Metal Free Reversible-Deactivation Radical Polymerizations: Advances, Challenges, and Opportunities


1
Department of Chemistry, Istanbul Technical University, Maslak, 34469 Istanbul, Turkey
2
Center of Excellence for Advanced Materials Research (CEAMR) and Chemistry Department, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia
*
Authors to whom correspondence should be addressed.
Polymers 2018, 10(1), 35; https://doi.org/10.3390/polym10010035
Submission received: 3 November 2017
/
Revised: 6 December 2017
/
Accepted: 7 December 2017
/
Published: 29 December 2017
(This article belongs to the Special Issue Living Polymerization)
Abstract
:A considerable amount of the worldwide industrial production of synthetic polymers is currently based on radical polymerization methods. The steadily increasing demand on high performance plastics and tailored polymers which serve specialized applications is driven by the development of new techniques to enable control of polymerization reactions on a molecular level. Contrary to conventional radical polymerization, reversible-deactivation radical polymerization (RDRP) techniques provide the possibility to prepare polymers with well-defined structures and functionalities. The review provides a comprehensive summary over the development of the three most important RDRP methods, which are nitroxide mediated radical polymerization, atom transfer radical polymerization and reversible addition fragmentation chain transfer polymerization. The focus thereby is set on the newest developments in transition metal free systems, which allow using these techniques for biological or biomedical applications. After each section selected examples from materials synthesis and application to biomedical materials are summarized.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Conventional radical polymerization was already a widespread technique when Swarcz introduced the concept of living anionic polymerization for the first time in 1956 [1] and demonstrated its viability for the fabrication of block copolymers [2]. Differing from ionic polymerization, radical polymerization suffers from bimolecular termination reactions and it was therefore considered impossible to transfer the concept of living anionic polymerization to radical systems.
In radical polymerization, continuous generation of radical chains is necessary to establish a steady radical concentration which should be very low to reduce termination processes and to achieve high molecular weight polymers. Thus, initiation proceeds slowly and at any time during the polymerization reaction while the rate of propagation proceeds fast when compared to the rate of initiation. Reaction between the propagating polymer chain and a monomer occurs approximately every 1 ms and the lifetime of a propagating polymer chain, until it undergoes irreversible termination is approximately 1 s. This short reaction time essentially disables chemical functionalization or chain extension and makes the preparation of well-defined polymers or block polymers impossible.
In reversible-deactivation radical polymerization (RDRP) different criteria must be met which allow the formation of polymers with narrow molecular weight distribution. Contrary to the conventional radical polymerization, initiation in RDRP must be fast to guarantee a uniform growth of the polymer chains starting from the beginning of the reaction. Long lifetime of growing chains allows chemical reactions, such as chain end-functionalization and chain extension. Furthermore, a low degree of termination reactions is necessary. Typically, these criteria are met by a dynamic equilibrium between a dormant and an active state which can be enabled by trapping radical chains with stable radicals or by reversible transfer processes. Different mechanisms for reversible activation-deactivation processes (see Scheme 1a) exist, leading to a manifold of different approaches to RDRP. In degenerative exchange chain transfer (DT, Scheme 1b) the dormant polymer is attracted by a propagating radical to form the active species and the dormant species, P–X. This mechanism can proceed in two ways: If the capping group is an atom or a simple group, it can be transferred from radical to radical without forming any kinetically important intermediate. Iodine mediated polymerization for example follows this mechanism. A second possibility opens up, if the capping group has a double bond which is accessible to the addition of a polymer. In this case the exchange reaction occurs with the addition of the polymer radical P∙ to the polymer P′–X to form a P…X…P′∙ intermediate, which can disproportionate to P–X and P′∙. Reversible addition fragmentation chain transfer polymerization (RAFT) is the most prominent example following this mechanism. In dissociation-combination (DC, see Scheme 1c) a capped polymer P–X dissociates in the individual radicals by thermal or photochemical cleavage. The capping radical must be stable and not undergo any other reactions except recombination with the polymer radical to give the dormant species again. A typical example for DC is nitroxide mediated radical polymerization (NMRP) where nitroxides act as a capping agent. Furthermore, two catalytic pathways exist to establish a dynamic equilibrium: In reversible chain transfer polymerization (RCTP, Scheme 1d) iodine complexes of Ge, Sn, N or P compounds (AI) mediate as catalysts the abstraction of capping iodine from dormant polymer species to form a complex A…I. Transfer of an iodine radical from this complex to the propagating polymer eventually leads to the formation of the dormant species P–I again. In atom transfer radical polymerization (ATRP, Scheme 1e), the P–X bond is activated by an activator complex (typically Cu or other transition metal (Mtn) complexes) or organic photo catalysts to release the polymer radical. Transfer of the halide group to the propagating polymer chain gives the dormant species again.
