Starting from the list of 5719 pre-selected CpG sites, we applied the evolutionary feature selection and/or GORevenge-based selection under the following four scenarios:
(1) Keep the dimensionality of selected features relatively low, but simultaneously with a high, robust, predictive efficiency. This aspect sets a grand challenge, as the effect of the epigenetic imprinting, especially in blood tissue (a heterogeneous collection of multiple cell types of different origins), is an extremely fuzzy one, with an intricate, indirect, accumulative mechanism of action, with respect to the prospective macroscopic biological outcome, which might take place many decades afterwards. We applied the evolutionary feature selection to the list of 5719 CpG sites with a dimensionality threshold equal to 150 for all the classification schemes,
i.e., controls
vs. BCCA
vs. LYCA (142 features were found), controls
vs. BCCA (129 features), controls
vs. LYCA (143 features), and BCCA
vs. LYCA (146 features). The derived subset of features were fed to the classifiers and tested either using a training set (leave-one-out resampling) or a totally unknown testing set. Results using leave-one-out resampling are presented in the
Supplementary Material (Tables S1–S4). Results for the four classification schemes mentioned above on the independent testing set are presented here in
Table 3,
Table 4,
Table 5 and
Table 6, respectively.
Results are reported and commented beneath, firstly on the basis of the performance obtained using resampling on the training set. Then, results are discussed and conclusions are made on the basis of performance we obtained when models constructed using the training set were applied to the totally unknown testing set, used as an independent set. Lists of CpG sites selected for the three-class problem (controls vs. BCCA vs. LYCA) were finally evaluated and compared in terms of their biological content (GO terms and KEGG pathways).
3.1. Performance Obtained Using Resampling
Results show that the provided subsets of features corresponding to equal in number CpG sites achieved moderate or high accuracies and class sensitivity measurements when the training set was solely used along with resampling. This was found both within the evolutionary selection process when applied (reported measurements for the embedded 12-nn classifier are shown in
Tables S1–S4,S8,S9 (Column 2), and additionally when features subsets were evaluated by the classifiers on a leave-one-out strategy using the training set (
Tables S1–S9, Columns 3–7). Almost all performance measurements were found better that 50%, however, some outliers (performing even down to 35%) existed for the case of the weakest classifiers. The classifiers perform a lot better in the two-class task BCCA
vs. LYCA, showing that this is an easier distinguishing task than that of distinguishing control from a case (either cancer type), or distinguishing controls from the two cancer types simultaneously. The most well performing classifier overall seems to be the ANN, with the 12-nn classifier following in most classification tasks (expected since it comprises the classifier embedded into the evolutionary feature selection process when applied). For the three-class problem controls
vs. BCCA
vs. LYCA the best performing ANN classifier, in particular, achieved performance measurements all in the range of 68% up to 91.4%.The rates achieved for the task BCCA
vs. LYCA by ANN, the best among all rates achieved for the two-class tasks, are equally to 100% (for all evaluation metrics).
