
Theoretical and Experimental Studies on the Crystal Structure, Electronic Structure and Optical Properties of SmTaO4


Abstract
:1. Introduction
2. Experimental Methods and Theoretical Calculations
2.1. Experimental Methods
2.2. Computational Details
3. Results and Discussion
3.1. Crystal Structure



{kind=link}
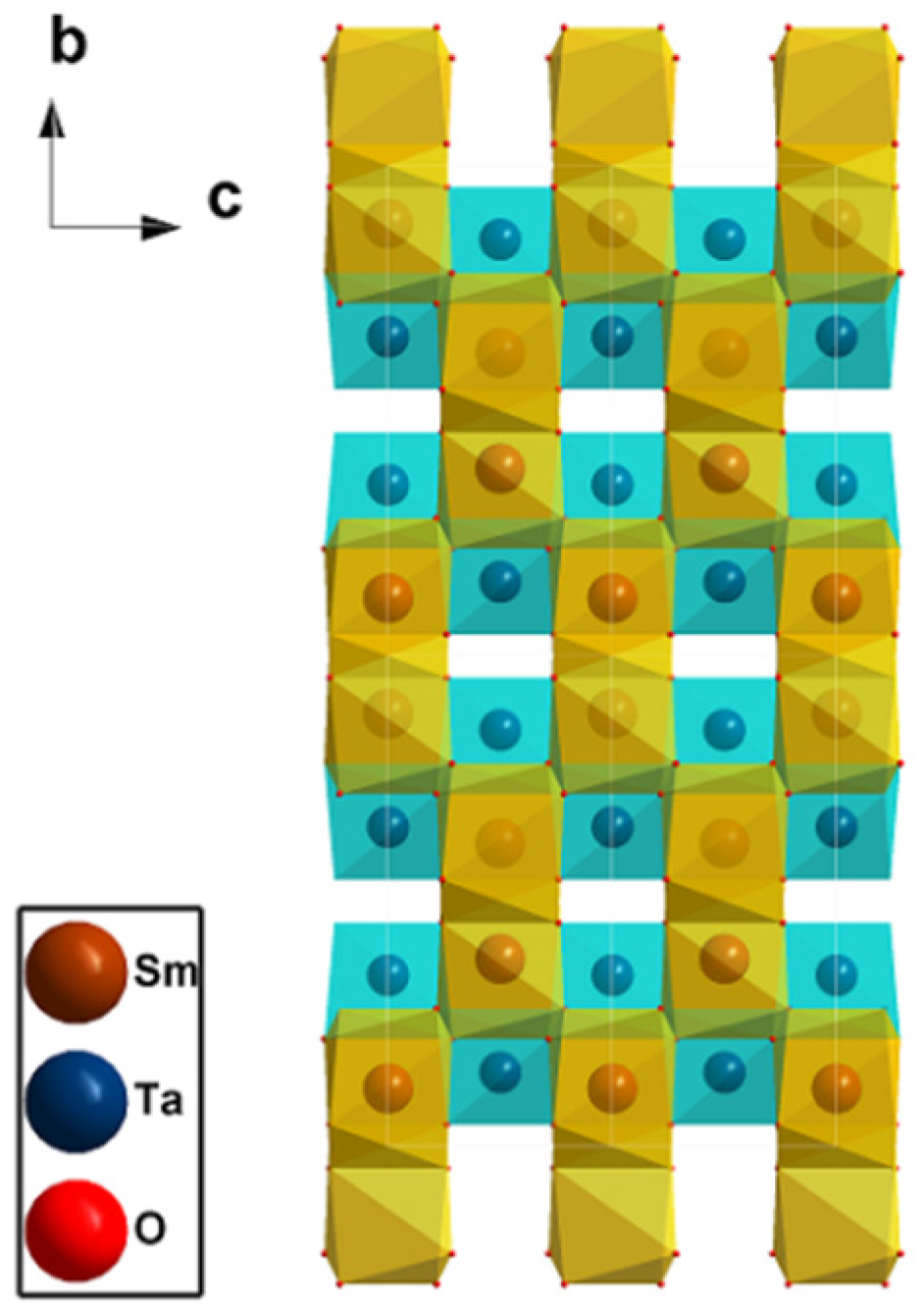{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice Constant | Experimental | LDA | GGA | Reported |
|---|---|---|---|---|
| a (Å) | 5.444 (±0.0085) | 5.393 | 5.514 | 5.455 a, 5.405 b |
| b (Å) | 11.115 (±0.0072) | 10.968 | 11.241 | 11.137 a, 11.063 b |
| c (Å) | 5.081 (±0.0079) | 5.094 | 5.161 | 5.091 a, 5.081 b |
| β (°) | 95.74 (±0.0027) | 96.06 | 95.47 | 94.73 a, 95.62 b |
| V (Å3) | 305.93 | 299.63 | 318.43 | 308.24 a |
| Density (g/cm3) | 8.55 | 8.76 | 8.25 | / |
| Compound | Ta–O | Sm–O | O–O |
|---|---|---|---|
| LDA | 1.9345 × 4 a 1.9955 × 4 2.2452 × 4 | 2.3002 × 4 2.3173 × 4 2.4774 × 4 | 2.6182 × 2 2.6952 × 4 2.7614 × 2 2.8789 × 4 |
| GGA | 1.9864 × 4 2.0482 × 4 2.3671 × 4 | 2.3570 × 4 2.3975 × 4 2.5567 × 4 | 2.7089 × 2 2.7779 × 4 2.8499 × 2 2.9822 × 4 |
| Experimental | 1.840 1.925 2.335 | 2.338 2.447 2.545 | / |
| Atom | LDA | GGA | Experimental | Reported a |
|---|---|---|---|---|
| Sm | (0.25, 0.116, 0) | (0.25, 0.115, 0) | (0.25, 0.112, 0) | (0.25, 0.118, 0) |
| Ta | (0.25, 0.652, 0) | (0.25, 0.651, 0) | (0.25, 0.649, 0) | (0.25, 0.651, 0) |
| O1 | (0.010, 0.722, 0.231) | (0.009, 0.722, 0.227) | (0.006, 0.717, 0.205) | (0.015, 0.719, 0.216) |
| O2 | (0.909, 0.465, 0.247) | (0.911, 0.453, 0.249) | (0.894, 0.454, 0.266) | (0.901, 0.456, 0.240) |
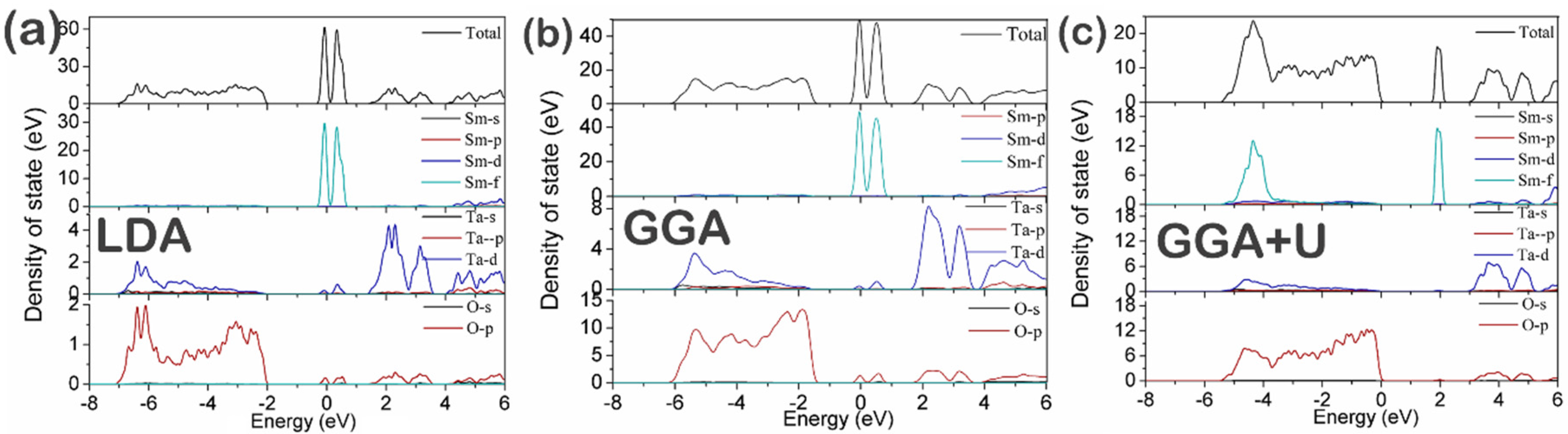
3.2. Electronic Structure

| Band Gap | LDA | GGA | GGA + U | Experiment | Reported [16] |
|---|---|---|---|---|---|
| O 2p–Ta 5d | 3.65 | 3.49 | 3.57 | 3.68 | 3.8 |
| O 2p–Sm 4f | 1.35 | 0.98 | 1.76 | / | / |
| Sm 4f–Ta 5d | 0.46 | 0.7 | 1.12 | / | / |

| Atom | Ion | Charge (e) | ||
|---|---|---|---|---|