DC, RCT and ATRP follow the persistent radical effect (PRE) which provides a self-regulating effect in RDRP. Persistent radicals, which are used as mediators in the reaction, cannot terminate with each other but only couple with the growing chain. Thus, every termination reaction between two propagating chains results in accumulation of persistent radicals, whose concentration steadily increases with time. This causes a decrease in the concentration of radicals and termination probability. Degenerative transfer is the system which shows the smallest perturbation of the free radical polymerization mechanism. Radicals are formed externally and polymerization stops when the source of radicals is consumed, polymerization proceeds under a steady state equilibrium as found in free radical polymerization.
Degenerative transfer does not obey the PRE and follows typically free radical polymerization kinetics with slow initiation and fast termination.
Termination in RDRP does occur, however, the risk is reduced by increasing the concentration of dormant species, establishing a fast exchange between active radicals and dormant species and reducing the polymerization rate. Self-termination can be quantified by determining the dead chain fraction (DCF) as given in Equation (1) and typically should not exceed 10% in RDRP [3].
DCF is defined as the ratio of the number of dead chains [T] to the number of all initiated chains [R − X]. Decreasing the degree of polymerization (DPT), conversion (p) or rate coefficients of termination (kt) to propagation (kp) as well as increasing monomer concentration [M]0 and reaction time (t) results in decreased DCF and increased chain end-functionality.
The lifetime of a growing chain is extended from ~1 s in free radical polymerization to 1 d in RDRP. Initiation happens fast and allows for nearly instantaneous growth of all chains. Furthermore, DCF in RDRP is below 10% while it is nearly 100% in free radical polymerization. Deactivation and activation reactions in RDRP, as well as the PRE in the case of NMRP and ATRP, allow the establishment of a steady radical concentration. In free radical polymerization and RAFT a steady radical concentration, however, is established by balancing the rate of initiation and termination. Termination reactions typically occur between newly generated short chains and long chains. In reactions following the PRE, initiation occurs fast at the beginning and all chains grow uniformly while the reaction proceeds. As a result, the probability of termination decreases with time.
Almost simultaneously with the emergence of RDRP, the concept of living polymers arose. Polymer chains formed by NMRP, ATRP or RAFT are end-functionalized and capable of initiating polymerization as macro initiators. Thus, synthesis of block copolymers became accessible. The synthesis of telechelic polymers by RDRP was summarized recently in a review article [4].
2. Nitroxide Mediated Radical Polymerization (NMRP)
NMRP became a widespread RDRP method during the last decades, particularly for styrene based monomers with various functionalities in the absence of expensive metal catalysts and under easily controllable reaction conditions. Polymers obtained by NMRP do not require additional purification steps.