It is encouraging that the different learning algorithms evaluated in the current study were all found to perform adequately well (most of the times far more than 50% for the two-class problems, and far more than 33% for the three-class problem corresponding to random classifiers), when evaluated on a resampling basis using the training set. It should be noted that two more learning algorithms were added to the k-nn weighted classifier, which was used internally by the evolutionary feature selection, when selected features and classifiers were evaluated using resampling. Thus, any doubts for a bias introduced by the use of an embedded classifier into the feature selection module could be eliminated.
The evolutionary feature selection method itself performed well when applied: it lowers the dimensionality of the features and selected features perform well based on resampling using the training set (
Tables S1–S4,S8,S9). This is due to the fact that the genetic algorithm could screen effectively the complete solution space given the initial population used. Actually, the setting of 500 chromosomes in the initial pool of candidate solutions (each corresponding to 150 or 400 features maximum) ensures that all features (out of 5719 or 352 features submitted to GA depending on the case) could be handled as candidate features for the final solution. GA showed a quick convergence even the low
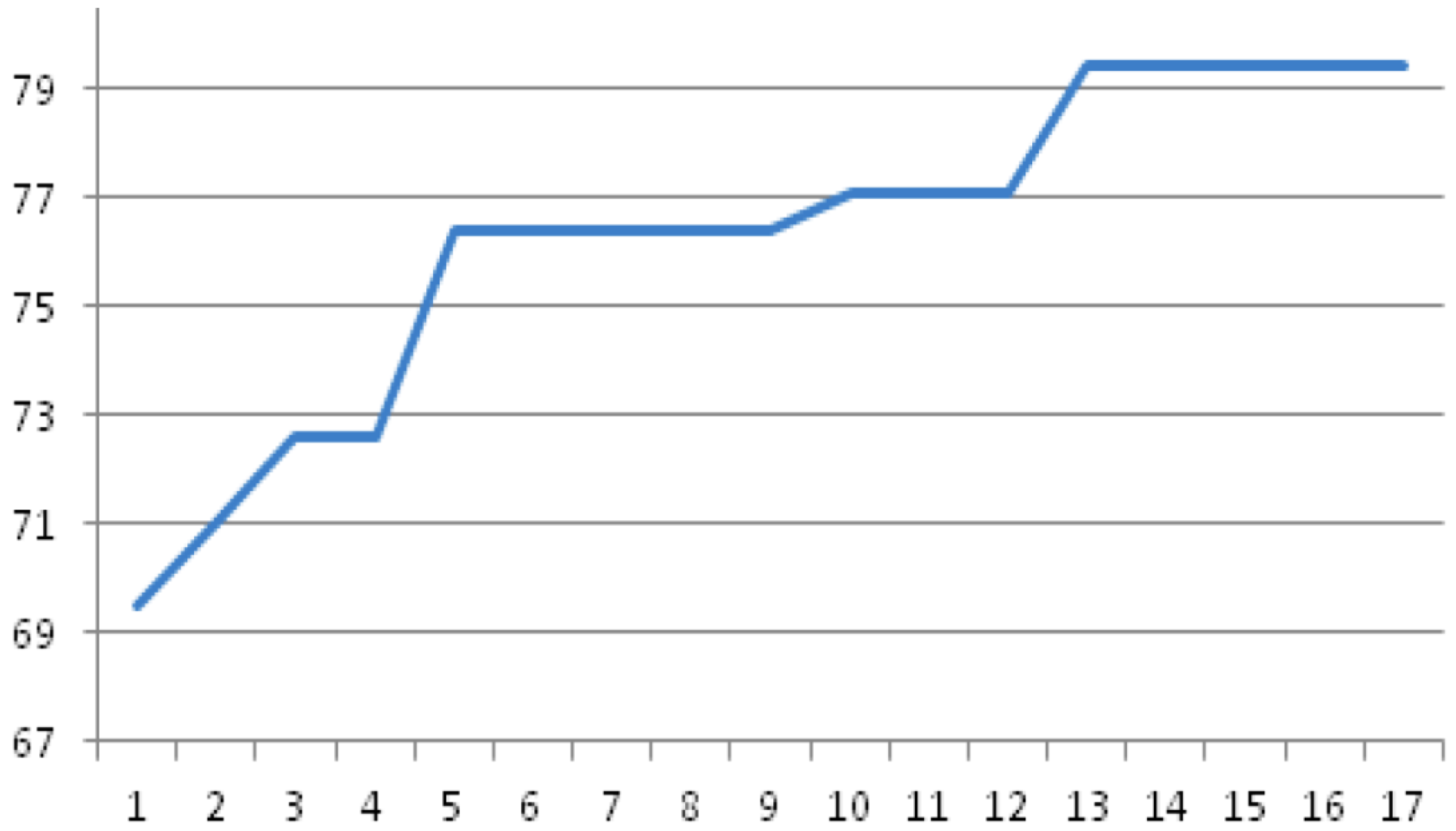
Pc (=0.5) value. The convergence of GA within feature selection for the three-class problem controls
vs. BCCA
vs. LYCA is shown in
Figure 1: the GA reaches the maximum performance for the embedded classifier already in the 13th generation (out of 50 generations).
Figure 1.
Total accuracy (%) of the embedded 12-nn classifier during GA evolution (three-fold cross-validation) for the three-class problem controls vs. BCCA vs. LYCA (% accuracy vs. number of generations completed).
Figure 1.
Total accuracy (%) of the embedded 12-nn classifier during GA evolution (three-fold cross-validation) for the three-class problem controls vs. BCCA vs. LYCA (% accuracy vs. number of generations completed).
On the other side, GORevenge as a feature selection method provides features that perform adequately well when fed to classifiers and tested using resampling on training set. We stress here anyway, that the comparative assessment of the different feature selection scenarios, using results based on resampling on training set, is not the goal of our work (hence results are presented as supplementary). However, we attempt to identify critical performance aspects of the decision mechanism, with respect to the topological structure of the given dataset, on the basis of results obtained using the independent testing set in the following subsection.
3.2. Performance Obtained Using the Independent Set—Functional Analysis—General Discussion
Regarding the application of the classifiers constructed by the training set and evaluated on the independent testing set, performance is lower than the performance obtained using resampling on the training set. This is a finding in conformance with our expectations and is shown in detail in
Table 2,
Table 3,
Table 4,
Table 5,
Table 6,
Table 7,
Table 8,
Table 9,
Table 10 and
Table 11 (comparing to
Tables S1–S9) where results obtained using all classification models for all feature selection scenarios and all classification tasks are reported. The results show that the ANN classifier is again here the best-performing classifier overall and probably its superiority is due to its high capability of capturing non-linear effects within the input set. Based on that we attempt to evaluate the feature selection scenarios followed using the performance that the ANN classifier could achieve in the independent testing set.
