Molecular Modeling to Study Dendrimers for Biomedical Applications

 and
and
Abstract
:1. Introduction

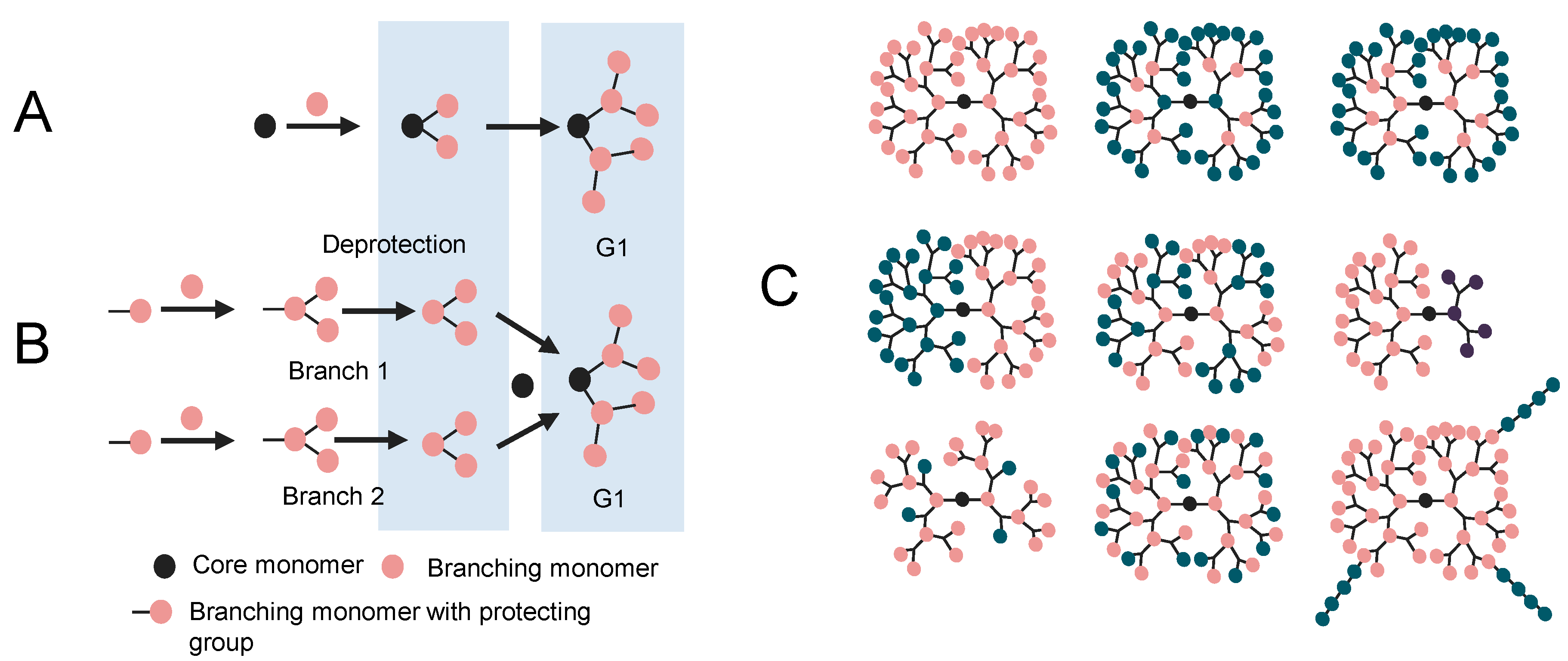
2. Molecular Modeling of Dendrimers


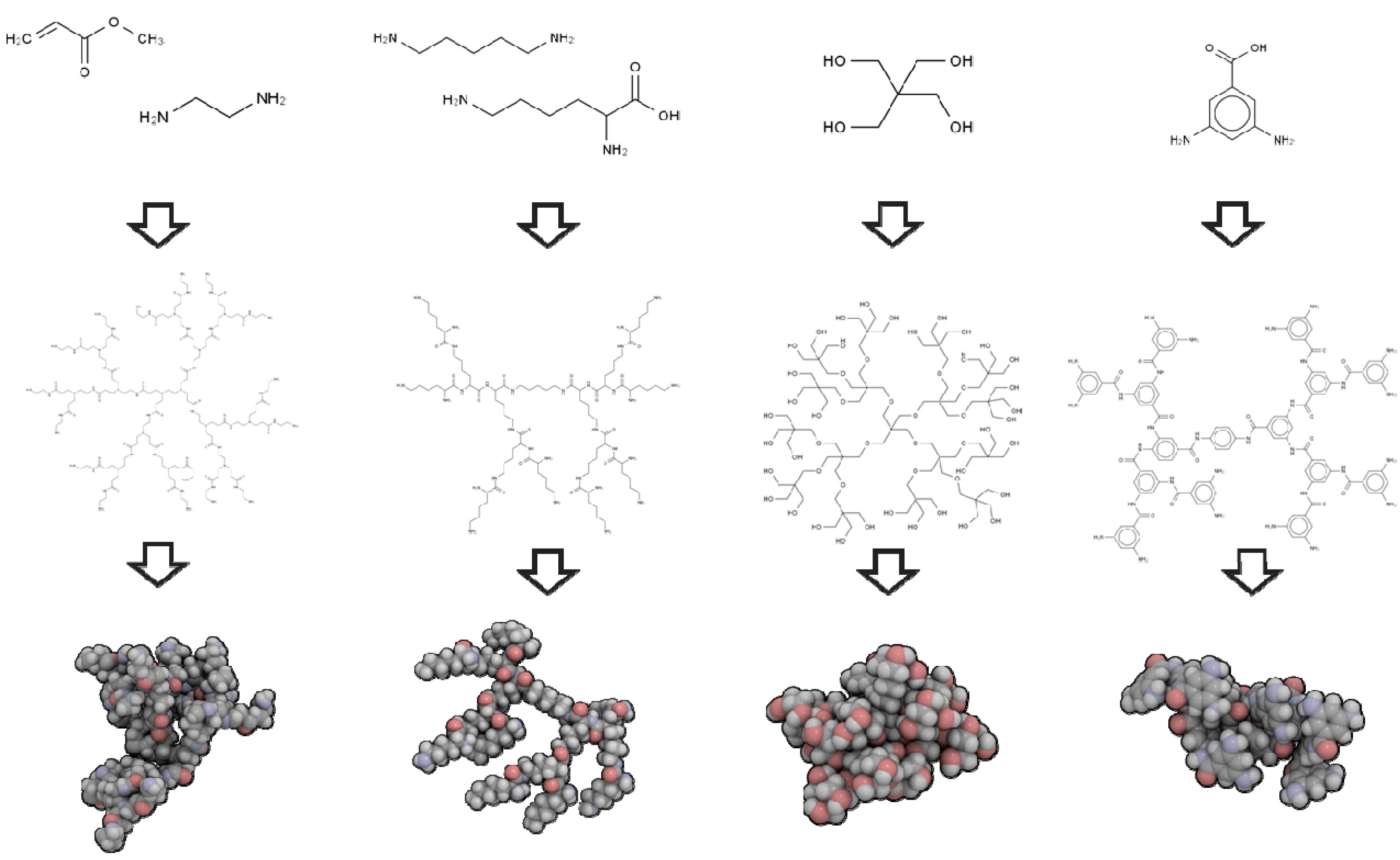
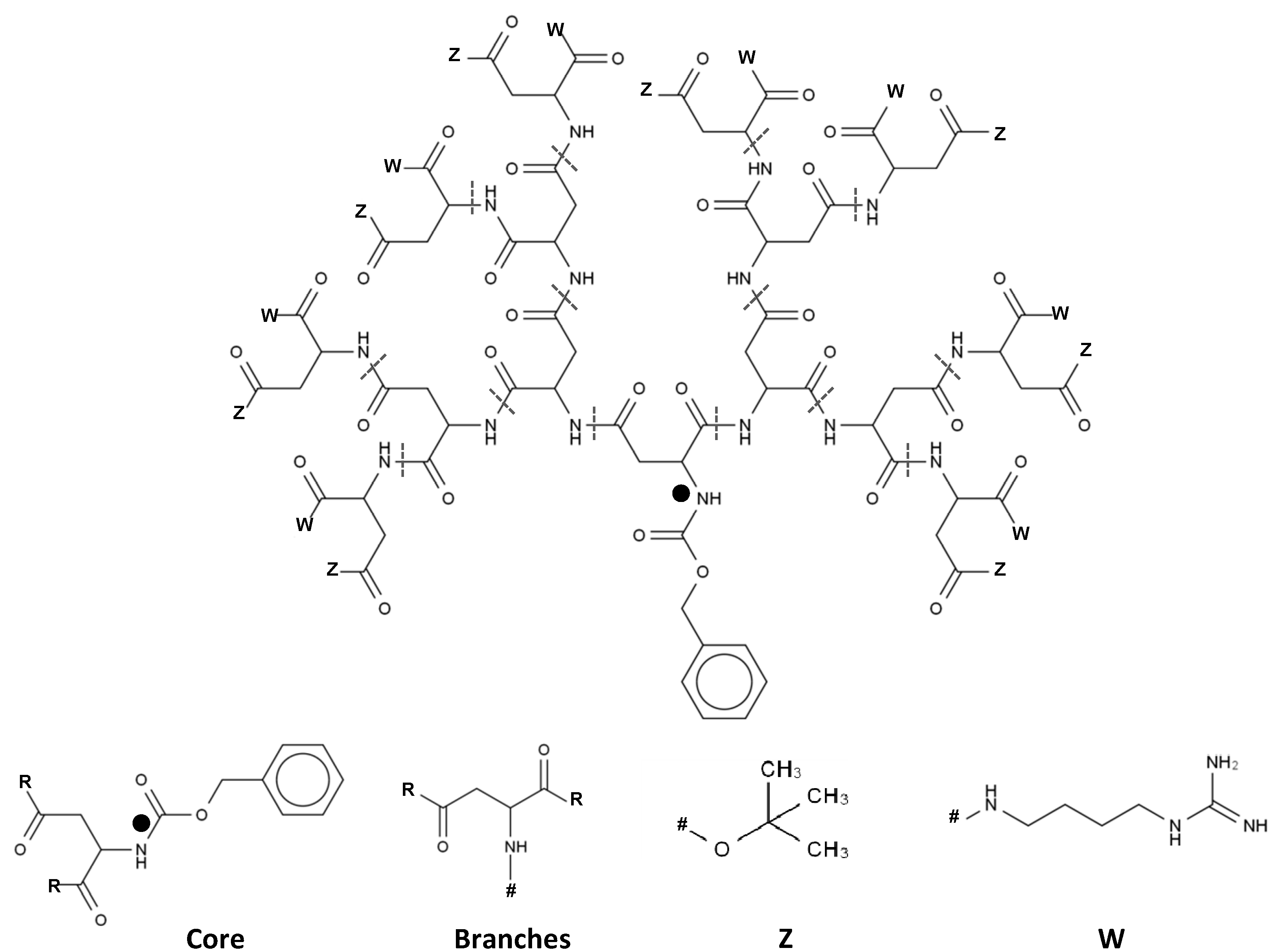
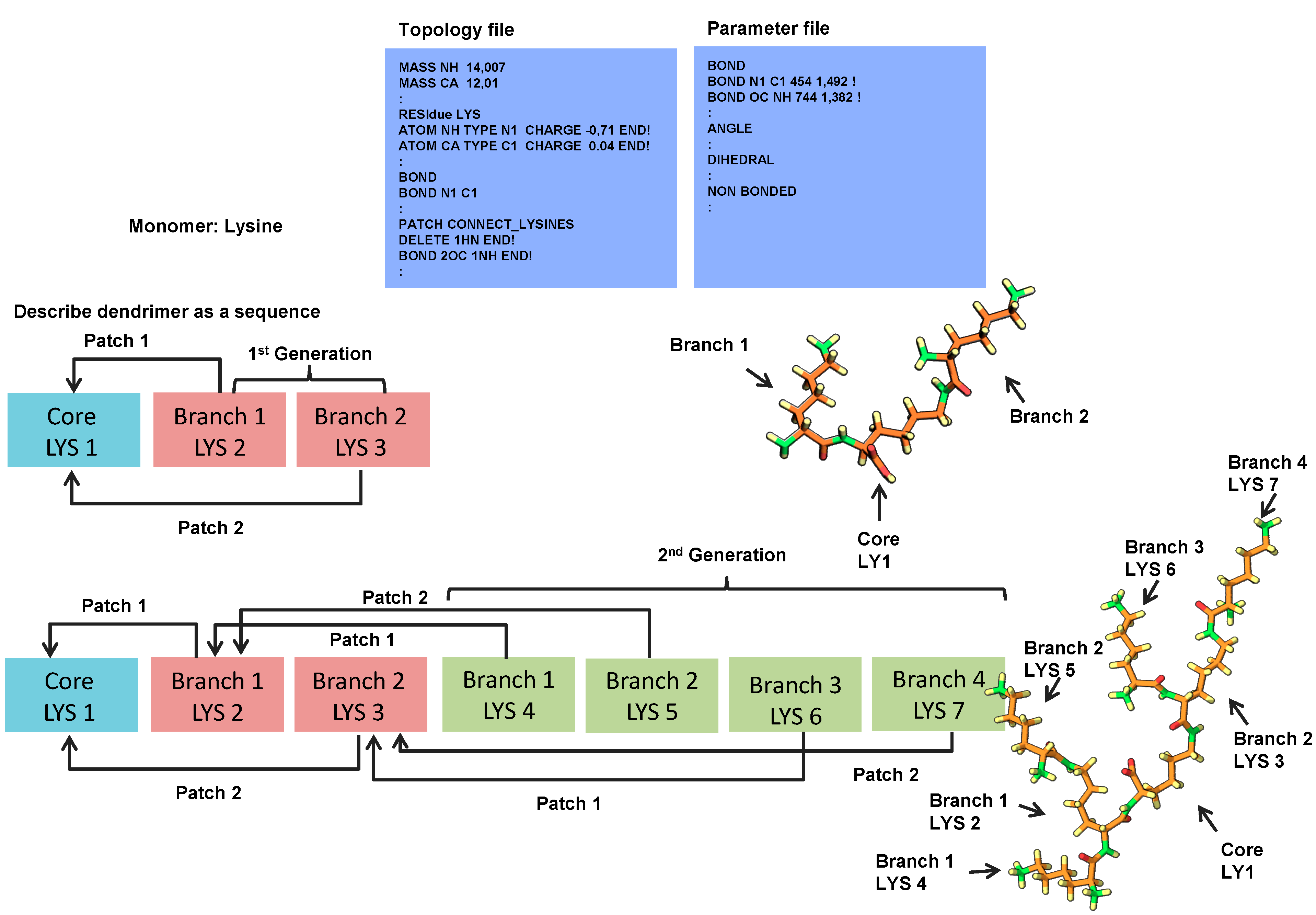
2.1. 3D Structure Generation


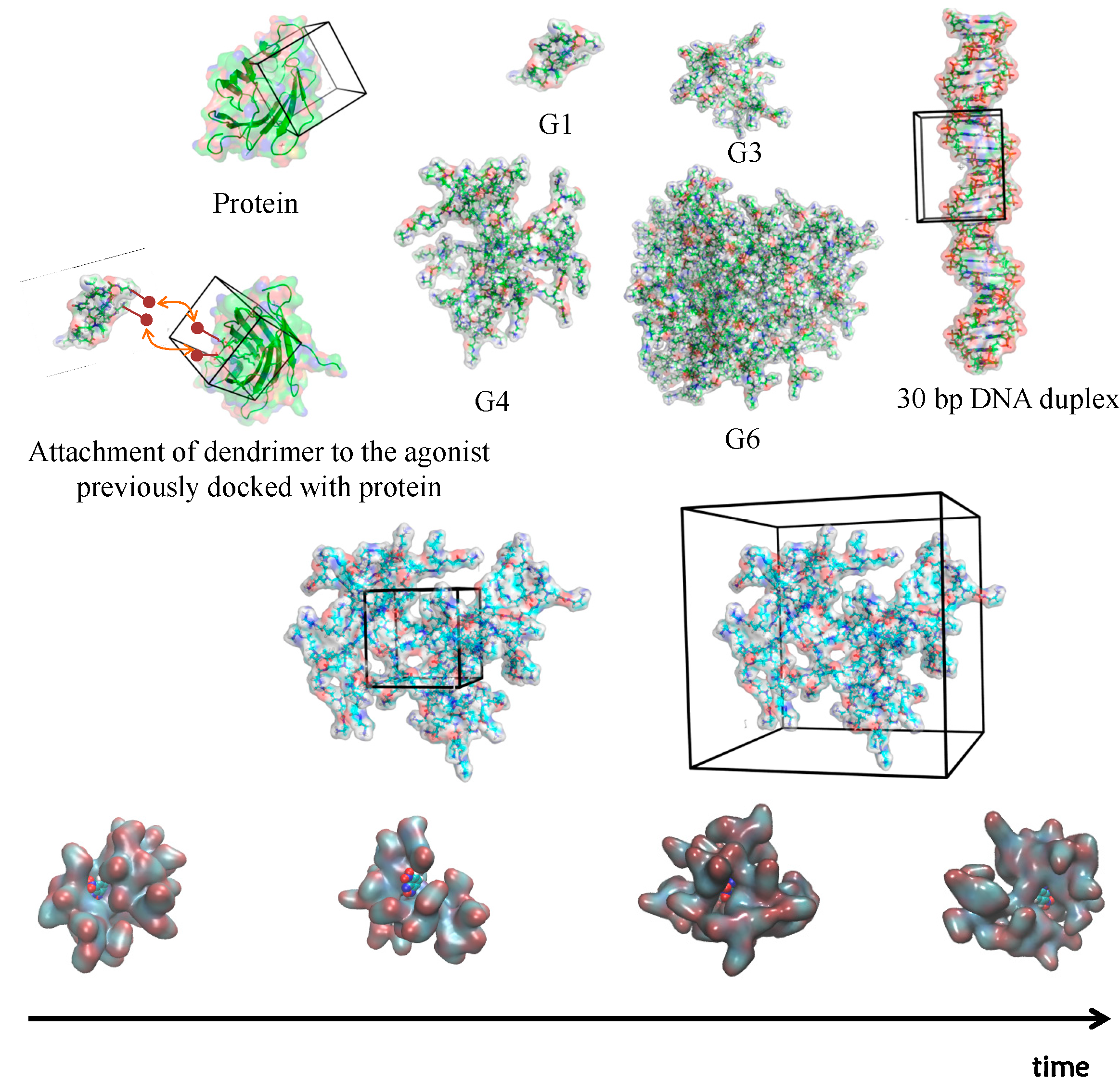
2.2. Simulation of Dendrimers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dendrimer Type | Software for Dendrimer Construct | Software Used for Simulations | Force Field | Aim of the Study |
|---|---|---|---|---|
| Conformational analysis | ||||
| PAMAM G2 to G6 [87] | Insight II | Insight II ƚ CHARMM ƚ | CVFF CHARMM | Structural features at different pH and generations |
| PAMAM G5 [88] | NA* | MPsim | DREIDING | Effect of pH to study the water dynamics on dendrimers |
| PAMAM G50% and 90% acetylated [89] | Insight II | AMBER8 ƚ | GAFF | Effect of acetylation on structural features |
| Glycosylated PAMAM G3.5 [90] | XPLOR | Desmond § | OPLS_2005 | Effect of glycosylation on structural features |
| Triazine G3 and G5 with DOTA terminals [91] | AMBER 11 | AMBER 11 ƚ | GAFF and parm99 | Location of DOTA groups complexed with Gd ions |
| PEgylated PAMAM G3 to G5 [92] | NA * | GROMACS § | MARTINI | Effect of PEGylation on the structure and interparticle interaction |
| Pegylated triazine dendrimers linked with paclitaxel [72] | Material Studio 5 | AMBER 11 ƚ | parm99 | Effect of PEGylation on availability of linkers |
| Carboxylic modified PAMAM G5 with gold, fluorescein isothiocyanate (FI) and folic acid (FA) [93] | Insight II | Insight II ƚ | CVFF | Conformation and location of FI and FA |
| PAMAM G5 with amine, carboxyl and acetamide groups linked to fluorescein and folic acid [80] | Insight II | Insight II ƚ | CVFF | Location of folic acid and its availability |
| PAMAM G5 with methotrexate [29] | CHARMM | CHARMM ƚ | CHARMM | Location of MTX when directly linked or with a spacer |
| Poly(l-lysine) and Poly(amide) G4 dendrimers [94] | Starmaker (Silico) | NAMD § | OPLS-AA | Comparison of dendrimer architectures in solution |
| Dendrimer-small molecule interactions | ||||
| PAMAM G5 with different terminal 81] | Insight II | Insight II ƚ | CVFF | Influence of terminal groups on the complexation |
| PAMAM G4 + polyphenols [55] | ChemOffice Ultra 6.0 | HyperChem Pro 7.0 ƚ | MM+ | Free binding energies |
| PAMAM G3 + nicotinic acid, nicotinate and 3-pyridiniumcarboxylate [95] | HyperChem | NAMD § | CHARMM27 | Free energy of binding and the effect of pH variation on binding |
| Peptide dendrimers + hydroxypyrene trisulfonate butyrate ester [38] | CORINA | GROMACS § | GROMOS-96 43a1 | Conformation and docking site location |
| PAMAM G5-Folic acid + Morphine and Tramadol [96] | ICM | NAMD § | CHARMM 27; ParamChem | Different pHs; Binding mechanism elucidadtion; Location of folic acid |
| PAMAM G5 + salicylic acid, L-alanine, phenylbutazone, primidone [97] | DBT/AMBER | AMBER9 ƚ | GAFF | Effect of pH on interaction and relation with drug release |
| Poly(l-lysine) G4 dendrimer + doxorubicin [83] | ChemBioOffice | Desmond § | OPLS-AA | Complex formation |
| Dendrimer-nucleic acid interaction | ||||
| Triazine G2dendrimers + siRNA or DNA [98] | AMBER 10 | AMBER 10 ƚ | Parm99 | Binding mechanism and energy contributions |
| PAMAM ssDNA [49] | AMBER 7 | AMBER 7 ƚ | AMBER 95 (DNA) DREIDING (dendrimer) | Binding interaction and energy contributions |
| PAMAM G3 DNA [75] | NA * | NAMD § | CHARMM 27 | Complexation mechanism |
| PAMAM G0 and G1 + siRNA [43] | Material Studio 5 | AMBER9 ƚ | Ff99 FF for RNA GAFF for dendrimers | Effects of pH on the complexation |
| PAMAM G7 + siRNA [99] | Material Studio 5 | AMBER10 ƚ | GAFF (non-standard residues); parm99 | Complex interaction |
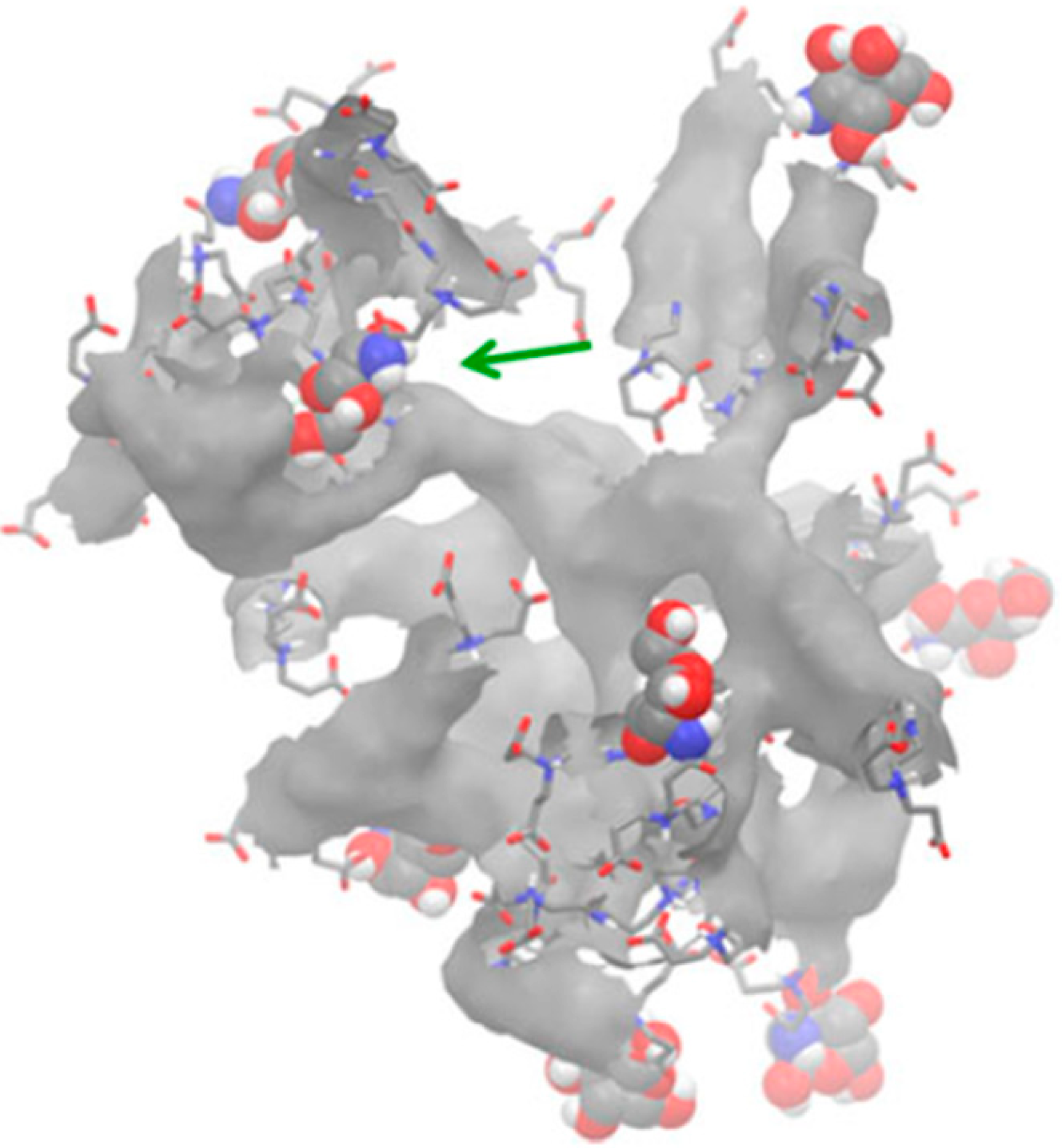
| Dendrimer-protein interaction | ||||
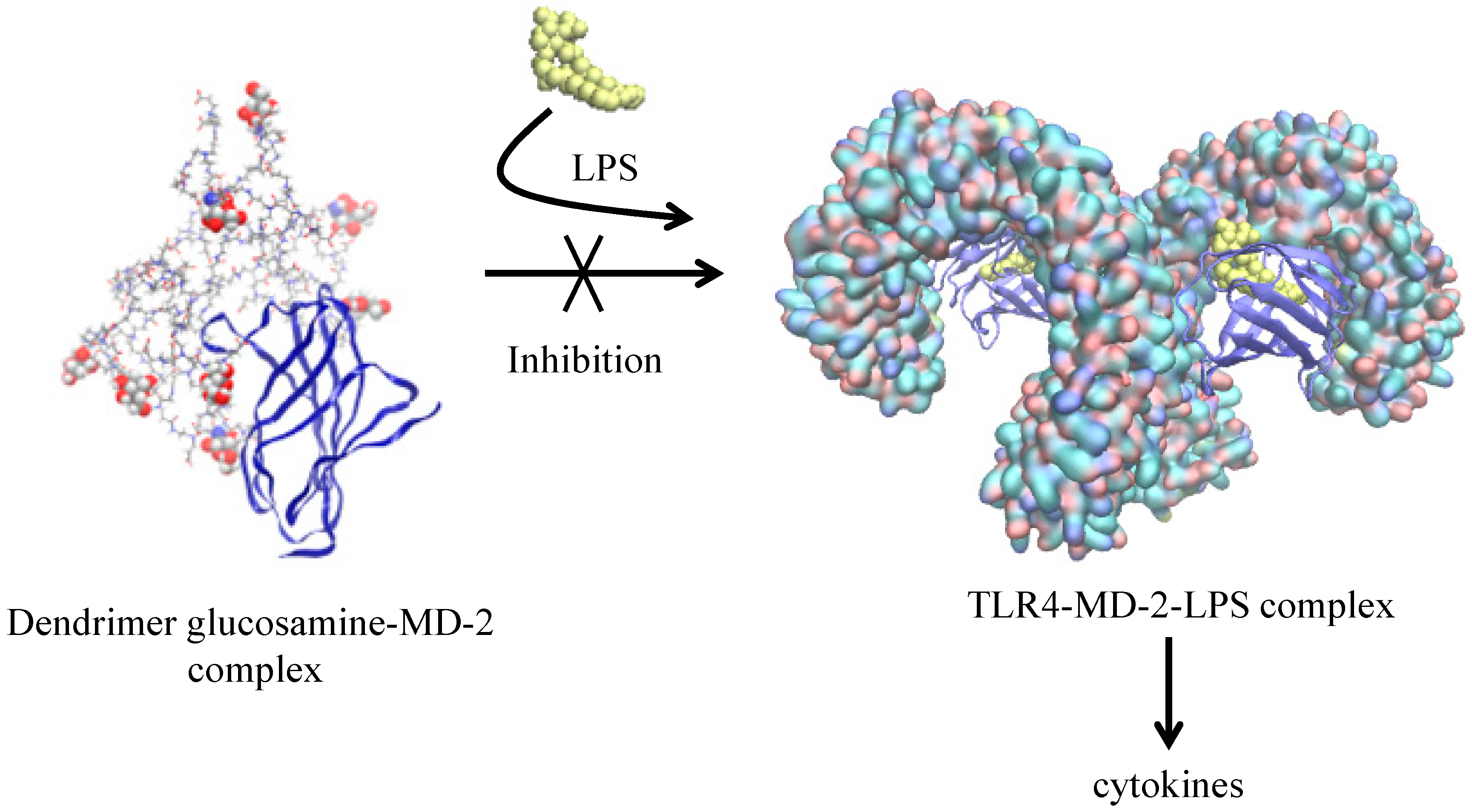
| Glycosylated PAMAM G3.5 + MD-2 protein [52] | XPLOR | Desmond § | OPLS_2005 | Docking and interaction between active and non-active forms |
| PAMAM G4 Albumin [47] | NA * | NA * | DREIDING | Contact points between dendrimer-albumin at physiological pH |
| PAMAM G0 with guanidinium terminal groups α-chymotrypsinogen A [73] | NA * | NAMD § | CHARMM 27 | Site of interaction with the protein and effect of salt types |
| Dendrimer-lipid bilayer interaction | ||||
| Acetylated and non-acetylated PAMAM G5 and G7 + DMPC [53] | Insight II | GROMACS § | MARTINI and adapted MARTINI | Effect of size, charge and concentration on dendrimer-membrane interaction |
| PAMAM G3 and G5 with different acetylation levels + DPPC [60] | Insight II | GROMACS § | MARTINI | Effect of size, charge and lipid phase on dendrimer-membrane interaction |
| PAMAM G3 with amine, acetyl and carboxyl terminals + DMPC [74] | CHARMM | CHARMM ƚ | CHARMM27 (lipid) and CHARMM 22 (dendrimer) | Effect of terminal groups and lipid phase |
2.3. Molecular Docking of Dendrimers

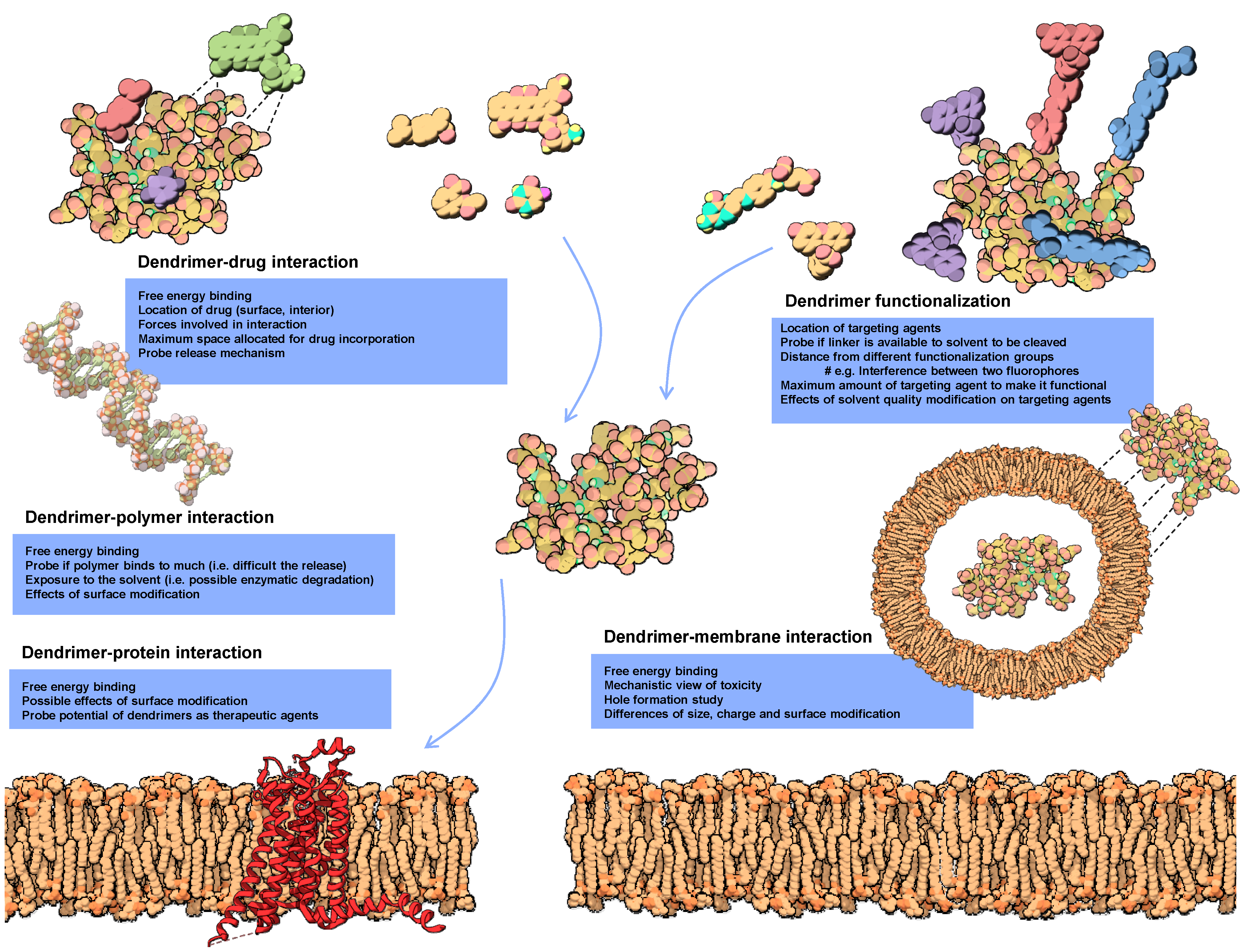
3. Modeling Dendrimers for Biomedical Applications
3.1. Impact of Solvent and Dendrimer Topology
| FF | Solvent | G0 | G1 | G2 | G3 | G4 | G5 | G6 | G7 | G8 | G9 | G10 | Source |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SAXS | Methanol | 15.8 | 17.1 | 24.1 | 26.3 | 31.9 | 40.3 | 49.2 | 57.4 | [86] | |||
