QSAR Models for CXCR2 Receptor Antagonists Based on the Genetic Algorithm for Data Preprocessing Prior to Application of the PLS Linear Regression Method and Design of the New Compounds Using In Silico Virtual Screening

Abstract
:1. Introduction
2. Data and Methods
2.1. Data Set
2.2. Computer Hardware and Software
2.3. Structural Descriptors
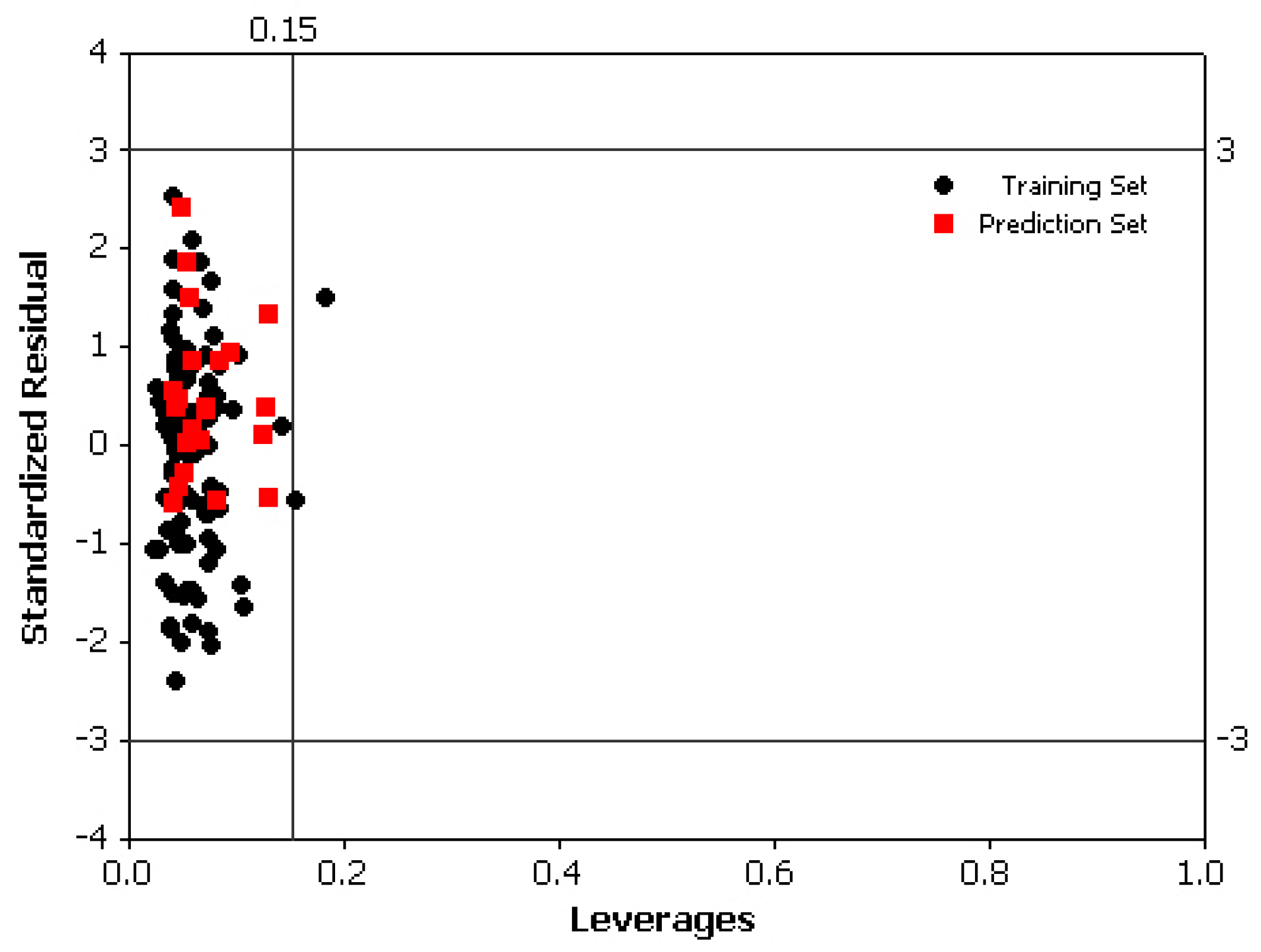
2.4. Model Validation
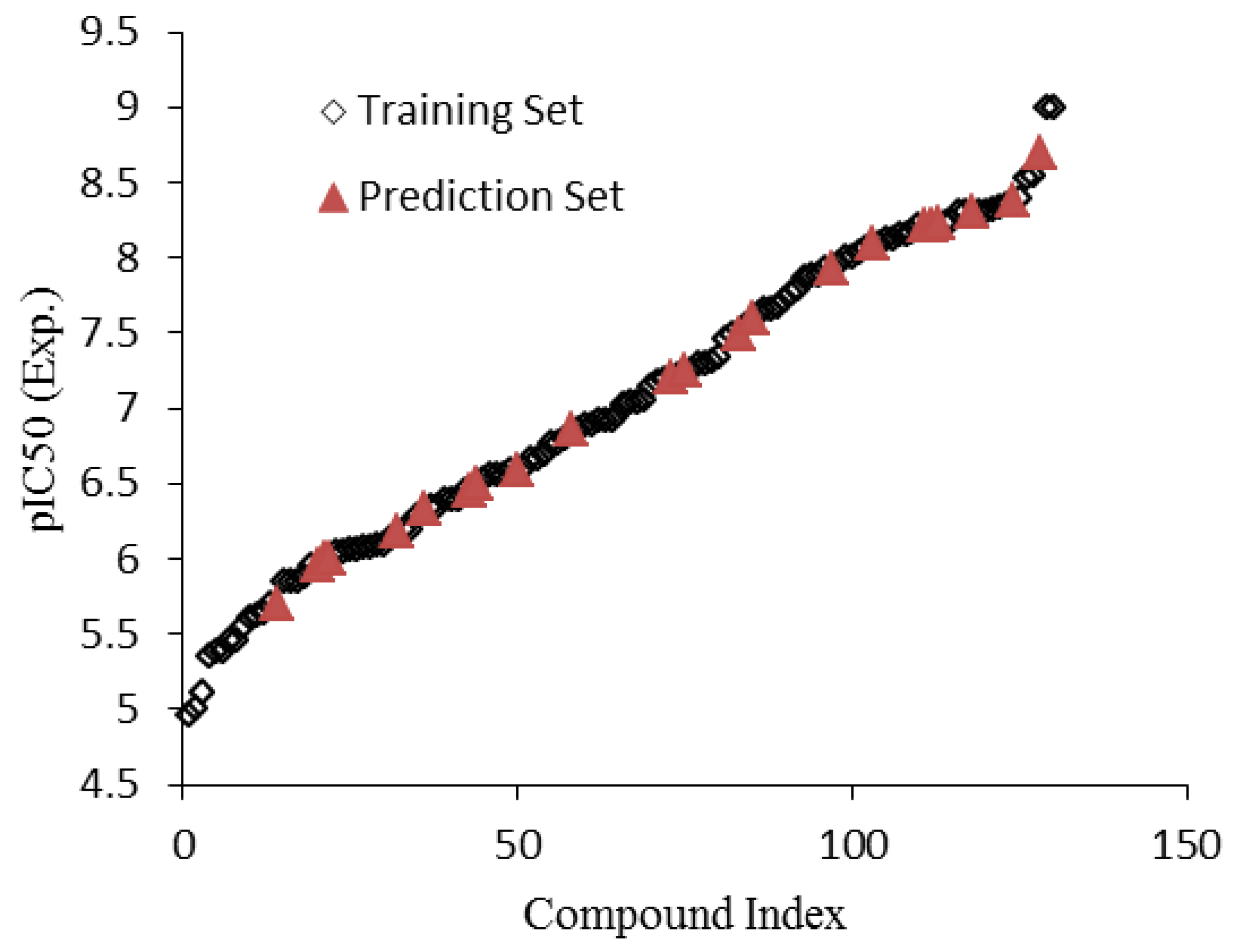
3. Results and Discussion
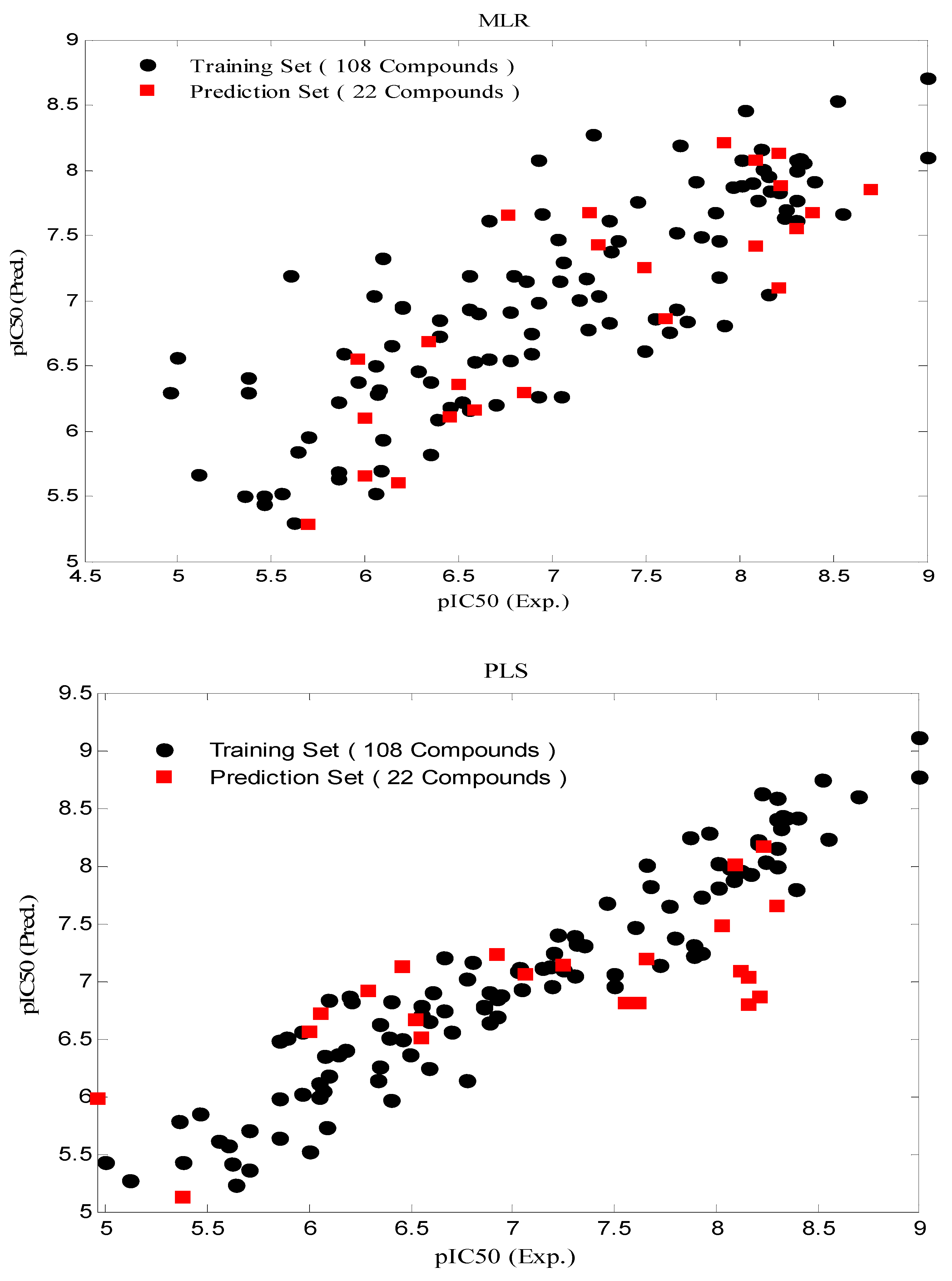
3.1. Stepwise Multiple Linear Regression (MLR)
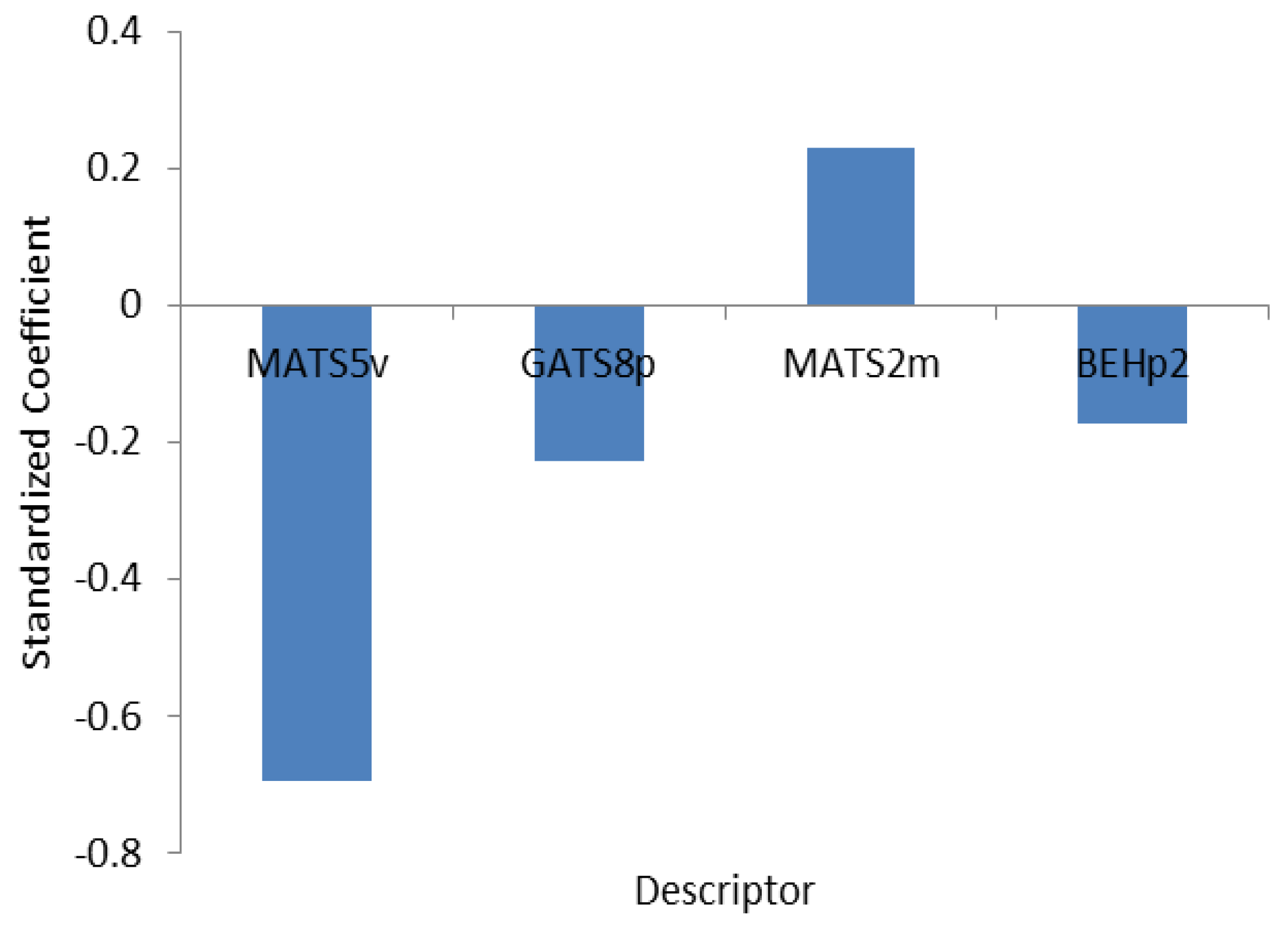
3.2. Interpretation of the Selected Descriptors
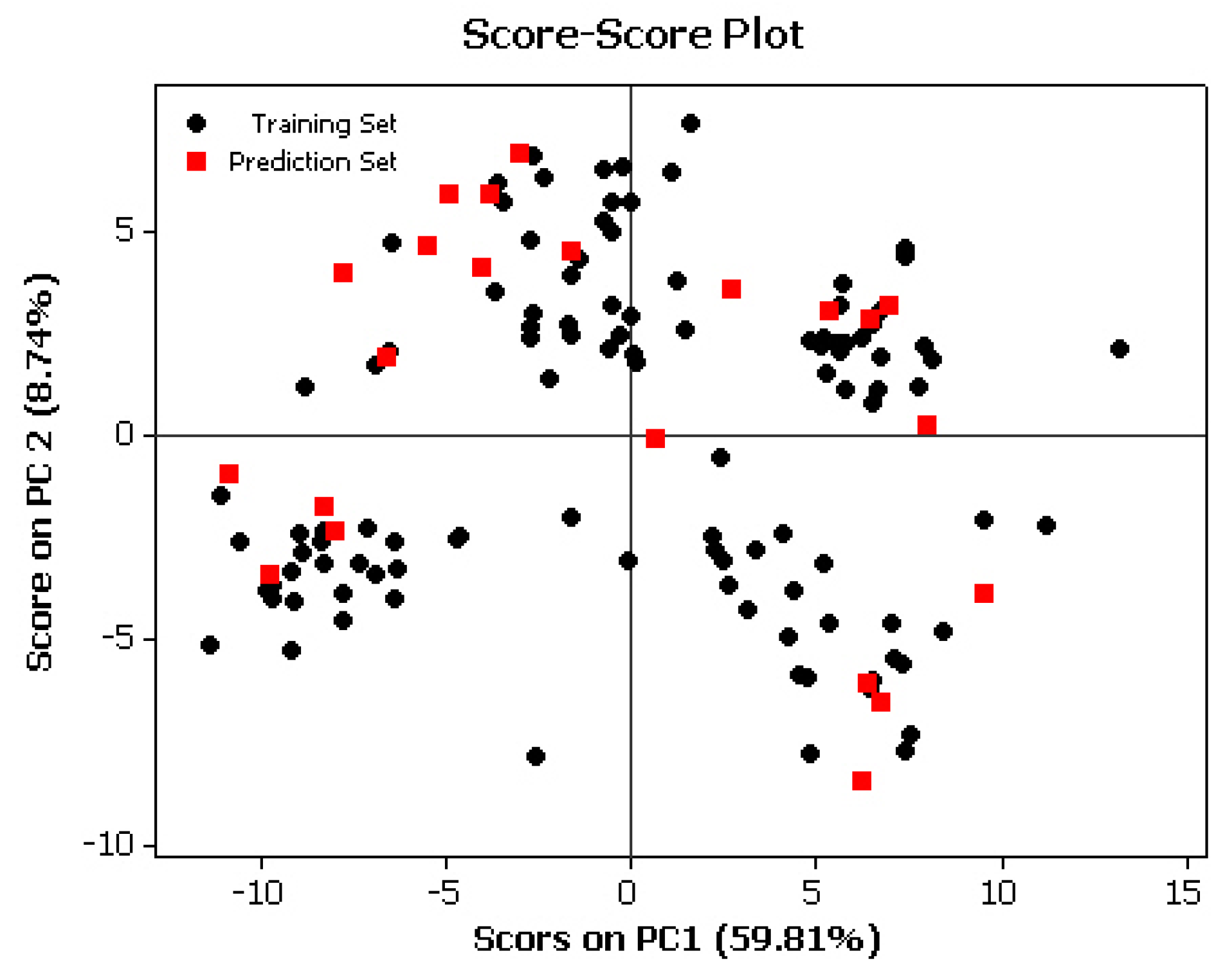
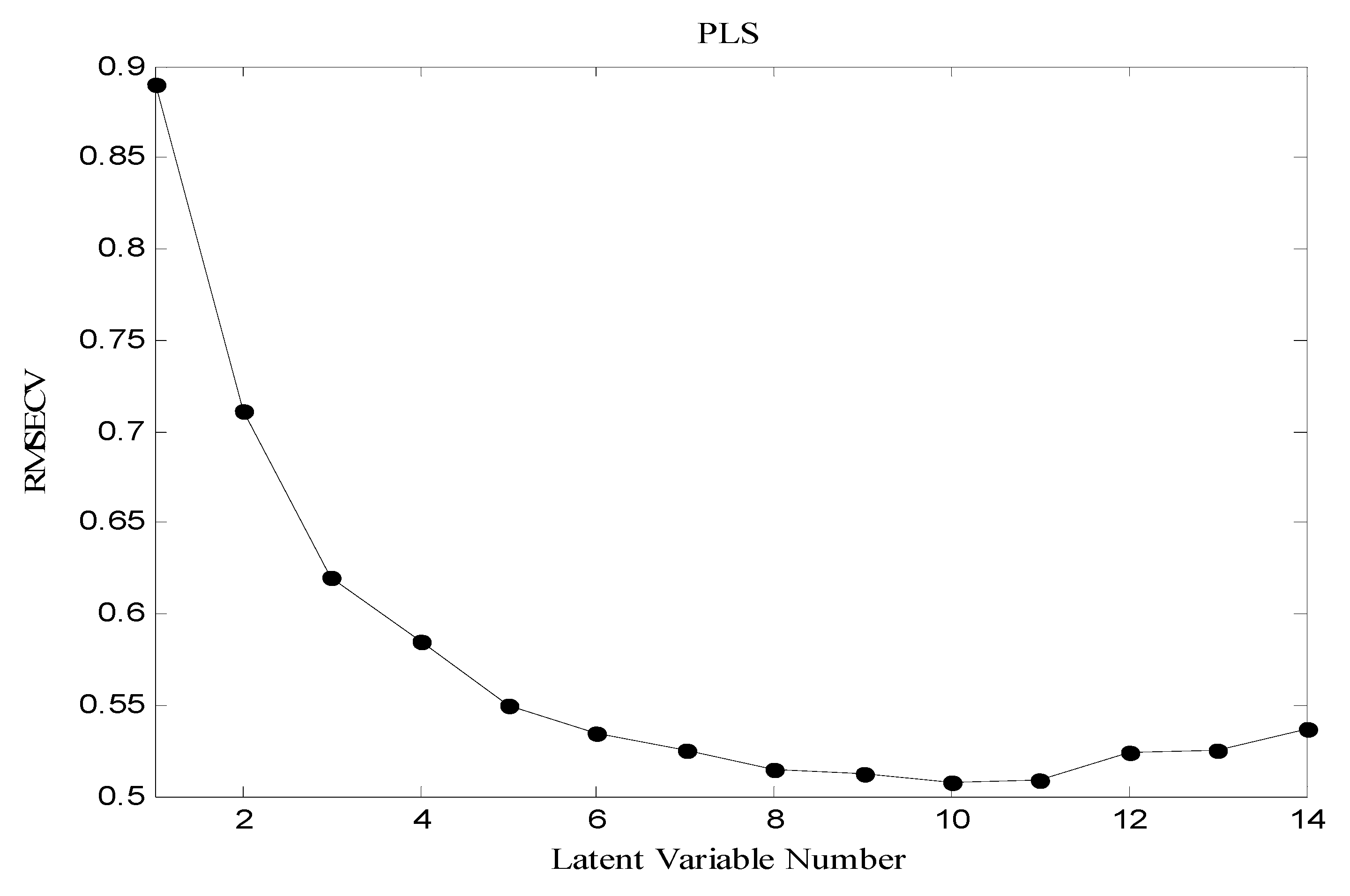
3.3. Partial Least Squares (PLS)
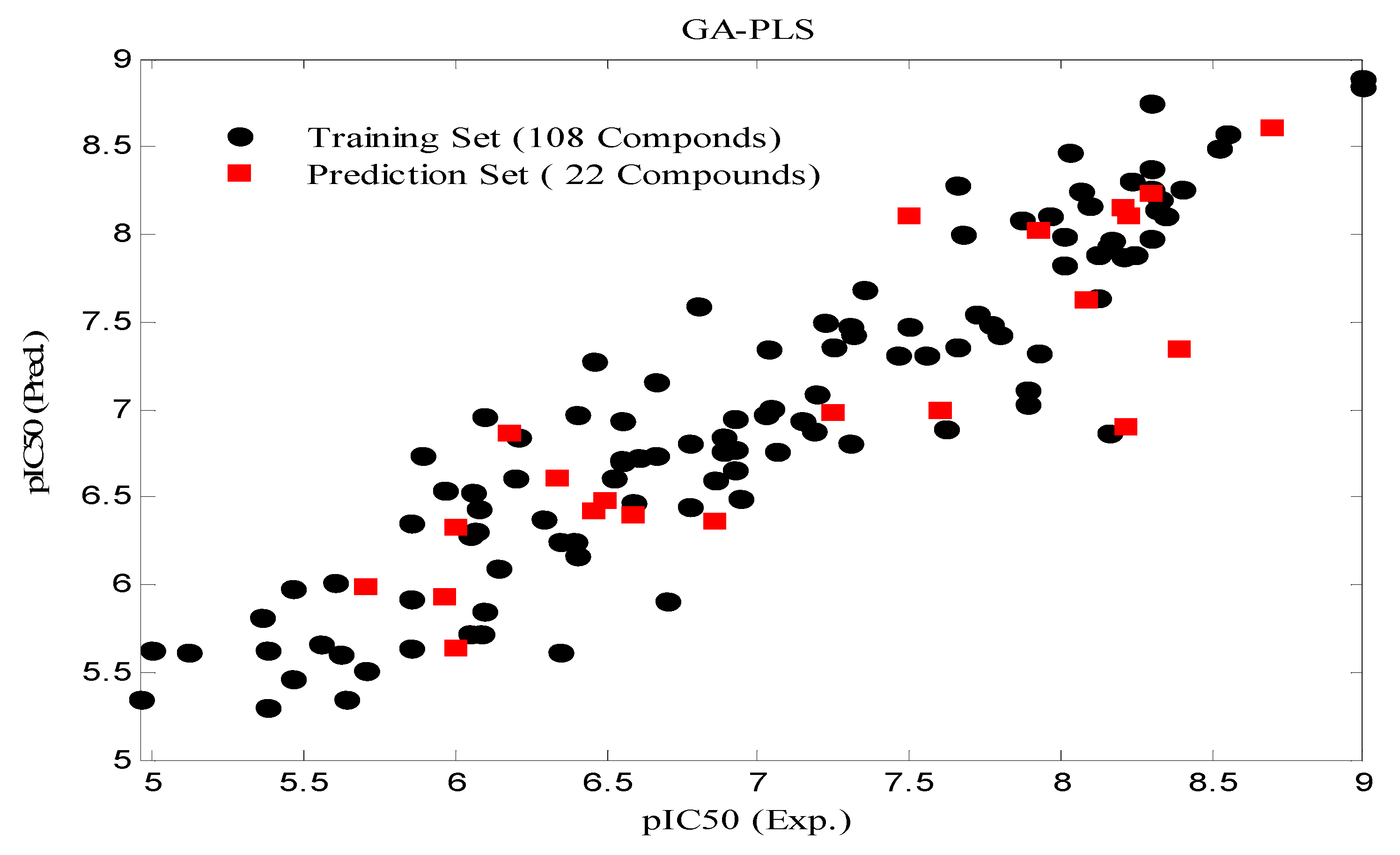
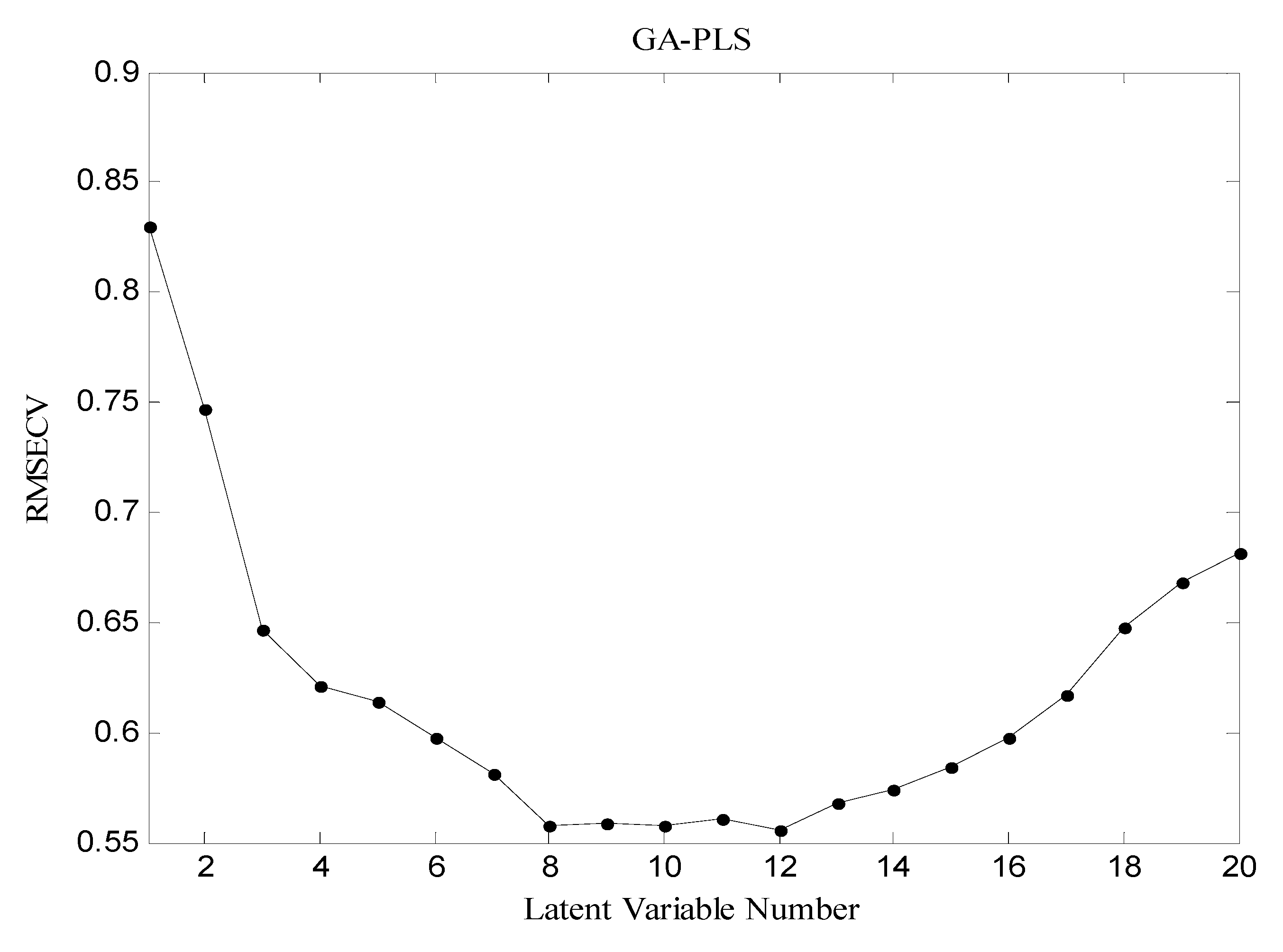
3.4. Partial Least Squares combined with Genetic Algorithm (GA-PLS)
3.5. In Silico Screening
4. Conclusions
References
- Holmes, W.E.; Lee, J.; Kuang, W.J.; Rice, G.C.; Wood, W.I. Structure and functional expression of a human interleukin-8 receptor. Science 1991, 253, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M.; Tiffany, H.L. Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 1991, 253, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M.; Baggiolini, M.; Charo, I.F.; Hebert, C.A.; Horuk, R.; Matsushima, K.; Miller, L.H.; Oppenheim, J.J.; Power, C.A. International Union of Pharmacology. XXII. Nomenclature for Chemokine Receptors. Pharmacol. Rev. 2000, 52, 145–176. [Google Scholar] [PubMed]