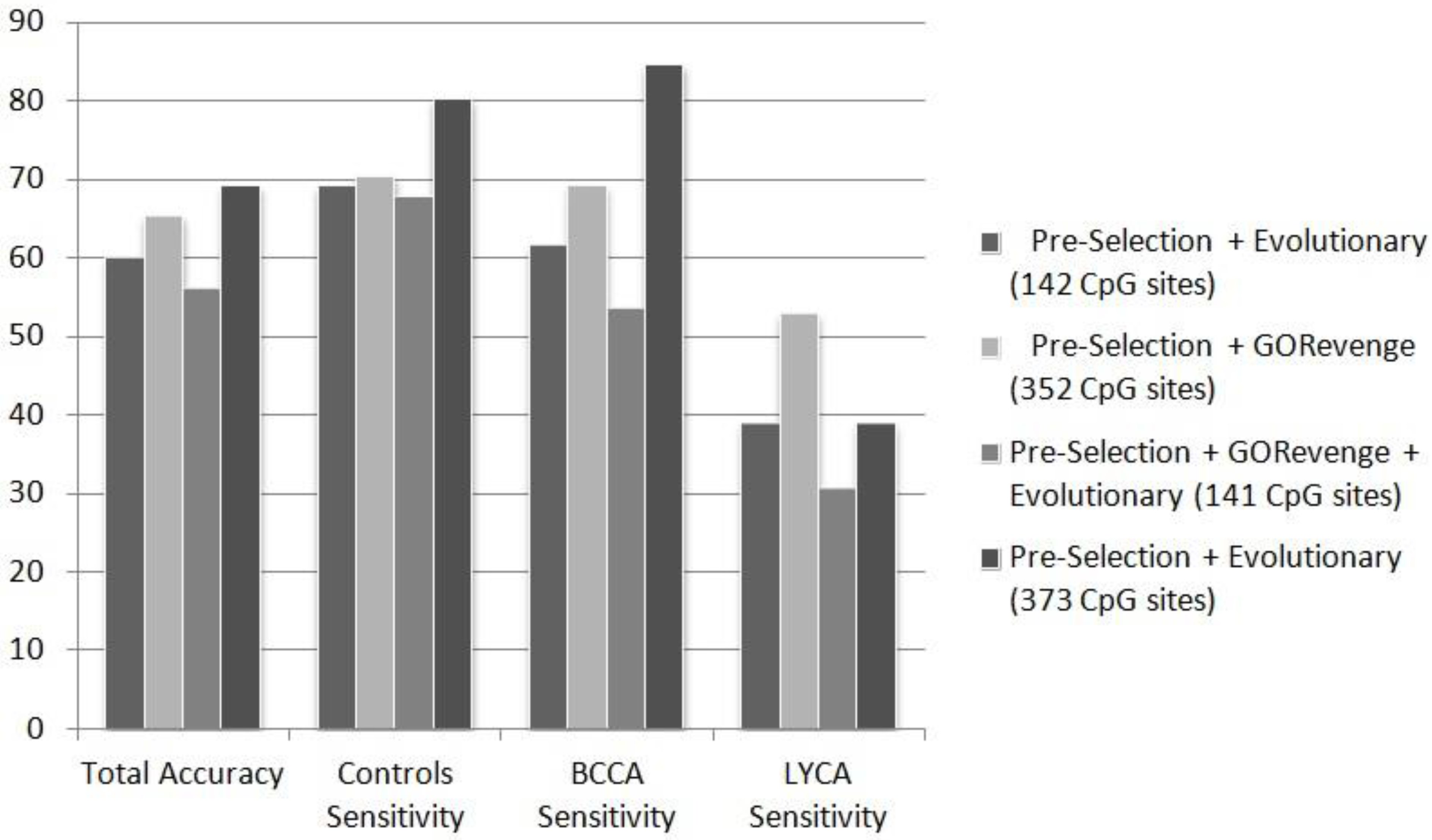
Figure 2 summarizes the results of the ANN classifier for each of the feature selection scenarios for the three-class problem controls
vs. BCCA
vs. LYCA. Perusing the epidemiological dataset as a three-class problem represents not only legitimacy, from the data-mining perspective, but also a plausible investigative strategy to be evaluated for its practical performance in real-life problem. The diversity of epidemiological phenotypes that should be inquired from blood tissue screening is immense, and there is a great need for a rapid, standard framework, for initial analysis, prior to more exhaustive investigations. Most of the accuracy or class sensitivity measurements in the three-class problem are greater than 50% far beyond the 33% random choice threshold, however the greater drawback is the sensitivity of B-cell lyphoma class, which samples are mislabeled as controls or breast cancer cases.
Figure 2.
Performance (%) of ANN in the totally unknown—independent testing set in the three-class problem (controls vs. BCCA vs. LYCA), fed by CpG sites subsets selected by the selection schemes applied.
Figure 2.
Performance (%) of ANN in the totally unknown—independent testing set in the three-class problem (controls vs. BCCA vs. LYCA), fed by CpG sites subsets selected by the selection schemes applied.