| SAXS | Methanol | 4.0 | 7.9 | 11.8 | 15.09 | 18.60 | 23.07 | 27.50 | 32.11 | 38.58 | - | - | [116] |
| SANS | D2O | - | - | - | - | 20.90–21.30 | - | - | - | - | - | - | [117] |
| SANS | D2O pH 4.97–10.25 | - | - | - | - | 20.64–21.58 | - | - | - | - | - | - | [111] |
| SANS | D2O pH 4.7–10.1 | - | - | - | - | - | - | - | - | 38.1–40.7 | - | - | [112] |
| SANS | Different solvents | - | - | - | - | - | 22.1 | - | - | 32.8–43.8 | - | - | [118] |
| DREIDING | Vacuum | 4.93 | 7.46 | 9.17 | 11.23 | 14.50 | 18.34 | 22.40 | 29.09 | 36.42 | 46.03 | 55.19 | [10] |
| DREIDING optimized QM | Water explicit | - | - | - | - | 21.07–22.11 | - | - | - | - | - | - | [85] |
| DREIDING | Water implicit | 4.97 | 7.03 | 9.77 | 13.01 | 16.36 | 21.67 | 27.62 | - | - | - | - | [35] |
| DREIDING | Water explicit High pH | - | 7.4 | 11.5 | 12.9 | 16.9 | 20.3 | 24.7 | 30.1 | - | - | - | [119] |
| DREIDING | Water explicit Low pH | - | 9.4 | 13.6 | 17.2 | 21.1 | 26.1 | 32.5 | 37.57 | - | - | - | [119] |
| CHARMM 27 | Water explicit | - | - | - | 15.33 | 21.04 | 25.50 | 30.18 | - | - | - | - | [95] |
| AMBER | Water explicit pH 7.4 | - | - | - | 16.25 | 18.8–20 | 22.43–22.9 | 27.2 | - | - | - | - | [50] |
| AMBER | Water explicit pH 5 | - | - | - | - | 21.0 | 24.2 | 28.9 | - | - | - | - | [50] |
| DREIDING | Water explicit pH 4–12 | - | - | - | - | - | - | - | 37.8–43.11 | - | - | [120] | |
| Coarse-Grained (MARTINI) | Water explicit | - | - | - | - | 20.1 | 25.6 | - | - | - | - | - | [59] |
| Coarse-Grained | Water explicit | - | - | - | 13.1 | - | 23.20 | - | - | - | - | - | [60] |
| DREIDING | Water explicit pH 12 | - | - | - | - | 16.78 | 20.67 | 26.76 | - | - | - | - | [110] |
| DREIDING | Water explicit pH 7 | - | - | - | - | 17.01 | 22.19 | 27.28 | - | - | - | - | [110] |
| DREIDING | Water explicit pH 4 | - | - | - | - | 19.01 | 24.76 | 30.89 | - | - | - | - | [110] |
| CVFF | Water implicit Low pH | - | - | 16.6 | 22.8 | 29.9 | 38.0 | 46.8 | - | - | - | - | [87] |
| CVFF | Water implicit Neutral pH | - | - | 14.5 | 19.7 | 26.7 | 32.8 | 41.3 | - | - | - | - | [87] |
| CVFF | Water implicit High pH | - | - | 8.4 | 11.6 | 14.8 | 18.3 | 24.2 | - | - | - | - | [87] |
| OPLS | Vacuum | - | - | 11.0 | 13.7 | - | - | - | - | - | - | - | [121] |
3.2. Impact and Versatility of the End Groups

3.3. Dendrimers Interaction with Lipid Membranes

3.4. Modeling Dendrimers for Drug Delivery Applications

3.5. Modeling Dendrimers as Therapeutic Agents

4. Challenges and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tomalia, D.A. Birth of a new macromolecular architecture: Dendrimers as quantized building blocks for nanoscale synthetic polymer chemistry. Prog. Polym. Sci. 2005, 30, 294–324. [Google Scholar]
- Ali, M.; Brocchini, S. Synthetic approaches to uniform polymers. Adv. Drug Deliv. Rev. 2006, 58, 1671–87. [Google Scholar] [CrossRef] [PubMed]
- Hodge, P. Polymer science branches out. Nature 1993, 362, 18–19. [Google Scholar] [CrossRef]
- Morefield, G.L.; Hawkins, L.D.; Ishizaka, S.T.; Kissner, T.L.; Ulrich, R.G. Synthetic Toll-like receptor 4 agonist enhances vaccine efficacy in an experimental model of toxic shock syndrome. Clin. Vaccine Immunol. 2007, 14, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Klajnert, B.; Bryszewska, M. Dendrimers: Properties and applications. Acta Biochim. Pol. 2001, 48, 199–208. [Google Scholar] [PubMed]
- Hawker, C.J.; Frechet, J.M.J. Preparation of Polymers with Controlled Molecular Architecture. A New Convergent Approach to Dendritic Macromolecules. J. Am. Chem. Soc. 1990, 32, 7638–7647. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Naylor, A.M.; Goddard, W.A. Starburst Dendrimers: Molecular-Level Control of Size, Shape, Surface Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter. Angew. Chem. Int. Ed. Eng. 1990, 29, 138–175. [Google Scholar] [CrossRef]
- Mourey, T.H.; Turner, S.R.; Rubinstein, M.; Frechet, J.M.J.; Hawker, C.J.; Wooley, K.L. Unique behavior of dendritic macromolecules: Intrinsic viscosity of polyether dendrimers. Macromolecules 1992, 25, 2401–2406. [Google Scholar] [CrossRef]
- Jackson, C.L.; Chanzy, H.D.; Booy, F.P.; Drake, B.J.; Tomalia, D.A.; Bauer, B.J.; Amis, E.J. Visualization of Dendrimer Molecules by Transmission Electron Microscopy (TEM): Staining Methods and Cryo-TEM of Vitrified Solutions. Macromolecules 1998, 31, 6259–6265. [Google Scholar] [CrossRef]
- Maiti, P.K.; Çaǧın, T.; Wang, G.; Goddard, W.A. Structure of PAMAM Dendrimers: Generations 1 through 11. Macromolecules 2004, 37, 6236–6254. [Google Scholar] [CrossRef]
- Zeng, F.; Zimmerman, S.C. Dendrimers in Supramolecular Chemistry: From Molecular Recognition to Self-Assembly. Chem. Rev. 1997, 97, 1681–1712. [Google Scholar] [CrossRef] [PubMed]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A new class of polymers starburst dendritic macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef]
- Svenson, S. Dendrimers as versatile platform in drug delivery applications. Eur. J. Pharm. Biopharm. 2009, 71, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Buhleier, E.; Wehner, W.; Vogtle, F. “Cascade”-and “Nonskid-Chain-like” Syntheses of Molecular Cavity Topologies. Synthesis 1978, 155–158. [Google Scholar]
- Fischer, M.; Appelhans, D.; Schwarz, S.; Klajnert, B.; Bryszewska, M.; Voit, B.; Rogers, M. Influence of surface functionality of poly(propylene imine) dendrimers on protease resistance and propagation of the scrapie prion protein. Biomacromolecules 2010, 11, 1314–1325. [Google Scholar] [CrossRef]