2.1. Reaction Mechanism of NMRP
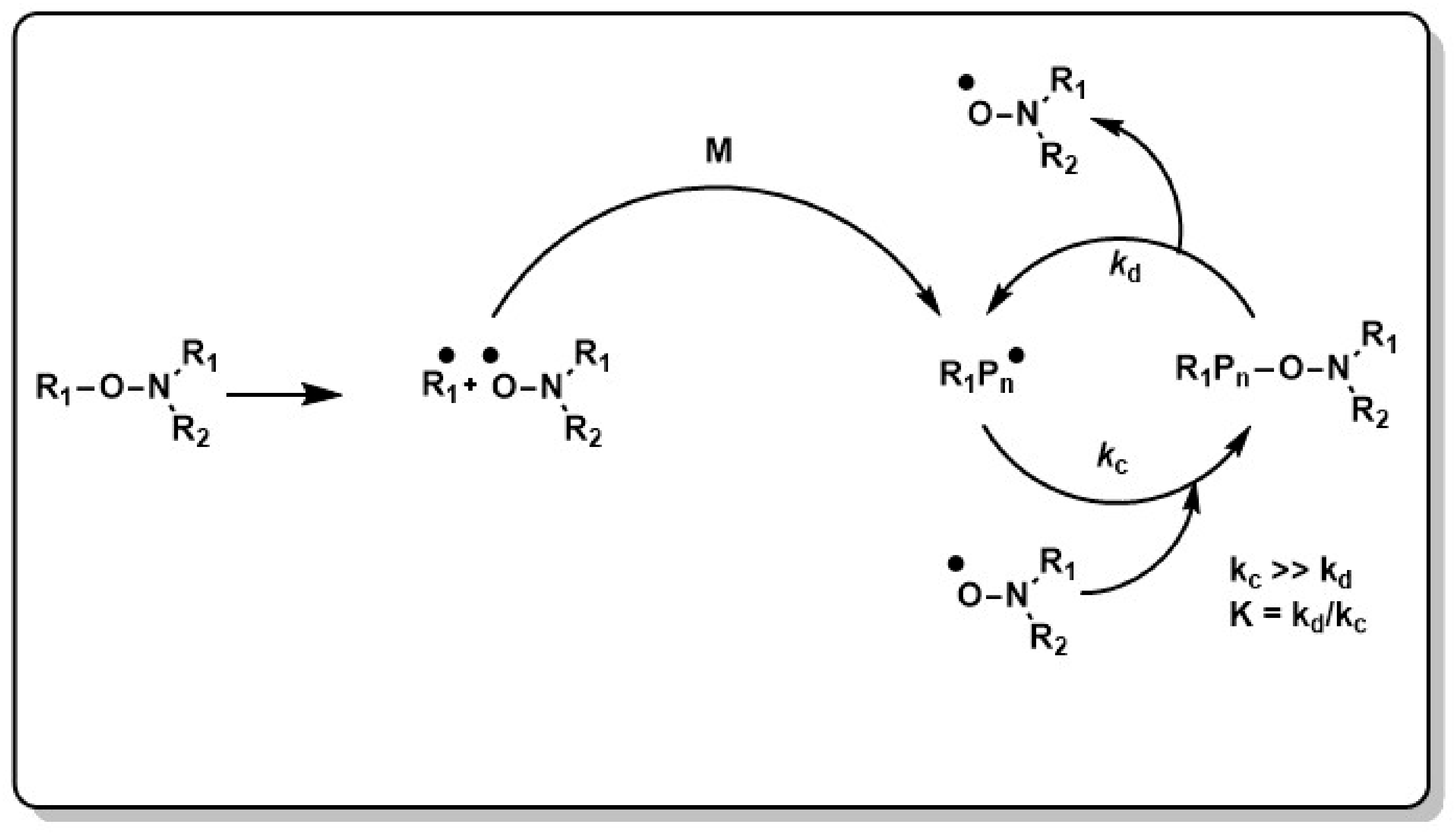
The control principle in NMRP is the reversible trapping of propagating polymer chains by stable nitroxide radicals and the formation of dormant species (see Scheme 2).
