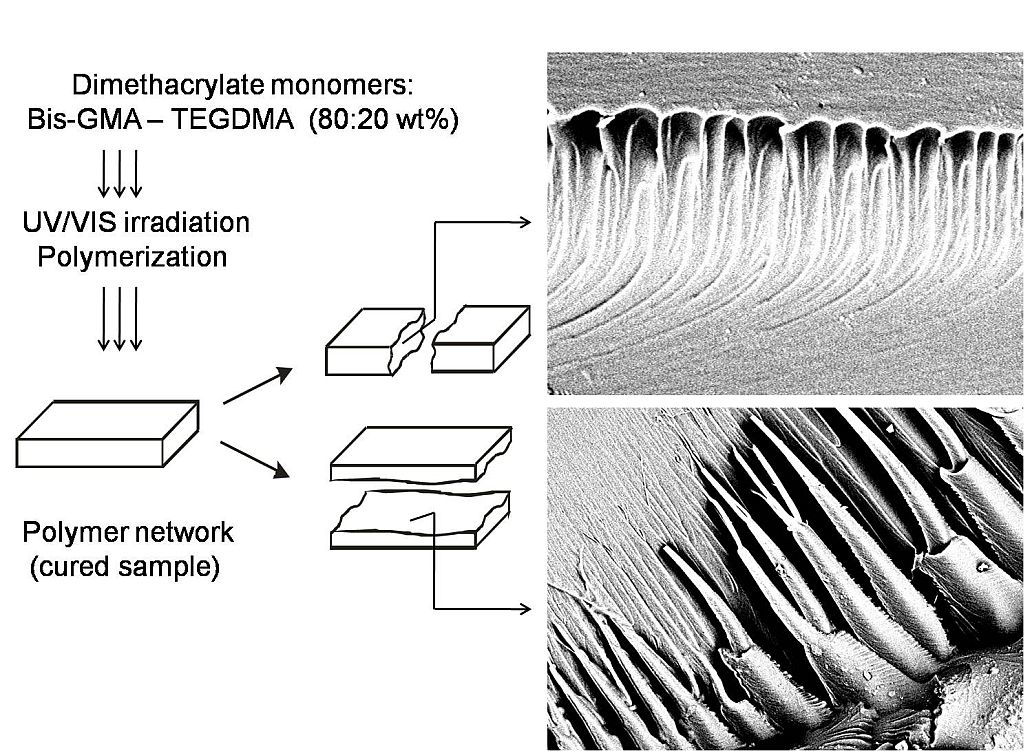
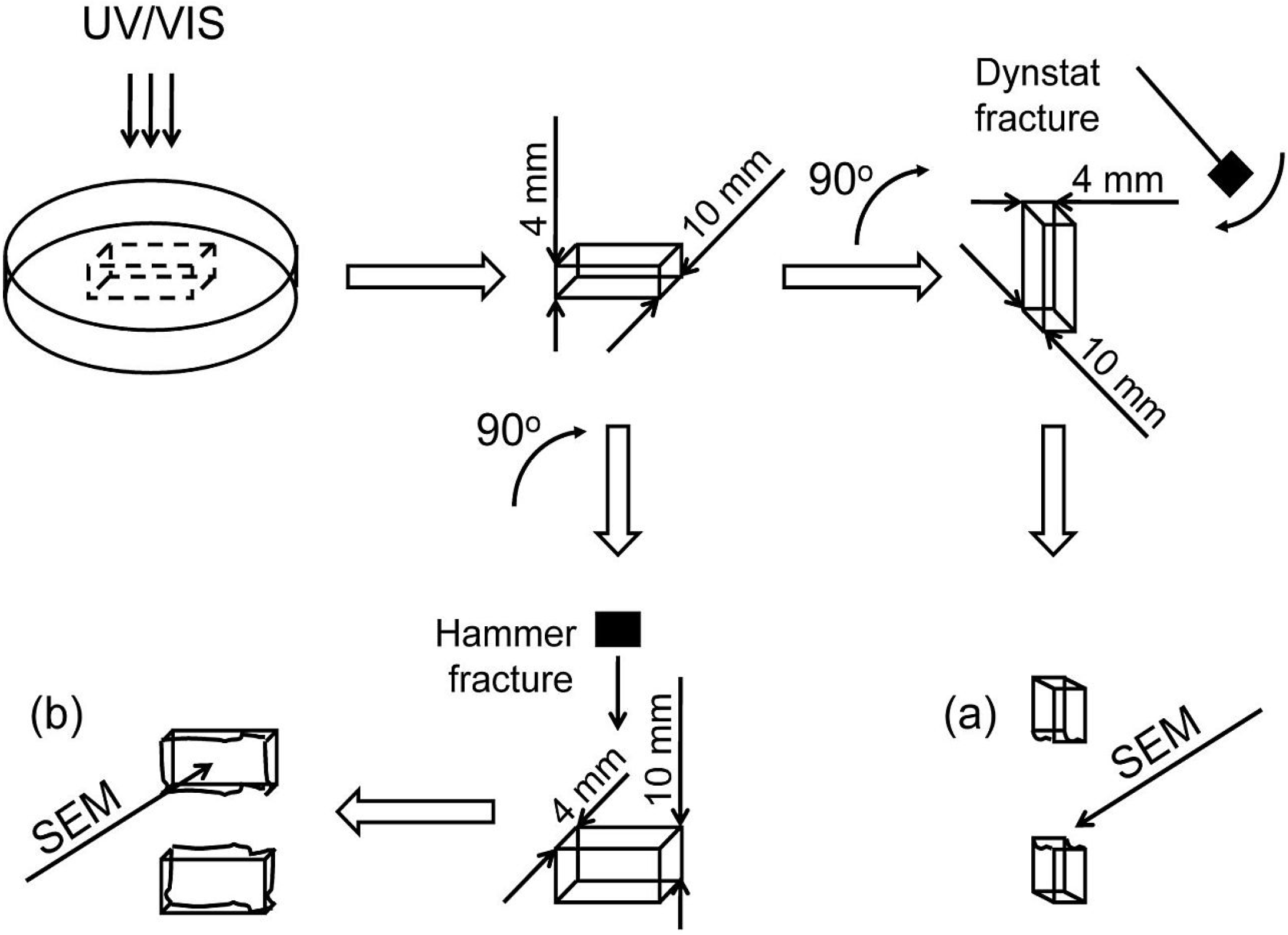
In this study, SEM was used for imaging the internal morphologies of polymer networks produced from Bis-GMA, HEMA/IPDI and TEGDMA.