From the phenotypic point of view the B-cell lymphoma class, represents an extremely broad family of various lymphatic cancers with diverse molecular mechanism and probably etiology. Given the utilization of 46 out of the 82 samples describing the B-cell lymphoma category, this number is actually meager in order to effectively address ranges of the phenotypic versatility of this category. The subset of CpG sites selected when evolutionary selection was applied to the statistically pre-selected CpG sites, includes a total of 373 CpG sites. This subset is the one to feed the best performing ANN classifier in terms of total accuracy, controls class sensitivity, and breast cancer class sensitivity. However, it does not perform well regarding the B-cell lymphoma class sensitivity (38.89%), for the reasons mentioned above. Regarding this measurement, the ANN fed by the 352 CpG sites proposed by selection based solely to GORevenge showed superior performance. This subset of CpG sites seems to feed an ANN with a balanced performance in terms of sensitivity across all three classes (70.37%, 69.23% and 52.78% for the classes of controls, breast cancer cases and B-cell lymphoma cases). It is worth noting that this subset has been derived solely by using a functional analysis approach, without embedding classification mechanisms within the selection of CpG sites. The subsets of CpG sites proposed either by the evolutionary selection (combined with a threshold=400 for the number of CpG returned) or by GORevenge followed by evolutionary selection seem to perform worse.
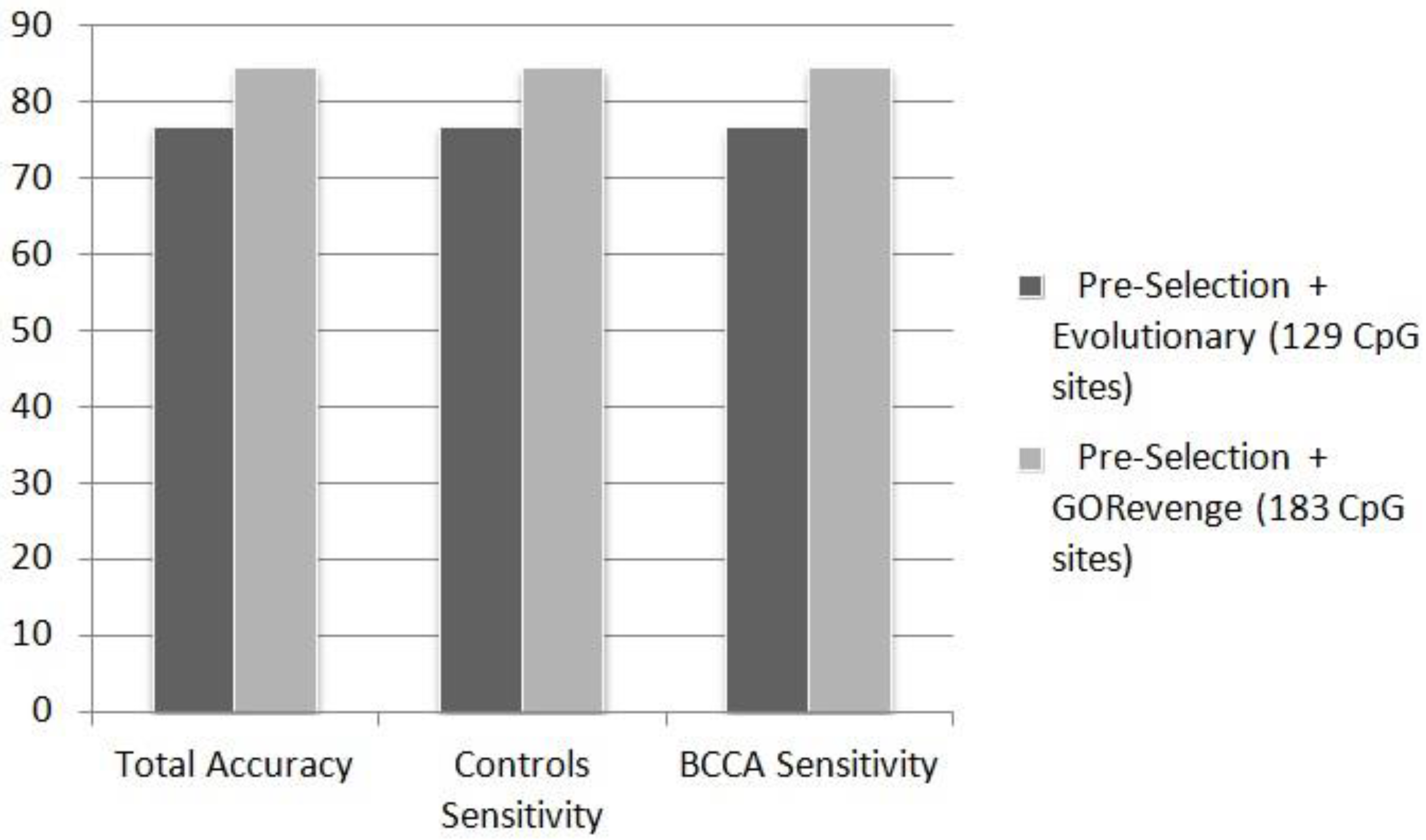
Figure 3 shows that the 183 CpG sites selected by GORevenge (accuracy: 84.62%, controls sensitivity: 84.62%, cases sensitivity: 84.62%) outperform the 129 CpG sites provided by the evolutionary selection (accuracy: 76.92%, controls sensitivity: 76.92%, cases sensitivity: 76.92%) for the two-class problem controls
vs. BCCA.
