“Head-to-Side-Chain” Cyclodepsipeptides of Marine Origin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. “Head-to-Side-Chain” Cyclodepsipeptides
3. Families of “Head-to-Side-Chain” Cyclodepsipetides












4. Conclusions
Acknowledgments
Conflict of Interest
References
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Takagi, M.; Shin-Ya, K. Construction of a natural product library containing secondary metabolites produced by actinomycetes. J. Antibiot. 2012, 65, 443–447. [Google Scholar] [CrossRef]
- Lam, K.S. New aspects of natural products in drug discovery. Trends Microbiol. 2007, 15, 279–289. [Google Scholar] [CrossRef]
- Baltz, R.H. Marcel Faber Roundtable: Is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J. Ind. Microbiol. Biot. 2006, 33, 507–513. [Google Scholar] [CrossRef]
- Von Nussbaum, F.; Anlauf, S.; Benet-Buchholz, J.; Häbich, D.; Köbberling, J.; Musza, L.; Telser, J.; Rübsamen-Waigmann, H.; Brunner, N.A. Structure and total synthesis of lysobactin (katanosin B). Angew. Chem. Int. Ed. 2007, 46, 2039–2042. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.; Oliveira, A.C.D.; Santoro, A.; Raoul, J.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Coriat, R.; Nicco, C.; Chéreau, C.; Mir, O.; Alexandre, J.; Ropert, S.; Weill, B.; Chaussade, S.; Goldwasser, F.; Batteux, F. Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 2284–2293. [Google Scholar] [CrossRef]
- Leeds, J.A.; Schmitt, E.K.; Krastel, P. Recent developments in antibacterial drug discovery: Microbe-derived natural products-from collection to the clinic. Expert Opin. Investig. Drugs 2006, 15, 211–226. [Google Scholar] [CrossRef]
- Butler, M.S.; Buss, A.D. Natural products-the future scaffolds for novel antibiotics? Biochem. Pharmacol. 2006, 71, 919–929. [Google Scholar] [CrossRef]
- Müller, M. Chemical diversity through biotransformations. Curr. Opin. Biotechnol. 2004, 15, 591–598. [Google Scholar] [CrossRef]
- Lamb, S.S.; Wright, G.D. Accessorizing natural products: Adding to nature’s toolbox. Proc. Natl. Acad. Sci. USA 2005, 102, 519–520. [Google Scholar] [CrossRef]
- Paterson, I.; Anderson, E.A. The renaissance of natural products as drug candidates. Science 2005, 310, 451–453. [Google Scholar] [CrossRef]
- Ortholand, J.-Y.; Ganesan, A. Natural products and combinatorial chemistry: Back to the future. Curr. Opin. Chem. Biol. 2004, 8, 271–280. [Google Scholar] [CrossRef]
- Ganesan, A. Natural products as a hunting ground for combinatorial chemistry. Curr. Opin. Biotech. 2004, 15, 584–590. [Google Scholar] [CrossRef]
- Sánchez, C.; Zhu, L.; Braña, A.F.; Salas, A.P.; Rohr, J.; Méndez, C.; Salas, J.A. Combinatorial biosynthesis of antitumor indolocarbazole compounds. Proc. Natl. Acad. Sci. USA 2005, 102, 461–466. [Google Scholar]
- Floss, H.G. Combinatorial biosynthesis-potential and problems. J. Biotechnol. 2006, 124, 242–257. [Google Scholar] [CrossRef]
- Baltz, R.H. Molecular engineering approaches to peptide, polyketide and other antibiotics. Nat. Biotechnol. 2006, 24, 1533–1540. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem. 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Kegler, C.; Gerth, K.; Müller, R. Establishment of a real-time PCR protocol for expression studies of secondary metabolite biosynthetic gene clusters in the G/C-rich myxobacterium Sorangium cellulosum So ce56. J. Biotechnol. 2006, 121, 201–212. [Google Scholar] [CrossRef]
- Marris, E. Drugs from the deep. Nature 2006, 443, 904–905. [Google Scholar]