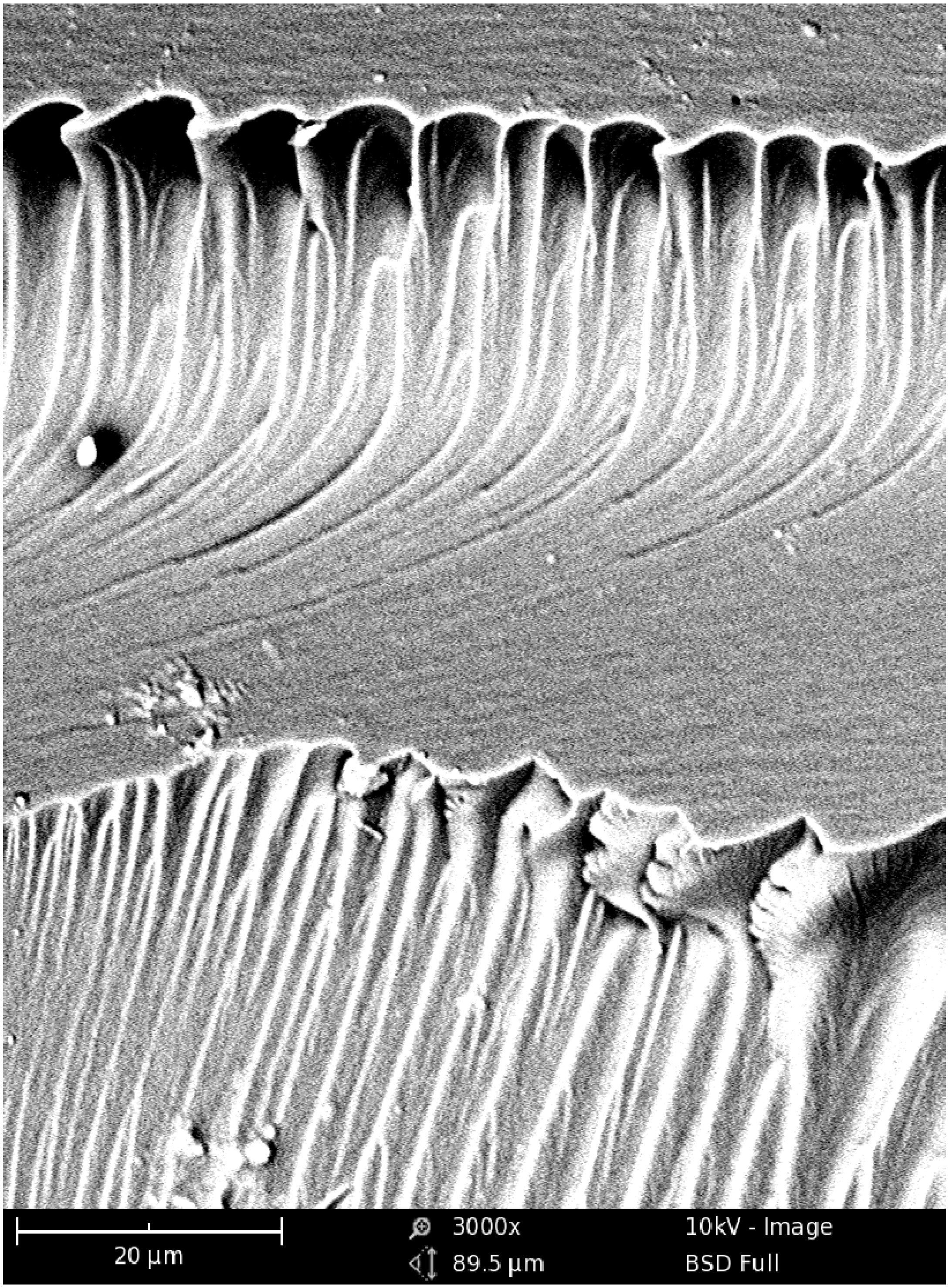
Figure 6 shows the exemplary micrograph of the Bis-GMA–TEGDMA (80:20) sample, fractured in impact tests. The observed surface was parallel to the direction of the UV/VIS irradiation (
Figure 7a). The SEM image revealed patterns, suggesting a unidirectional orientation of microgel agglomerates, perpendicular to this fracture,
i.e., perpendicular to the light direction. This finding was confirmed by SEM analysis of the fractures, performed during controlled crack propagation, presenting surfaces perpendicular to the direction of irradiation (
Figure 8,
Figure 7b). The copolymers revealed a morphology, consisting of nodular objects arranged in a regular array, perpendicular to the UV/VIS light. The Bis-GMA copolymers revealed sharper-edged morphological objects in comparison to the HEMA/IPDI copolymers. It suggests that the chemical crosslinking and the agglomerate dimensions determine the fracture pattern. However, impact strength values are dependent on the overall energy of interactions between clusters in poly(dimethacrylate)s, coming from chemical and physical crosslinks.
Figure 6.
The SEM image of the Bis-GMA–TEGDMA (80:20) fractured surface; a scale bar represents 20 μm.
Figure 6.
The SEM image of the Bis-GMA–TEGDMA (80:20) fractured surface; a scale bar represents 20 μm.
Figure 7.
The representation of the fracture directions: (a) from impact tests, fracture surface was parallel to the direction of irradiation; (b) made with a hammer, fracture surface was perpendicular to the direction of irradiation.
Figure 7.
The representation of the fracture directions: (a) from impact tests, fracture surface was parallel to the direction of irradiation; (b) made with a hammer, fracture surface was perpendicular to the direction of irradiation.