Figure 3.
Performance (%) of ANN in the totally unknown—independent testing set in the two-class problem (controls vs. BCCA), fed by CpG sites subsets selected by the selection schemes.
Figure 3.
Performance (%) of ANN in the totally unknown—independent testing set in the two-class problem (controls vs. BCCA), fed by CpG sites subsets selected by the selection schemes.
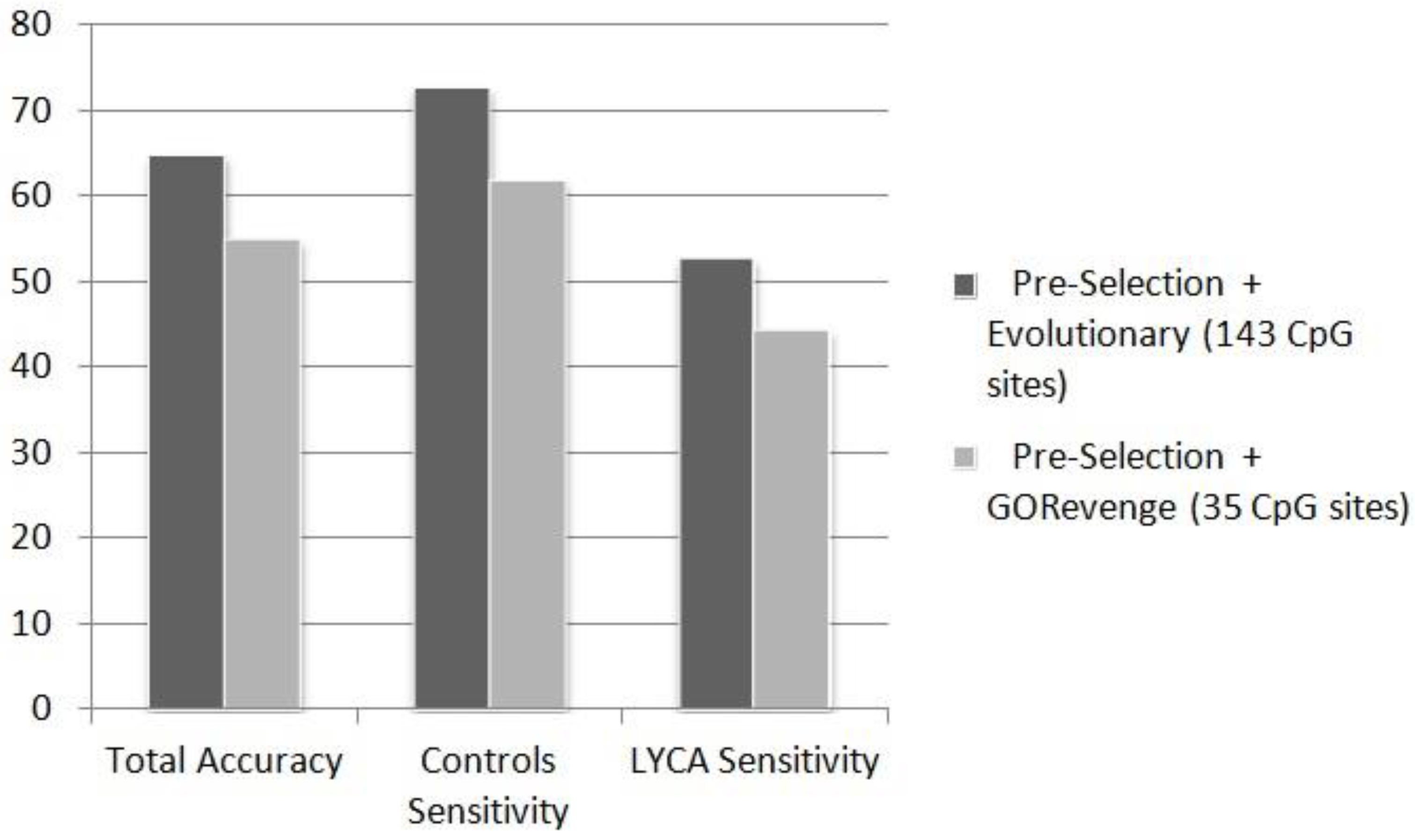
Figure 4 shows that for the case of the task controls
vs. LYCA, the subset of 143 CpGs provided by the evolutionary scheme (accuracy: 64.84%, controls sensitivity: 72.73%, cases sensitivity: 52.78%) outperforms the subset of 35 CpG sites, much lower in dimensionality though, provided by GORevenge (accuracy: 54.95%, controls sensitivity: 61.82%, cases sensitivity: 44.44%). The task of recognizing B-cell lymphoma samples from controls prove quite more difficult than discriminating breast cancer samples from controls. This can be, again, attributed to the variety of lymphatic cancers with diverse molecular mechanism compared to the small number of B-cell lymphoma samples exploited to train the constructed classifiers. The two-class task of discriminating breast cancer from B-cell lyphoma in the unknown testing set shows that this task is of moderate complexity and classifiers provide an exceptional performance, almost equal to 100% (
Table 6). This is very important as regards the practical utility of the use of this signature for diagnostic purposes, because it manages to mark off the fundamentally different, underlying, molecular underpinnings of these two cancer pathologies, confirming the profoundly different histological nature which, indeed, sets wide apart breast from hematological neoplastic pathologies.