| LDA | GGA | GGA + U | ||
| O | 1 | −0.63 | −0.67 | −0.68 |
| O | 2 | −0.58 | −0.63 | −0.64 |
| O | 3 | −0.63 | −0.67 | −0.68 |
| O | 4 | −0.58 | −0.63 | −0.64 |
| O | 5 | −0.63 | −0.67 | −0.68 |
| O | 6 | −0.58 | −0.63 | −0.64 |
| O | 7 | −0.63 | −0.67 | −0.68 |
| O | 8 | −0.58 | −0.63 | −0.64 |
| Sm | 1 | 1.39 | 1.44 | 1.75 |
| Sm | 2 | 1.39 | 1.44 | 1.75 |
| Ta | 1 | 1.02 | 1.17 | 0.90 |
| Ta | 2 | 1.02 | 1.17 | 0.90 |
3.3. Optical Properties

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dorenbos, P.; Rogers, E.G. Vacuum referred binding energies of the lanthanides in transition metal oxide compounds. J. Solid State Sci. Technol. 2014, 3, R150–R158. [Google Scholar]
- Porter, S.H. Perovskite and pyrochlore Tantalum Oxide Nitrides: Synthesis and characterization. Master’s Thesis, Ohio State University, Columbus, OH, USA, December 2012. [Google Scholar]
- Voloshyna, O.; Neicheva, S.V.; Starzhinskiy, N.G.; Zeny, I.M.; Gridin, S.S.; Baumer, V.N.; Sidletskiy, O.Ts. Luminescent and scintillation properties of orthotantalates with common formulae RETaO4 (RE = Y, Sc, La, Lu and Gd). Mater. Sci. Eng. B 2013, 178, 1491–1496. [Google Scholar] [CrossRef]
- Wu, M.; Liu, X.; Gu, M.; Li, C.; Liu, B.; Huang, S. Characterization and luminescence properties of sol-gel derived M’-type LuTaO4:Ln3+ (Ln = Pr, Sm, Dy) phosphors. Mater. Res. Bull. 2014, 60, 652–658. [Google Scholar] [CrossRef]
- Sych, A.M.; Golub, A.M. Niobates and tantalates of tervalent elements. Russ. Chem. Rev. 1977, 46, 210–2250. [Google Scholar] [CrossRef]
- Shimizu, K.; Itoh, S.; Hatamachi, T.; Kodama, T.; Sato, M.; Toda, K. Photocatalytic water splitting on Ni-intercalated Ruddlesden-Popper Tantalate H2La2/3Ta2O7. Chem. Mater. 2005, 17, 5161–5166. [Google Scholar] [CrossRef]
- Osterloh, F.E. Inorganic materials as catalysts for photochemical splitting of water. Chem. Mater. 2008, 20, 35–54. [Google Scholar] [CrossRef]