- Newman, D.J.; Cragg, G.M. A selective and sensitive fluorescence probe for imaging endogenous zinc in living cells. Curr. Drug Targets 2006, 7, 279–304. [Google Scholar] [CrossRef]
- Zotchev, S.B. Marine actinomycetes as an emerging resource for the drug development pipelines. J. Biotechnol. 2012, 158, 168–175. [Google Scholar] [CrossRef]
- Hill, R.A. Marine natural products. Annu. Rep. Prog. Chem. B 2012, 108, 131–146. [Google Scholar] [CrossRef]
- Salomon, C.E.; Magarvey, N.A.; Sherman, D.H. Merging the potential of microbial genetics with biological and chemical diversity: An even brighter future for marine natural product drug discovery. Nat. Prod. Rep. 2004, 21, 105–121, and references therein. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Williams, A.J.; Day, M.; Heavner, J.E. Ziconotide: An update and review. Expert Opin. Pharmacother. 2008, 9, 1575–1583. [Google Scholar] [CrossRef]
- Cuevas, C.; Pérez, M.; Martín, M.J.; Chicharro, J.L.; Fernández-Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; de La Calle, F.; et al. Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Butler, M.S. Natural products to drugs: Natural product derived compounds in clinical trials. Nat. Prod. Rep. 2005, 22, 162–195. [Google Scholar] [CrossRef]
- Lemmens-Gruber, R.; Kamyar, M.R.; Dornetshuber, R. Cyclodepsipeptides—Potential drugs and lead compounds in the drug development process. Curr. Med. Chem. 2009, 16, 1122–1137. [Google Scholar] [CrossRef]
- Andavan, G.S.B.; Lemmens-Gruber, R. Cyclodepsipeptides from marine sponges: Natural agents for drug research. Mar. Drugs 2010, 8, 810–834. [Google Scholar] [CrossRef]
- Suarez-Jimenez, G.-M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.-M. Bioactive peptides and depsipeptides with anticancer potential: Sources from marine animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef]
- Sarabia, F.; Chammaa, S.; Sanchez Ruiz, A.; Martin Ortiz, L.; Lopez Herrera, F.J. Chemistry and biology of cyclic depsipeptides of medicinal and biological interest. Curr. Med. Chem. 2004, 11, 1309–1332. [Google Scholar] [CrossRef]
- Banker, R.; Carmeli, S. Inhibitors of serine proteases from a waterbloom of the cyanobacterium Microcystis sp. Tetrahedron 1999, 55, 10835–10844. [Google Scholar] [CrossRef]
- Reshef, V.; Carmeli, S. Protease inhibitors from a water bloom of the cyanobacterium Microcystis aeruginosa. Tetrahedron 2001, 57, 2885–2894. [Google Scholar] [CrossRef]
- Itou, Y.; Ishida, K.; Shin, H.J.; Murakami, M. Oscillapeptins A to F, serine protease inhibitors from the three strains of Oscillatoria agardhii. Tetrahedron 1999, 55, 6871–6882. [Google Scholar] [CrossRef]
- Fujii, K.; Sivonen, K.; Naganawa, E.; Harada, K. Non-toxic peptides from toxic cyanobacteria, Oscillatoria agardhii. Tetrahedron 2000, 56, 725–733. [Google Scholar] [CrossRef]
- Ploutno, A.; Carmeli, S. Modified peptides from a water bloom of the cyanobacterium Nostoc sp. Tetrahedron 2002, 58, 9949–9957. [Google Scholar] [CrossRef]
- Von Elert, E.; Oberer, L.; Merkel, P.; Huhn, T.; Blom, J.F. Cyanopeptolin 954, a chlorine-containing chymotrypsin inhibitor of Microcystis aeruginosa NIVA Cya 43. J. Nat. Prod. 2005, 68, 1324–1327. [Google Scholar] [CrossRef]
- Harada, K.; Mayumi, T.; Shimada, T.; Fujii, K.; Kondo, F.; Watanabe, M.F. Synthesis and thermophysical properties of ionic liquids: Cyclopropyl moieties versus olefins as Tm-reducing elements in lipid-inspired ionic liquids. Tetrahedron Lett. 1993, 34, 6091–6094. [Google Scholar] [CrossRef]