- Al-Jamal, K.T.; Al-Jamal, W.T.; Akerman, S.; Podesta, J.E.; Yilmazer, A.; Turton, J.A.; Bianco, A.; Vargesson, N.; Kanthou, C.; Florence, A.T.; et al. Systemic antiangiogenic activity of cationic poly-l-lysine dendrimer delays tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 3966–3971. [Google Scholar] [CrossRef] [PubMed]
- Gillies, E.R.; Dy, E.; Fréchet, J.M.J.; Szoka, F.C. Biological evaluation of polyester dendrimer: Poly(ethylene oxide) “bow-tie” hybrids with tunable molecular weight and architecture. Mol. Pharm. 2004, 2, 129–138. [Google Scholar] [CrossRef]
- Lim, J.; Pavan, G.M.; Annunziata, O.; Simanek, E.E. Experimental and computational evidence for an inversion in guest capacity in high-generation triazine dendrimer hosts. J. Am. Chem. Soc. 2012, 134, 1942–1945. [Google Scholar] [CrossRef]
- Ciepluch, K.; Katir, N.; El Kadib, A.; Felczak, A.; Zawadzka, K.; Weber, M.; Klajnert, B.; Lisowska, K.; Caminade, A.-M.; Bousmina, M.; et al. Biological properties of new viologen-phosphorus dendrimers. Mol. Pharm. 2012, 9, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.P.; Patri, A.K.; Andrzej, M.; Myaing, M.T.; Ye, J.Y.; Norris, T.B.; Baker, J.R., Jr. In Vitro Targeting of Synthesized Antibody-Conjugated Dendrimer Nanoparticles. Biomacromolecules 2004, 5, 2269–2274. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-Y.; Lv, B.; Luo, Y. The effects of an RGD-PAMAM dendrimer conjugate in 3D spheroid culture on cell proliferation, expression and aggregation. Biomaterials 2013, 34, 2665–2673. [Google Scholar] [CrossRef] [PubMed]
- Hedden, R.C.; Bauer, B.J. Structure and Dimensions of PAMAM/PEG Dendrimer—Star Polymers. Macromolecules 2003, 36, 1829–1835. [Google Scholar] [CrossRef]
- Twyman, L.J.; King, A.S.H.; Martin, I.K. Catalysis inside dendrimers. Chem. Soc. Rev. 2002, 31, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Degoricija, L.; Johnson, C.S.; Wathier, M.; Kim, T.; Grinstaff, M.W. Photo Cross-linkable Biodendrimers as Ophthalmic Adhesives for Central Lacerations and Penetrating Keratoplasties. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2037–2042. [Google Scholar] [CrossRef]
- Šebestík, J.; Reiniš, M.; Ježek, J. Dendrimers as Biosensors and Imaging Tools. In Biomedical Applications of Peptide-, Glyco- and Glycopeptide Dendrimers, and Analogous Dendrimeric Structures; Springer Science & Business Media: Wien, Austria, 2012; pp. 191–195. [Google Scholar]
- Grinstaff, M.W. Biodendrimers: New Polymeric Biomaterials for Tissue Engineering. Chem. A Eur. J. 2002, 8, 2838–2846. [Google Scholar] [CrossRef]
- Shaunak, S.; Brocchini, S. Dendrimer-Based Drugs as Macromolecular Medicines. Biotechnol. Genet. Eng. Rev. 2006, 23, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Shcharbin, D.G.; Klajnert, B.; Bryszewska, M. Dendrimers in gene transfection. Biochemistry 2009, 74, 1070–1079. [Google Scholar] [PubMed]
- Tyssen, D.; Henderson, S.A.; Johnson, A.; Sterjovski, J.; Moore, K.; La, J.; Zanin, M.; Sonza, S.; Karellas, P.; Giannis, M.P.; et al. Structure Activity Relationship of Dendrimer Microbicides with Dual Action Antiviral Activity. PLoS One 2010, 5, e12309. [Google Scholar] [CrossRef] [PubMed]
- Darbre, T.; Reymond, J.-L. Peptide dendrimers as artificial enzymes, receptors, and drug-delivery agents. Acc. Chem. Res. 2006, 39, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Kesharwani, P.; Gupta, U.; Jain, N.K. Dendrimer toxicity: Let’s meet the challenge. Int. J. Pharm. 2010, 394, 122–142. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Izzo, L. Dendrimer biocompatibility and toxicity. Adv. Drug Deliv. Rev. 2005, 57, 2215–2237. [Google Scholar] [CrossRef] [PubMed]
- McNerny, D.Q.; Leroueil, P.R.; Baker, J.R. Understanding specific and nonspecific toxicities: A requirement for the development of dendrimer-based pharmaceuticals. Nanomed. Nanobiotechnol. 2010, 2, 249–259. [Google Scholar] [CrossRef]
- Caminade, A.-M.; Laurent, R.; Majoral, J.-P. Characterization of dendrimers. Adv. Drug Deliv. Rev. 2005, 57, 2130–2146. [Google Scholar] [CrossRef] [PubMed]
- Cagin, T.; Wang, G.; Martin, R.; Zamanakos, G.; Vaidehi, N.; Mainz, T.; Iii, W.A.G. Multiscale modeling and simulation methods with applications to dendritic polymers. Comput. Theor. Polym. Sci. 2001, 11, 345–356. [Google Scholar] [CrossRef]
- Çagin, T.; Wang, G.; Martin, R.; Breen, N.; Goddard, W. A Molecular modeling of dendrimers for nanoscale applications. Nanotechnology 2000, 11, 77–84. [Google Scholar] [CrossRef]
- Lee, H.; Larson, R.G. Multiscale modeling of dendrimers and their interactions with bilayers and polyelectrolytes. Molecules 2009, 14, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Javor, S.; Reymond, J.-L. Molecular dynamics and docking studies of single site esterase peptide dendrimers. J. Org. Chem. 2009, 74, 3665–3674. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, D.; Kurur, S. Synthesis of totally chiral, multiple armed, poly Glu and poly Asp scaffoldings on bifunctional adamantane core. Tetrahedron Lett. 1997, 38, 1265–1268. [Google Scholar] [CrossRef]