- Loetscher, P.; Seitz, M.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. Both interleukin-8 receptors independently mediate chemotaxis: Jurkat cells transfected with IL-8R1 or IL-8R2 migrate in response to IL-8, GROα and NAP-2. FEBS Lett. 1994, 341, 187–192. [Google Scholar] [CrossRef]
- Ahuja, S.K.; Lee, J.C.; Murphy, P.M. The CXC chemokines growth-regulated oncogene (GRO) α, GROβ, GROγ, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human Interleukin-8 Receptor. J. Biol. Chem. 1996, 271, 20545–20550. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Allegretti, M.; Bitondo, R. Di; Cervellera, M. N.; Collota, F.; Bertini, R. Pharmacological inhibition of Interleukin-8 (CXCL8) as a new approach for the prevention and treatment of several human diseases. Curr. Med. Chem. Anti-inflamm. Anti-Allergy Agents 2003, 2, 67–79. [Google Scholar] [CrossRef]
- Busch-Petersen, J. Small molecule antagonists of the CXCR2 and CXCR1 chemokine receptors as therapeutic agents for the treatment of inflammatory diseases. Curr. Med. Chem. 2006, 6, 1345–1352. [Google Scholar] [CrossRef]
- Ribeiro, F.A.L.; Ferreira, M.M.C. QSPR models of boiling point, octanol-water partition coefficient and retention time index of polycyclic aromatic hydrocarbons. J. Mol. Struct. Theochem. 2003, 663, 109–126. [Google Scholar] [CrossRef]
- Molfetta, F.A.; Bruni, A.T.; Rosseli, F.P.; Silva, A.B.F. A partial least squares and principal component regression study of quinone compounds with trypanocidal activity. Struct. Chem. 2007, 18, 49–57. [Google Scholar] [CrossRef]
