
Alkaloids with Cardiovascular Effects from the Marine-Derived Fungus Penicillium expansum Y32

Abstract
:
1. Introduction

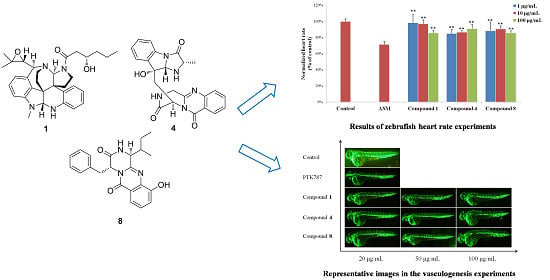
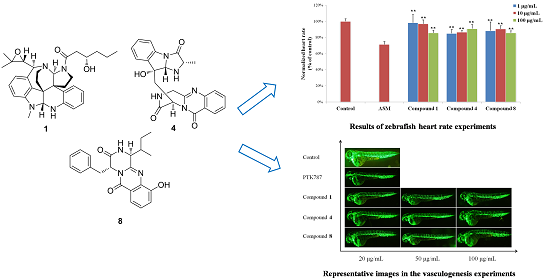
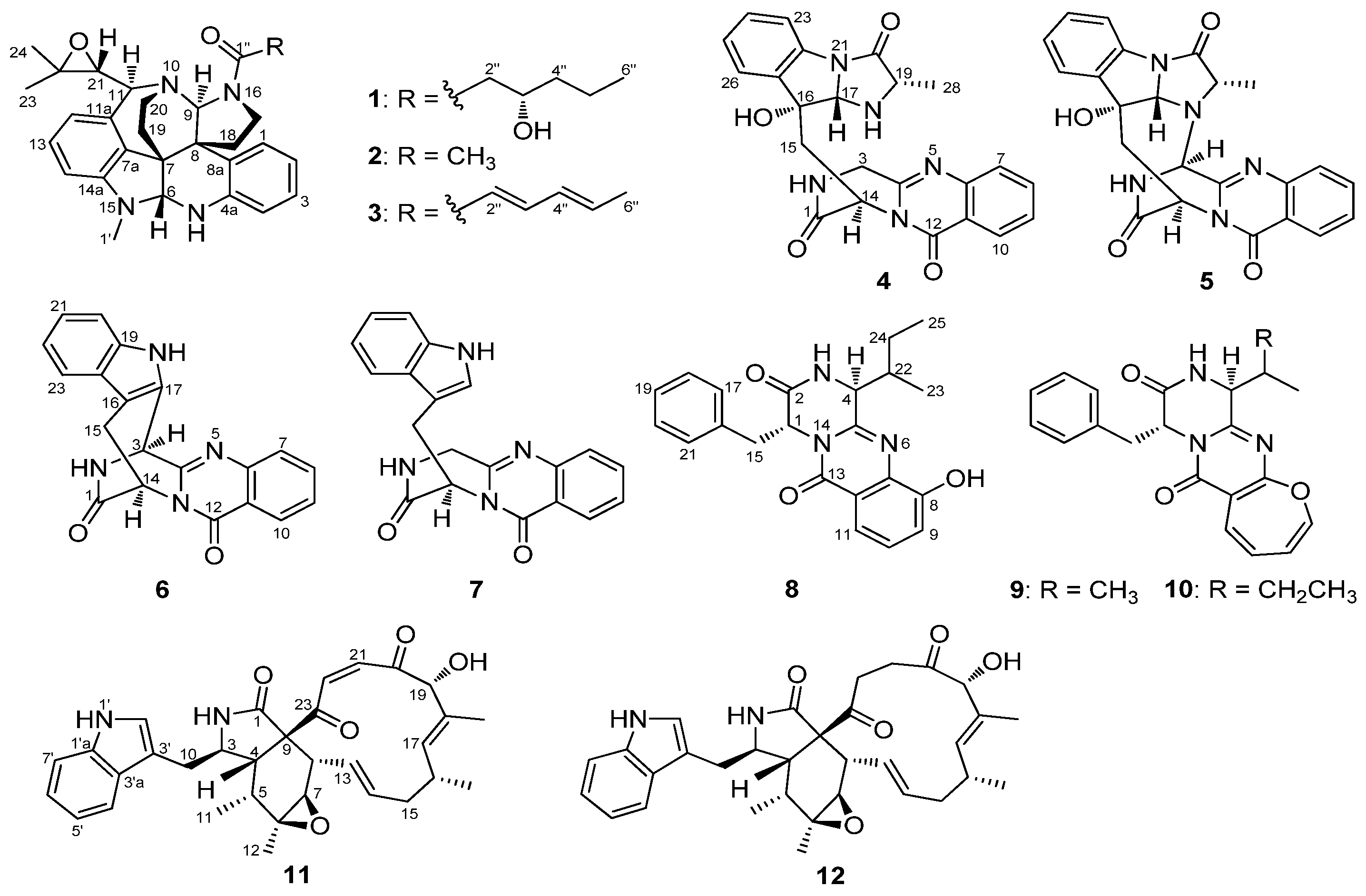
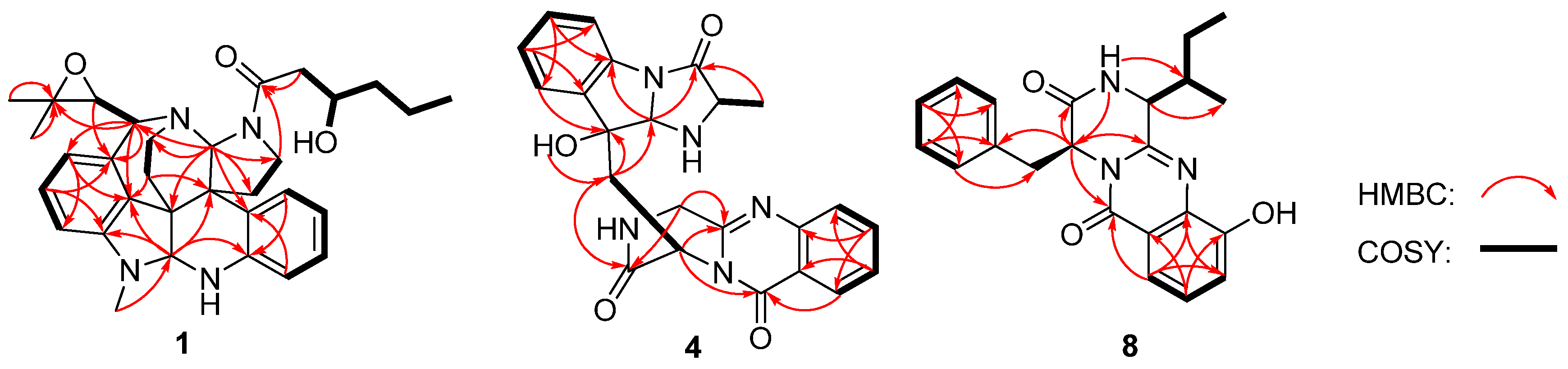
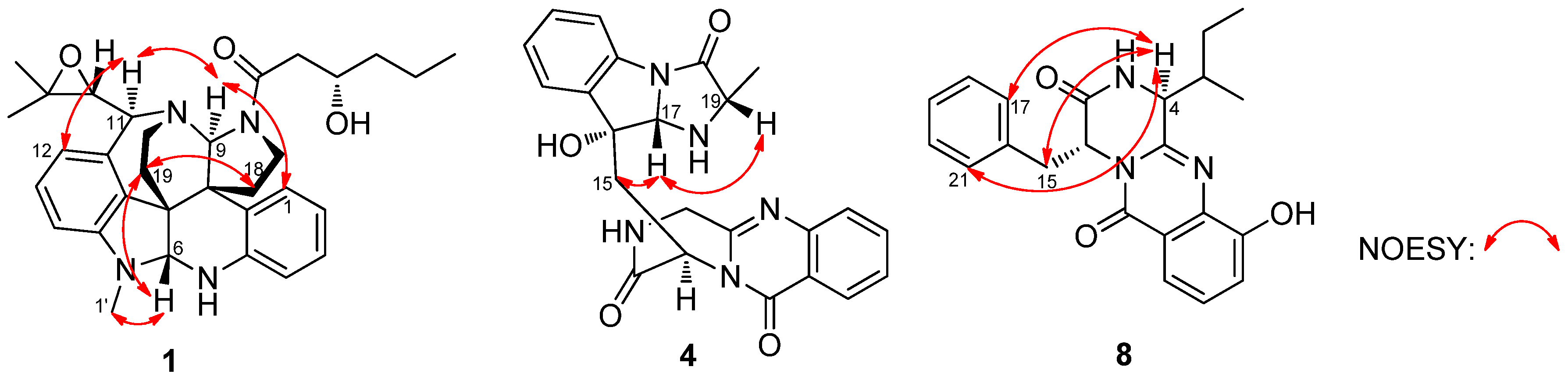
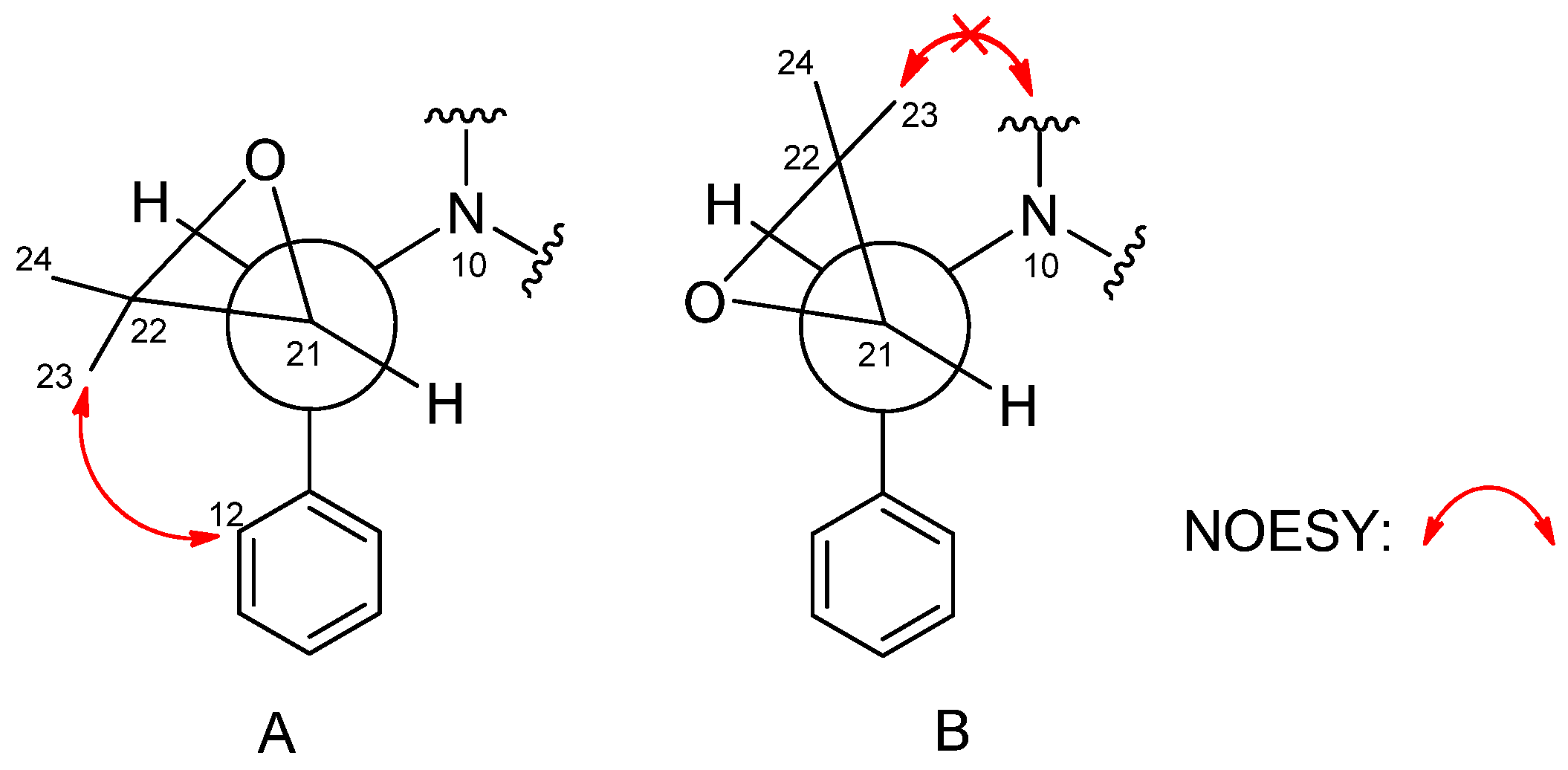
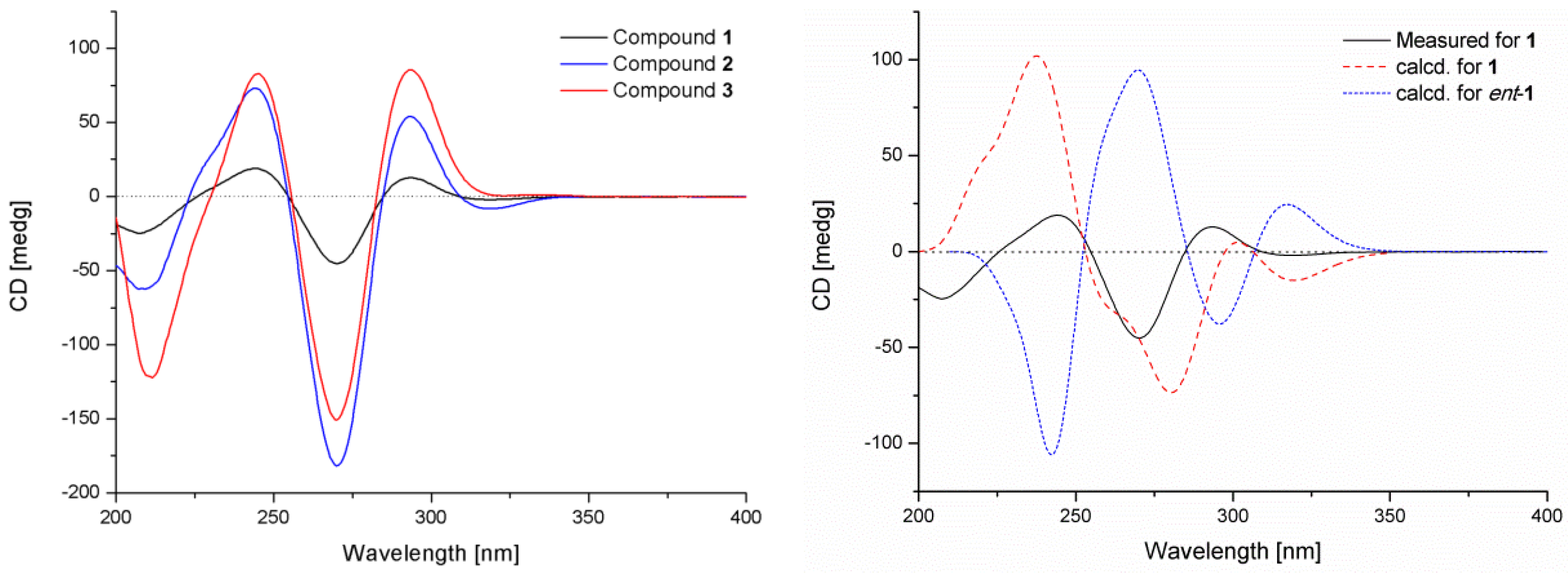
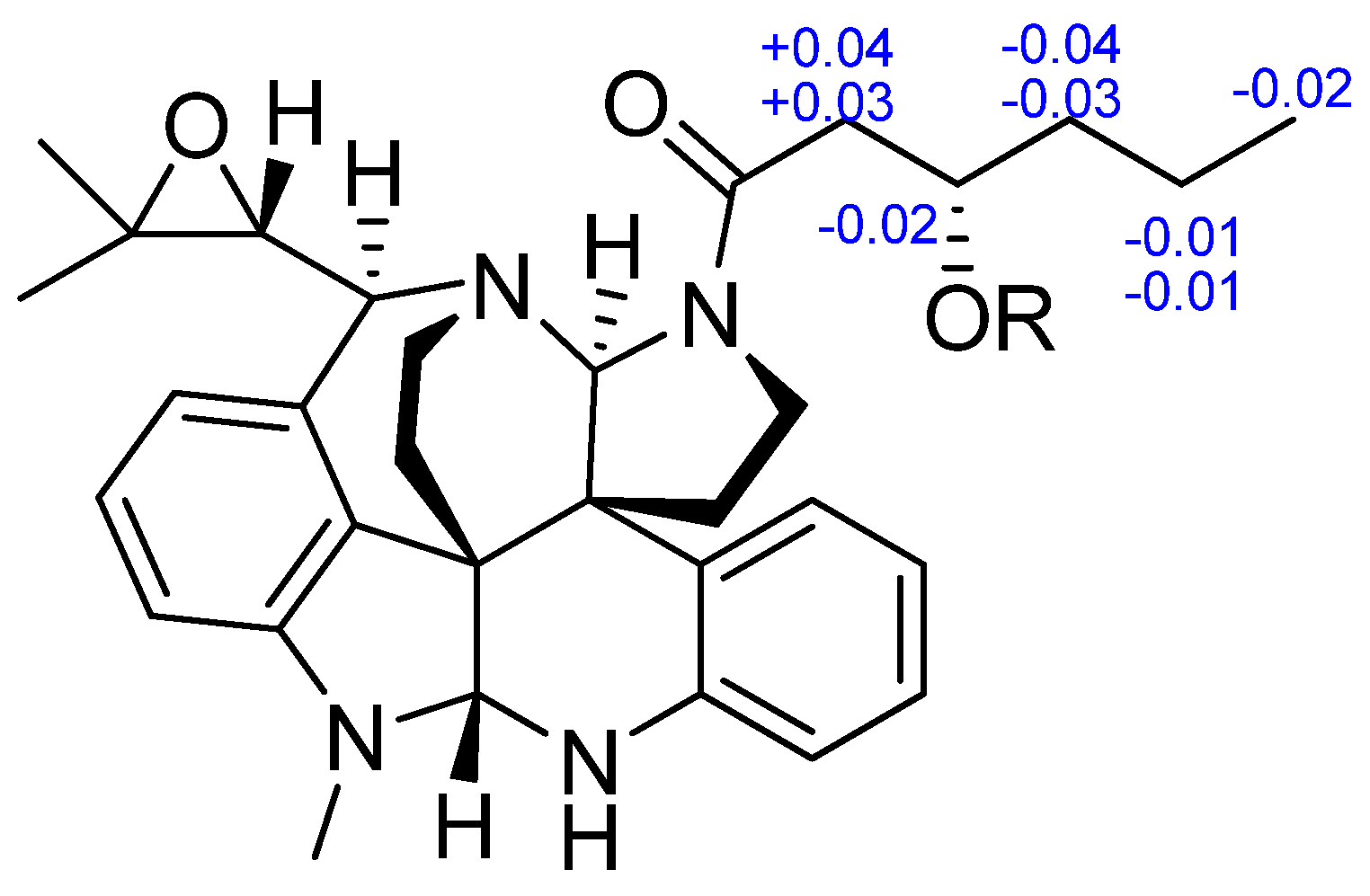
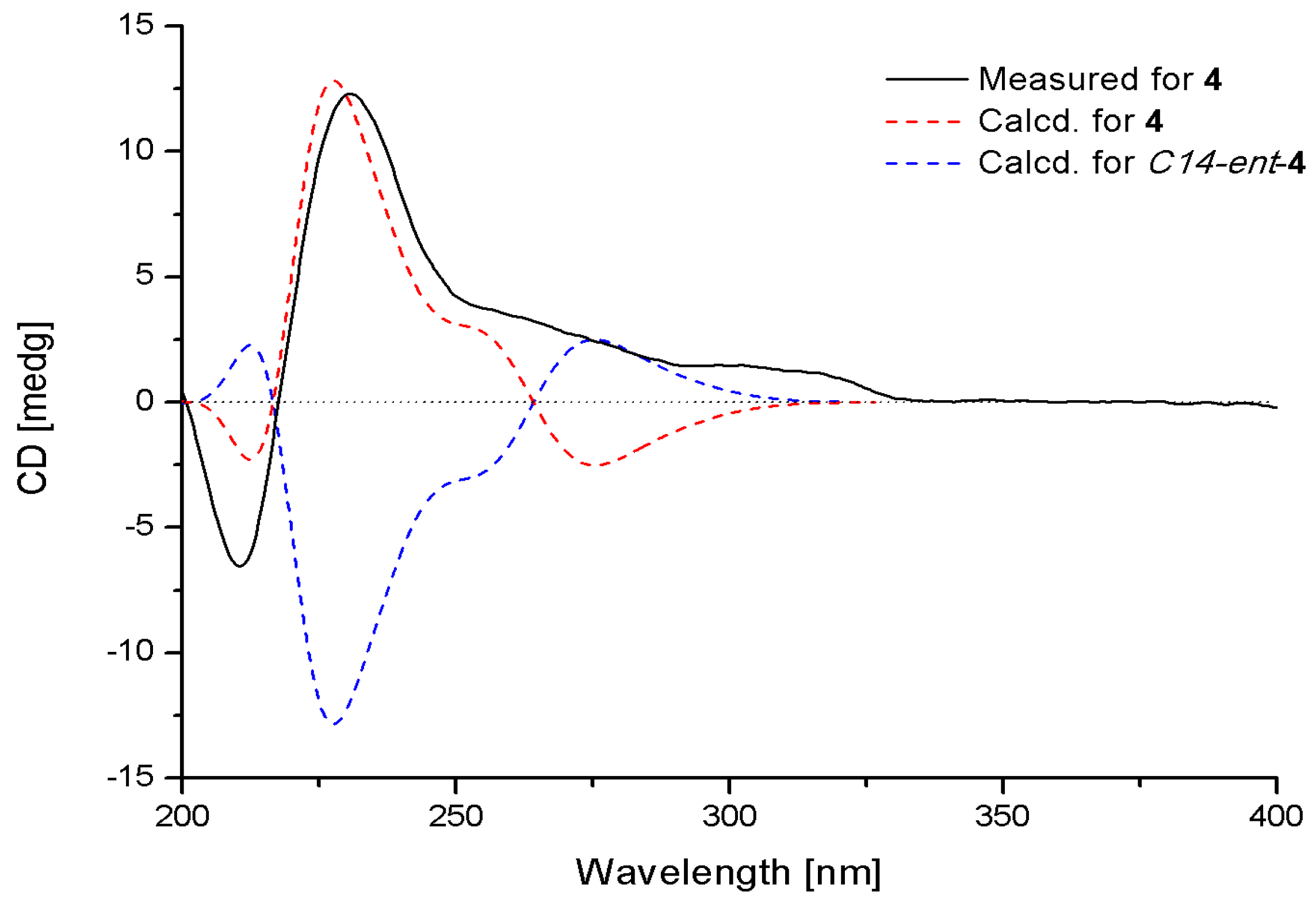
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 a | 8 b | ||||
|---|---|---|---|---|---|
| Position | δC | δH, Mult. (J in Hz) | Position | δC | δH, Mult. (J in Hz) |