- Zloh, M.; Ramaswamy, C.; Sakthivel, T.; Wilderspin, A.; Florence, A.T. Investigation of the association and flexibility of cationic lipidic peptide dendrons by NMR spectroscopy. Magn. Reson. Chem. 2005, 43, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Naylor, A.M.; Goddard, W.A.; Kiefer, G.E.; Tomalia, D.A. Starburst Dendrimers. 5. Molecular Shape Control. J. Am. Chem. Soc 1989, 111, 2339–2341. [Google Scholar] [CrossRef]
- Tian, W.; Ma, Y. Molecular dynamics simulations of a charged dendrimer in multivalent salt solution. J. Phys. Chem. B 2009, 113, 13161–13170. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, D.; Zhang, H.; Parekh, H.S.; Smith, S.C. The effect of pH on PAMAM dendrimer-siRNA complexation: Endosomal considerations as determined by molecular dynamics simulation. Biophys. Chem. 2011, 158, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Maingi, V.; Maiti, P.K.; Bharatam, P.V. Molecular dynamics simulations of PPI dendrimer–drug complexes. Soft Matter 2013, 9, 6482–6496. [Google Scholar] [CrossRef]
- Barata, T.S.; Teo, I.; Lalwani, S.; Simanek, E.E.; Zloh, M.; Shaunak, S. Computational design principles for bioactive dendrimer based constricts as antagonists of the TLR4-MD-2-LPS complex. Biomaterials 2012, 32, 8702–8711. [Google Scholar] [CrossRef]
- Barnard, A.; Posocco, P.; Pricl, S.; Calderon, M.; Haag, R.; Hwang, M.E.; Shum, V.W.T.; Pack, D.W.; Smith, D.K. Degradable self-assembling dendrons for gene delivery: Experimental and theoretical insights into the barriers to cellular uptake. J. Am. Chem. Soc. 2011, 133, 20288–20300. [Google Scholar] [CrossRef] [PubMed]
- Giri, J.; Diallo, M.S.; Simpson, A.J.; Liu, Y.; Goddard, W.A.; Kumar, R.; Woods, G.C. Interactions of poly(amidoamine) dendrimers with human serum albumin: Binding constants and mechanisms. ACS Nano 2011, 5, 3456–3468. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Jacobson, K.A. Molecular modeling of a PAMAM-CGS21680 dendrimer bound to an A2A adenosine receptor homodimer. Bioorg. Med. Chem. Lett. 2008, 18, 4312–4315. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.K.; Bagchi, B. Structure and dynamics of DNA-dendrimer complexation: Role of counterions, water, and base pair sequence. Nano Lett. 2006, 6, 2478–2485. [Google Scholar] [CrossRef] [PubMed]
- Pavan, G.M.; Albertazzi, L.; Danani, A. Ability to adapt: Different generations of PAMAM dendrimers show different behaviors in binding siRNA. J. Phys. Chem. B 2010, 114, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- Vasumathi, V.; Maiti, P.K. Complexation of siRNA with Dendrimer: A Molecular Modeling Approach. Macromolecules 2010, 43, 8264–8274. [Google Scholar] [CrossRef]
- Barata, T.S.; Teo, I.; Brocchini, S.; Zloh, M.; Shaunak, S. Partially Glycosylated Dendrimers Block MD-2 and Prevent TLR4-MD-2-LPS Complex Mediated Cytokine Responses. PLoS Comput. Biol. 2011, 7, e1002095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Larson, R.G. Coarse-grained molecular dynamics studies of the concentration and size dependence of fifth- and seventh-generation PAMAM dendrimers on pore formation in DMPC bilayer. J. Phys. Chem. B 2008, 112, 7778–7784. [Google Scholar] [CrossRef] [PubMed]
- Tanis, I.; Karatasos, K. Association of a weakly acidic anti-inflammatory drug (ibuprofen) with a poly(amidoamine) dendrimer as studied by molecular dynamics simulations. J. Phys. Chem. B 2009, 113, 10984–10993. [Google Scholar] [CrossRef] [PubMed]
- Abderrezak, A.; Bourassa, P.; Mandeville, J.-S.; Sedaghat-Herati, R.; Tajmir-Riahi, H.-A. Dendrimers bind antioxidant polyphenols and cisplatin drug. PLoS One 2012, 7, e33102. [Google Scholar] [CrossRef] [PubMed]
- Brocchini, S.; Godwin, A.; Balan, S.; Choi, J.; Zloh, M.; Shaunak, S. Disulfide bridge based PEGylation of proteins. Adv. Drug Deliv. Rev. 2008, 60, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Uhlich, N.A.; Darbre, T.; Reymond, J.-L. Peptide dendrimer enzyme models for ester hydrolysis and aldolization prepared by convergent thioether ligation. Org. Biomol. Chem. 2011, 9, 7071–7084. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- He, X.; Qu, Z.; Xu, F.; Lin, M.; Wang, J.; Shi, X.; Lu, T. Molecular analysis of interactions between dendrimers and asymmetric membranes at different transport stages. Soft Matter 2014, 10, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Larson, R.G. Molecular dynamics simulations of PAMAM dendrimer-induced pore formation in DPPC bilayers with a coarse-grained model. J. Phys. Chem. B 2006, 110, 18204–18211. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Lee, H. Self-assembly of mixtures of a dendrimer and lipids : Effects of hydrophobicity and electrostatics. Mol. Simul. 2012, 38, 534–539. [Google Scholar] [CrossRef]
- Lozac’h, N.; Goodson, A.L.; Powell, W.H. Nobel Nomenclature—General Principles. Angew. Chem. Int. Ed. Engl. 1979, 18, 887–899. [Google Scholar] [CrossRef]
- Roberts, B.P.; Scanlon, M.J.; Krippner, G.Y.; Chalmers, D.K. The Dotted Cap Notation: A concise notation for describing variegated dendrimers. New J. Chem. 2008, 32, 1543–1554. [Google Scholar] [CrossRef]
- Florida, S.; Rouge, B. Systematic Nomenclature for Cascade Polymers. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 641–651. [Google Scholar]
- Friedhofen, J.H.; Vögtle, F. Detailed nomenclature for dendritic molecules. New J. Chem. 2006, 30, 32–43. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4 : Algorithms for Highly Efficient , Load-Balanced , and Scalable Molecular Simulation. J. Chem. Theory Comput. 2007, 4, 436–447. [Google Scholar]