- Harada, K.; Mayumi, T.; Shimada, T.; Fujii, K.; Kondo, F.; Park, H.D.; Watanabe, M.F. Co-production of microcystins and aeruginopeptins by natural cyanobacterial bloom. Environ. Toxicol. 2001, 16, 298–305. [Google Scholar] [CrossRef]
- Plaza, A.; Bewley, C.A. Largamides A–H, unusual cyclic peptides from the marine cyanobacterium Oscillatoria sp. J. Org. Chem. 2006, 71, 6898–6907. [Google Scholar] [CrossRef]
- Matthew, S.; Ross, C.; Rocca, J.R.; Paul, V.J.; Luesch, H. Lyngbyastatin 4, a dolastatin 13 analogue with elastase and chymotrypsin inhibitory activity from the marine cyanobacterium Lyngbya confervoides. J. Nat. Prod. 2007, 70, 124–127. [Google Scholar] [CrossRef]
- Taori, K.; Matthew, S.; Rocca, J.R.; Paul, V.J.; Luesch, H. Lyngbyastatins 5–7, potent elastase inhibitors from Floridian marine cyanobacteria, Lyngbya spp. J. Nat. Prod. 2007, 70, 1593–1600. [Google Scholar] [CrossRef]
- Ploutno, A.; Shoshan, M.; Carmeli, S. Three novel protease inhibitors from a natural bloom of the cyanobacterium Microcystis aeruginosa. J. Nat. Prod. 2002, 65, 973–978. [Google Scholar] [CrossRef]
- Lee, A.Y.; Smitka, T.A.; Bonjouklian, R.; Clardy, J. Atomic structure of the trypsin-A90720A complex: A unified approach to structure and function. Chem. Biol. 1994, 1, 113–117. [Google Scholar] [CrossRef]
- Matern, U.; Oberer, L.; Falchetto, R.A.; Erhard, M.; König, W.A.; Herdman, M.; Weckesser, J. Scyptolin A and B, cyclic depsipeptides from axenic cultures of Scytonema hofmanni PCC 7110. Phytochemistry 2001, 58, 1087–1095. [Google Scholar] [CrossRef]
- Yamaki, H.; Sitachitta, N.; Sano, T.; Kaya, K. Two new chymotrypsin inhibitors isolated from the cyanobacterium Microcystis aeruginosa NIES-88. J. Nat. Prod. 2005, 68, 14–18. [Google Scholar] [CrossRef]
- Nakanishi, I.; Kinoshita, T.; Sato, A.; Tada, T. Structure of porcine pancreatic elastase complexed with FR901277, a novel macrocyclic inhibitor. Biopolymers 2000, 53, 434–445. [Google Scholar] [CrossRef]
- Matern, U.; Schleberger, C.; Jelakovic, S.; Weckesser, J.; Schulz, G.E. Binding structure of elastase inhibitor scyptolin A. Chem. Biol. 2003, 10, 997–1001. [Google Scholar] [CrossRef]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27. [Google Scholar] [CrossRef]
- Okino, T.; Qi, S.; Matsuda, H.; Murakami, M.; Yamaguchi, K. Nostopeptins A and B, elastase inhibitors from the cyanobacterium Nostoc minutum. J. Nat. Prod. 1997, 60, 158–161. [Google Scholar] [CrossRef]
- Grach-Pogrebinsky, O.; Sedmak, B.; Carmeli, S. Protease inhibitors from a Slovenian Lake Bled toxic waterbloom of the cyanobacterium Planktothrix rubescens. Tetrahedron 2003, 59, 8329–8336. [Google Scholar] [CrossRef]
- Yokokawa, F.; Shioiri, T. Total synthesis of somamide A, an Ahp (3-amino-6-hydroxy-2-piperidone)-containing cyclic depsipeptide. Tetrahedron Lett. 2002, 43, 8673–8677. [Google Scholar] [CrossRef]
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Solid phase total synthesis of the 3-amino-6-hydroxy-2-piperidone (Ahp) cyclodepsipeptide and protease inhibitor Symplocamide A. Chem. Commun. 2010, 46, 8857–8859. [Google Scholar] [CrossRef]
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Development of a solid-phase approach to the natural product class of Ahp-containing cyclodepsipeptides. Eur. J. Org. Chem. 2012, 2012, 1616–1625. [Google Scholar] [CrossRef]
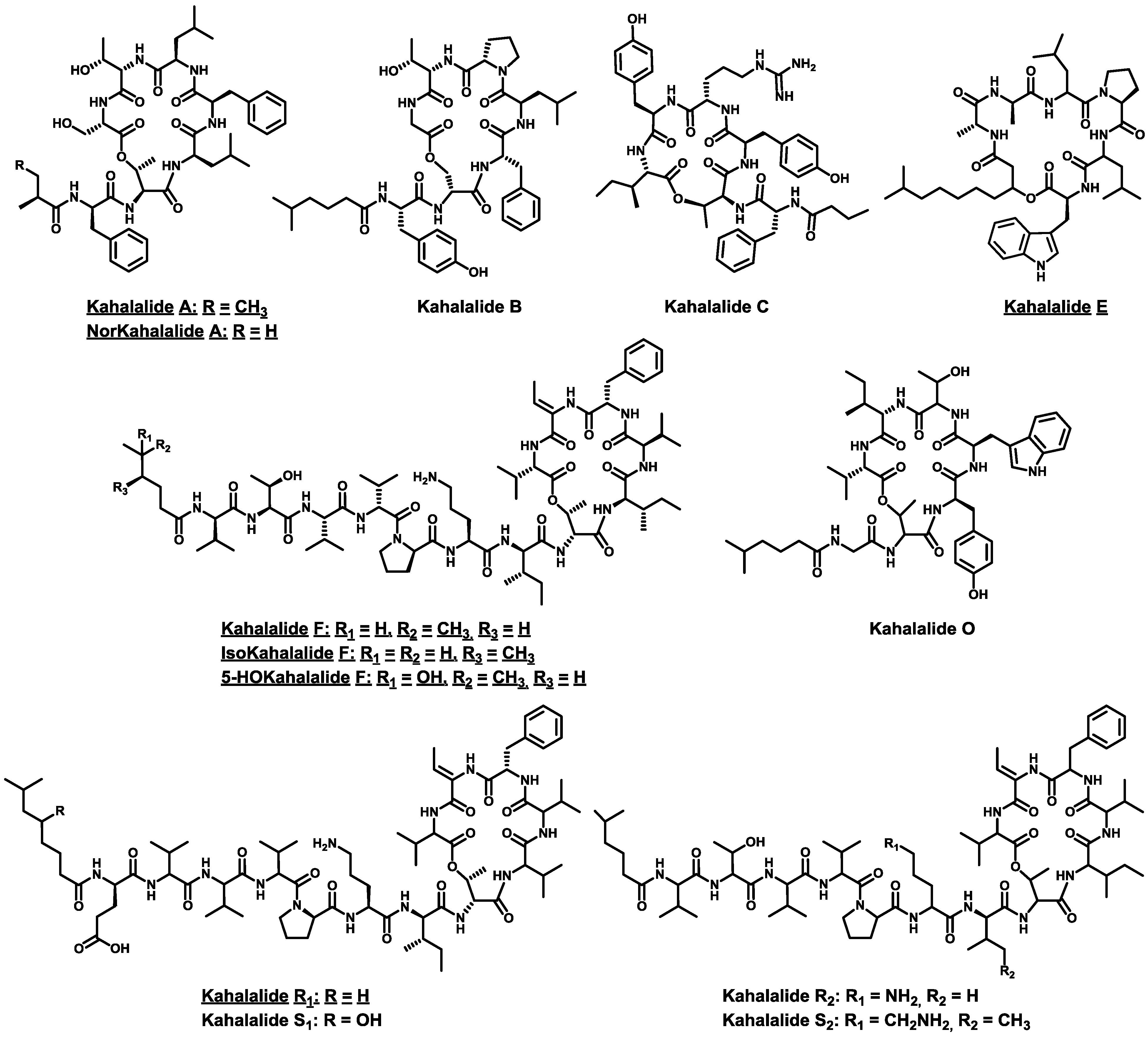