Figure 8.
The SEM images of: (a) and (b) the Bis-GMA–TEGDMA (80:20); (c) HEMA/IPDI–TEGDMA (80:20) fractured surfaces; a scale bars represents 30 μm.
Figure 8.
The SEM images of: (a) and (b) the Bis-GMA–TEGDMA (80:20); (c) HEMA/IPDI–TEGDMA (80:20) fractured surfaces; a scale bars represents 30 μm.
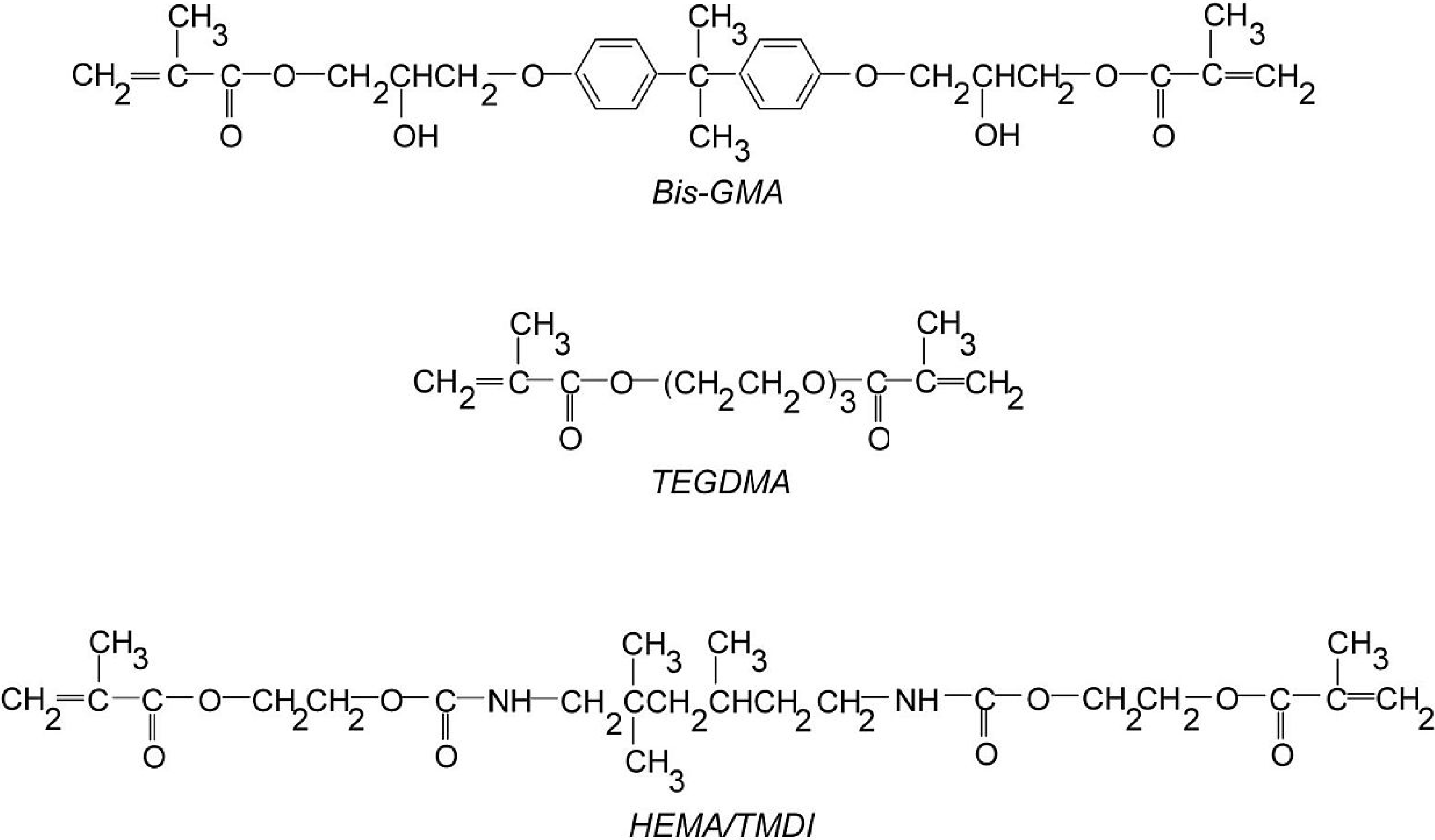
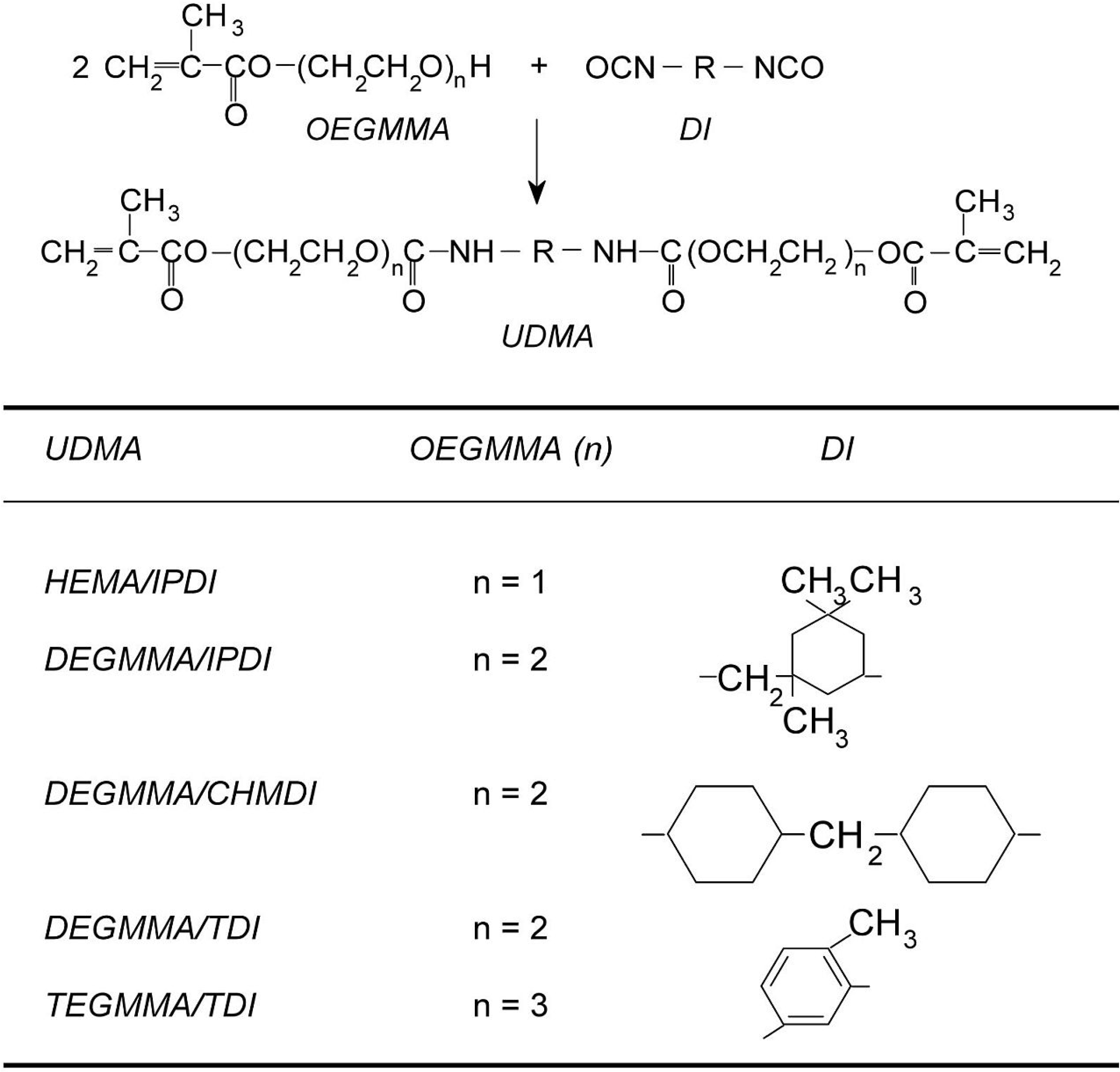
2.2. Alternative UDMA Monomers as HEMA/TMDI Replacement in Copolymers with Bis-GMA and TEGDMA
The second group of copolymers was tested for the influence of various UDMA monomers on the properties of networks, produced by their copolymerization with Bis-GMA and TEGDMA. This was achieved by mixing HEMA/IPDI, DEGMMA/IPDI, DEGMMA/CHMDI, DEGMMA/TDI, as well as TEGMMA/TDI with Bis-GMA and TEGDMA, in a 38:42:20 wt% ratio, and photopolymerized. The popular dentistry copolymer of HEMA/TMDI–Bis-GMA–TEGDMA (38:42:20) was produced as the precursor. For comparative purposes, the homopolymers of all UDMAs were obtained.
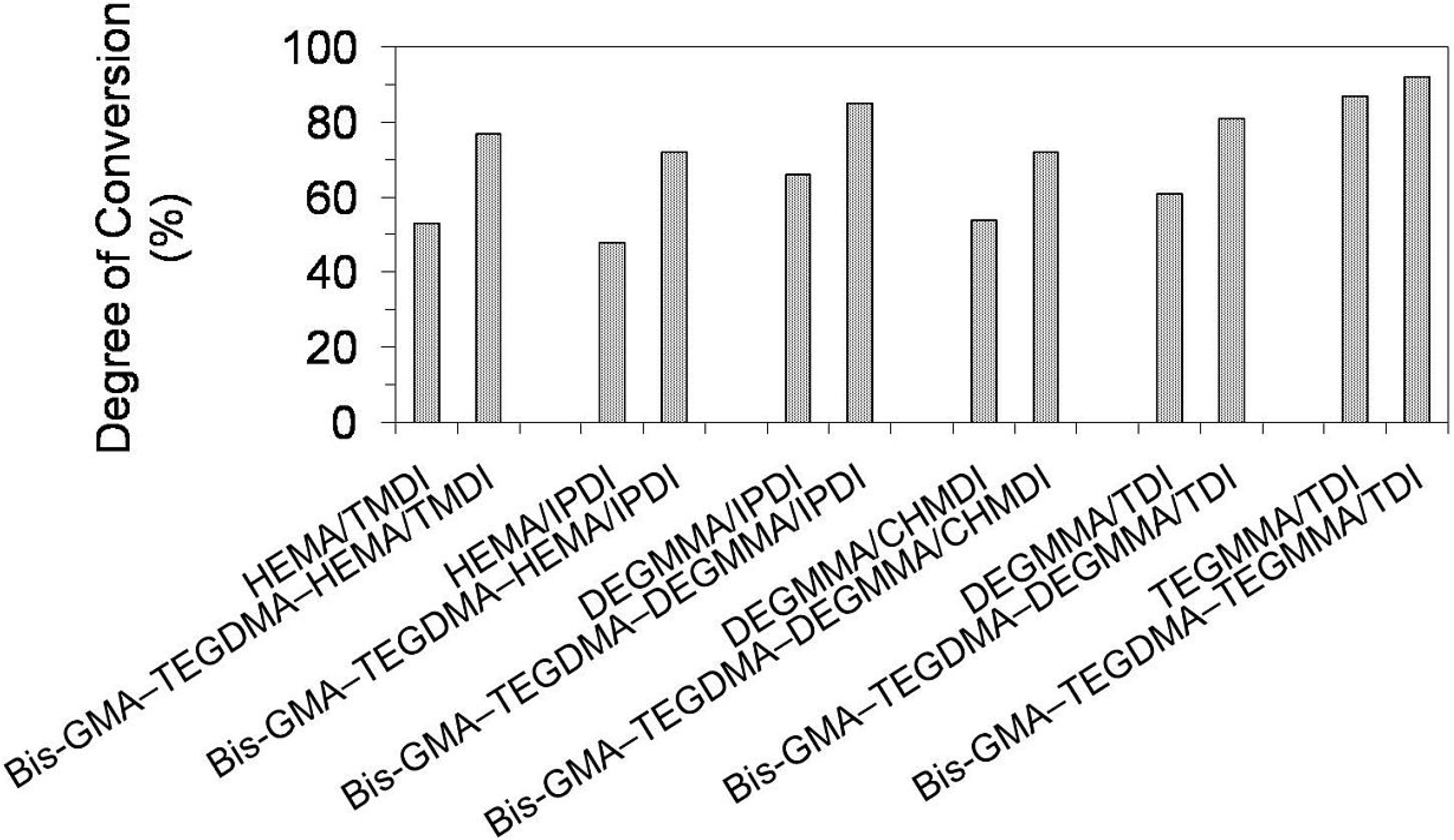
Figure 9 presents results for the degree of conversion (
DC) in homopolymer and copolymer networks. The
DC in homopolymers ranged from 48% to 87% and followed the order:
DCpoly(HEMA/IPDI) <
DCpoly(HEMA/TMDI) <
DCpoly(DEGMMA/CHMDI) <
DCpoly(DEGMMA/TDI) <
DCpoly(DEGMMA/IPDI) <