- Tong, W.; Hong, H.; Xie, Q.; Shi, L.; Fang, H.; Perkins, R. Assessing QSAR limitations-A regulatory perspective. Curr. Comput. Aided Drug Des. 2005, 1, 195–205. [Google Scholar] [CrossRef]
- He, L.; Jurs, P.C. Assessing the reliability of a QSAR model’s predictions. J. Mol. Graphics Model. 2005, 23, 503–523. [Google Scholar] [CrossRef] [PubMed]
- Ghafourian, T.; Cronin, M.T.D. The impact of variable selection on the modelling of oestrogenicity. SAR QSAR Environ. Res. 2005, 16, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Shahlaei, M.; Fassihi, A.; Saghaie, L. Application of PC-ANN and PC-LS-SVM in QSAR of CCR1 antagonist compounds: A comparative study. Eur. J. Med. Chem. 2010, 45, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Aher, Y.D.; Agrawal, A.; Bharatam, P.V.; Garg, P. 3D-QSAR studies of substituted1-(3,3-diphenylpropyl)-piperidinyl amides and ureas as CCR5 receptor antagonists. J. Mol. Model. 2007, 13, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Afantitis, A.; Melagraki, G.; Sarimveis, H.; Koutentis, P.A.; Markopoulosd, J.; Igglessi-Markopoulou, O. Investigation of substituent effect of 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides on CCR5 binding affinity using QSAR and virtual screening techniques. J. Comput. Aided Mol. Des. 2006, 20, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Afantitis, A.; Melagraki, G.; Sarimveis, H.; Igglessi-Markopoulou, O.; Kollias, G. A novel QSAR model for predicting the inhibition of CXCR3 receptor by 4-N-aryl-[1,4] diazepane ureas. Eur. J. Med. Chem. 