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A bioactive depsipeptide from the Sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Hamann, M.T.; Otto, C.S.; Scheuer, P.J.; Dunbar, D.C. Kahalalides: Bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996, 61, 6594–6600. [Google Scholar] [CrossRef]
- Goetz, G.; Nakao, Y.; Scheuer, P.J. Two acyclic kahalalides from the Sacoglossan mollusk Elysia rufescens. J. Nat. Prod. 1997, 60, 562–567. [Google Scholar] [CrossRef]
- Horgen, F.D.; delos Santos, D.B.; Goetz, G.; Sakamoto, B.; Kan, Y.; Nagai, H.; Scheuer, P.J. A new depsipeptide from the sacoglossan mollusk Elysia ornata and the green alga Bryopsis species. J. Nat. Prod. 2000, 63, 152–154. [Google Scholar] [CrossRef]
- Gao, J.; Hamann, M.T. Chemistry and biology of kahalalides. Chem. Rev. 2011, 111, 3208–3235. [Google Scholar] [CrossRef]
- Ashour, M.; Edrada, R.; Ebel, R.; Wray, V.; Wätjen, W.; Padmakumar, K.; Müller, W.E.G.; Lin, W.H.; Proksch, P. Kahalalide derivatives from the Indian sacoglossan mollusk Elysia grandifolia. J. Nat. Prod. 2006, 69, 1547–1553. [Google Scholar] [CrossRef]
- Gao, J.; Caballero-George, C.; Wang, B.; Rao, K.V.; Shilabin, A.G.; Hamann, M.T. 5-OHKF and NorKA, depsipeptides from a Hawaiian collection of Bryopsis pennata: Binding properties for NorKA to the human neuropeptide Y Y1 receptor. J. Nat. Prod. 2009, 72, 2172–2176. [Google Scholar] [CrossRef]
- Cruz, L.J.; Luque-ortega, J.R.; Rivas, L.; Albericio, F. Kahalalide F, an antitumor depsipeptide in clinical trials, and its analogues as effective antileishmanial agents. Mol. Pharm. 2009, 6, 813–824. [Google Scholar] [CrossRef]
- Shilabin, A.G.; Kasanah, N.; Wedge, D.E.; Hamann, M.T. Lysosome and HER3 (ErbB3) selective anticancer agent kahalalide F: Semisynthetic modifications and antifungal lead-exploration studies. J. Med. Chem. 2007, 50, 4340–4350. [Google Scholar] [CrossRef]
- Jiménez, J.C.; López-Macià, A.; Gracia, C.; Varón, S.; Carrascal, M.; Caba, J.M.; Royo, M.; Francesch, A.M.; Cuevas, C.; Giralt, E.; et al. Structure-activity relationship of kahalalide F synthetic analogues. J. Med. Chem. 2008, 51, 4920–4931. [Google Scholar]
- Bourel-Bonnet, L.; Rao, K.V.; Hamann, M.T.; Ganesan, A. Solid-phase total synthesis of kahalalide A and related analogues. J. Med. Chem. 2005, 48, 1330–1335. [Google Scholar] [CrossRef]
- López-Macià, À.; Jiménez, J.C.; Royo, M.; Giralt, E.; Albericio, F. Kahalalide B. Synthesis of a natural cyclodepsipeptide. Tetrahedron Lett. 2000, 41, 9765–9769. [Google Scholar] [CrossRef]
- López-Macià, A.; Jiménez, J.C.; Royo, M.; Giralt, E.; Albericio, F. Synthesis and structure determination of kahalalide F. J. Am. Chem. Soc. 2001, 123, 11398–11401. [Google Scholar] [CrossRef]
- López, P.E.; Isidro-Llobet, A.; Gracia, C.; Cruz, L.J.; García-Granados, A.; Parra, A.; Álvarez, M.; Albericio, F. Use of p-nitrobenzyloxycarbonyl (pNZ) as a permanent protecting group in the synthesis of Kahalalide F analogs. Tetrahedron Lett. 2005, 46, 7737–7741. [Google Scholar] [CrossRef]
- Gracia, C.; Isidro-Llobet, A.; Cruz, L.J.; Acosta, G.A.; Alvarez, M.; Cuevas, C.; Giralt, E.; Albericio, F. Convergent approaches for the synthesis of the antitumoral peptide, kahalalide F. Study of orthogonal protecting groups. J. Org. Chem. 2006, 71, 7196–7204. [Google Scholar] [CrossRef]
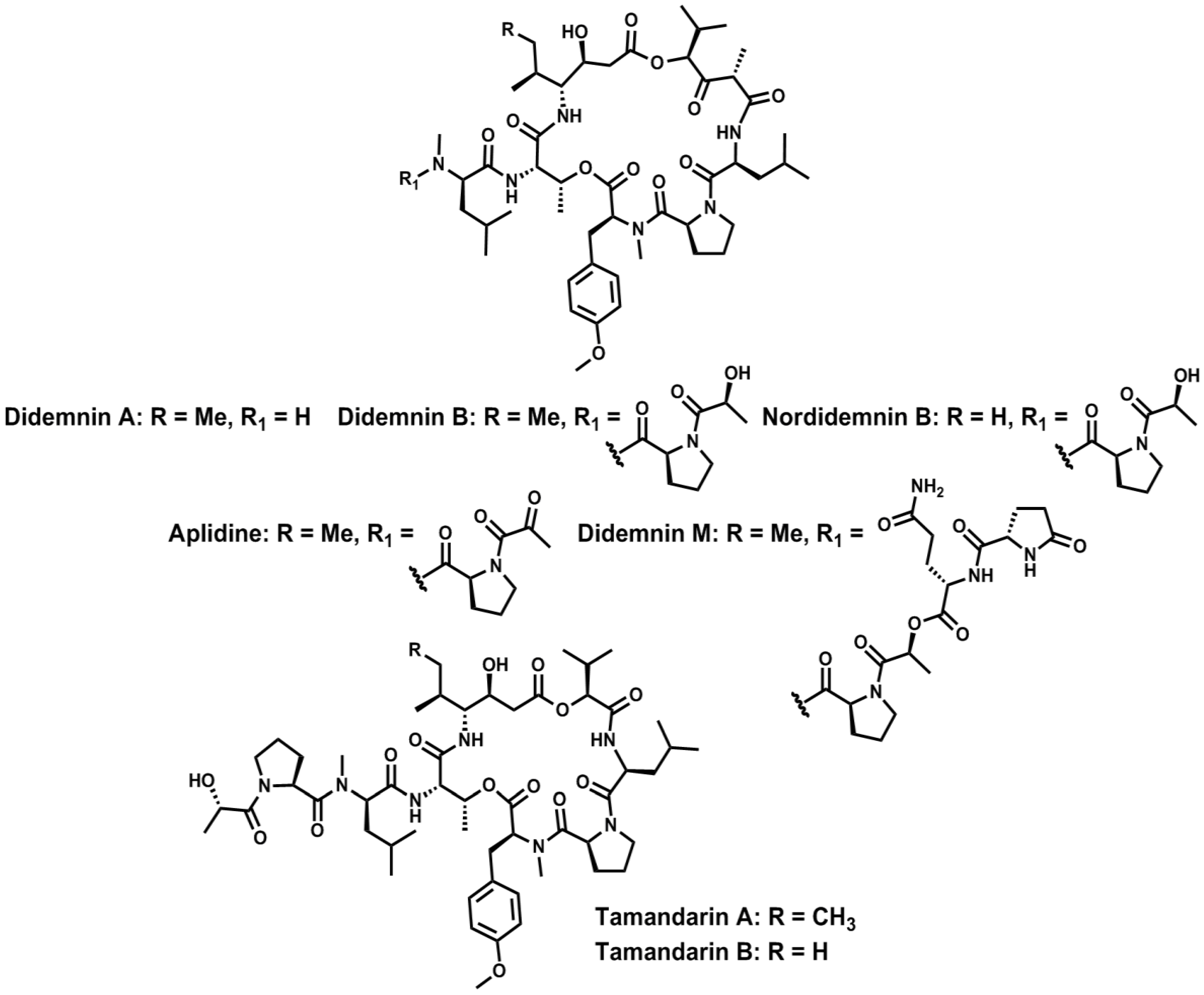
- Rinehart, K.L.; Gloer, J.B.; Carter Cook, J. Structures of the didemnins, antiviral and cytotoxic depsipeptides from a Caribbean tunicate. J. Am. Chem. Soc. 1981, 103, 1857–1859. [Google Scholar] [CrossRef]