DCpoly(TEGMMA/TDI). As can be seen, increasing oligooxyethylene chain length caused an increase of the UDMA elasticity and mobility, giving rise to an increase in the
DC. The diisocyanate chemical character also influenced the homopolymer
DC, which increased in the following way: cycloaliphatic symmetrical (CHMDI) < aromatic asymmetrical (TDI) < cycloaliphatic asymmetrical (IPDI) < aliphatic (TMDI). The lowest
DC in the HEMA/IPDI homopolymer, of 48%, can be explained by the spacious and stiff cycloaliphatic ring, asymmetrically substituted with two short HEMA wings. HEMA/TMDI, which is sometimes used alone in dental formulations [
4] when homopolymerized, achieved a
DC of 53% slightly exceeding 50%. The
DC in homopolymers, having DEGMMA was found within the range of 54%–66%, whereas in poly(TEGMMA/TDI) it equaled 87%.
Figure 9.
The degree of conversion in homopolymers and copolymers composed of UDMA, Bis-GMA and TEGDMA.
Figure 9.
The degree of conversion in homopolymers and copolymers composed of UDMA, Bis-GMA and TEGDMA.
Copolymerization resulted in an increase in the degree of conversion in polymer networks, when compared to UDMA homopolymers. The DC in copolymers ranged from 72% to 92%. The differences in DCs between corresponding homo- and copolymers decreased with increasing oligooxyethylene chain length. The increases of around 50% in the DC