- Schwieters, C.D.; Kuszewski, J.J.; Tjandra, N.; Clore, G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Schwieters, C.; Kuszewski, J.; Mariusclore, G. Using Xplor–NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. [Google Scholar] [CrossRef]
- Charlmers, D.; Roberts, B. Silico—A Perl Molecular Modeling Toolkit. Available online: http://silico.sourceforge.net/Silico/Home.html (accessed on 1 August 2014).
- Maingi, V.; Jain, V.; Bharatam, P.V.; Maiti, P.K. Dendrimer building toolkit: Model building and characterization of various dendrimer architectures. J. Comput. Chem. 2012, 33, 1997–2011. [Google Scholar] [CrossRef] [PubMed]
- Barata, T.S.; Brocchini, S.; Teo, I.; Shaunak, S.; Zloh, M. From sequence to 3D structure of hyperbranched molecules: Application to surface modified PAMAM dendrimers. J. Mol. Model. 2011, 17, 2741–2749. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Lo, S.-T.; Hill, S.; Pavan, G.M.; Sun, X.; Simanek, E.E. Antitumor activity and molecular dynamics simulations of paclitaxel-laden triazine dendrimers. Mol. Pharm. 2012, 9, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.P.; Shukla, D.; Trout, B.L. Effects of solute-solute interactions on protein stability studied using various counterions and dendrimers. PLoS One 2011, 6, e27665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, C.V.; Leroueil, P.R.; Orr, B.G.; Banaszak Holl, M.M.; Andricioaei, I. Poly(amidoamine) dendrimers on lipid bilayers II: Effects of bilayer phase and dendrimer termination. J. Phys. Chem. B 2008, 112, 9346–9353. [Google Scholar] [CrossRef] [PubMed]
- Mills, M.; Orr, B.G.; Banaszak Holl, M.M.; Andricioaei, I. Attractive hydration forces in DNA-dendrimer interactions on the nanometer scale. J. Phys. Chem. B 2013, 117, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Filipe, C.S.; Machuqueiro, M.; Darbre, T.; Baptista, M. Unraveling the Conformational Determinants of Peptide Dendrimers Using Molecular Dynamics Simulations. Macromolecules 2013, 46, 9427–9436. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Lu, Z.-Y.; Laaksonen, A. Specific binding structures of dendrimers on lipid bilayer membranes. Phys. Chem. Chem. Phys. 2012, 14, 8348–8359. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Ma, Y. Insights into the endosomal escape mechanism via investigation of dendrimer–membrane interactions. Soft Matter 2012, 8, 6378–6384. [Google Scholar] [CrossRef]
- Zhong, T.; Ai, P.; Zhou, J. Structures and properties of PAMAM dendrimer: A multi-scale simulation study. Fluid Phase Equilib. 2011, 302, 43–47. [Google Scholar] [CrossRef]
- Quintana, A.; Raczka, E.; Piehler, L.; Lee, I.; Myc, A.; Majoros, I.; Patri, A.K.; Thomas, T.; Mulé, J.; Baker, J.R. Design and function of a dendrimer-based therapeutic nanodevice targeted to tumor cells through the folate receptor. Pharm. Res. 2002, 19, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Lee, I.; Chen, X.; Shen, M.; Xiao, S.; Zhu, M.; Baker, J.R., Jr.; Wang, S.H. Influence of dendrimer surface charge on the bioactivity of 2-methoxyestradiol complexed with dendrimers. Soft Matter 2010, 6, 20–27. [Google Scholar] [CrossRef]
- Stach, M.; Maillard, N.; Kadam, R.U.; Kalbermatter, D.; Meury, M.; Page, M.G.P.; Fotiadis, D.; Darbre, T.; Reymond, J.-L. Membrane disrupting antimicrobial peptide dendrimers with multiple amino termini. Med. Chem. Comm. 2012, 3, 86–89. [Google Scholar] [CrossRef]
- Al-Jamal, K.T.; Al-Jamal, W.T.; Wang, J.T.-W.; Rubio, N.; Buddle, J.; Gathercole, D.; Zloh, M.; Kostarelos, K. Cationic poly-l-lysine dendrimer complexes doxorubicin and delays tumor growth in vitro and in vivo. ACS Nano 2013, 7, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Miklis, P.; Tahir, C.; Iii, W.A.G. Dynamics of Bengal Rose Encapsulated in the Meijer Dendrimer Box. J. Am. Chem. Soc. 1997, 7863, 7458–7462. [Google Scholar] [CrossRef]
- Liu, Y.; Bryantsev, V.S.; Diallo, M.S.; Goddard, W. A PAMAM dendrimers undergo pH responsive conformational changes without swelling. J. Am. Chem. Soc. 2009, 131, 2798–2799. [Google Scholar] [CrossRef] [PubMed]
- Prosa, T.J.; Bauer, B.J.; Amis, E.J.; Tomalia, D.A.; Scherrenberg, R. A SAXS study of the internal structure of dendritic polymer systems. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 2913–2924. [Google Scholar] [CrossRef]
- Lee, I.; Athey, B.D.; Wetzel, A.W.; Meixner, W.; Baker, J.R. Structural Molecular Dynamics Studies on Polyamidoamine Dendrimers for a Therapeutic Application: Effects of pH and Generation. Macromolecules 2002, 35, 4510–4520. [Google Scholar] [CrossRef]
- Lin, S.-T.; Maiti, P.K.; Goddard, W.A. Dynamics and thermodynamics of water in PAMAM dendrimers at subnanosecond time scales. J. Phys. Chem. B 2005, 109, 8663–8672. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Baker, J.R.; Larson, R.G. Molecular dynamics studies of the size, shape, and internal structure of 0% and 90% acetylated fifth-generation polyamidoamine dendrimers in water and methanol. J. Phys. Chem. B 2006, 110, 4014–4019. [Google Scholar] [CrossRef] [PubMed]