2009, 44, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Bhonsle, J.B.; Wang, Z.X.; Tamamura, H.; Fujii, N.; Peiper, S.C.; Trent, J.O. A simple, automated Quasi-4D-QSAR, Quasi-multi way PLS approach to develop highly predictive QSAR models for highly flexible CXCR4 inhibitor cyclic pentapeptide ligands using scripted common molecular modeling tools. QSAR Comb. Sci. 2005, 24, 620–630. [Google Scholar] [CrossRef]
- Khelebnikov, A.I.; Schepetkin, I.A.; Quinn, M.T. Quantitative structure activity relationships for small non- peptide antagonistsof CXCR2: Indirect 3D approach using the frontal polygon method. Bioorg. Med. Chem. Lett. 2006, 14, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, J.B.; Zohrabi, P.; Khajehsharifi, H. Quantitative structure-activity relationship study of nonpeptide antagonists of CXCR2 using stepwise multiple linear regression analysis. Monatsh. Chem. 2010, 141, 111–118. [Google Scholar] [CrossRef]
- Yu, Y.; Dwyer, M.P.; Chao, J.; Aki, C.; Chao, J.; Purakkattle, B.; Rindgen, D.; Bond, R.; Mayer-Ezel, R.; kway, J.; et al. Synthesis and structure-activity relationships of heteroaryl substituted-3,4-diamino-3-cyclobut-3-ene-1,2-dione CXCR2/CXCR1 receptor antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Winters, M.P.; Crysler, C.; Subasinghe, N.; Ryan, D.; Leong, L.; Zhao, S.; Donatelli, R.; Yurkow, E.; Mazzulla, M.; Boczon, L.; et al. Carboxylic acid bioisosteres acylsulfonamides, acylsulfamides, and sulfonylureas as novel antagonists of the CXCR2 receptor. Bioorg. Med. Chem. Lett. 2008, 18, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Walters, I.; Austin, C.; Austin, R.; Bonnert, R.; Cage, P.; Christie, M.; Ebden, M.; Gardiner, S.; Grahames, C.; Hill, S.; et al. Evaluation of a series of bicyclic CXCR2 antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Kennard, R.W.; Stone, L.A. Computer aided design of experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Melagraki, G.; Afantitis, A.; Makridima, K.; Sarimveis, H.; Igglessi-Markopoulou, O. Prediction of toxicity using a novel RBF neural network training methodology. J. Mol. Model. 2006, 12, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Walczak, B.; Massart, D.L.; Heuerding, S.; Erni, F.; Last, I.R.; Prebble, K.A. Artificial neural networks in classification of NIR spectral data: design of the training set. Chemometr. Intell. Lab. Syst. 1996, 33, 35–46. [Google Scholar] [CrossRef]
- Ghosh, P.; Ghosh, M.; Bagchi, M.C. On an aspect of calculated molecular descriptors in QSAR studies of quinolone antibacterials. Mol. Divers. 2006, 10, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Chakraborti, A.K.; Gopalakrishnan, B.; Sobhia, M.E.; Malde, A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors. Eur. J. Med. Chem. 2003, 38, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, V.K.; Sohgaura, R.; Khadikar, P.V. QSAR studies on biological activity of piritrexim analogues against pc DHFR. Bioorg. Med. Chem. 2002, 10, 2919–2926. [Google Scholar] [CrossRef]
- Todeschini, R. Milano Chemometrics, QSPR Group. http://michem.disat.unimib.it/chm/.