- Mokkelbost, T.; Andersen, Ø.; Strøm, R.A.; Wiik, K.; Grande, T.; Einarsrud, M.-A. High-temperature proton-conducting LaNbO4-based materials: Powder synthesis by spray pyrolysis. J. Am. Ceram. Soc. 2007, 90, 3395–3400. [Google Scholar] [CrossRef]
- Shen, Y.; Leckie, R.M.; Levi, C.G.; Clarke, D.R. Low thermal conductivity without oxygen vacancies in equimolar YO1.5 + TaO2.5− and YbO1.5 + TaO2.5− stabilized tetragonal zirconia ceramics. Acta Mater. 2010, 58, 4424–4431. [Google Scholar]
- Siqueira, K.P.F.; Carvalho, G.B.; Dias, A. Influence of the processing conditions and chemical environment on the crystal structures and phonon modes of lanthanide orthotantalates. Dalton Trans. 2011, 40, 9454–9460. [Google Scholar] [CrossRef] [PubMed]
- Nyman, M.; Rodriguez, M.A.; Rohwer, L.E.S.; Martin, J.E.; Waller, M.; Osterloh, F.E. Unique LaTaO4 polymorph for multiple energy applications. Chem. Mater. 2009, 21, 4731–4737. [Google Scholar] [CrossRef]
- Forbes, T.Z.; Nyman, M.; Rodriguez, M.A.; Navrotsky, A. The energetics of lanthanum tantalate materials. J. Solid State Chem. 2010, 183, 2516–2521. [Google Scholar] [CrossRef]
- Machida, M.; Yabunaka, J.; Kijima, T. Synthesis and photocatalytic property of layered perovskite tantalates, RbLnTa2O7 (Ln = La, Pr, Nd, and Sm). Chem. Mater. 2000, 12, 812–817. [Google Scholar] [CrossRef]
- Gillen, R.; Clark, S.J.; Robertson, J. Nature of the electronic band gap in lanthanide oxides. Phys. Rev. B 2013, 87, 125116. [Google Scholar] [CrossRef] [Green Version]
- Machida, M.; Yabunaka, J.; Kijima, T.; Matsushima, S.; Arai, M. Electronic structure of layered tantalates photocatalysts, RbLnTa2O7 (Ln = La, Pr, Nd, and Sm). Int. J. Inorg. Mater. 2001, 3, 545–550. [Google Scholar] [CrossRef]
- Machida, M.; Murakami, S.; Kijima, T. Photocatalytic property and electronic structure of lanthanide tantalates, LnTaO4 (Ln = La, Ce, Pr, Nd, and Sm). J. Phys. Chem. B 2001, 105, 3289–3294. [Google Scholar] [CrossRef]
- Gao, L.H.; Ma, Z.; Fan, Q.B. First-principle studies of the electronic structure and reflectivity of LaTiO3 and Sr doped LaTiO3 (La1−xSrxTiO3). J. Electroceram. 2011, 27, 114–119. [Google Scholar] [CrossRef]