| 1 | 123.2, CH | 6.68, d (7.6) | 1 | 56.5, CH | 5.29, t (5.0) |
| 2 | 120.6, CH | 6.71, t (7.6) | 2 | 167.4, C | |
| 3 | 127.5, CH | 7.01, t (7.6) | 3 | 8.32, brs | |
| 4 | 117.0, CH | 6.69, d (7.6) | 4 | 57.8, CH | 3.19, d (1.7) |
| 4a | 142.6, C | 5 | 149.1, C | ||
| 6 | 82.4, CH | 4.70, s | 7 | 135.7, C | |
| 7 | 51.3, C | 8 | 152.6, C | ||
| 7a | 132.1, C | 9 | 118.7, CH | 7.25, d (7.9) | |
| 8 | 52.1, C | 10 | 127.3, CH | 7.37, t ( 7.9) | |
| 8a | 132.1, C | 11 | 116.0, CH | 7.59, d (7.9) | |
| 9 | 79.1, CH | 5.05, s | 12 | 120.5, C | |
| 11 | 65.2, CH | 4.11, d (9.0) | 13 | 160.0, C | |
| 11a | 136.1, C | 15 | 36.4, CH2 | 3.27, dd (9.2, 5.0) 3.32 c, m | |
| 12 | 113.1, CH | 6.05, d (7.7) | 16 | 135.4, C | |
| 13 | 129.0, CH | 6.88, t (7.7) | 17 | 129.5, CH | 6.92, d (7.0) |
| 14 | 101.9, CH | 5.95, d (7.7) | 18 | 128.5, CH | 7.20, t (7.0) |
| 14a | 150.5, C | 19 | 127.5, CH | 7.24, m | |
| 17a | 44.1, CH2 | 3.92, dd (12.2, 8.7); 3.00, dt (12.2, 7.1) | 20 | 128.5, CH | 7.20, t (7.0) |
| 17b | |||||
| 18a | 30.6, CH2 | 2.71, dt (12.2, 8.7); 2.00, dd (12.2, 7.1) | 21 | 129.5, CH | 6.92, d (7.0) |
| 18b | |||||
| 19a | 37.7, CH2 | 2.37, dd (12.8, 8.5); 2.28, dt (12.8, 9.5) | 22 | 35.0, CH | 2.69, m |
| 19b | |||||
| 20a | 36.1, CH2 | 3.46, dd (15.8, 9.5); 3.36, dt (15.8, 8.5) | 23 | 15.4, CH3 | 0.91, d (7.3) |