were recorded for UDMAs, having HEMA, of around 30% for those having DEGMMA, and 8% in the case of TEGDMA/TDI. It can be concluded, that copolymerization is beneficial for the structural heterogeneity of dimethacrylate networks from the perspective of the degree of conversion.
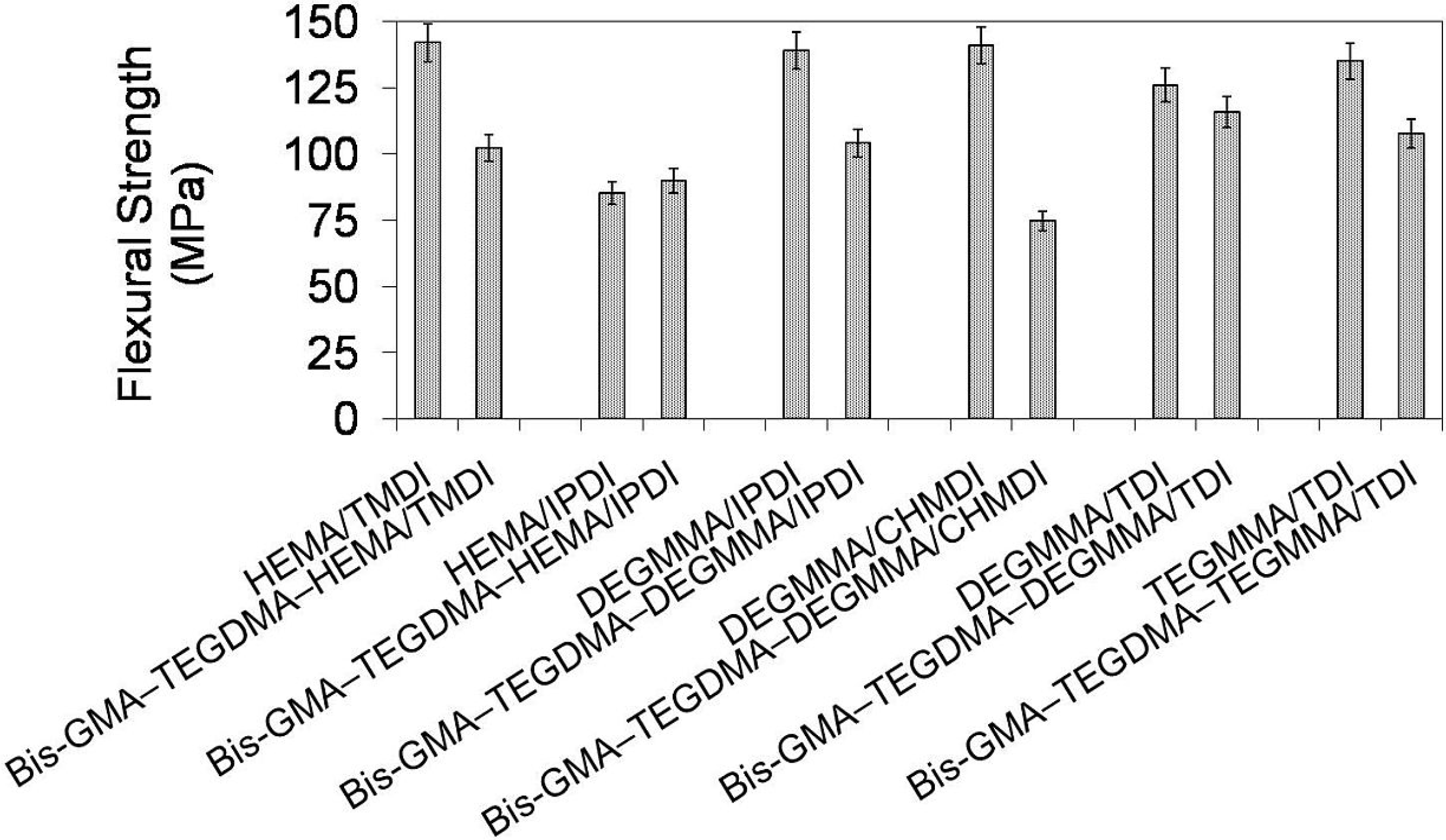
In
Figure 10 the relationship between flexural strength and polymer composition is depicted. It is seen, that flexural strengths of the investigated homopolymers ranged from 85 to 142 MPa and followed the order: σ
poly(HEMA/IPDI) < σ
poly(DEGMMA/TDI) < σ
poly(TEGMMA/TDI) < σ
poly(DEGMMA/IPDI) < σ
poly(DEGMMA/CHMDI) = σ
poly(HEMATMDI). The flexural strengths of their corresponding copolymers were lower, except HEMA/IPDI, and ranged from 75 to 116 MPa, following this order: σ
poly(DEGMMA/CHMDI) < σ
poly(HEMA/IPDI) < σ
poly(HEMATMDI) < σ
poly(DEGMMA/IPDI) < σ
poly(DEGMMA/TDI) < σ
poly(TEGMMA/TDI). The HEMA/IPDI copolymer had a flexural strength of 90 MPa, which was slightly higher than σ of the homopolymer. This could be explained by the TEGDMA and Bis-GMA influence. The flexural strength values of poly(TEGDMA) (87 MPa), and poly(Bis-GMA) (112 MPa) were higher than σ corresponding to poly(HEMA/IPDI) (85 MPa) and lower than σ corresponding to the remaining UDMA homopolymers, starting from 126 MPa. The copolymers of the following alternative UDMA monomers: DEGMMA/IPDI, DEGMMA/TDI and TEGMMA/TDI had a higher flexural strength than the HEMA/TMDI–Bis-GMA–TEGDMA copolymer.