- Rinehart, K.L.J.; Gloer, J.B.; Hughes, R.G.J.; Renis, H.E.; McGroven, J.P.; Swynenberg, E.B.; Stringfellow, D.A.; Kuentzel, S.L.; Li, L.H. Didemnins: Antiviral and antitumor depsipeptides from a Caribbean tunicate. Science 1981, 212, 933–935. [Google Scholar]
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullié, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424. [Google Scholar] [CrossRef]
- Vervoort, H.; Fenical, W.; Epifanio, R.A. Tamandarins A and B: New cytotoxic depsipeptides from a Brazilian ascidian of the family Didemnidae. J. Org. Chem. 2000, 65, 782–792. [Google Scholar] [CrossRef]
- Rawat, D.S.; Joshi, M.C.; Joshi, P.; Atheaya, H. Marine peptides and related compounds in clinical trials. Anti-Cancer Agents Med. Chem. 2006, 6, 33–40. [Google Scholar] [CrossRef]
- Schwartsmann, G.; Brondani, A.; Mattei, J.; Martins, R. Marine-derived anticancer agents in clinical trials. Expert. Opin. Investig. Drugs 2003, 12, 1367–1383. [Google Scholar]
- Rinehart, K.L. Antitumor compounds from tunicates. Med. Res. Rev. 2000, 20, 1–27. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Kishore, V.; Nagarajan, S.; Lake, R.J.; Gloer, J.B.; Bozich, F.A.; Li, K.; Maleczka, R.E.; Todsen, W.L.; Munro, M.H.G.; et al. Total synthesis of didemnins A, B, and C. J. Am. Chem. Soc. 1987, 109, 6846–6848. [Google Scholar] [CrossRef]
- Hamada, Y.; Kondo, Y.; Shibata, M.; Shioiri, T. Efficient total synthesis of didemnins A and B. J. Am. Chem. Soc. 1989, 111, 669–673. [Google Scholar] [CrossRef]
- Li, W.-R.; Ewing, W.R.; Harris, B.D.; Joullié, M.M. Total synthesis and structural investigations of didemnins A, B, and C. J. Am. Chem. Soc. 1990, 112, 7659–7672. [Google Scholar] [CrossRef]
- Jou, G.; González, I.; Albericio, F.; Lloyd-Williams, P.; Giralt, E. Total synthesis of dehydrodidemnin B. Use of uronium and phosphonium salt coupling reagents in peptide synthesis in solution. J. Org. Chem. 1997, 62, 354–366. [Google Scholar] [CrossRef]
- Jouin, P.; Poncet, J.; Dufour, M.-N.; Pantaloni, A.; Castro, B. Synthesis of the cyclodepsipeptide nordidemnin B, a cytotoxic minor product isolated from the sea tunicate Trididemnum cyanophorum. J. Org. Chem. 1989, 54, 617–627. [Google Scholar] [CrossRef]
- Vera, M.D.; Joullié, M.M. Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 2002, 22, 102–145. [Google Scholar] [CrossRef]
- Liang, B.; Portonovo, P.; Vera, M.D.; Xiao, D.; Joullié, M.M. The first total synthesis of (−)-tamandarin A. Org. Lett. 1999, 1, 1319–1322. [Google Scholar] [CrossRef]
- Liang, B.; Richard, D.J.; Portonovo, P.S.; Joullié, M.M. Total syntheses and biological investigations of tamandarins A and B and tamandarin A analogs. J. Am. Chem. Soc. 2001, 123, 4469–4474. [Google Scholar] [CrossRef]
- Joullié, M.M.; Portonovo, P.; Liang, B.; Richard, D.J. Total synthesis of (−)-tamandarin B. Tetrahedron Lett. 2000, 41, 9373–9376. [Google Scholar] [CrossRef]
- Lassen, K.M.; Lee, J.; Joullié, M.M. Synthetic studies of tamandarin B side chain analogues. J. Org. Chem. 2010, 75, 3027–3036. [Google Scholar] [CrossRef]
- Adrio, J.; Cuevas, C.; Manzanares, I.; Joullié, M.M. Synthesis and biological evaluation of tamandarin B analogues. Org. Lett. 2006, 8, 511–514. [Google Scholar] [CrossRef]
- Adrio, J.; Cuevas, C.; Manzanares, I.; Joullié, M.M. Total synthesis and biological evaluation of tamandarin B analogues. J. Org. Chem. 2007, 72, 5129–5138. [Google Scholar] [CrossRef]