| 20b | |||||
| 21 | 64.1, CH | 2.87, d (9.0) | 24 | 22.8, CH2 | 1.17, m |
| 22 | 60.4, C | 25 | 12.2, CH3 | 0.73, t (7.4) | |
| 23 | 24.9, CH3 | 1.39, s | 8-OH | 9.50, s | |
| 24 | 20.3, CH3 | 1.57, s | |||
| 1ʹ | 29.6, CH3 | 2.84, s | |||
| 1″ | 173.9, C | ||||
| 2″ | 42.1, CH2 | 2.82, dd (14.6, 3.4); 2.48, dd (14.6, 3.4) | |||
| 3″ | 69.0, CH | 4.06, brs | |||
| 4″ | 39.5, CH2 | 1.60, m; 1.53, m | |||
| 5ʹʹ | 18.9, CH2 | 1.54, m; 1.44, m | |||
| 6ʹʹ | 14.1, CH3 | 0.96, t (7.0) | |||
| OH | 4.14, brs | ||||





| Position | 4 a | 5 a | ||
|---|---|---|---|---|
| δC | δH, Mult. (J in Hz) | δC | δH, Mult. (J in Hz) | |
| 1 | 169.9, C | 170.1, C | ||
| 2 | 7.01, brs | 8.26, brs | ||
| 3a | 45.3, CH2 | 4.70, d (15.6) | 66.2, CH | 5.31, d (3.1) |
| 3b | 4.49, d (15.6) | |||
| 4 | 148.0, C | 146.8, C | ||
| 6 | 147.1, C | 146.4, C | ||
| 7 | 127.1, CH | 7.63, d (8.0) | 127.8, CH | 7.64, d (7.9) |
| 8 | 135.1, CH | 7.78, t (8.0) | 135.3, CH | 7.72, t (7.9) |
| 9 | 127.6, CH | 7.50, t (8.0) | 127.0, CH | 7.45, t (7.9) |
| 10 | 127.3, CH | 8.30, d (8.0) | 125.6, CH | 8.18, d (7.9) |
| 11 | 120.5, C | 120.5, C | ||
| 12 | 160.4, C | 159.8, C | ||
| 14 | 51.5, CH | 5.81, dd (8.6, 3.8) | 54.1, CH | 5.62, dd (4.7, 2.3) |
| 15a | 39.7, CH2 | 2.37, dd (14.5, 8.9) | 37.2, CH2 | 2.49, dd (15.3, 2.1) |