Figure 10.
The flexural strength of homopolymers and copolymers composed of UDMA, Bis-GMA and TEGDMA.
Figure 10.
The flexural strength of homopolymers and copolymers composed of UDMA, Bis-GMA and TEGDMA.
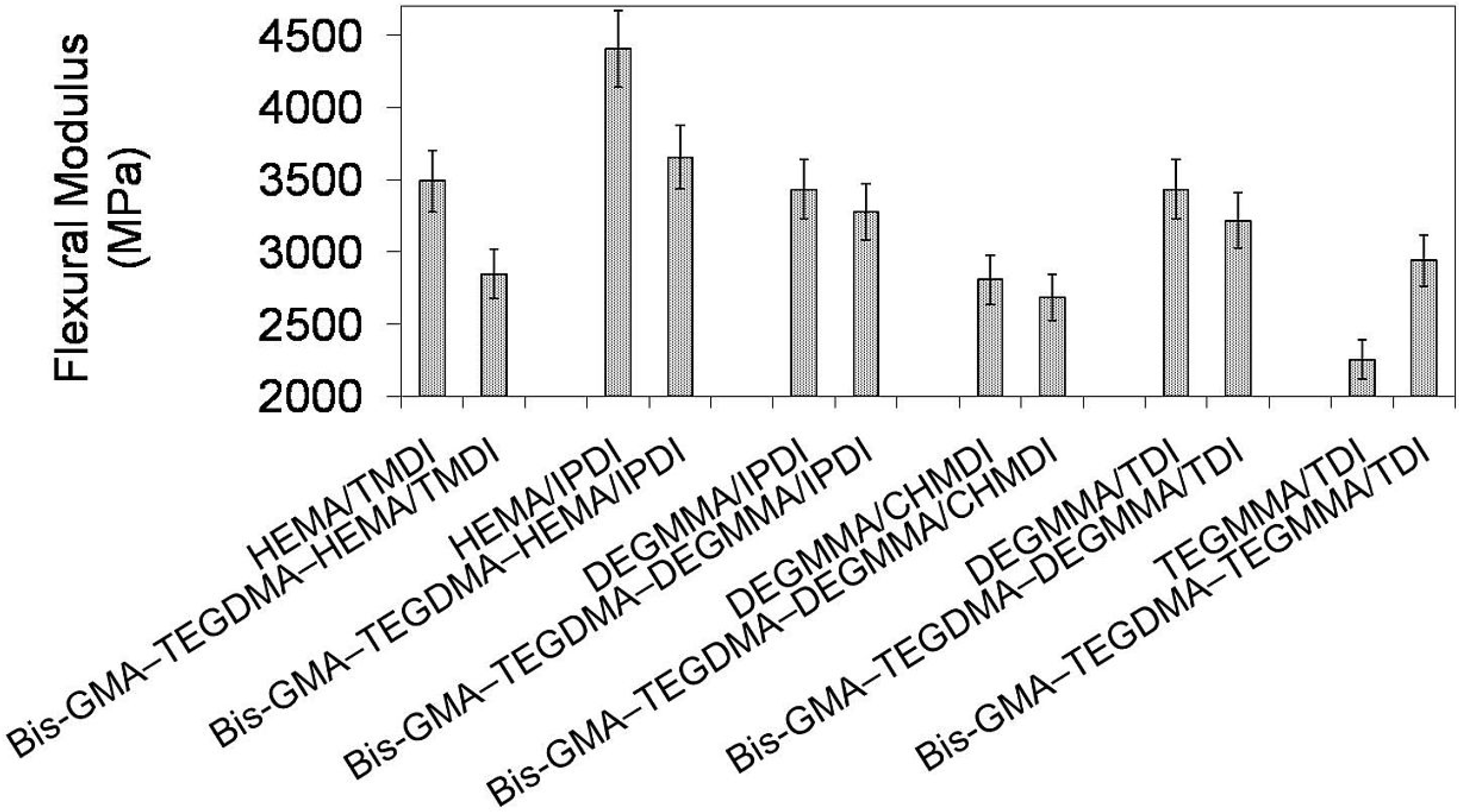
In
Figure 11 the relationship between flexural modulus and polymer compositions is depicted. One can observe that the modulus of investigated homopolymers followed the order:
Epoly(TEGMMA/TDI) <
Epoly(DEGMMA/CHMDI) <
Epoly(DEGMMA/TDI) =
Epoly(DEGMMA/IPDI) <
Epoly(HEMA/TMDI) <
Epoly(HEMA/IPDI) and ranged from 2252 to 4406 MPa. The modulus of copolymers ranged from 2681 to 3653 MPa and increased in this order:
Epoly(DEGMMA/CHMDI) <
Epoly(HEMA/TMDI) <
Epoly(TEGMMA/TDI) <
Epoly(DEGMMA/TDI) <
Epoly(DEGMMA/IPDI) <
Epoly(HEMA/IPDI). The modulus of the copolymer was always lower than
E of the corresponding UDMA homopolymer, except TEGMMA/TDI. In this case, the increase in modulus resulted from significantly higher stiffness of Bis-GMA and TEGDMA than TEGMMA/TDI. The copolymers of the following alternative UDMA monomers: HEMA/IPDI, DEGMMA/IPDI, DEGMMA/TDI and TEGMMA/TDI had a modulus higher than the HEMA/TMDI–Bis-GMA–TEGDMA copolymer.