- Sakai, R.; Rinehart, K.L.; Kishore, V.; Kundu, B.; Faircloth, G.; Gloer, J.B.; Carney, J.R.; Namikoshi, M.; Sun, F.; Hughes, R.G.; et al. Structure-activity relationships of the didemnins. J. Med. Chem. 1996, 39, 2819–2834. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fusetani, N.; Konosu, S. Bioactive marine metabolites VI. Structure elucidation of discodermin A, an antimicrobial peptide from the marine sponge Discodermia kiiensis. Tetrahedron Lett. 1984, 25, 5165–5168. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fusetani, N.; Konosu, S. Bioactive marine metabolites IV. Isolation and the amino acid composition of discodermin A, an antimicrobial peptide, from the marine sponge Discodermia kiiensis. J. Nat. Prod. 1985, 48, 236–241. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fusetani, N.; Konosu, S. Bioactive marine metabolites VII. Structures of discodermins B, C and D, antimicrobial peptides from the marine sponge Discodermia kiiensis. Tetrahedron Lett. 1985, 26, 855–856. [Google Scholar]
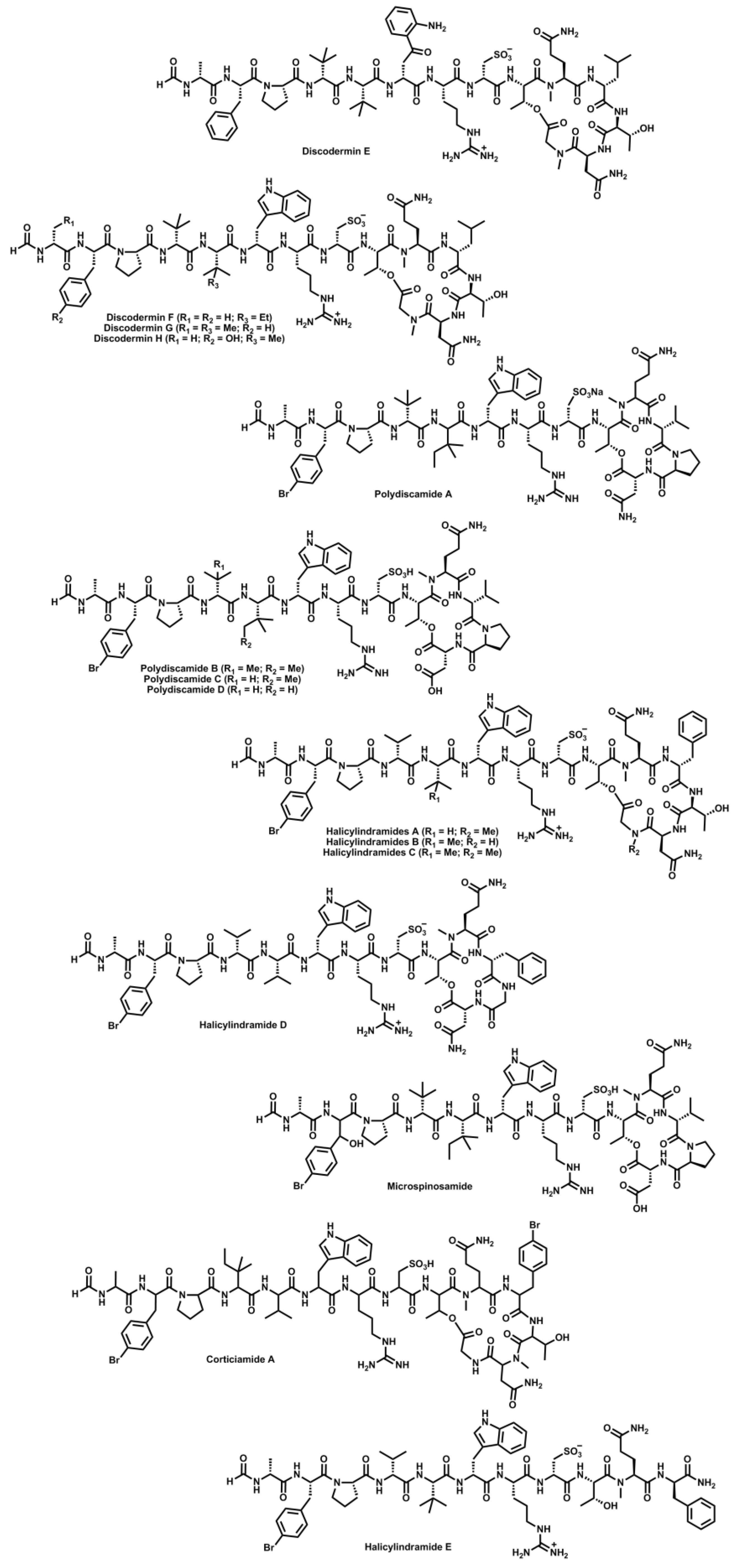
- Ryu, G.; Matsunaga, S.; Fusetani, N. Discodermin E, a cytotoxic and antimicrobial tetradecapeptide, from the marine sponge Discodermia kiiensis. Tetrahedron Lett. 1994, 35, 8251–8254. [Google Scholar]
- Ryu, G.; Matsunaga, S.; Fusetani, N. Discodermins F–H, cytotoxic and antimicrobial tetradecapeptides from the marine sponge Discodermia kiiensis: Structure revision of discodermins A–D. Tetrahedron 1994, 50, 13409–13416. [Google Scholar]
- Gulavita, N.K.; Gunasekera, S.P.; Pomponi, S.A.; Robinson, E.V. Polydiscamide A: A new bioactive depsipeptide from the marine sponge Discodermia sp. J. Org. Chem. 1992, 57, 1767–1772. [Google Scholar]
- Feng, Y.; Carroll, A.R.; Pass, D.M.; Archbold, J.K.; Avery, V.M.; Quinn, R.J. Polydiscamides B–D from a marine sponge Ircinia sp. as potent human sensory neuron-specific G protein coupled receptor agonists. J. Nat. Prod. 2008, 71, 8–11. [Google Scholar]
- Li, H.; Matsunaga, S.; Fusetani, N. Halicylindramides A–C, antifungal and cytotoxic depsipeptides from the marine sponge Halichondria cylindrata. J. Med. Chem. 1995, 38, 338–343. [Google Scholar]
- Li, H.; Matsunaga, S.; Fusetani, N. Halicylindramides D and E, antifungal peptides from the marine sponge Halichondria cylindrata. J. Nat. Prod. 1996, 59, 163–166. [Google Scholar] [CrossRef]
- Rashid, M.A.; Gustafson, K.R.; Cartner, L.K.; Shigematsu, N.; Pannell, L.K.; Boyd, M.R. Microspinosamide, a new HIV-inhibitory cyclic depsipeptide from the marine sponge Sidonops microspinosa. J. Nat. Prod. 2001, 64, 117–121. [Google Scholar] [CrossRef]
- Laird, D.W.; LaBarbera, D.V.; Feng, X.; Bugni, T.S.; Harper, M.K.; Ireland, C.M. Halogenated cyclic peptides isolated from the sponge Corticium sp. J. Nat. Prod. 2007, 70, 741–746. [Google Scholar] [CrossRef]
- Seo, H.; Lim, D. Total synthesis of Halicylindramide A. J. Org. Chem. 2009, 74, 906–909. [Google Scholar] [CrossRef]
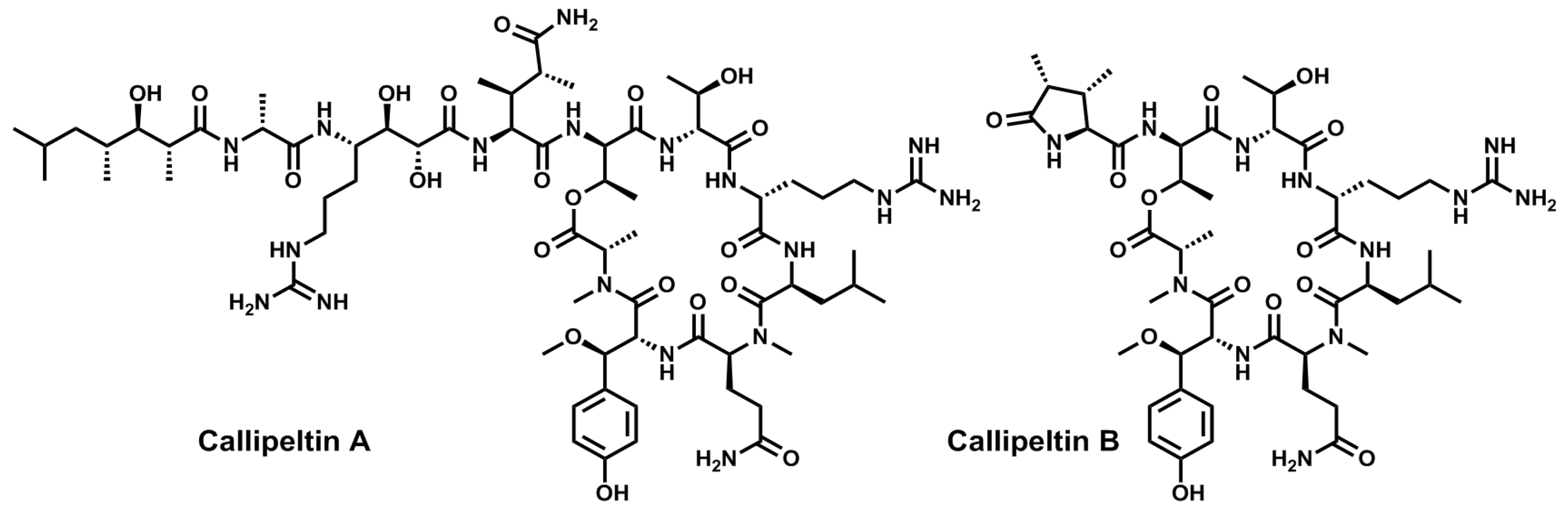
- Zampella, A.; D’Auria, M.V.; Gomez Paloma, L.; Casapullo, A.; Minale, L.; Debitus, C.; Henin, Y. Callipeltin A, an anti-HIV cyclic depsipeptide from the New Caledonian Lithistida sponge Callipelta sp. J. Am. Chem. Soc. 1996, 118, 6202–6209. [Google Scholar] [CrossRef]
- D’Auria, M.V.; Zampella, A.; Paloma, L.G.; Minale, L.; Debitus, C.; Roussakis, C.; Le Bert, V. Callipeltins B and C; bioactive peptides from a marine Lithistida sponge Callipelta sp. Tetrahedron 1996, 52, 9589–9596. [Google Scholar] [CrossRef]
- Zampella, A.; Randazzo, A.; Borbone, N.; Luciani, S.; Trevisi, L.; Debitus, C.; D’Auria, M.V. Isolation of callipeltins A–C and of two new open-chain derivatives of callipeltin A from the marine sponge Latrunculia sp. A revision of the stereostructure of callipeltins. Tetrahedron Lett. 2002, 43, 6163–6166. [Google Scholar]