Figure 4.
Performance (%) of ANN in the totally unknown-independent testing set in the two-class problem (controls vs. LYCA), fed by CpG sites subsets selected by the selection schemes applied.
Figure 4.
Performance (%) of ANN in the totally unknown-independent testing set in the two-class problem (controls vs. LYCA), fed by CpG sites subsets selected by the selection schemes applied.
The fact that the performances of the evolutionary selection scheme and GORevenge-based selection are close suggests that the fusion of these orthogonal methodologies might be a promising avenue, in order that one derives biomarkers sets, with both a high classification performance and a biological interpretive value. Experiments not reported here showed that cluster analysis could not provide a visually satisfying separation of the various samples classes (See
Figure S1 for clustering controls, BCCA, and LYCA samples using the 352 CpG sites selected by GORevenge). This can be considered an asset for the more sophisticated artificial intelligence methods applied here, that can capture more complex relations.Another approach that we plan to adopt in the near future towards improving the classification performances is the incorporation of bagging schemes, through which we will endeavor to integrate different top-performing classifiers.
Several subsets of CpG sites were provided here by the selection schemes applied that can be used to distinguish (one or two) cancer types from control samples, or one cancer type from another.The subsets of 373 and 352 CpG sites as proposed by the evolutionary feature selection and the GORevenge-based selection to be used for discriminating all three phenotypic classes used here, i.e., controls, breast cancer, and B-cell lymphoma, comprise promising candidates for DNA methylation biomarkers for the two cancer types studied here. These two subsets share a common part of 25 CpG sites whose biological impact is to be studied. Since selected subsets of CpG sites are currently undergoing further biological investigation, in order to highlight putative, causative, theoretical, molecular networks predisposing for disease onset and progression, CpGs sites, corresponding CpG sites are not reported here.
The aim of this study is to examine the suitability, from an epidemiological perspective, of the goal to predict manifestation of two cancer types (breast cancer and B-cell lymphoma) through examination of genome-scalemeasurements of the DNA methylation observed in blood samples of human donors who, at the time of collection, were healthy. In this sense, the derivation of early molecular predictive disease biomarkers, probably years before the macroscopic observation of the two globally-serious epidemiological threats represents a grand challenge. Despite the fact that these diseases possess a diverse phenotypic profile, encompassing very diverse combinations of molecular phenotypes (groups of related genes), the proposed biologically-inspired strategy of selecting biomarkers based on functional analysis yields promising results. This was shown even in the case of the blindfold validation, thus suggesting its expediency as a reliable avenue for robust biomarker discovery.
Towards enlightening the biological content beneath the selected lists of CpG sites for the three-class problem, we have initially performed GO enrichment analysis using the StRAnGER2 algorithm (Available online:
www.grissom.gr/stranger2, [
43]). Top ten results of the enrichment analysis for the Biological Process category for genes corresponding to the 352 CpG sites derived from the GORevenge-based selection and genes corresponding to the 373 CpGs selected by the evolutionary selectionare presented in
Table 12a,b, respectively. In spite of the small overlap between the two CpG lists, both sets of results suggest an epigenetic impact at the level of growth and development-related genes and, more particularly, related to axon guidance and the development of the neuronal system. However, the GORevenge selection captures more biologically-enriched ontological content, which is highlighted by the significantly lower hypergeometric
p-values and the higher gene enrichments. This finding provides adequate evidence for the plausibility of discovery strategies, which are shoring up in the diligent exploration of the wealth of functional information, regarding the role of genes in specific molecular mechanisms. For instance, GORevenge promoted CpG sites corresponding to 29 out of 345 axon guidance-related genes, whereas the GA derived only 15. Interestingly, the role of axon guidance molecules has been described in tumorigenesis, cancer progression and metastasis [
44] In this context, it has been shown that the role of Slit and Robo signaling in various cancer progressions (including breast cancer) is mediated through hypermethylation of their promoters (epigenetic inactivation) [
45,
46,
47]. Remark that the evolutionary selection promoted only Robo4-related CpG sites, whereas the GORevenge selection promoted both Robo and Slit genes (Robo2, Slit3) (see
Table S10 for full lists of GO terms and related genes resulting from the two lists of CpG sites).