| 15b | 2.74, dd (14.5, 3.9) | 3.28, dd (15.3, 5.1) | ||
| 16 | 74.2, C | 74.8, C | ||
| 17 | 79.8, CH | 5.25, s | 80.9, CH | 4.58, d (1.3) |
| 19 | 59.0, CH | 4.25, q (12.8, 6.2) | 64.3, CH | 4.21, q (11.9, 6.5) |
| 20 | 172.9, C | 165.8, C | ||
| 22 | 137.4, C | 136.1, C | ||
| 23 | 115.8, CH | 7.52, d (7.4) | 115.2, CH | 7.39, d (7.6) |
| 24 | 125.3, CH | 7.08, t (7.4) | 128.1, CH | 7.24, t (7.6) |
| 25 | 130.3, CH | 7.30, t (7.4) | 130.5, CH | 7.08, t (7.6) |
| 26 | 124.2, CH | 7.31, d (7.4) | 124.4, CH | 7.34, d (7.6) |
| 27 | 138.0, C | 138.1, C | ||
| 28 | 18.2, CH3 | 1.39, d (6.6) | 14.9, CH3 | 1.51, d (6.5) |
| 16-OH | 5.00, brs | 4.64, brs | ||



3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material and Fermentation
3.3. Extraction and Purification
3.4. Preparation of the (S)-and (R)-MTPA Esters of 1a and 1b by Modified Mosher’s Method
3.5. Marfey’s Analysis
3.6. Bioassays
3.6.1. Zebrafish Embryos
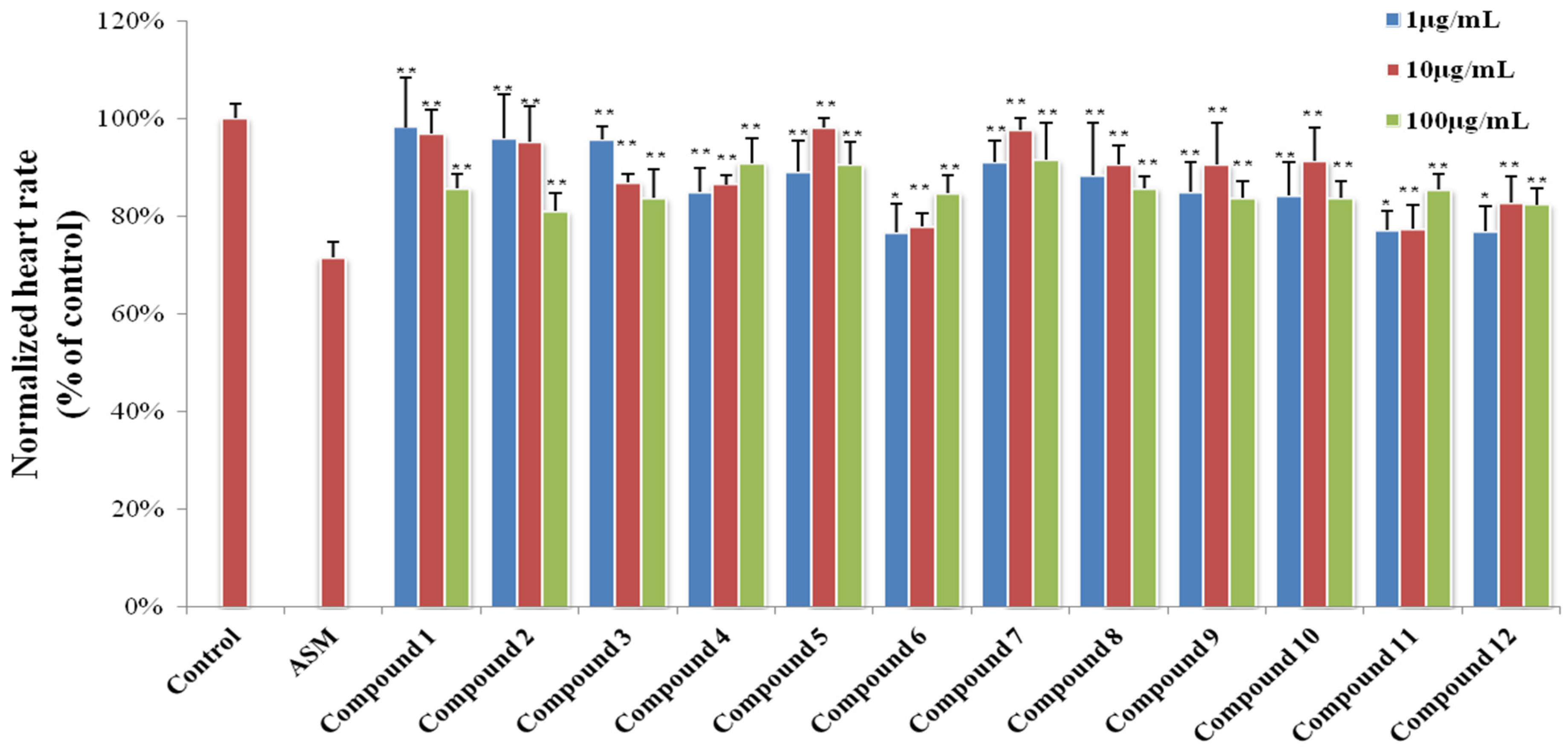
3.6.2. Heart Rate Experiments
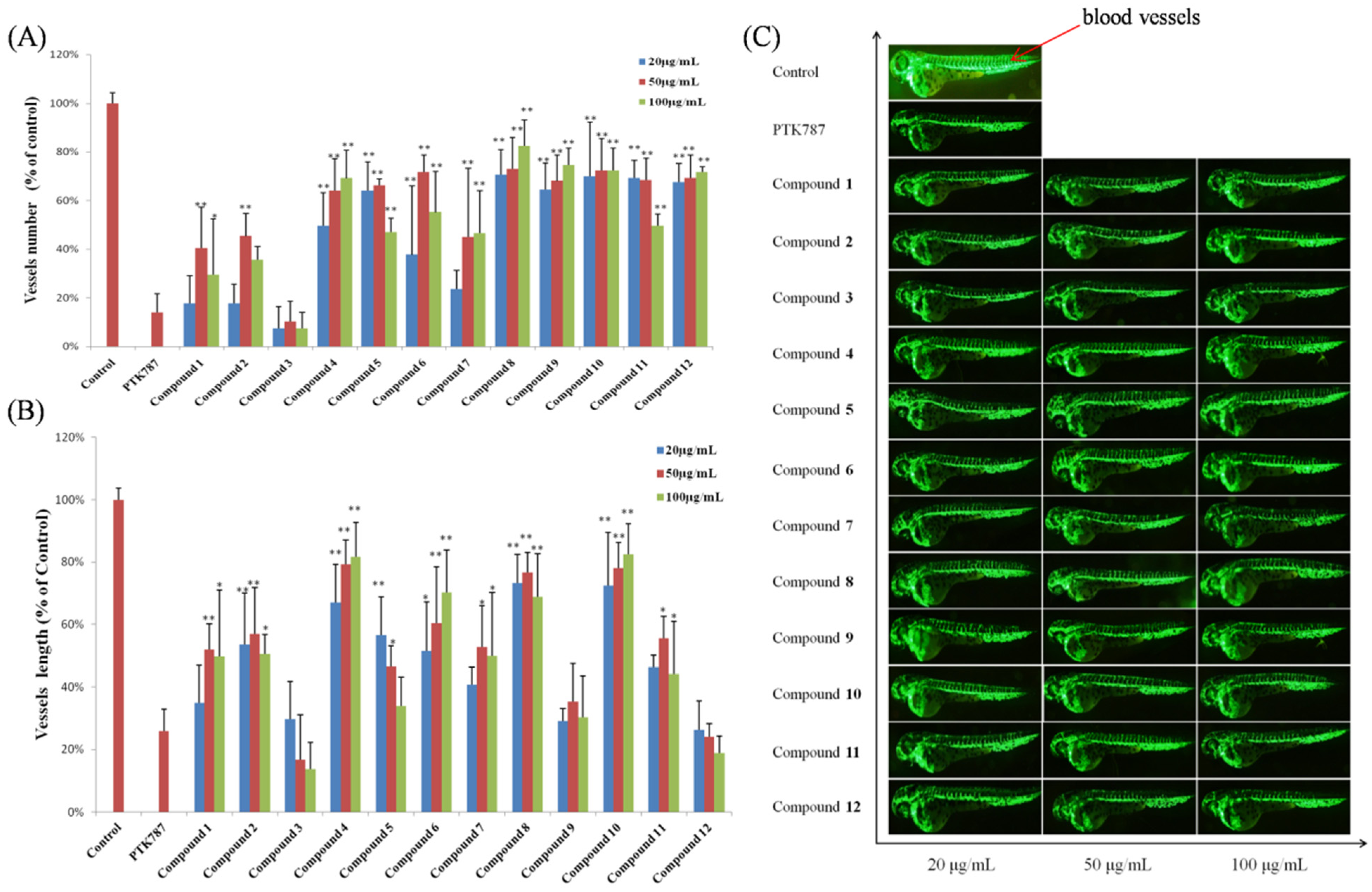
3.6.3. Vasculogenesis Experiments
3.6.4. Antiangiogenic Vessel Growth Experiments
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Cardiovascular diseases (CVDs). Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 27 September 2015).