- Barata, T.S.; Shaunak, S.; Teo, I.; Zloh, M.; Brocchini, S. Structural studies of biologically active glycosylated polyamidoamine (PAMAM) dendrimers. J. Mol. Model. 2011, 17, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Turkbey, B.; Bernardo, M.; Bryant, L.H.J.; Garzoni, M.; Pavan, G.M.; Nakajima, T.; Choyke, P.L.; Simanek, E.E.; Kobayashi, H. Gadolinium MRI contrast agents based on triazine dendrimers: Relaxivity and in vivo pharmacokinetics. Bioconjug. Chem. 2012, 23, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Larson, R.G. A molecular dynamics study of the structure and inter-particle interactions of polyethylene glycol-conjugated PAMAM dendrimers. J. Phys. Chem. B 2009, 113, 13202–13207. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, S.; Meshinchi, S.; van Antwerp, M.E.; Bi, X.; Lee, I.; Baker, J.R. Dendrimer-Entrapped Gold Nanoparticles as a Platform for Cancer-Cell Targeting and Imaging. Small 2007, 3, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.P.; Krippner, G.Y.; Scanlon, M.J.; Chalmers, D.K. Molecular Dynamics of Variegated Polyamide Dendrimers. Macromolecules 2009, 42, 2784–2794. [Google Scholar] [CrossRef]
- Caballero, J.; Poblete, H.; Navarro, C.; Alzate-Morales, J.H. Association of nicotinic acid with a poly(amidoamine) dendrimer studied by molecular dynamics simulations. J. Mol. Graph. Model. 2013, 39, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Sánchez, V.; Vergara-Jaque, A.; Zuñiga, M.; Comer, J.; John, A.; Nachtigall, F.M.; Valdes, O.; Duran-Lara, E.F.; Sandoval, C.; Santos, L.S. In situ and in silico evaluation of amine- and folate-terminated dendrimers as nanocarriers of anesthetics. Eur. J. Med. Chem. 2014, 73, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Maingi, V.; Kumar, M.V.S.; Maiti, P.K. PAMAM dendrimer-drug interactions: Effect of pH on the binding and release pattern. J. Phys. Chem. B 2012, 116, 4370–4376. [Google Scholar] [CrossRef] [PubMed]
- Pavan, G.M.; Mintzer, M.A.; Simanek, E.E.; Merkel, O.M.; Kissel, T.; Danani, A. Computational insights into the interactions between DNA and siRNA with “rigid” and “flexible” triazine dendrimers. Biomacromolecules 2010, 11, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.B.; Mortensen, K.; Pavan, G.M.; Kasimova, M.; Jensen, D.K.; Gadzhyeva, V.; Nielsen, H.M.; Foged, C. Molecular Characterization of the Interaction between siRNA and PAMAM G7 Dendrimers by SAXS, ITC, and Molecular Dynamics Simulations. Biomacromolecules 2010, 11, 3571–3577. [Google Scholar] [CrossRef] [PubMed]
- Huißmann, S.; Likos, C.N.; Blaak, R. Explicit vs Implicit Water Simulations of Charged Dendrimers. Macromolecules 2012, 45, 2562–2569. [Google Scholar] [CrossRef]
- Antosiewicz, J.M.; Shugar, D. Poisson-Boltzmann continuum-solvation models: Applications to pH-dependent properties of biomolecules. Mol. Biosyst. 2011, 7, 2923–2949. [Google Scholar] [CrossRef] [PubMed]
- Ballauff, M.; Likos, C.N. Dendrimers in solution: Insight from theory and simulation. Angew. Chem. Int. Ed. Engl. 2004, 43, 2998–3020. [Google Scholar] [CrossRef] [PubMed]
- Lyulin, S.V.; Darinskii, A.A.; Lyulin, A.V. Computer Simulation of Complexes of Dendrimers with Linear Polyelectrolytes. Macromolecules 2005, 38, 3990–3998. [Google Scholar] [CrossRef]
- Sadanobu, J.; Goddard, W.A. The continuous configurational Boltzmann biased direct Monte Carlo method for free energy properties of polymer chains. J. Chem. Phys. 1997, 106, 6722–6729. [Google Scholar] [CrossRef]
- Filipe, L.C.S.; Machuqueiro, M.; Baptista, A.M. Unfolding the conformational behavior of peptide dendrimers: Insights from molecular dynamics simulations. J. Am. Chem. Soc. 2011, 133, 5042–5052. [Google Scholar] [CrossRef] [PubMed]
- Barra, P.A.; Barraza, L.; Jiménez, V.A.; Gavín, J.A.; Alderete, J.B. Complexation of Mefenamic Acid by Low- Generation PAMAM Dendrimers: Insight from NMR Spectroscopy Studies and Molecular Dynamics Simulations. Macromol. Chem. Phys. 2014, 215, 372–383. [Google Scholar] [CrossRef]
- Evangelista-Lara, A.; Guadarrama, P. Theoretical evaluation of the nanocarrier properties of two families of functionalized dendrimers. Int. J. Quantum Chem. 2005, 103, 460–470. [Google Scholar] [CrossRef]
- Cao, J.; Zhang, H.; Wang, Y.; Yang, J.; Jiang, F. Investigation on the interaction behavior between curcumin and PAMAM dendrimer by spectral and docking studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 108, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Vergara-Jaque, A.; Comer, J.; Monsalve, L.; González-Nilo, F.D.; Sandoval, C. Computationally efficient methodology for atomic-level characterization of dendrimer-drug complexes: A comparison of amine- and acetyl-terminated PAMAM. J. Phys. Chem. B 2013, 117, 6801–6813. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.K.; Çaǧın, T.; Lin, S.-T.; Goddard, W.A. Effect of Solvent and pH on the Structure of PAMAM Dendrimers. Macromolecules 2005, 38, 979–991. [Google Scholar] [CrossRef]
- Chen, W.; Porcar, L.; Liu, Y.; Butler, P.D.; Magid, L.J. Small Angle Neutron Scattering Studies of the Counterion Effects on the Molecular Conformation and Structure of Charged G4 PAMAM Dendrimers in Aqueous Solutions. Macromolecules 2007, 40, 5887–5898. [Google Scholar] [CrossRef]
- Nisato, G.; Ivkov, R.; Amis, E.J. Size Invariance of Polyelectrolyte Dendrimers. Macromolecules 2000, 33, 4172–4176. [Google Scholar] [CrossRef]
- Gorman, C.B.; Smith, J.C. Effect of repeat unit flexibility on dendrimer conformation as studied by atomistic molecular dynamics simulations. Polymer 2000, 41, 675–683. [Google Scholar] [CrossRef]