Figure 11.
The flexural modulus of homopolymers and copolymers composed of UDMA, Bis-GMA and TEGDMA.
Figure 11.
The flexural modulus of homopolymers and copolymers composed of UDMA, Bis-GMA and TEGDMA.
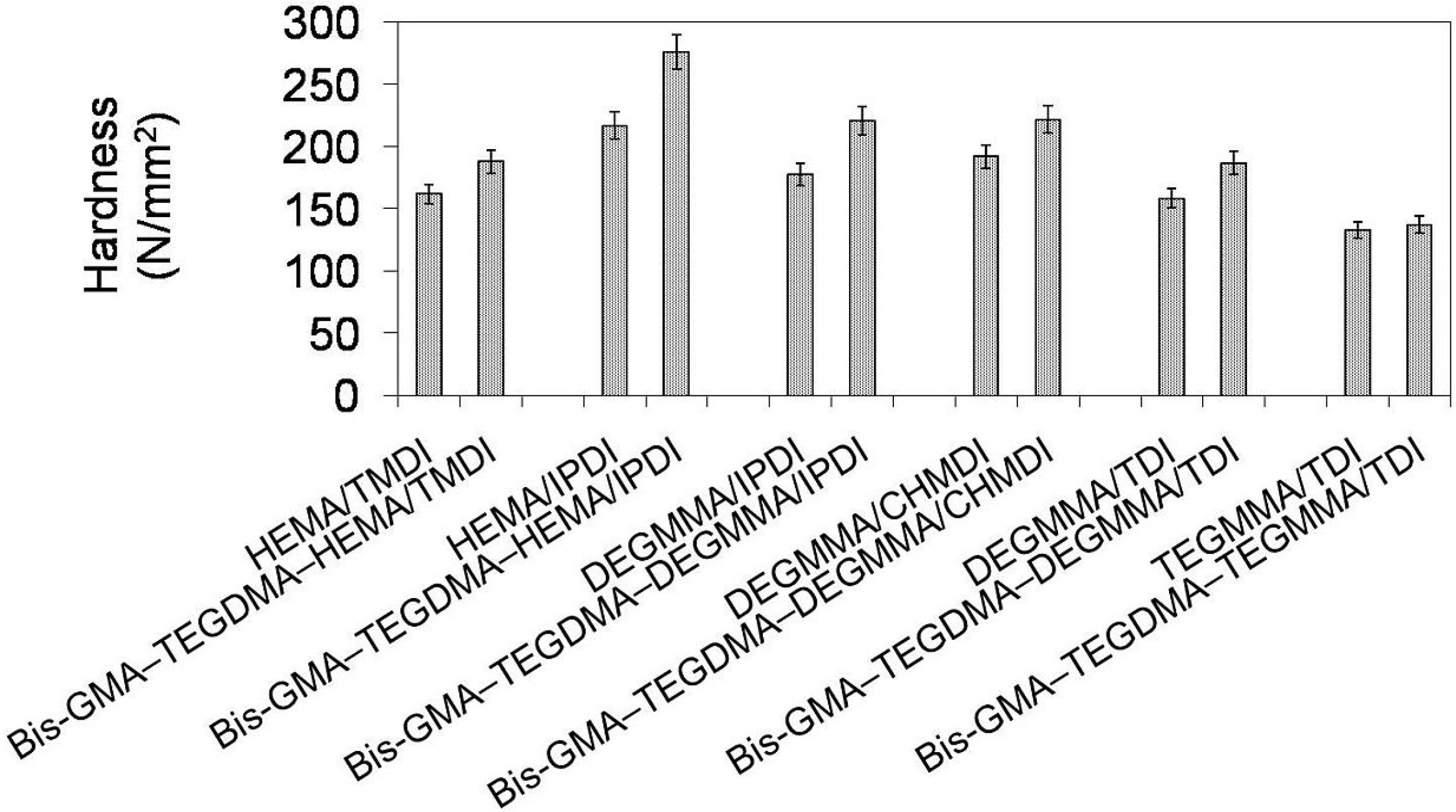
In
Figure 12 the relationship between Brinell hardness and monomer composition is depicted. One can note that the hardness of homopolymers as well as copolymers followed the same order:
HBpoly(TEGMMA/TDI) <
HBpoly(DEGMMA/TDI) <
HBpoly(HEMA/TMDI) <
HBpoly(DEGMMA/IPDI) <
HBpoly(DEGMMA/CHMDI) <
HBpoly(HEMA/IPDI). Hardness values of homopolymers ranged from 133 to 217 N/mm
2, whereas values of corresponding copolymers were higher and ranged from 137 to 276 N/mm
2. These increases might be explained by the increases in the degree of conversion. HEMA/IPDI demonstrated a very positive effect on the copolymer hardness, which was the highest among those studied. DEGMMA/IPDI and DEGMMA/CHMDI also improved the copolymer hardness, in relation to the HEMA/TMDI–Bis-GMA–TEGDMA copolymer.