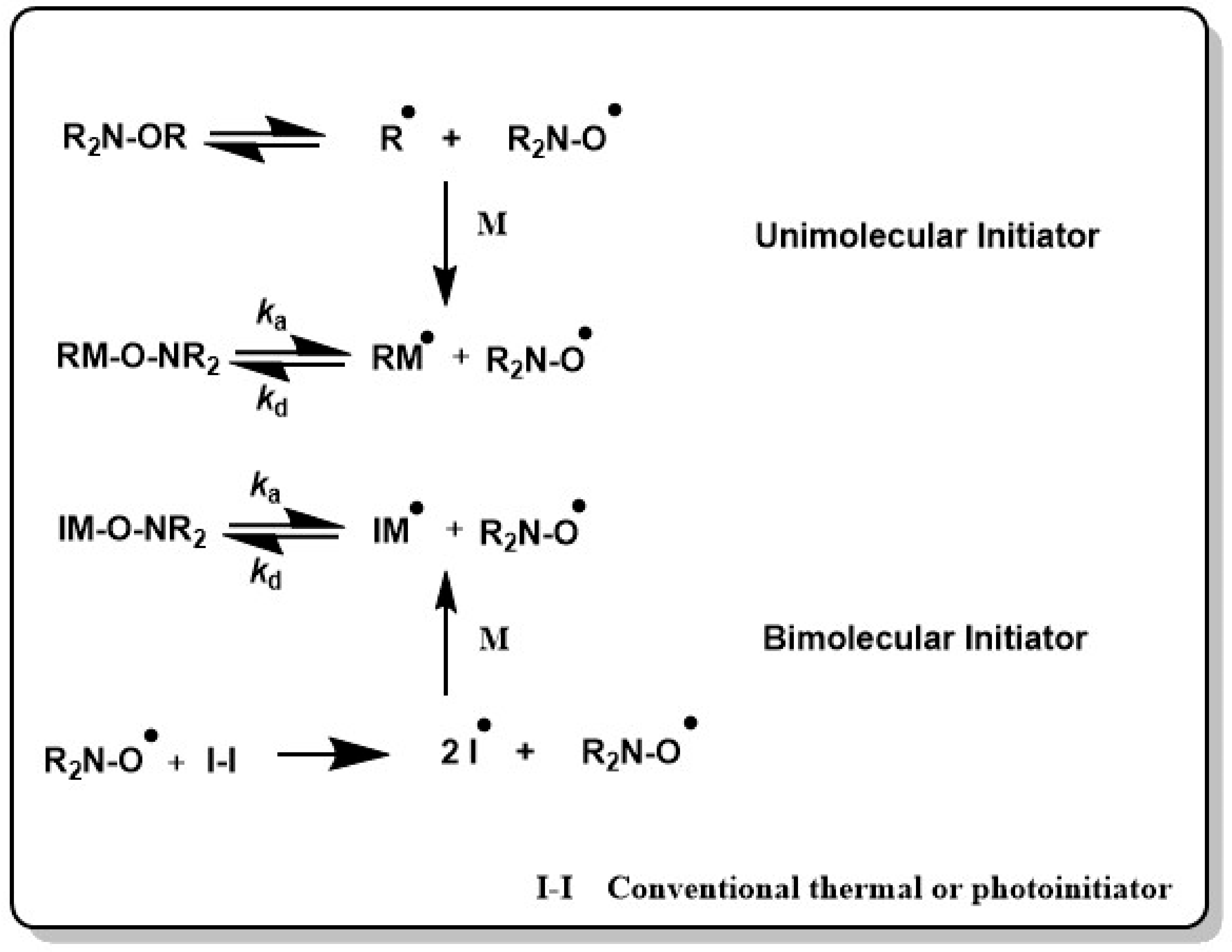
Shifting this equilibrium to the dormant form (kc >> kd, e.g., excess of nitroxide) keeps the concentration of the active chains low, which minimizes irreversible chain termination. Initiation can be either performed in a bicomponent system, where a thermal initiator such as 2,2′-azobis(2-methylpropionitril) (AIBN) or benzoylperoxide (BPO) starts the polymerization reaction in the presence of a nitroxide radical, or in a unimolecular initiation process, where the nitroxide radical is generated through chemical, thermal or photochemical cleavage of an alkoxyamine (see Scheme 3). The nitroxide can then recombine with the propagating active chain to give a nitroxide terminated radical as dormant species. This macroalkoxyamine can undergo reversible C–ON bond homolysis at typically elevated reaction temperatures and releases the stable nitroxide radical as well as the active polymer chain, which continues to propagate.