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiely-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Wold, S.; Eriksson, L. Chemometric Methods in Molecular Design; van de Waterbeemd, H., Ed.; VCH: Weinheim, Germany, 1995; pp. 312–317. [Google Scholar]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Leardi, R.; Boggia, R.; Terrile, M. Genetic algorithms as a strategy for feature selection. J. Chemometr. 1992, 6, 267–281. [Google Scholar] [CrossRef]
- Jalali, H.M.; Konuze, E. Use of quantitative structure property relationships in predicting the Kraft point of anionic surfactants. Int. Electron. J. Mol. Des. 2002, 1, 410–417. [Google Scholar]
- Acevedo-Martínez, J.; Escalona-Arranz, J.C.; Villar-Rojas, A.; Téllez-Palmero, F.; Pérez-Rosés, R.; González, L.; Carrasco-Velar, R. Quantitative study of the structure-retention index relationship in the imine family. J. Chromatogr. A 2006, 1102, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.J.; Wang, J.M.; Liao, N.; Xu, X.J. Application of Genetic algorithms on the structure-activity relationship analysis of some cinnamaides. J. Chem. Inf. Comput. Sci. 1999, 39, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K. GA strategy for variable selection in QSAR studies: application of GA-Based region selection to a 3D-QSAR study of acetylcholinesterase inhibitors. J. Chem. Inf. Comput. Sci. 1999, 39, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, H.C.; Olivieri, A.C. Wavelength selection for multivariate calibration using a genetic algorithm: A novel initialization strategy. J. Chem. Inf. Comput. Sci. 2001, 42, 1146–1153. [Google Scholar] [CrossRef]
- van de Waterbeemd, H. Chemometric Methods in Molecular Design, Methods and Principles in Medicinal Chemistry; Verlag Chemie: Weinheim, Germany, 1995; Volume 2. [Google Scholar]
- Rogers, D.; Hopfinger, A.J. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Hasegawa, K.; Kimura, T.; Funatsu, K. GA strategy for variable selection in QSAR studies: Enhancement of comparative molecular binding energy analysis by GA-based PLS method. Quant. Struct. Act. Relat. 1999, 18, 262–272. [Google Scholar] [CrossRef]
- Sagradoa, S.; Cronin, M.T.D. Application of the modelling power approach to variable subset selection for GA-PLS QSAR models. Anal. Chim. Acta 2008, 609, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A.; Golbraikh, A. Predictive QSAR modeling workflow model applicability domains and virtual screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Oloff, S. Advances in virtual screening. Drug Discov. Today Technol. 2006, 3, 405–411. [Google Scholar] [CrossRef]
- Melagraki, G.; Afantitis, A.; Sarimveis, H.; Koutentis, P.A.; Markopoulos, J.; Igglessi-Markopoulou, O. Optimization of biaryl piperidine and 4-amino-2-biarylurea MCH1 receptor antagonists using QSAR modeling, classification techniques and virtual screening. J. Comput. Aided Mol. Des. 2007, 21, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Melagraki, G.; Afantitis, A.; Sarimveis, H.; Koutentis, P. A.; Kollias, G. A.; Igglessi-Markopoulou, O. Predictive QSAR workflow for the in silico identification and screening of novel HDAC inhibitors. Mol. Divers. 2009, 13, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. Principles of QSAR models validation: internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Jaworska, J.S.; Nikolova, J.N.; Aldenberg, T. QSAR applicability domain estimation by projection of the training set in descriptor space: a review. ATLA Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | R3 | R4 | IC50 for CXCR2 (µM) | pIC50 |
|---|---|---|---|---|---|---|
| 1 | Me | CN | H | H | 0.07 | 7.14 |
| 2 | Me | Br | H | H | 0.17 | 6.77 |
| 3 | Et | CN | H | H | 0.06 | 7.19 |
| 4 | n-Pr | CN | H | H | 1.30 | 5.89 |
| 5 | Bn | CN | H | H | 1.40 | 5.85 |
| 6 | i-Pr | CN | H | H | 0.22 | 6.66 |
| 7 | Ph | CN | H | H | 0.26 | 6.58 |
| 8 | CF3 | CN | H | H | 0.09 | 7.06 |
| 9 | Me | CN | OMe | H | 0.16 | 6.80 |
| 10 | Me | CN | Me | H | 0.02 | 7.72 |
| 11 | Me | Br | - | - | 0.25 | 6.60 |
| 12 | Me | CN | - | - | 0.64 | 6.19 |
| 13 | Ph | Br | - | - | 0.12 | 6.92 |
| 14 | Ph | CN | - | - | 0.14 | 6.85 |
| 15 | o-Cl-Phenyl | CN | - | - | 0.40 | 6.40 |
| 16 | p-F-Phenyl | CN | - | - | 0.52 | 6.28 |
| 17 | Me | Me | H | - | 0.05 | 7.30 |
| 18 | Me | H | H | - | 0.12 | 6.92 |
| 19 | H | H | H | - | 0.07 | 7.18 |
| 20 | Et | Et | H | - | 1.10 | 5.96 |
| 21 | n-Butyl | H | H | - | 1.10 | 5.96 |
| 22 | Ph | H | H | - | 0.88 | 6.05 |
| 23 | -CH2CH2OMe | H | H | - | 0.26 | 6.58 |
| 24 | Me | Me | OMe | - | 0.06 | 7.24 |
| 25 | Me | Me | Me | - | 0.02 | 7.62 |

| Compound | R | IC50 CXCR2 (nM) | pIC50 |
|---|---|---|---|
| 26 | 5-H | 0.005 | 8.3 |
| 27 | 5-Me | 0.006 | 8.24 |
| 28 | 5-Et | 0.004 | 8.39 |
| 29 | 5-Br | 0.005 | 8.33 |
| 30 | 5-Cl | 0.005 | 8.32 |
| 31 | 5-CF3 | 0.017 | 7.76 |
| 32 | 5-CF2H | 0.007 | 8.17 |
| 33 | 5-CH2OH | 0.003 | 8.55 |