Figure 12.
The Brinell hardness of homopolymers and copolymers composed of UDMA, Bis-GMA, and TEGDMA.
Figure 12.
The Brinell hardness of homopolymers and copolymers composed of UDMA, Bis-GMA, and TEGDMA.
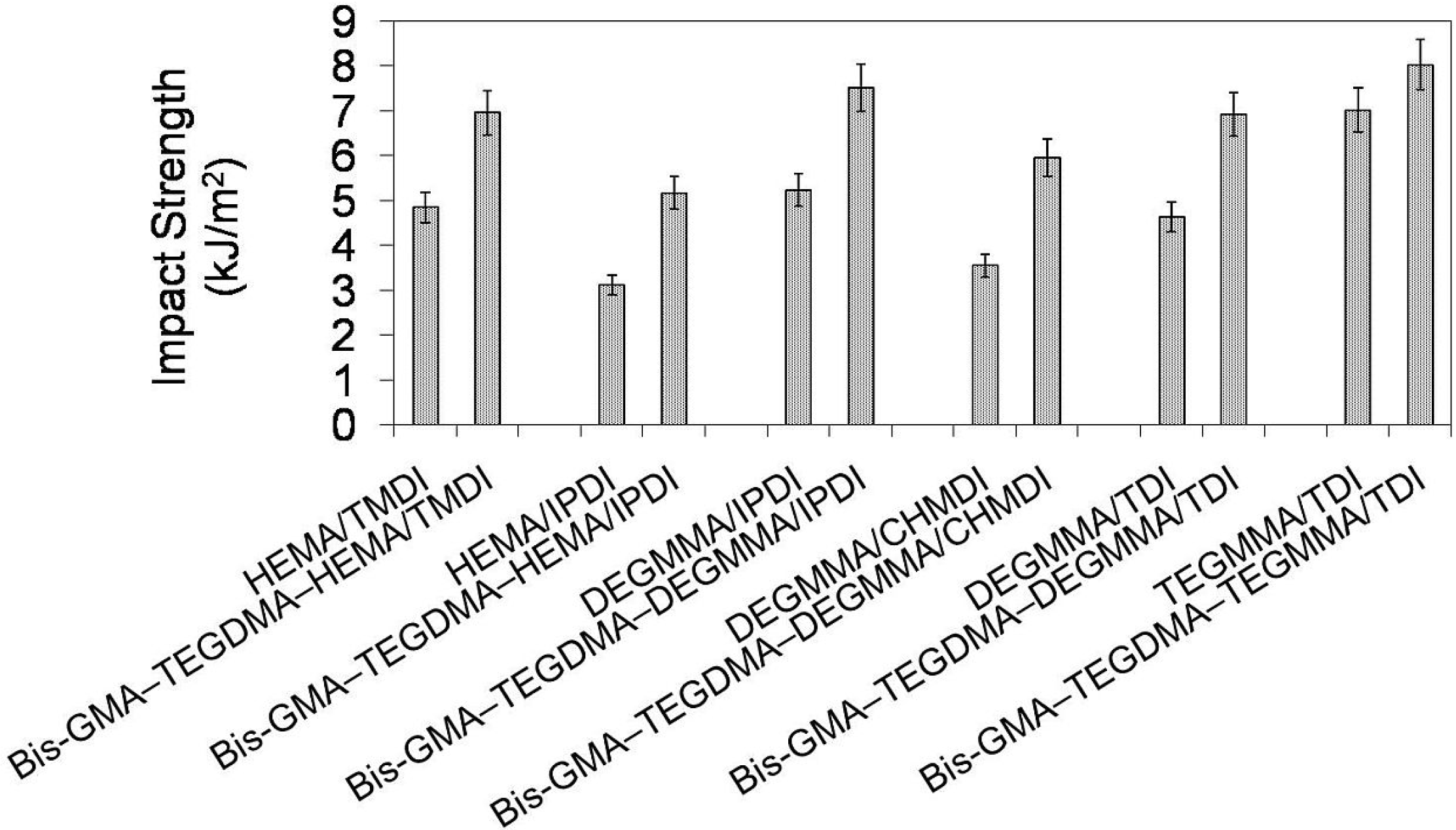
In
Figure 13 the relationship between impact strength and monomer composition is depicted. It may be seen that the impact resistance of homopolymers as well as copolymers followed the same order:
an poly(HEMA/IPDI) <
an poly(DEGMMA/CHMDI) <
an poly(DEGMMA/TDI) <
an poly(HEMA/TMDI) <
an poly(DEGMMA/IPDI) <
an poly(TEGMMA/TDI). Impact strength for UDMA homopolymers could be found within 3.12–7.02 kJ/m
2, whereas for corresponding copolymers from 5.17 to 8.03 kJ/m
2. The improvement of fracture resistance resulting from copolymerization can be explained by the TEGDMA properties. The TEGDMA homopolymer had the highest impact strength, of 8.83 kJ/m
2 among the polymers studied here. When comparing the HEMA/TMDI–Bis-GMA–TEGDMA copolymer to the copolymers with alternative UDMAs, the improvement of impact resistance was achieved when DEGMMA/IPDI and TEGMMA/TDI were used.
Figure 13.
The impact strength of homopolymers and copolymers composed of UDMA, Bis-GMA, and TEGDMA.
Figure 13.
The impact strength of homopolymers and copolymers composed of UDMA, Bis-GMA, and TEGDMA.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