In the unimolecular approach, the concentration of the initiating radical, typically an alkyl radical which is formed after cleavage of the alkoxyamine, equals the nitroxide radical concentration which facilitates the control of the polymerization reaction.
NMRP shows all typical features of a living polymerization reaction and tuning the alkyl as well as the nitroxide fragment in the unimolecular initiator allows for preparation of α- and ω-end-functionalized polymers. Extension of the alkoxyamine systems to bifunctional alkoxyamines allows for in situ “inside-out” or “outside-in” formation of block copolymers. Due to the living character of the reaction, block polymers are accessible and a vast amount of different complex polymer structures were lately prepared by NMRP.
2.2. Development of Nitroxides for NMRP
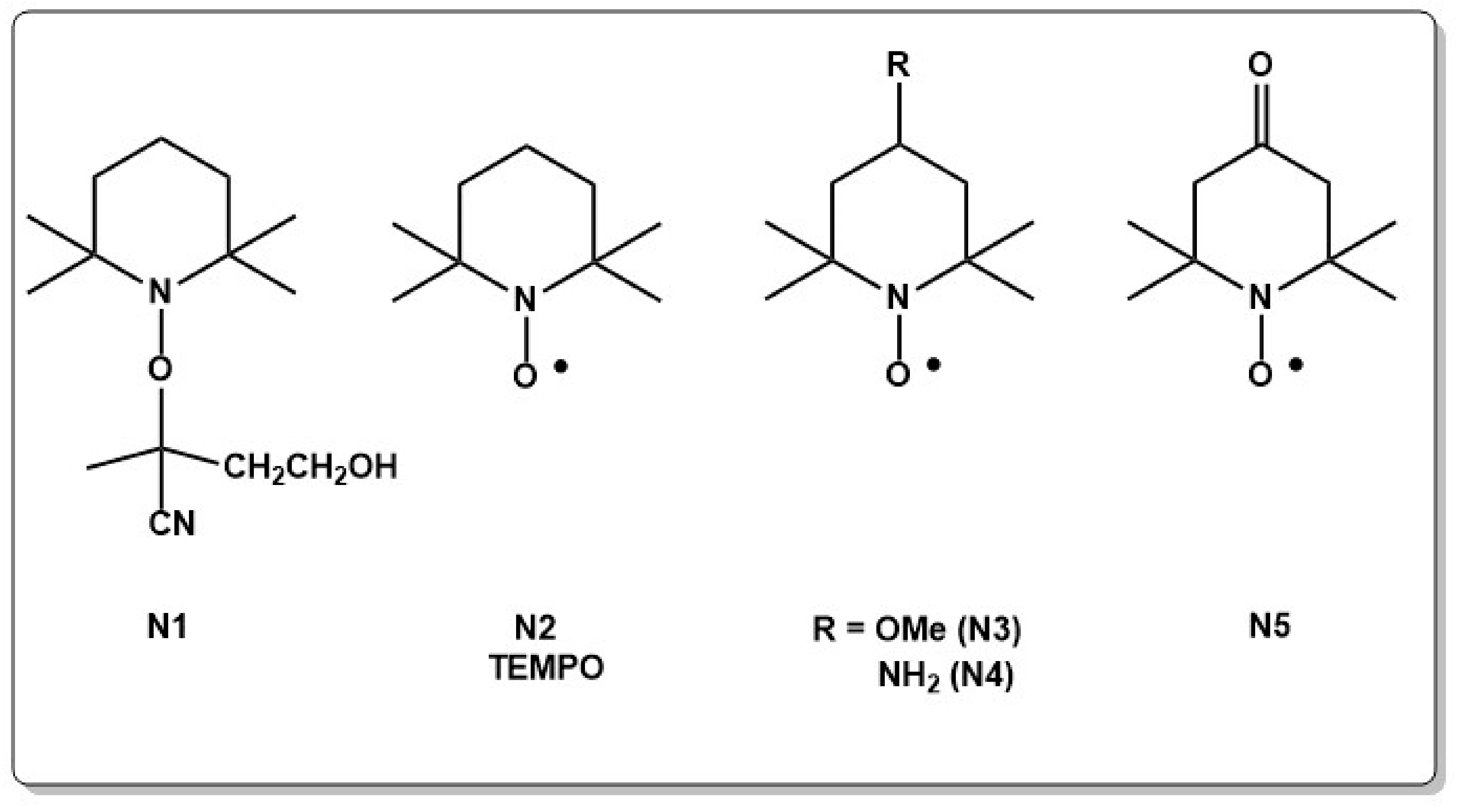
The development of NMRP dates back to research efforts at CSIRO in the seventies to reveal the chemistry of initiating polymerization reactions by radical trapping methods using nitroxides [5]. At this time, it was well known that nitroxide radicals favorably scavenge carbon centered radicals at near diffusion controlled rates and at low temperatures e.g., 40–60 °C which are typical for free radical polymerization reactions [6,7,8,9,10,11,12]. Important results from the trapping experiments, which led to the development of NMRP later on, can be summarized as: (i) trapping experiments with some nitroxides resulted in the formation of unstable products [13,14], (ii) some nitroxides showed isomerization upon heating or ageing [15] and (iii) small amounts of oligomeric products were found [13,16,17,18]. From these observations it was concluded, that nitroxides are able to undergo reversible dissociation under free radical polymerization conditions, a prerequisite for a RDRP approach. Based on the work of radical trapping experiments, the first successful NMRP polymerization was carried out in 1982 by thermally activating the trapping/dissociation of 1-(1-cyano-1-methylethoxy)-2,2,5,5-tetramethylpyrrolidine (N1 in Chart 1) as the nitroxide in the presence of methyl acrylate (MA) at 80 °C. In this attempt, shorter MA oligomers which were end-functionalized with the nitroxide fragment, could be isolated [19]. Further experiments to improve the technique were then carried out based on a bi-component system using a thermal initiator such as AIBN or BPO in combination with a