Table 13 reports the genomic distribution of the 352 CpGs sites derived from the GORevenge-based selection in different groups: promoter, body, 3′UTR, and intergenic. It is shown that the majority of selected CpG sites are located in the promoter region
Furthermore, the genes related to selected CpG sites were mapped to canonical pathways by employing KEGG Pathway enrichment analysis using StRAnGER2. Results presented in
Table 14 clearly show that the CpG promoted by GORevenge are strongly mapped to well-established cancer-related pathways, in contrast to the evolutionary selection, which yielded poor enrichments and not obviously related to cancer molecular phenotypes. For instance, deregulated Hippo signaling is frequently observed in human cancers as its deregulation leads to a concurrent combination of uncontrolled cellular proliferation and inhibition of apoptosis [
48]. In addition, the Wnt and TGFb signaling pathways have been shown to cross-talk with Hippo [
48,
49] and their signaling is deregulated in many human cancers [
50,
51]. Lastly, the PI3K-Akt signaling pathway has been shown to be inhibited by Robo1 signaling [
46] with a positive impact in breast cancer survival outcomes (see
Table S11 in Supplementary Material for lists of KEGG pathways and related genes resulting from the two lists of CpG sites).
Table 12.
Top enriched GO terms (analysis using the biological process category) derived by genes corresponding to CpG sites selected by GoRevenge-based selection (a) and genes corresponding to CpG sites selected by evolutionary selection (b). (a) Using GORevenge results; and (b) using evolutionary selection results.
Table 12.
Top enriched GO terms (analysis using the biological process category) derived by genes corresponding to CpG sites selected by GoRevenge-based selection (a) and genes corresponding to CpG sites selected by evolutionary selection (b). (a) Using GORevenge results; and (b) using evolutionary selection results.
| | GO ID | GO Description | p-Value | Enrichment |
|---|
| (a) |
| 1 | GO:0048709 | oligodendrocyte differentiation | 5.52 × 10−13 | 9/21 |
| 2 | GO:0044281 | small molecule metabolic process | 1.50 × 10−12 | 66/1530 |
| 3 | GO:0007411 | axon guidance | 1.92 × 10−12 | 29/345 |
| 4 | GO:0046777 | protein amino acid autophosphorylation | 1.93 × 10−12 | 21/171 |
| 5 | GO:0051216 | cartilage development | 3.58 × 10−12 | 14/90 |
| 6 | GO:0030900 | forebrain development | 3.86 × 10−12 | 15/87 |
| 7 | GO:0009790 | embryo development | 3.97 × 10−12 | 24/154 |
| 8 | GO:0007420 | brain development | 4.82 × 10−12 | 27/225 |
| 9 | GO:0001701 | in utero embryonic development | 5.75 × 10−12 | 36/256 |
| 10 | GO:0048011 | nerve growth factor receptor signaling pathway | 5.89 × 10−12 | 36/286 |
| (b) |
| 1 | GO:0033603 | positive regulation of dopamine secretion | 3.44 × 10−7 | 3/6 |
| 2 | GO:0016458 | gene silencing | 7.95 × 10−7 | 3/7 |
| 3 | GO:0007411 | axon guidance | 7.50 × 10−6 | 15/345 |
| 4 | GO:0035249 | synaptic transmission, glutamatergic | 8.03 × 10−6 | 4/23 |
| 5 | GO:0045662 | negative regulation of myoblast differentiation | 1.07 × 10−5 | 3/12 |
| 6 | GO:0030900 | forebrain development | 1.29 × 10−5 | 7/87 |
| 7 | GO:0043523 | regulation of neuron apoptosis | 1.85 × 10−5 | 4/27 |
| 8 | GO:0001501 | skeletal system development | 2.14 × 10−5 | 9/152 |
| 9 | GO:0031069 | hair follicle morphogenesis | 2.23 × 10−5 | 4/28 |
| 10 | GO:0048663 | neuron fate commitment | 2.66 × 10−5 | 4/29 |
Table 13.
Genomic distribution of 352 CpGs derived from the GORevenge-based selection, classified in different groups: promoter, body, 3′UTR, and intergenic.
Table 13.
Genomic distribution of 352 CpGs derived from the GORevenge-based selection, classified in different groups: promoter, body, 3′UTR, and intergenic.
| CpG Location | CpGs | Subgroup | CpGs |
|---|
| Promoter | 186 | TSS200 | 41 |
| TSS1500 | 82 |
| 5′UTR | 36 |
| 1stExon | 27 |
| Body | 147 | - | - |
| 3′UTR | 19 | - | - |
| Intergenic | - | - | - |
Table 14.