- Hideo, H.; Hirotaka, M.; Kohki, A. New insecticidal compounds, communesins C, D and E, from Penicillium expansum link MK-57. Biosci. Biotechnol. Biochem. 2004, 68, 753–756. [Google Scholar]
- Dalsgaard, P.W.; Blunt, J.W.; Munro, M.H.G.; Frisvad, J.C.; Christophersen, C. Communesins G and H, new alkaloids from the psychrotolerant fungus Penicillium rivulum. J. Nat. Prod. 2005, 68, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.W.; Ma, D. Enantioselective Total Syntheses of Communesins A and B. Angew. Chem. Int. Ed. 2011, 50, 12008–12011. [Google Scholar] [CrossRef] [PubMed]
- Fremlin, L.J.; Piggott, A.M.; Lacey, E.; Capon, R.J. Cottoquinazoline A and Cotteslosins A and B, metabolites from an Australian marine-derived strain of Aspergillus versicolor. J. Nat. Prod. 2009, 72, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.X.; Lin, T.; Wang, W.; Xin, Z.H.; Zhu, T.J.; Gu, Q.Q.; Li, D.H. Antiviral alkaloids produced by the mangrove-derived fungus Cladosporium sp. PJX-41. J. Nat. Prod. 2013, 76, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Asami, Y.; Lee, D.; Jang, J.H.; Ahn, J.S.; Oh, H. Protuboxepins A and B and protubonines A and B from the marine-derived fungus Aspergillus sp. SF-5044. J. Nat. Prod. 2011, 74, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, H.; Murakami, Y.; Ichihara, A. 20-Ketoreductase Activity of Chaetoglobosin A and prochaetoglobosins in a cell-free system of chaetomium subaffine and the isolation of new chaetoglobosins. Biosci. Biotech. Biochem. 1993, 57, 628–631. [Google Scholar] [CrossRef]
- Iwamoto, C.; Yamada, T.; Ito, Y.; Minoura, K.; Numata, A. Cytotoxic cytochalasans from a Penicillium species separated from a marine alga. Tetrahedron 2001, 57, 2997–3004. [Google Scholar] [CrossRef]
- Tran, T.C.; Sneed, B.; Haider, J.; Blavo, D.; White, A.; Aiyejorun, T.; Baranowski, T.C.; Rubinstein, A.L.; Doan, T.N.; Dingledine, R.; et al. Automated, quantitative screening assay for antiangiogenic compounds using transgenic zebrafish. Cancer Res. 2007, 67, 11386–11392. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Ueoka, R.; Nakao, Y.; Kawatsu, S.; Yaegashi, J.; Matsumoto, Y.; Matsunaga, S.; Furihata, K.; van Soest, R.W.; Fusetani, N. Gracilioethers A–C, antimalarial metabolites from the marine sponge Agelas gracilis. J. Org. Chem. 2009, 74, 4203–4207. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.B.; Teng, X.C.; Wang, Y.; Liu, P.P.; Li, G.Q.; Zhu, W.M. New quinazolinone alkaloids within rare amino acid residue from coral-associated fungus, Aspergillus versicolor LCJ-5-4. Org. Lett. 2011, 13, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Matsushita, T.; Doi, M.; Minoura, K.; Shingu, T.; Kumeda, Y.; Numata, A. Fumiquinazolines A–G, novel metabolites of a fungus separated from a Pseudolabrus marine fish. J. Chem. Soc. Perkin Trans. 1995, 1, 2345–2353. [Google Scholar] [CrossRef]
- Marfey, P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commum. 1984, 49, 591–596. [Google Scholar] [CrossRef]
- Li, G.Y.; Li, L.M.; Yang, T.; Chen, X.Z.; Fang, D.M.; Zhang, G.L. Four new alkaloids, brevianamides O–R, from the fungus Aspergillus versicolor. Helv. Chim. Acta 2010, 93, 2075–2080. [Google Scholar] [CrossRef]
- Chan, J.; Bayliss, P.E.; Wood, J.M.; Roberts, T.M. Dissection of angiogenic signaling in zebrafish using a chemical genetic approach. Cancer Cell 2002, 1, 257–267. [Google Scholar] [CrossRef]
- Cross, L.M.; Cook, M.A.; Lin, S.; Chen, J.N.; Rubinstein, A.L. Rapid analysis of angiogenesis drugs in a live fluorescent zebrafish assay. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Seng, W.L.; Eng, K.; Lee, J.; McGrath, P. Use of a monoclonal antibody specific for activated endothelial cells to quantitate angiogenesis in vivo in zebrafish after drug treatment. Angiogenesis 2004, 7, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, J.P.; Qian, J.Q.; Hu, C.Q. Cardiotoxicity evaluation of anthracyclines in zebrafish (Danio rerio). J. Appl. Toxicol. 2015, 35, 241–252. [Google Scholar] [CrossRef] [PubMed]
- McKinley, E.T.; Baranowski, T.C.; Blavo, D.O.; Cato, C.; Doan, T.N.; Rubinstein, A.L. Neuroprotection of MPTP-induced toxicity in zebrafish dopaminergic neurons. Mol. Brain Res. 2005, 141, 128–137. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.-Q.; Li, P.-H.; Chao, Y.-X.; Chen, H.; Du, N.; He, Q.-X.; Liu, K.-C. Alkaloids with Cardiovascular Effects from the Marine-Derived Fungus Penicillium expansum Y32. Mar. Drugs 2015, 13, 6489-6504. https://doi.org/10.3390/md13106489
Fan Y-Q, Li P-H, Chao Y-X, Chen H, Du N, He Q-X, Liu K-C. Alkaloids with Cardiovascular Effects from the Marine-Derived Fungus Penicillium expansum Y32. Marine Drugs. 2015; 13(10):6489-6504. https://doi.org/10.3390/md13106489
Chicago/Turabian StyleFan, Ya-Qin, Pei-Hai Li, Ya-Xi Chao, Hao Chen, Ning Du, Qiu-Xia He, and Ke-Chun Liu. 2015. "Alkaloids with Cardiovascular Effects from the Marine-Derived Fungus Penicillium expansum Y32" Marine Drugs 13, no. 10: 6489-6504. https://doi.org/10.3390/md13106489