- Mccusker, L.B.; Vonele, R.B.; Cox, D.E.; Louër, D.; Scardi, P. Rietveld refinement guidelines. J. Appl. Cryst. 1999, 32, 36–50. [Google Scholar] [CrossRef]
- Tauc, J.; Grigorovici, R.; Vancu., A. Optical Properties and Electronic Structure of Amorphous Germanium. Phys. Status Solidi 1966, 15, 627–637. [Google Scholar] [CrossRef]
- Liu, Y.J.; Zhu, N. Precise determination of band gap naturally via absorption/reflectance/transmission spectra. Appl. Mech. Mater. 2014, 509, 3–8. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Perdew, P.J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Khadraoui, Z.; Horchani-Naifer, K.; Ferhi, M. Electronic structure and optical properties of TbPO4: Experiment and density functional theory calculations. Opt. Mater. 2015, 47, 484–489. [Google Scholar] [CrossRef]
- Pang, M.J.; Zhan, Y.Z.; Wang, H.Z.; Jiang, W.P.; Du, Y. Structural, electronic, elastic and thermodynamic properties of AlSi2RE (RE = La, Ce, Pr and Nd) from first-principle calculations. Comput. Mater. Sci. 2011, 50, 3303–3310. [Google Scholar]
- Khadraoui, Z.; Horchani-Naifer, K.; Ferhi, M.; Ferid, M. Synthesis, characterization and DFT calculations of electronic and optical properties of YbPO4. Chem. Phys. 2015, 457, 37–42. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Zhou, P.; Zhang, Q.; Ning, K.; Yang, H.; Sun, D.; Luo, J.; Yin, S. Structural and spectral investigations on heavily Er3+ doped RETaO4 (RE = Sc, Y, Gd, Lu) polycrystalline powders. In Proceedings of 1st Pacific Rim Laser Damage Symposium on Optical Materials for High Power Lasers (PLD), Shanghai, China, 6–9 November 2011.
- Keller, C. Über ternäre Oxide des Niobs und Tantals vom Typ ABO4. Z. Anorg. Allg. Chem. 1962, 318, 89–106. (In German) [Google Scholar] [CrossRef]
- Wuilloud, E.; Delley, B.; Schneider, W.-D.; Bear, Y. Spectroscopic evidence for localized and extended f-symmetry States in CeO2. Phys. Rev. Lett. 1984, 53, 202–205. [Google Scholar] [CrossRef]