- Blaak, R.; Lehmann, S.; Likos, C.N. Charge-induced conformational changes of dendrimers. Macromolecules 2008, 41, 4452–4458. [Google Scholar] [CrossRef]
- Gitsov, I.; Fre, J.M.J. Stimuli-Responsive Hybrid Macromolecules: Novel Amphiphilic Star Copolymers With Dendritic Groups at the Periphery. J. Am. Chem. Soc. 1996, 118, 3785–3786. [Google Scholar] [CrossRef]
- Rathgeber, S.; Monkenbusch, M.; Kreitschmann, M.; Urban, V.; Brulet, A. Dynamics of star-burst dendrimers in solution in relation to their structural properties. J. Chem. Phys. 2002, 117, 4047–4062. [Google Scholar] [CrossRef]
- Porcar, L.; Hong, K.; Butler, P.D.; Herwig, K.W.; Smith, G.S.; Liu, Y.; Chen, W.-R. Intramolecular structural change of PAMAM dendrimers in aqueous solutions revealed by small-angle neutron scattering. J. Phys. Chem. B 2010, 114, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Topp, A.; Bauer, B.J.; Tomalia, D.A.; Amis, E.J. Effect of Solvent Quality on the Molecular Dimensions of PAMAM Dendrimers. Macromolecules 1999, 32, 7232–7237. [Google Scholar] [CrossRef]
- Maiti, P.K.; Messina, R. Counterion Distribution and ζ-Potential in PAMAM Dendrimer. Macromolecules 2008, 41, 5002–5006. [Google Scholar] [CrossRef]
- Maiti, P.K.; Goddard, W.A. Solvent quality changes the structure of G8 PAMAM dendrimer, a disagreement with some experimental interpretations. J. Phys. Chem. B 2006, 110, 25628–25632. [Google Scholar] [CrossRef] [PubMed]
- Carbone, P.; Müller-Plathe, F. Molecular dynamics simulations of polyaminoamide (PAMAM) dendrimer aggregates: Molecular shape, hydrogen bonds and local dynamics. Soft Matter 2009, 2638–2647. [Google Scholar]
- Li, T.; Hong, K.; Porcar, L.; Verduzco, R.; Butler, P.D.; Smith, G.S.; Liu, Y.; Chen, W.-R. Assess the Intramolecular Cavity of a PAMAM Dendrimer in Aqueous Solution by Small-Angle Neutron Scattering. Macromolecules 2008, 41, 8916–8920. [Google Scholar] [CrossRef]
- Huißmann, S.; Likos, C.N.; Blaak, R. Conformations of high-generation dendritic polyelectrolytes. J. Mater. Chem. 2010, 20, 10486–10494. [Google Scholar] [CrossRef]
- Tian, W.; Ma, Y. Theoretical and computational studies of dendrimers as delivery vectors. Chem. Soc. Rev. 2013, 42, 705–727. [Google Scholar] [CrossRef]
- Zhang, Y.; Thomas, T.P.; Lee, K.-H.; Li, M.; Zong, H.; Desai, A.M.; Kotlyar, A.; Huang, B.; Holl, M.M.B.; Baker, J.R. Polyvalent saccharide-functionalized generation 3 poly(amidoamine) dendrimer-methotrexate conjugate as a potential anticancer agent. Bioorg. Med. Chem. 2011, 19, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Albertazzi, L.; Brondi, M.; Pavan, G.M.; Sato, S.S.; Signore, G.; Storti, B.; Ratto, G.M.; Beltram, F. Dendrimer-based fluorescent indicators: In vitro and in vivo applications. PLoS One 2011, 6, e28450. [Google Scholar] [CrossRef] [PubMed]
- Alcala, M.A.; Kwan, S.Y.; Shade, C.M.; Lang, M.; Uh, H.; Wang, M.; Weber, S.G.; Bartlett, D.L.; Petoud, S.; Lee, Y.J. Luminescence targeting and imaging using a nanoscale generation 3 dendrimer in an in vivo colorectal metastatic rat model. Nanomedicine 2011, 7, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Mecke, A.; Uppuluri, S.; Sassanella, T.M.; Lee, D.-K.; Ramamoorthy, A.; Baker, J.R.; Orr, B.G.; Banaszak Holl, M.M. Direct observation of lipid bilayer disruption by poly(amidoamine) dendrimers. Chem. Phys. Lipids 2004, 132, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mecke, A.; Majoros, I.J.; Patri, A.K.; Baker, J.R.; Holl, M.M.B.; Orr, B.G. Lipid bilayer disruption by polycationic polymers: The roles of size and chemical functional group. Langmuir 2005, 21, 10348–10354. [Google Scholar] [CrossRef] [PubMed]
- Gardikis, K.; Hatziantoniou, S.; Viras, K.; Wagner, M.; Demetzos, C. A DSC and Raman spectroscopy study on the effect of PAMAM dendrimer on DPPC model lipid membranes. Int. J. Pharm. 2006, 318, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Ionov, M.; Gardikis, K.; Wróbel, D.; Hatziantoniou, S.; Mourelatou, H.; Majoral, J.-P.; Klajnert, B.; Bryszewska, M.; Demetzos, C. Interaction of cationic phosphorus dendrimers (CPD) with charged and neutral lipid membranes. Colloids Surf. B. Biointerfaces 2011, 82, 8–12. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.E.H.; Ghandehari, H.; Ginski, M.; Rhodes, C.A. Influence of Surface Chemistry of Poly (Amidoamine) Dendrimers on Caco-2 Cell Monolayers. J. Bioact. Compat. Polym. 2003, 18, 7–22. [Google Scholar] [CrossRef]
- Hong, S.; Leroueil, P.R.; Janus, E.K.; Peters, J.L.; Kober, M.-M.; Islam, M.T.; Orr, B.G.; Baker, J.R.; Banaszak Holl, M.M. Interaction of polycationic polymers with supported lipid bilayers and cells: Nanoscale hole formation and enhanced membrane permeability. Bioconjug. Chem. 2006, 17, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Domański, D.M.; Klajnert, B.; Bryszewska, M. Influence of PAMAM dendrimers on human red blood cells. Bioelectrochemistry 2004, 63, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Kitchens, K.M.; Foraker, A.B.; Kolhatkar, R.B.; Swaan, P.W.; Ghandehari, H. Endocytosis and interaction of poly (amidoamine) dendrimers with Caco-2 cells. Pharm. Res. 2007, 24, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Kitchens, K.M.; El-Sayed, M.E.H.; Ghandehari, H. Transepithelial and endothelial transport of poly (amidoamine) dendrimers. Adv. Drug Deliv. Rev. 2005, 57, 2163–2176. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, M.K.; Kumar, A.; McDermott, A.M.; Cai, C. Antibacterial activities of poly(amidoamine) dendrimers terminated with amino and poly(ethylene glycol) groups. Biomacromolecules 2007, 8, 1807–1811. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.V.; Leroueil, P.R.; Nett, E.K.; Wereszczynski, J.M.; Baker, J.R.; Orr, B.G.; Banaszak Holl, M.M.; Andricioaei, I. Poly(amidoamine) dendrimers on lipid bilayers I: Free energy and conformation of binding. J. Phys. Chem. B 2008, 112, 9337–9345. [Google Scholar] [CrossRef] [PubMed]