wide range of nitroxides [20]. It soon became clear that the major drawback of this system is the poor reproducibility due to primary radical initiation reactions which are hard to control. Therefore, a new concept was introduced by Rize and Hawker [19,21,22,23,24], who used so-called unimolecular initiators. Upon heating, such unimolecular initiators decompose to form alkyl and nitroxyl radical fragments. The unimolecular approach led not only to a better control of the polymerization due to an exact stoichiometric ratio of initiating alkyl radical and mediating nitroxide radical but pathed also the way to α- and ω-end-functionalized polymers as will be outlined below [21]. Hawker elegantly showed with labelling experiments that the nitroxide moiety does not necessarily stay on one particular polymer chain end but migrates from one chain to another during the polymerization reaction [25]. During the last decades, development of new initiating systems [26,27], improvement of nitroxides [20,28,29,30,31], accelerating the polymerization rates by using additives [32,33,34,35,36,37] or additionally added initiators [38,39,40] and the possibility to synthesize complex polymer structures [41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57] in combination with the easiness of the method, which does not need high purity chemicals or water free systems further subsidized the success of NMRP. While first experiments which were carried out with 2,2,6,6,-tetramethylpiperidin-1-oxyl (TEMPO) (N2 in Chart 1) as the nitroxide and styrene (St) as the monomer were successful, it became soon evident that tuning the electronic structure and the steric demand of the nitroxide is a crucial point in further developing NMRP [28,58,59,60,61,62]. TEMPO was mainly limited to polymerization of St at high temperatures and long reaction times [20]. Thus, a demand in improving the performance of nitroxides in NMRP led to increased efforts to understand the electronic and steric effects of nitroxides and alkoxyamines. First attempts made use of the molecular structure of TEMPO and soon derivatives with substituents in para position (N3–N5 in Chart 1) were developed. Changing the substituent in para position allowed not only for the introduction of functional groups but also showed influence on the reactivity and 4-oxo TEMPO (N5 in Chart 1) rapidly became the first nitroxide which allowed polymerization of a wide range of monomers [63].