- Khadraoui, Z.; Bouzidi, C.; Horchani-Naifer, K.; Ferid, M. Crystal structure, energy band and optical properties of dysprosium monophosphate DyPO4. J. Alloys Compd. 2014, 617, 281–286. [Google Scholar] [CrossRef]
- Bouhemadou, A.; Djabi, F.; Khenata, R. First principles study of structural, elastic, electronic and optical properties of the cubic perovskite BaHfO3. Phys. Lett. A 2008, 372, 4527–4531. [Google Scholar] [CrossRef]
- Fujimori, A.; Minami, F. Valence-band photoemission and optical absorption in nickel compounds. Phys. Rev. B 1984, 30, 957–971. [Google Scholar] [CrossRef]
- Smith, N.V. Photoelectron energy spectra and the band structures of the noble metals. Phys. Rev. B 1971, 3, 1862–1878. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef]
- Hopfield, J.J. Theory of the contribution of excitons to the complex dielectric constant of crystals. Phys. Rev. 1958, 112, 1555. [Google Scholar] [CrossRef]
- Pang, M.; Zhan, Y.; Ling, M.; Wei, S.; Liu, Y.; Du, Y. First-principles study on the crystal, electronic structure and mechanical properties of hexagonal Al3Re (Re = La, Ce, Pr, Nd, Sm, Gd) intermetallic compounds. Solid State Commun. 2011, 151, 1135–1140. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Jiang, M.; Gao, L.; Ma, Z.; Wang, F. Theoretical and Experimental Studies on the Crystal Structure, Electronic Structure and Optical Properties of SmTaO4. Materials 2016, 9, 55. https://doi.org/10.3390/ma9010055
Wang S, Jiang M, Gao L, Ma Z, Wang F. Theoretical and Experimental Studies on the Crystal Structure, Electronic Structure and Optical Properties of SmTaO4. Materials. 2016; 9(1):55. https://doi.org/10.3390/ma9010055
Chicago/Turabian StyleWang, Song, Miao Jiang, Lihong Gao, Zhuang Ma, and Fuchi Wang. 2016. "Theoretical and Experimental Studies on the Crystal Structure, Electronic Structure and Optical Properties of SmTaO4" Materials 9, no. 1: 55. https://doi.org/10.3390/ma9010055