| 34 | 5-CH2N(Me)2 | 0.094 | 7.03 |
| 35 | 5-CON(Me)2 | 0.171 | 6.77 |
| 36 | 5-(20Cl)Ph | 0.049 | 7.31 |
| 37 | 5-(2-CF3)Ph | 0.15 | 6.82 |
| 38 | 5-(3-Cl)Ph | 0.058 | 7.24 |
| 39 | 5-(3-CF3)Ph | 0.087 | 7.06 |
| 40 | 4-Cl | 0.0045 | 8.35 |
| 41 | 4-Br | 0.005 | 8.30 |
| 42 | 4-(4-Pyridyl) | 0.009 | 8.02 |
| 43 | 4-(3-Thienyl) | 0.008 | 8.09 |
| 44 | 4-(3,5-Dimethyl-4-isoxazoyl) | 0.008 | 8.12 |
| 45 | 2,3-Benzofuran | 0.003 | 8.46 |
| 46 | 3-Br | 0.016 | 7.78 |
| 47 |  | 8.6 | 8.06 |
| 48 |  | 10.9 | 7.96 |
| 49 |  | 9.8 | 8.01 |
| 50 |  | 9.8 | 8.01 |
| 51 |  | 7.5 | 8.12 |
| 52 |  | 8.2 | 8.10 |
| 53 |  | 8.0 | 8.10 |
| 54 |  | 5.8 | 8.24 |
| 55 |  | 6.2 | 8.21 |
| 56 |  | 6.2 | 8.21 |
| 57 |  | 21 | 7.68 |
| 58 |  | 50 | 7.30 |

| Compound | R1 | R2 | R3 | R4 | R5 | R6 | IC50 for CXCR2 (nM) | pIC50 |
|---|---|---|---|---|---|---|---|---|
| 59 | OH | H | Cl | H | Br | H | 906 | 6.04 |
| 60 | OH | Cl | Cl | H | Br | H | 63 | 7.20 |
| 61 | OH | CONH2 | Cl | H | Br | H | 10 | 8.00 |
| 62 | OH | CH2NH2 | Cl | H | Br | H | 114 | 6.94 |
| 63 | OH | SO2NH2 | Cl | H | Br | H | 7 | 8.15 |
| 64 | OH | SO2NMe2 | Cl | H | Br | H | 12 | 7.92 |
| 65 | OH | H | CN | H | Br | H | 25 | 7.60 |
| 66 | OH | Br | CN | H | Br | H | 6 | 8.22 |
| 67 | OH | Cl | CN | H | Br | H | 22 | 7.66 |
| 68 | OH | CN | Cl | H | Br | H | 57 | 7.24 |
| 69 | OH | H | NO2 | H | Br | H | 22 | 7.66 |
| 70 | OH | H | NO2 | H | H | H | 320 | 6.49 |
| 71 | OH | NO2 | H | H | H | H | 860 | 6.07 |
| 72 | OH | H | H | NO2 | H | H | 10900 | 4.96 |
| 73 | OH | H | CN | H | H | H | 200 | 6.70 |
| 74 | OH | SO2NH2 | Cl | H | Cl | Cl | 9.3 | 8.03 |
| 75 | –N=N–NH– | CN | H | Br | H | 39 | 7.49 |

| Compound | R | IC50 for CXCR2 (nM) | pIC50 |
|---|---|---|---|
| 76 | -SO2C2H5 | 130 | 6.87 |
| 77 | -SO2CH(CH3)2 | 400 | 6.40 |
| 78 |  | 460 | 6.34 |
| 79 | -SO2C6H5 | 90 | 7.05 |
| 80 |  | 32 | 7.49 |
| 81 | -SO2CH2C6H5 | 280 | 6.55 |
| 82 | Cl | 1000 | 6.00 |

| Compound | R1 | R2 | IC50 for CXCR2 (nM) | pIC50 |
|---|---|---|---|---|
| 83 | C6H5CH2 | C6H5 | 2400 | 5.62 |
| 84 | 3-OHC6H4CH2 | C6H5 | 4400 | 5.36 |
| 85 | C6H5CH2 | 4-Pyridinyl | 7700 | 5.11 |
| 86 | C6H5CH2 | 2-Furanyl | 4200 | 5.38 |
| 87 | C6H5CH2 | 4-CNC6H4 | 3500 | 5.46 |
| 88 | C6H5CH2 | 3-CF3C6H4 | 3500 | 5.46 |
| 89 | C6H5CH2 | 4-CF3C6H4 | 2800 | 5.55 |
| 90 | C6H5CH2 | 4-CH3OC6H4 | 2300 | 5.64 |
| 91 | C6H5CH2 | 3,5-diClC6H3 | 2000 | 5.70 |
| 92 | C6H5CH2 | 2-Thienyl | 2000 | 5.70 |
| 93 | C6H5CH2 | 2-CH3C6H4 | 1400 | 5.85 |
| 94 | C6H5CH2 | 2-CH3OC6H4 | 1400 | 5.85 |
| 95 | C6H5CH2 | 3-ClC6H4 | 1000 | 6.00 |
| 96 | C6H5CH2 | 2-FC6H4 | 890 | 6.05 |
| 97 | C6H5CH2 | 4-ClC6H4 | 830 | 6.08 |
| 98 | C6H5CH2 | 3,4-diClC6H3 | 800 | 6.10 |
| 99 | C6H5CH2 | 2,5-diClC6H3 | 670 | 6.17 |
| 100 | C6H5CH2 | 2-ClC6H4 | 450 | 6.35 |
| 101 | C6H5CH2 | 2,4-diClC6H3 | 410 | 6.39 |
| 102 | C6H5CH2 | 2-BrC6H4 | 350 | 6.46 |
| 103 | C6H5CH2 | 2,3-diClC6H3 | 350 | 6.46 |
| 104 | 4- CH3OC6H4CH2 | 2,4-diClC6H3 | 10000 | 5.00 |
| 105 | 3-CH3OC6H4CH2 | 2,4-diClC6H3 | 4200 | 5.38 |
| 106 | 3-CH3C6H4CH2 | 2,4-diClC6H3 | 730 | 6.14 |
| 107 | 4-Cl C6H4CH2 | 2,4-diClC6H3 | 300 | 6.52 |
| 108 | 3-C6H5O C6H4CH2 | 2,4-diClC6H3 | 170 | 6.77 |
| 109 | 3-Cl C6H4CH2 | 2,4-diClC6H3 | 92 | 7.04 |
| 110 | 3-Cl C6H4CH2 | 2-ClC6H4 | 28 | 7.55 |
| Compound | IC50 for CXCR2 (nM) | pIC50 | |
|---|---|---|---|
| 111 |  | 160 | 6.80 |
| 112 |  | 4 | 8.40 |
| 113 |  | 13 | 7.89 |
| 114 |  | 630 | 6.20 |
| 115 |  | 7 | 8.15 |
| 116 |  | 280 | 6.55 |
| 117 |  | 140 | 6.85 |
| 118 |  | 280 | 6.55 |
| 119 |  | 850 | 6.07 |
| 120 |  | 5 | 8.30 |
| 121 |  | 350 | 6.46 |
| 122 |  | 16 | 7.80 |
| 123 |  | 2 | 8.70 |
| 124 |  | 45 | 7.35 |
| 125 |  | 2500 | 5.60 |
| 126 |  | 220 | 6.66 |

| Compound | R | IC50 for CXCR2 (nM) | pIC50 |
|---|---|---|---|
| 1a 10a |  | 3 1 | 8.52 9 |
| 1b 10b |  | 4 1 | 8.40 9.00 |
| 1c 10c |  | 13 2 | 7.89 8.70 |
| 1d 10d |  | 13 5 | 7.89 8.30 |
| 1e 10e |  | 35 5 | 7.46 8.30 |
| 1f 10f |  | 120 60 | 6.92 7.22 |
| Parameter | PLS | GA-PLS | SMLR |
|---|---|---|---|
| RMSEP | 0.50 | 0.51 | 0.56 |
| AREPred. | 5.98 | 5.53 | 1.3 |
| R2 | 0.748 | 0.779 | 0.78 |
| R2Training Set | 0.727 | 0.88 | 0.68 |
| Q2 | 0.68 | 0.713 | 0.66 |
| SEP | 0.50 | 0.51 | 0.53 |
| R2 − Ro2/R2 | −0.291 | −0.254 | −0.254 |
| K | 1.019 | 1.035 | 0.962 |
| Iteration | PLS | GA-PLS | ||
|---|---|---|---|---|
| R2 | Q2 | R2 | Q2 | |
| 1 | 0.0047 | −0.949 | 0.010 | −0.577 |
| 2 | 0.005 | −0.423 | 0.010 | −0.919 |
| 3 | 0.039 | −0.467 | 0.036 | −0.417 |
| 4 | 0.12 | −0.198 | 0.019 | −0.506 |
| 5 | 0.005 | −0.955 | 0.006 | −0.878 |
| 6 | 0.005 | −0.955 | 0.153 | −0.063 |
| 7 | 0.006 | −0.967 | 0.084 | −0.245 |
| 8 | 0.186 | −1.601 | 0.001 | −0.699 |
| 9 | 0.002 | −0.753 | 0.073 | −1.21 |
| 10 | 0.171 | −1.57 | 0.147 | −0.41 |
| pIC50 | MATS5v | GATS8p | MATS2m | BEHp2 | |
|---|---|---|---|---|---|
| pIC50 | 1 | ||||
| MATS5v | −0.26863 | 1 | |||