- Sepe, V.; D’Orsi, R.; Borbone, N.; Valeria D’Auria, M.; Bifulco, G.; Monti, M.C.; Catania, A.; Zampella, A. Callipeltins F–I: New antifungal peptides from the marine sponge Latrunculia sp. Tetrahedron 2006, 62, 833–840. [Google Scholar] [CrossRef]
- D’Auria, M.V.; Sepe, V.; D’Orsi, R.; Bellotta, F.; Debitus, C.; Zampella, A. Isolation and structural elucidation of callipeltins J–M: Antifungal peptides from the marine sponge Latrunculia sp. Tetrahedron 2007, 63, 131–140. [Google Scholar] [CrossRef]
- Krishnamoorthy, R.; Vazquez-Serrano, L.D.; Turk, J.A.; Kowalski, J.A.; Benson, A.G.; Breaux, N.T.; Lipton, M.A. Solid-phase total synthesis and structure proof of callipeltin B. J. Am. Chem. Soc. 2006, 128, 15392–15393. [Google Scholar] [CrossRef]
- Krishnamoorthy, R.; Richardson, B.L.; Lipton, M.A. Synthesis and cytotoxicity of desmethoxycallipeltin B: Lack of a quinone methide for the cytotoxicity of callipeltin B. Bioorg. Med. Chem. Lett. 2007, 17, 5136–5138. [Google Scholar] [CrossRef]
- Kikuchi, M.; Watanabe, Y.; Tanaka, M.; Akaji, K.; Konno, H. Synthesis and cytotoxicity of the depsipeptides analogues of callipeltin B. Bioorg. Med. Chem. Lett. 2011, 21, 4865–4868. [Google Scholar] [CrossRef]
- Cranfill, D.C.; Morales-Ramos, A.I.; Lipton, M.A. Solid-phase synthesis of callipeltin D. Stereochemical confirmation of the unnatural amino acid AGDHE. Org. Lett. 2005, 7, 5881–5883. [Google Scholar] [CrossRef]
- Çalimsiz, S.; Morales Ramos, A.I.; Lipton, M.A. Solid-phase synthesis and configurational reassignment of callipeltin E. Implications for the structures of callipeltins A and B. J. Org. Chem. 2006, 71, 6351–6356. [Google Scholar] [CrossRef]
- Kikuchi, M.; Nosaka, K.; Akaji, K.; Konno, H. Solid phase total synthesis of callipeltin E isolated from marine sponge Latrunculia sp. Tetrahedron Lett. 2011, 52, 3872–3875. [Google Scholar] [CrossRef]
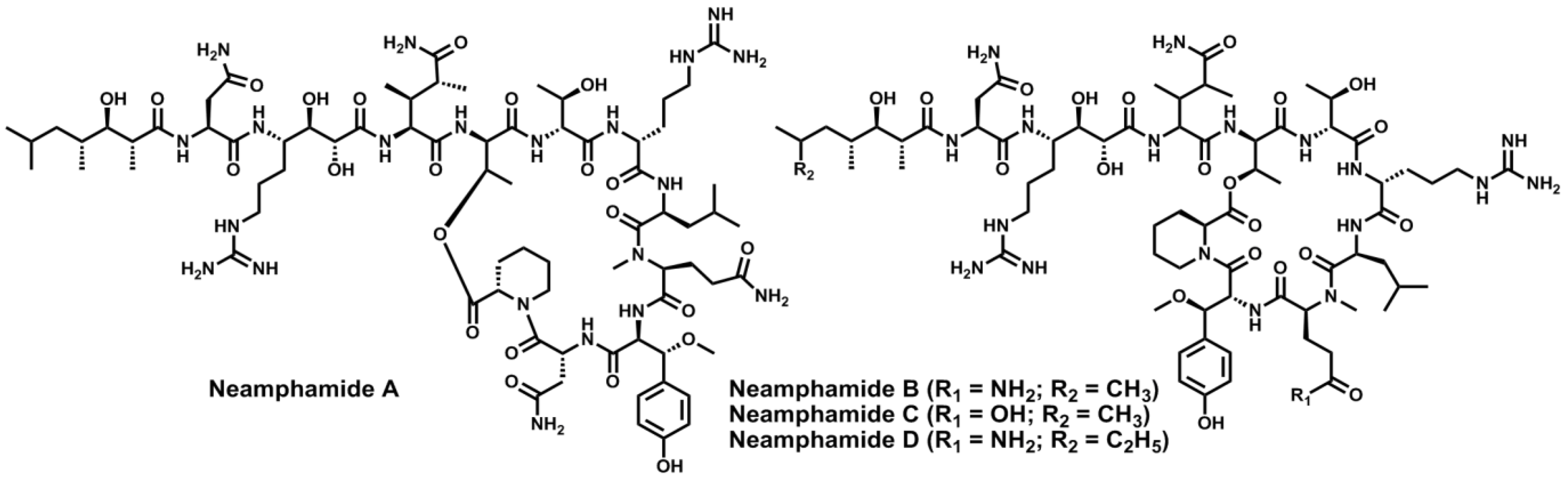
- Oku, N.; Krishnamoorthy, R.; Benson, A.G.; Ferguson, R.L.; Lipton, M.A.; Phillips, L.R.; Gustafson, K.R.; McMahon, J.B. Complete stereochemistry of neamphamide A and absolute configuration of the β-methoxytyrosine residue in papuamide B. J. Org. Chem. 2005, 70, 6842–6847. [Google Scholar] [CrossRef]
- Oku, N.; Gustafson, K.R.; Cartner, L.K.; Wilson, J.A.; Shigematsu, N.; Hess, S.; Pannell, L.K.; Boyd, M.R.; McMahon, J.B. Neamphamide A, a new HIV-inhibitory depsipeptide from the Papua New Guinea marine sponge Neamphius huxleyi. J. Nat. Prod. 2004, 67, 1407–1141. [Google Scholar] [CrossRef]
- Yamano, Y.; Arai, M.; Kobayashi, M. Neamphamide B, new cyclic depsipeptide, as an anti-dormant mycobacterial substance from a Japanese marine sponge of Neamphius sp. Bioorg. Med. Chem. Lett. 2012, 22, 4877–4881. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.D.; Pham, N.B.; Fechner, G.; Zencak, D.; Vu, H.T.; Hooper, J.N.A.; Quinn, R.J. Cytotoxic cyclic depsipeptides from the Australian marine sponge Neamphius huxleyi. J. Nat. Prod. 2012, 75, 2200–2208. [Google Scholar] [CrossRef]
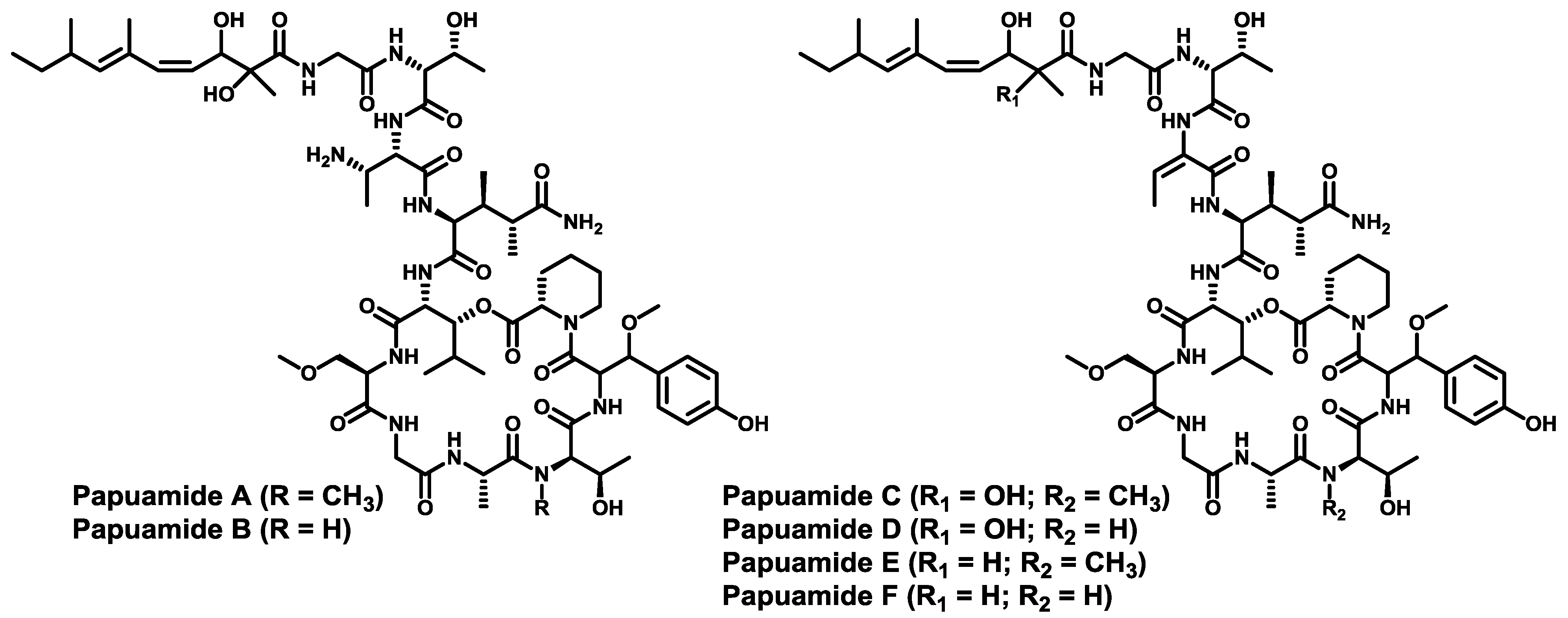
- Ford, P.W.; Gustafson, K.R.; Mckee, T.C.; Shigematsu, N.; Maurizi, L.K.; Pannell, L.K.; Williams, D.E.; Silva, E.D.D.; Lassota, P.; Allen, T.M.; et al. Papuamides A–D , HIV-inhibitory and cytotoxic depsipeptides from the sponges Theonella mirabilis and Theonella swinhoei collected in Papua New Guinea. J. Am. Chem. Soc. 1999, 121, 5899–5909. [Google Scholar] [CrossRef]