Enriched KEGG pathways derived by genes corresponding to CpG sites selected by GoRevenge-based selection (a) and genes corresponding to CpG sites selected by evolutionary selection (b). (a) Using GoRevenge results; and (b) using evolutionary selection results.
Table 14.
Enriched KEGG pathways derived by genes corresponding to CpG sites selected by GoRevenge-based selection (a) and genes corresponding to CpG sites selected by evolutionary selection (b). (a) Using GoRevenge results; and (b) using evolutionary selection results.
| | Pathway ID | Pathway Description | p-Value | Enrichment |
|---|
| (a) |
| 1 | hsa05205 | Proteoglycans in cancer | 1.37 × 10−12 | 24/222 |
| 2 | hsa05215 | Prostate cancer | 2.58 × 10−12 | 17/88 |
| 3 | hsa04390 | Hippo signaling pathway | 5.86 × 10−12 | 20/154 |
| 4 | hsa04910 | Insulin signaling pathway | 9.91 × 10−12 | 20/136 |
| 5 | hsa05200 | Pathways in cancer | 2.39 × 10−11 | 44/326 |
| 6 | hsa05166 | HTLV-I infection | 3.52 × 10−11 | 25/263 |
| 7 | hsa04020 | Calcium signaling pathway | 4.52 × 10−11 | 25/181 |
| 8 | hsa04916 | Melanogenesis | 5.73 × 10−11 | 15/99 |
| 9 | hsa04010 | MAPK signaling pathway | 6.98 × 10−11 | 27/257 |
| 10 | hsa04722 | Neurotrophin signaling pathway | 1.18 × 10−10 | 16/118 |
| 11 | hsa04151 | PI3K-Akt signaling pathway | 1.22 × 10−10 | 27/341 |
| 12 | hsa04310 | Wnt signaling pathway | 2.82 × 10−10 | 17/143 |
| 13 | hsa05217 | Basal cell carcinoma | 3.93 × 10−10 | 11/55 |
| 14 | hsa04510 | Focal adhesion | 3.97 × 10−10 | 20/204 |
| 15 | hsa04350 | TGF-beta signaling pathway | 4.18 × 10−10 | 13/81 |
| 16 | hsa05202 | Transcriptional misregulation in cancer | 4.78 × 10−10 | 18/165 |
| 17 | hsa00053 | Ascorbate and aldarate metabolism | 7.52 × 10−10 | 8/26 |
| 18 | hsa05030 | Cocaine addiction | 2.11 × 10−9 | 10/50 |
| 19 | hsa00500 | Starch and sucrose metabolism | 3.30 × 10−9 | 10/52 |
| (b) |
| 1 | hsa04260 | Cardiac muscle contraction | 9.81 × 10−5 | 6/76 |
| 2 | hsa04974 | Protein digestion and absorption | 2.29 × 10−4 | 6/87 |
| 3 | hsa05410 | Hypertrophic cardiomyopathy (HCM) | 1.28 × 10−3 | 5/85 |
| 4 | hsa05414 | Dilated cardiomyopathy | 1.72 × 10−3 | 5/90 |
| 5 | hsa00061 | Fatty acid biosynthesis | 2.86 × 10−3 | 1/6 |
| 6 | hsa05412 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 3.70 × 10−3 | 4/73 |
| 7 | hsa00460 | Cyanoamino acid metabolism | 3.97 × 10−3 | 1/7 |
| 8 | hsa04961 | Endocrine and other factor-regulated calcium reabsorption | 4.98 × 10−3 | 3/49 |
| 9 | hsa05030 | Cocaine addiction | 5.36 × 10−3 | 3/50 |
| 10 | hsa04512 | ECM-receptor interaction | 7.41 × 10−3 | 4/86 |
| 11 | hsa04730 | Long-term depression | 0.010 | 3/60 |
Overall, the results of the enrichment analysis clearly show that the semantics-driven selection by GORevenge promotes CpG sites with enriched biological content, while it preserves the accuracy of the classification. Whereas this approach is limited by the depth of ontological annotation, it enables the derivation of more relevant biomarkers with respect to underlying molecular phenotypes. The performance of this methodology is expected to be constantly improving by the expansion of ontological gene annotations. Thus, the proposed methodology may provide a tool for more effective, semantics-driven targeting of molecular candidates for clinical validation, bridging the gap between the genome-scale studies on large cohorts and the smaller, more focused projects for specific clinical applications (ex. subtyping cancers). In this scope, the proposed methodology could also enhance the exploitation of large publicly available databases (The Cancer Genome Atlas, the Encyclopedia of DNA Elements Consortium and the NIH Roadmap Epigenomics Mapping Consortium) which comprise, in addition to methylation data, gene expression and pathway data.These publicly-available data have only partially been exploited and the application of advanced mining techniques is required for their adaptation to answer more specific questions, in regard to diagnostic and prognostic applications [
52].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