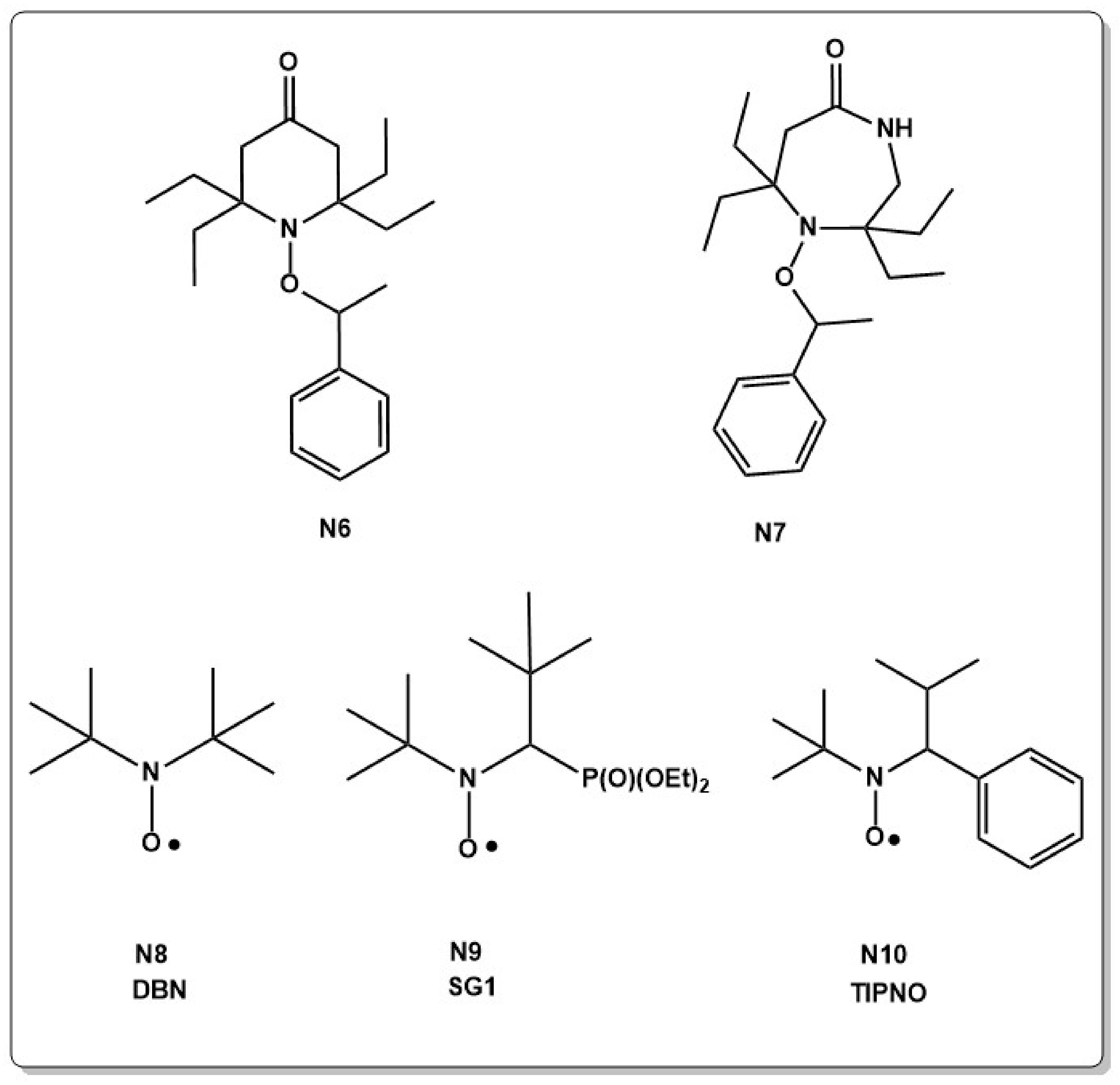
High polymerization rates and molecular weight distribution rely on facile bond homolysis of the alkoxyamine [58]. However, if the C–ON bond is too labile, the reaction does not proceed in a controlled fashion. Strong bonds on the other side can inhibit the polymerization. Thus, a lot of work was devoted to adjusting the bond homolysis behavior of different alkoxyamines. Experimental and theoretical work showed that the stabilization of the radical after homolysis, or the destabilization of the alkoxyamine before homolysis facilitates a bond homolysis [58,59,64,65,66,67,68,69,70,71,72,73,74,75,76,77]. Thereby, steric effects have a more important influence on the bond dissociation energy than electronic effects [58,78,79,80]. Spiro type alkoxyamines are, for example, sterically crowded close to the C–ON bond and allow for polymerization at temperatures as low as 50 °C. Ring enlargement of the well investigated 2,2,6,6-tetraethyl-1-(1-phenylethoxy) piperidin-4-one (N6 in Chart 2) to the seven membered alkoxamine 2,2,7,7-tetraethyl-1-(1-phenylethoxy)-1,4-diazepan-5-one (N7 in Chart 2) on the other side resulted in a three times higher equilibrium constant and increased conversion at lower temperatures [58,59,60,81].
Overcrowding, however, can introduce negative effects on the polymerization control. In 2006 Studer and co-workers started an attempt to investigate in detail the steric effects and to estimate a possible maximum in steric crowding at TEMPO based alkoxyamines. The results made clear that steric influence mainly effects the recombination between the nitroxide radical and the living chain end. Very bulky substituents decrease the possibility of a trapping reaction, which results in loss of control during the polymerization [82].