- Prasad, P.; Aalbersberg, W.; Feussner, K.-D.; van Wagoner, R.M. Papuamides E and F, cytotoxic depsipeptides from the marine sponge Melophlus sp. Tetrahedron 2011, 67, 8529–8531. [Google Scholar] [CrossRef]
- Xie, W.; Ding, D.; Zi, W.; Li, G.; Ma, D. Total synthesis and structure assignment of papuamide B, a potent marine cyclodepsipeptide with anti-HIV properties. Angew. Chem. Int. Ed. 2008, 47, 2844–2848. [Google Scholar] [CrossRef]
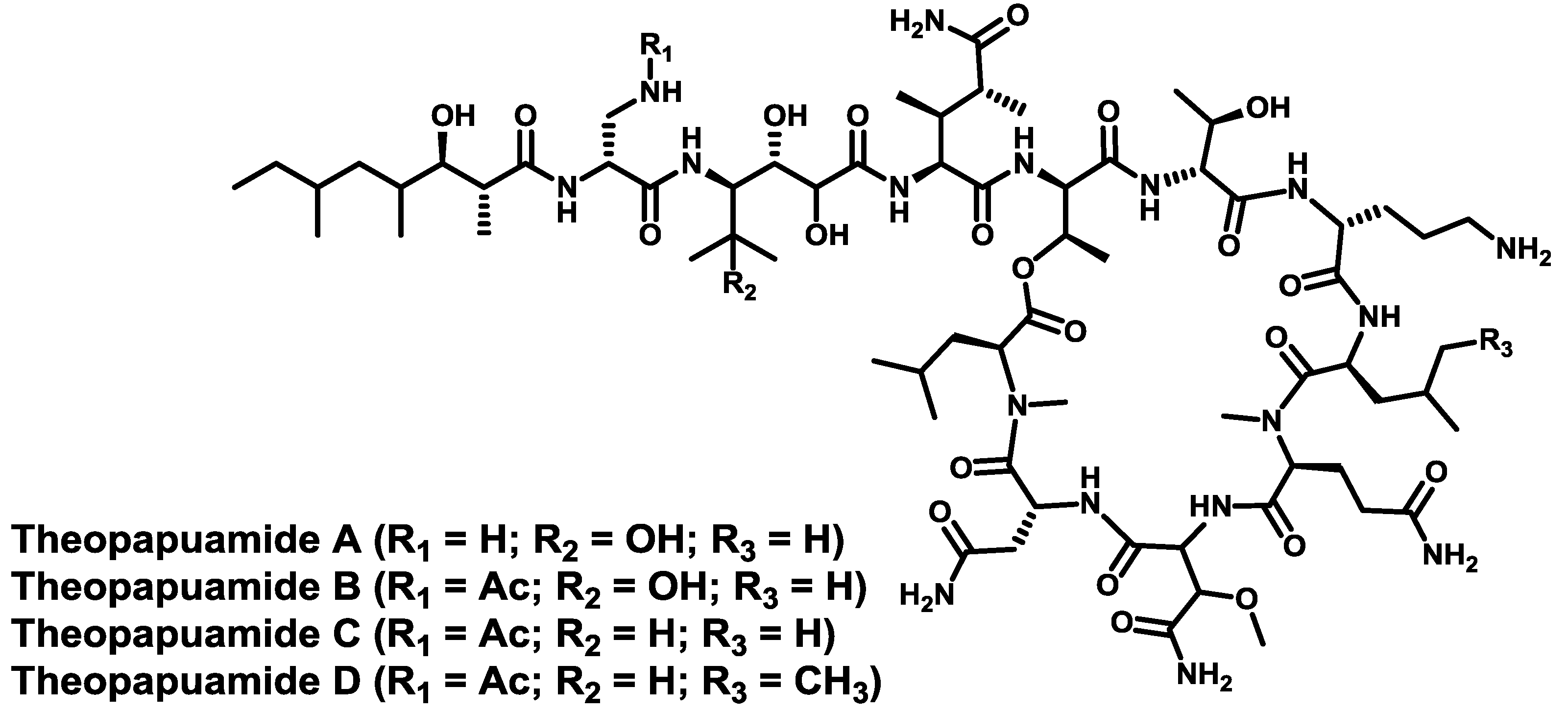
- Ratnayake, A.S.; Bugni, T.S.; Feng, X.; Harper, M.K.; Skalicky, J.J.; Mohammed, K.A.; Andjelic, C.D.; Barrows, L.R.; Ireland, C.M. Theopapuamide, a cyclic depsipeptide from a Papua New Guinea lithistid sponge Theonella swinhoei. J. Nat. Prod. 2006, 69, 1582–1586. [Google Scholar] [CrossRef]
- Plaza, A.; Bifulco, G.; Keffer, J.L.; Lloyd, J.R.; Baker, H.L.; Bewley, C.A. Celebesides A–C and theopapuamides B–D, depsipeptides from an Indonesian sponge that inhibit HIV-1 entry. J. Org. Chem. 2009, 74, 504–512. [Google Scholar] [CrossRef]
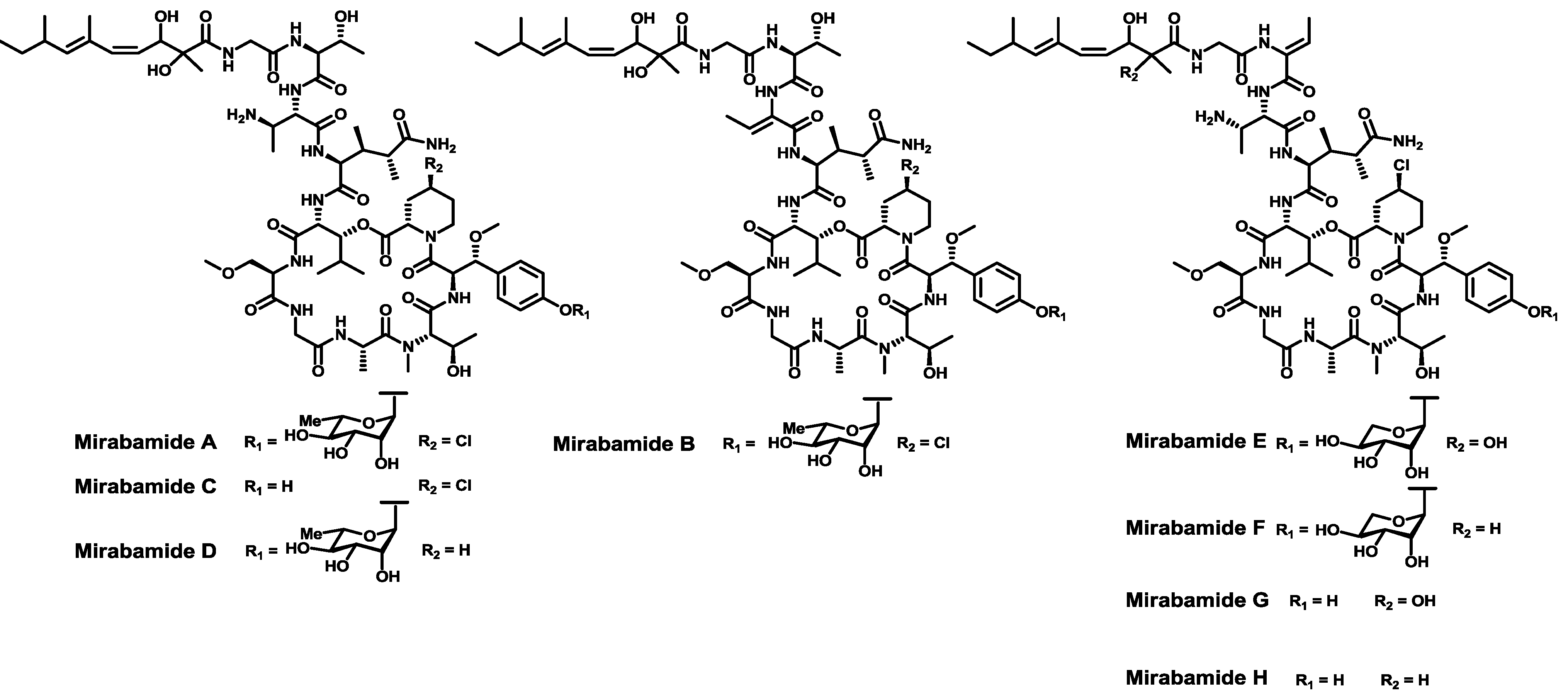
- Plaza, A.; Gustchina, E.; Baker, H.L.; Kelly, M.; Bewley, C.A. Mirabamides A–D, depsipeptides from the sponge Siliquariaspongia mirabilis that inhibit HIV-1 fusion. J. Nat. Prod. 2007, 70, 1753–1760. [Google Scholar] [CrossRef]
- Lu, Z.; Wagoner, R.M.V.; Harper, M.K.; Baker, H.L.; Hooper, J.N.A.; Bewley, C.A.; Ireland, C.M. Mirabamides E–H, HIV-inhibitory depsipeptides from the sponge Stelletta clavosa. J. Nat. Prod. 2011, 74, 185–193. [Google Scholar] [CrossRef]
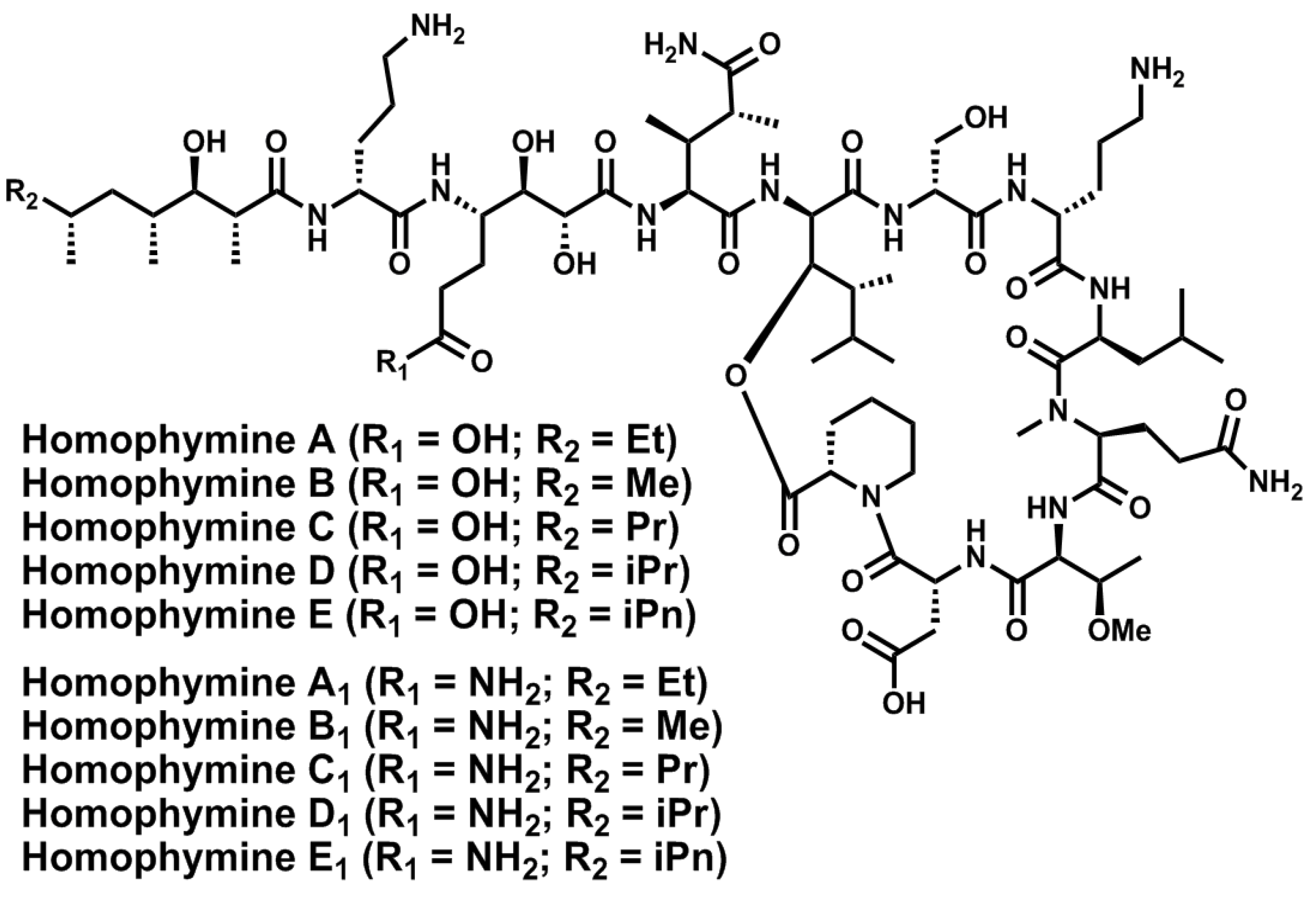
- Zampella, A.; Sepe, V.; Luciano, P.; Bellotta, F.; Monti, M.C.; D’Auria, M.V.; Jepsen, T.; Petek, S.; Adeline, M.-T.; Laprévôte, O.; et al. Homophymine A, an anti-HIV cyclodepsipeptide from the sponge Homophymia sp. J. Org. Chem. 2008, 73, 5319–5327. [Google Scholar] [CrossRef]
- Zampella, A.; Sepe, V.; Bellotta, F.; Luciano, P.; D’Auria, M.V.; Cresteil, T.; Debitus, C.; Petek, S.; Poupat, C.; Ahond, A. Homophymines B–E and A1–E1, a family of bioactive cyclodepsipeptides from the sponge Homophymia sp. Org. Biomol. Chem. 2009, 7, 4037–4044. [Google Scholar]
- Bellotta, F.; D’Auria, M.V.; Sepe, V.; Zampella, A. Synthetic studies on homophymine A: Stereoselective synthesis of (2R,3R,4R,6R)-3-hydroxy-2,4,6-trimethyloctanoic acid. Tetrahedron 2009, 65, 3659–3663. [Google Scholar] [CrossRef]
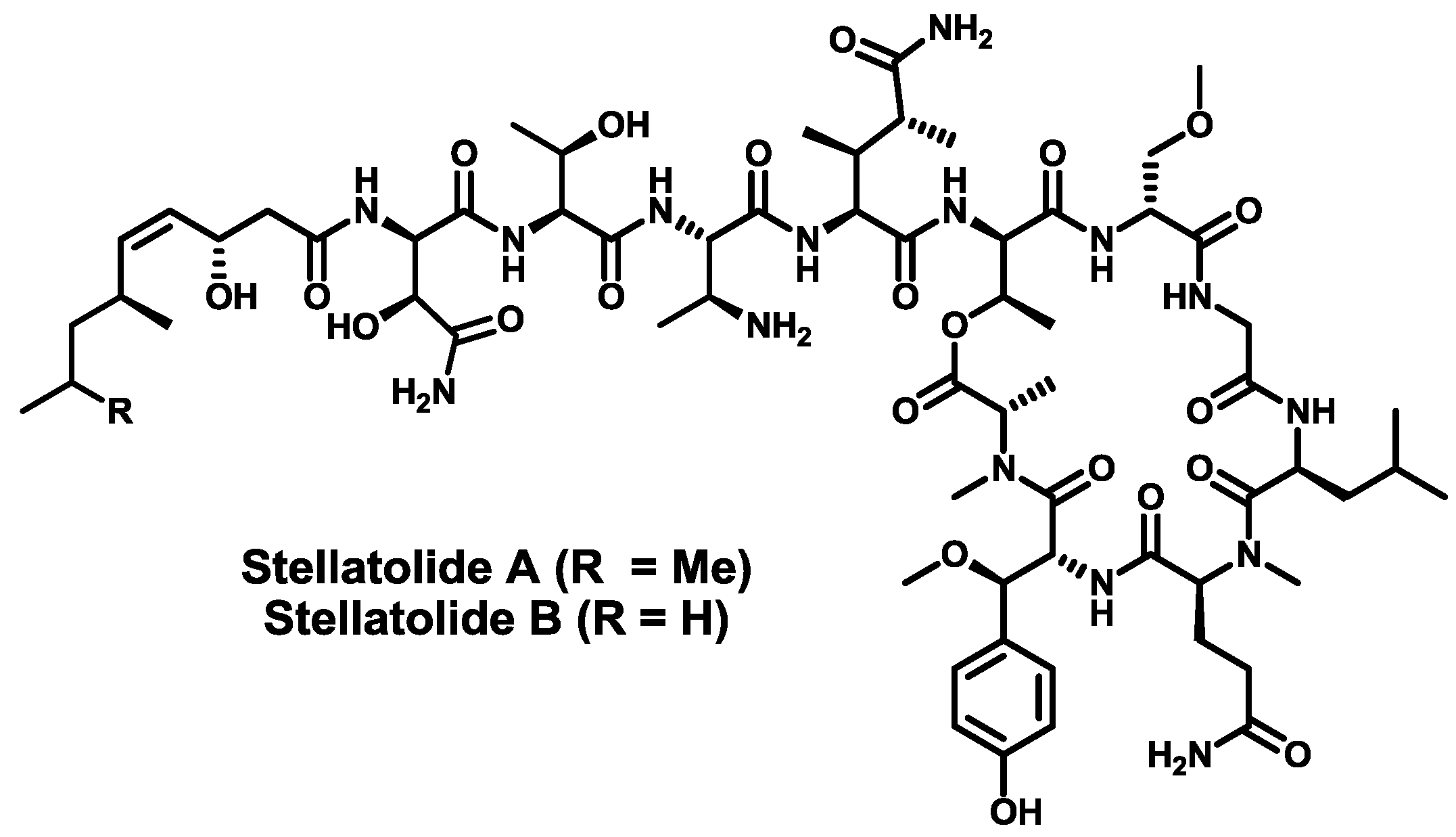
- Rodríguez, R.; Fernández, R.; Reyes, J.F.; Martín, M.J.; Marco, I.; Digón, I.; Francesch, A.; Cuevas, M.C. Stellatolide analogs as anticancer compounds. Int. Appl. Pat. WO 2010/007147 A1, 20 January 2010. [Google Scholar]
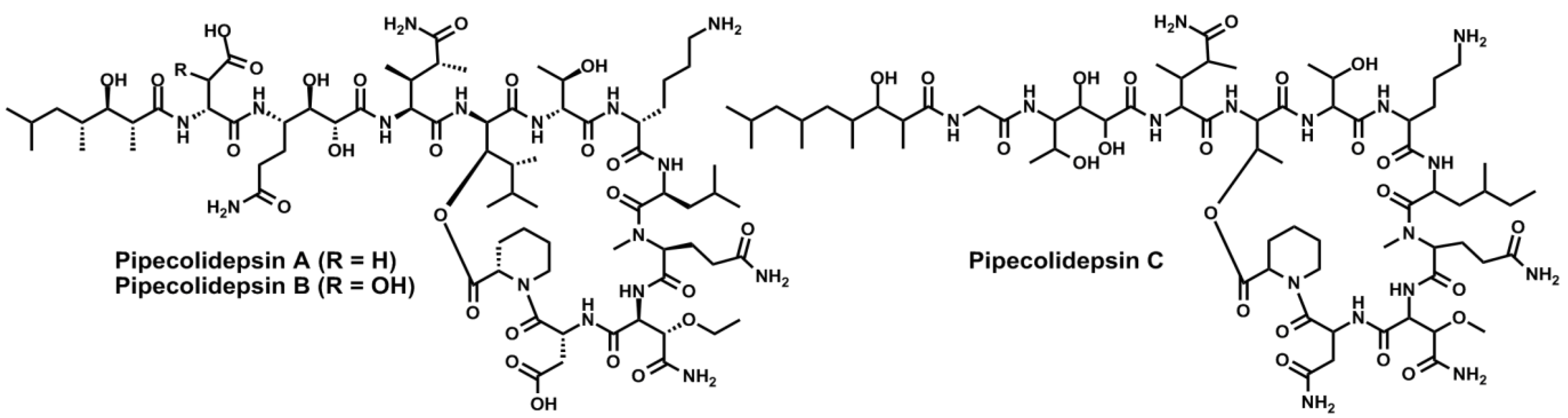
- Coello, L.; Fernández, R.; Reyes, J.F.; Francesch, A.; Cuevas, M.D.C. Anticancer pipecolidepsins from marine organisms. Int. Appl. Pat. WO 2010/070078 A1, 24 June 2010. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pelay-Gimeno, M.; Tulla-Puche, J.; Albericio, F. “Head-to-Side-Chain” Cyclodepsipeptides of Marine Origin. Mar. Drugs 2013, 11, 1693-1717. https://doi.org/10.3390/md11051693
Pelay-Gimeno M, Tulla-Puche J, Albericio F. “Head-to-Side-Chain” Cyclodepsipeptides of Marine Origin. Marine Drugs. 2013; 11(5):1693-1717. https://doi.org/10.3390/md11051693
Chicago/Turabian StylePelay-Gimeno, Marta, Judit Tulla-Puche, and Fernando Albericio. 2013. "“Head-to-Side-Chain” Cyclodepsipeptides of Marine Origin" Marine Drugs 11, no. 5: 1693-1717. https://doi.org/10.3390/md11051693