| GATS8P | −0.16055 | −0.00856 | 1 | ||
| MATS2m | 0.001149 | −0.08958 | −0.0286 | 1 | |
| BEHp2 | 0.214723 | −0.04342 | −05904 | 0.000615 | 1 |
| Descriptora | Coefficient | MFb |
|---|---|---|
| MATS5v | −8.9918 (±8.729) | −0.254 |
| GATS8P | −5.409 (±0.463) | −0.063 |
| MATS2m | −1.337 (±0.349) | 1.484 |
| BEHp2 | 31.527 (±7.936) | −0.166 |
| Constant | −3.539 (±1.156) |
| No. | pIC50 (Exp.) | PLS | GA-PLS | SMLR | |||
|---|---|---|---|---|---|---|---|
| pIC50 (Pred.) | Residual | pIC50 (Pred.) | Residual | pIC50 (Pred.) | Residual | ||
| 10 | 7.24 | 7.34 | 0.10 | 6.79 | −0.45 | 7.42 | 0.18 |
| 12 | 6.50 | 6.32 | −0.17 | 6.71 | 0.22 | 6.35 | −0.14 |
| 17 | 7.50 | 7.44 | −0.06 | 7.82 | 0.32 | 7.26 | −0.24 |
| 2 | 7.20 | 7.80 | 0.60 | 8.31 | 1.11 | 7.67 | 0.47 |
| 21 | 6.34 | 6.64 | 0.30 | 6.67 | 0.33 | 6.68 | 0.35 |
| 25 | 6.00 | 6.51 | 0.51 | 6.52 | 0.52 | 6.10 | 0.10 |
| 25a | 8.70 | 7.81 | −0.89 | 8.72 | 0.02 | 7.85 | −0.84 |
| 37b | 6.58 | 6.46 | −0.13 | 6.57 | −0.01 | 6.16 | −0.43 |
| 40 | 5.70 | 5.73 | 0.03 | 6.00 | 0.30 | 5.28 | −0.42 |
| 43 | 6.00 | 5.52 | −0.48 | 5.78 | −0.22 | 5.65 | −0.35 |
| 45b | 5.96 | 5.22 | −0.73 | 5.60 | −0.36 | 6.55 | 0.59 |
| 47 | 6.14 | 6.80 | 0.62 | 6.70 | 0.52 | 5.60 | −0.57 |
| 51 | 6.45 | 6.58 | 0.12 | 6.30 | −0.15 | 6.10 | −0.35 |
| 53b | 6.85 | 6.45 | −0.41 | 6.61 | −0.24 | 6.30 | −0.56 |
| 58c | 8.39 | 7.60 | −0.79 | 7.31 | −1.08 | 7.67 | −0.71 |
| 6 | 7.92 | 8.50 | 0.58 | 7.64 | −0.28 | 8.21 | 0.29 |
| ID | Definition | Group |
|---|---|---|
| 1 | RBN, RBF | Constitutional |
| 2 | D/D, J, MAXDN, MAXDP, X5, X0v, X1v, X3v, X4Av, X5Av, X0sol, X0sol, X1sol, X2sol, X3sol, X4sol, X5sol, S0K, S1K, IDDE, IVDE, SIC0, CIC0, IC1, SIC1, CIC1,IC2, BIC4, BIC5, D/Dr05, D/dr06, T(N..O), T(N..S), T(O..O) | Topological |
| 3 | BEHm1, BEHm2, BEHm3, BEHm4, BEHm5, BEHm6, BEHv6, BEHv7, BEHe3, BEHe4, BELe5, BELe6 | BUCUT |
| 4 | GGI2,GGI3,GGI10, JGI1 | Galvez topol. Charge indices |
| 5 | ATS8m, ATS8v, MATS5e, MTAS6e, GATS4e, GATS5e | 2D Autocorrelations |
| 6 | qnmax, Qpos | Charge descriptors |
| 7 | FDI, PJI3, DISPv, QYYv | Geometrical |
| 8 | RDF06u, RDF065u, RDF120u, RDF125u, RDF130u, RDF135u, RDF030m, RDF035m, RDF080m, RDF085m, RDF120m, RDF125m, RDF105v, RDF110v | RDF |
| 9 | Mor17u, Mor18u, Mor29u, Mor30u, Mor08m, Mor09m, Mor14m, Mor15m, Mor22m, Mor23m, Mor24m, Mor25m, Mor30m, Mor31m, Mor17v, Mor18v, Mor19v, Mor20v, Mor21v, Mor22v, Mor27v, Mor28v, Mor18e, Mor28e, Mor11p, Mor12p | 3D-MoRSE |
| 10 | E2u, E3u, E3e, G1p, G2p, E1p, L2s, L3s, G1s, G2s, Au, Am | WHIM |
| 11 | HIC, HGM, H3u, H4u, H3m, H4m, H7m, H8m, HATS2m, HATS3m, HATS1e, HATS2e, HATS7p, HATS8p, RARS, REIG, R5u, R6u, R3u+, R4u+, RTu+, R2m, RTm, R1m+, R8m+, RTm+, R1v, R2v, RTv, R1v+, R2e, R3e, RTp,R1p+ | GETAWAY |
| 12 | MR, PSA, MLOGP | Properties |
| Cross validation | Random subset |
| Number of subset | 4 |
| Window width | 2 |
| Initial term % | 20% |
| Maximum generation | 100 |
| Convergence (%) | 80 |
| Cross-over | Double |

| ID | X | Y | GA-PLS (pIC50 predicted) | Leverage-limit |
|---|---|---|---|---|
| 1c | H | Br | 7.10 | 0.07 |
| 2c | H | Cl | 5.63 | 0.05 |
| 3c | H | NO2 | 6.17 | 0.05 |
| 4c | H | OMe | 6.01 | 0.04 |
| 5c | H | Me | 5.50 | 0.03 |
| 6c | H | Et | 5.50 | 0.04 |
| 7c | Br | NO2 | 5.48 | 0.04 |
| 8c | Br | Me | 7.20 | 0.05 |
| 9c | Br | OMe | 6.67 | 0.04 |
| 10c | Br | Et | 8.50 | 0.06 |
| 11c | H | H | 6.49 | 0.04 |

| ID | X | GA-PLS (pIC50 predicted) | Leverage-limit |
|---|---|---|---|
| 10c | O | 8.50 | 0.04 |
| 2d | NH | 7.74 | 0.07 |
| 3d | NMe | 8.82 | 0.05 |
| 4d | NOH | 7.91 | 0.07 |
| 5d | NOMe | 8.42 | 0.06 |
| 6d | NNH2 | 7.99 | 0.06 |
| 7d | NNHMe | 8.39 | 0.05 |
| 8d | NNMe2 | 8.10 | 0.08 |
| 9d | S | 8.98 | 0.05 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Asadollahi, T.; Dadfarnia, S.; Shabani, A.M.H.; Ghasemi, J.B.; Sarkhosh, M. QSAR Models for CXCR2 Receptor Antagonists Based on the Genetic Algorithm for Data Preprocessing Prior to Application of the PLS Linear Regression Method and Design of the New Compounds Using In Silico Virtual Screening. Molecules 2011, 16, 1928-1955. https://doi.org/10.3390/molecules16031928
Asadollahi T, Dadfarnia S, Shabani AMH, Ghasemi JB, Sarkhosh M. QSAR Models for CXCR2 Receptor Antagonists Based on the Genetic Algorithm for Data Preprocessing Prior to Application of the PLS Linear Regression Method and Design of the New Compounds Using In Silico Virtual Screening. Molecules. 2011; 16(3):1928-1955. https://doi.org/10.3390/molecules16031928
Chicago/Turabian StyleAsadollahi, Tahereh, Shayessteh Dadfarnia, Ali Mohammad Haji Shabani, Jahan B. Ghasemi, and Maryam Sarkhosh. 2011. "QSAR Models for CXCR2 Receptor Antagonists Based on the Genetic Algorithm for Data Preprocessing Prior to Application of the PLS Linear Regression Method and Design of the New Compounds Using In Silico Virtual Screening" Molecules 16, no. 3: 1928-1955. https://doi.org/10.3390/molecules16031928