- Ainalem, M.-L.; Campbell, R.A.; Khalid, S.; Gillams, R.J.; Rennie, A.R.; Nylander, T. On the Ability of PAMAM Dendrimers and Dendrimer/DNA Aggregates To Penetrate POPC Model Biomembranes. J. Phys. Chem. B 2010, 114, 7229–7244. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.L.; Wang, Z.-G. Interactions of a charged nanoparticle with a lipid membrane: Implications for gene delivery. Biophys. J. 2011, 100, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Lipid, D.; Kelly, C.V.; Liroff, M.G.; Triplett, Ќ.L.D.; Leroueil, P.R.; Mullen, Ќ.D.G.; Wallace, J.M.; Meshinchi, Ќ. S.; Baker, J.R.; Orr, B.G.; et al. Stoichiometry and Structure of Poly(amidoamine) Dendrimer-Lipid Complexes. ACS Nano 2009, 3, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kwak, Y.; Chang, R. Free energy of PAMAM dendrimer adsorption onto model biological membranes. J. Phys. Chem. B 2014, 118, 6792–6802. [Google Scholar] [CrossRef] [PubMed]
- Mecke, A.; Lee, D.-K.; Ramamoorthy, A.; Orr, B.G.; Holl, M.M.B. Synthetic and natural polycationic polymer nanoparticles interact selectively with fluid-phase domains of DMPC lipid bilayers. Langmuir 2005, 21, 8588–8590. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Ma, Y. pH-responsive dendrimers interacting with lipid membranes. Soft Matter 2012, 8, 2627–2632. [Google Scholar] [CrossRef]
- Tu, C.; Chen, K.; Tian, W.; Ma, Y. Computational Investigations of a Peptide- Modified Dendrimer Interacting with Lipid Membranes. Macromol. Rapid Commun. 2013, 34, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Tian, W.; Ma, Y. Computer simulations of the interactions of high-generation polyamidoamine dendrimers with electronegative membranes. Soft Matter 2013, 9, 9319–9325. [Google Scholar] [CrossRef]
- Milhem, O.M.; Myles, C.; McKeown, N.B.; Attwood, D.; D’Emanuele, A. Polyamidoamine Starburst dendrimers as solubility enhancers. Int. J. Pharm. 2000, 197, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Kolhe, P. Drug complexation, in vitro release and cellular entry of dendrimers and hyperbranched polymers. Int. J. Pharm. 2003, 259, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Avila-Salas, F.; Sandoval, C.; Caballero, J.; Guiñez-Molinos, S.; Santos, L.S.; Cachau, R.E.; González-Nilo, F.D. Study of Interaction Energies between the PAMAM Dendrimer and Nonsteroidal Anti-Inflammatory Drug Using a Distributed Computational Strategy and Experimental Analysis by ESI-MS/MS. J. Phys. Chem. B 2012, 116, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Radhika, R.; Rohith, V.; Anil Kumar, N.C.; Varun Gopal, K.; Krishnan Namboori, P.K.; Deepak, O.M. Insilico Analysis of Nano Polyamidoamine ( PAMAM ) Dendrimers for Cancer Drug Delivery. Int. J. Recent Trends Eng. Technol. 2010, 4, 142–144. [Google Scholar]
- Satish Kumar, M.V.; Maiti, P.K. Structure of DNA-functionalized dendrimer nanoparticles. Soft Matter 2012, 8, 1893–1900. [Google Scholar] [CrossRef]
- Nandy, B.; Maiti, P.K. DNA compaction by a dendrimer. J. Phys. Chem. B 2011, 115, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Zheng, M.; Mintzer, M.A.; Pavan, G.M.; Librizzi, D.; Maly, M.; Höffken, H.; Danani, A.; Simanek, E.E.; Kissel, T. Molecular modeling and in vivo imaging can identify successful flexible triazine dendrimer-based siRNA delivery systems. J. Control. Release 2011, 153, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Shaunak, S.; Thomas, S.; Gianasi, E.; Godwin, A.; Jones, E.; Teo, I.; Mireskandari, K.; Luthert, P.; Duncan, R.; Patterson, S.; et al. Polyvalent dendrimer glucosamine conjugates prevent scar tissue formation. Nature 2004, 22, 977–984. [Google Scholar]
- Teo, I.; Toms, S.M.; Marteyn, B.; Barata, T.S.; Simpson, P.; Johnston, K.A.; Schnupf, P.; Puhar, A.; Bell, T.; Tang, C.; et al. Preventing acute gut wall damage in infectious diarrhoeas with glycosylated dendrimers. EMBO Mol. Med. 2012, 4, 866–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isea, R.; Hoebeke, J.; Mayo-García, R. Designing a Peptide-dendrimer for Use as a Synthetic Vaccine Against Plasmodium falciparum. Am. J. Bioinforma. Comput. Biol. 2013, 1, 1–8. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinho, N.; Florindo, H.; Silva, L.; Brocchini, S.; Zloh, M.; Barata, T. Molecular Modeling to Study Dendrimers for Biomedical Applications. Molecules 2014, 19, 20424-20467. https://doi.org/10.3390/molecules191220424
Martinho N, Florindo H, Silva L, Brocchini S, Zloh M, Barata T. Molecular Modeling to Study Dendrimers for Biomedical Applications. Molecules. 2014; 19(12):20424-20467. https://doi.org/10.3390/molecules191220424
Chicago/Turabian StyleMartinho, Nuno, Helena Florindo, Liana Silva, Steve Brocchini, Mire Zloh, and Teresa Barata. 2014. "Molecular Modeling to Study Dendrimers for Biomedical Applications" Molecules 19, no. 12: 20424-20467. https://doi.org/10.3390/molecules191220424