NO–C vs. N–OC bond cleavage was theoretically investigated on a large set of alkoxyamines and showed that the free energies of the NO–C homolysis depends on the properties of the alkyl fragment, while the free energies of the N–OC bond homolysis depends more on the properties of the nitroxide fragment. N–OC bond homolysis is only favored when heteroatoms can be found in α-position of the NO–C carbon. Furthermore, acyclic and indoline type alkoxyamines have a higher tendency to C–ON homolysis than other cyclic alkoxyamines [83]. A big step forward in alkoxyamine design was therefore the development of acyclic nitroxides such as the nowadays commercially available di-t-butyl nitroxide (DBN, N8 in Chart 2). Acyclic alkoxyamines showed to have much higher dissociation constants than the cyclic nitroxides. It was therefore not surprising that the breakthrough in the development of nitroxides was the design of acyclic alkoxyamines, such as N-t-butyl-1-diethoxyphosphoryl-N-oxidanyl-2,2-dimethylpropan-1-amine (SG1, N9 in Chart 2) and 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (TIPNO, N10 in Chart 2). The latter, which was developed by the groups of Tordo [61] and Hawker [28], was the first nitroxide that effectively polymerized a wide range of monomers in a controlled fashion. SG1 and TIPNO are still one of the most effective and widely used nitroxides so far [62].
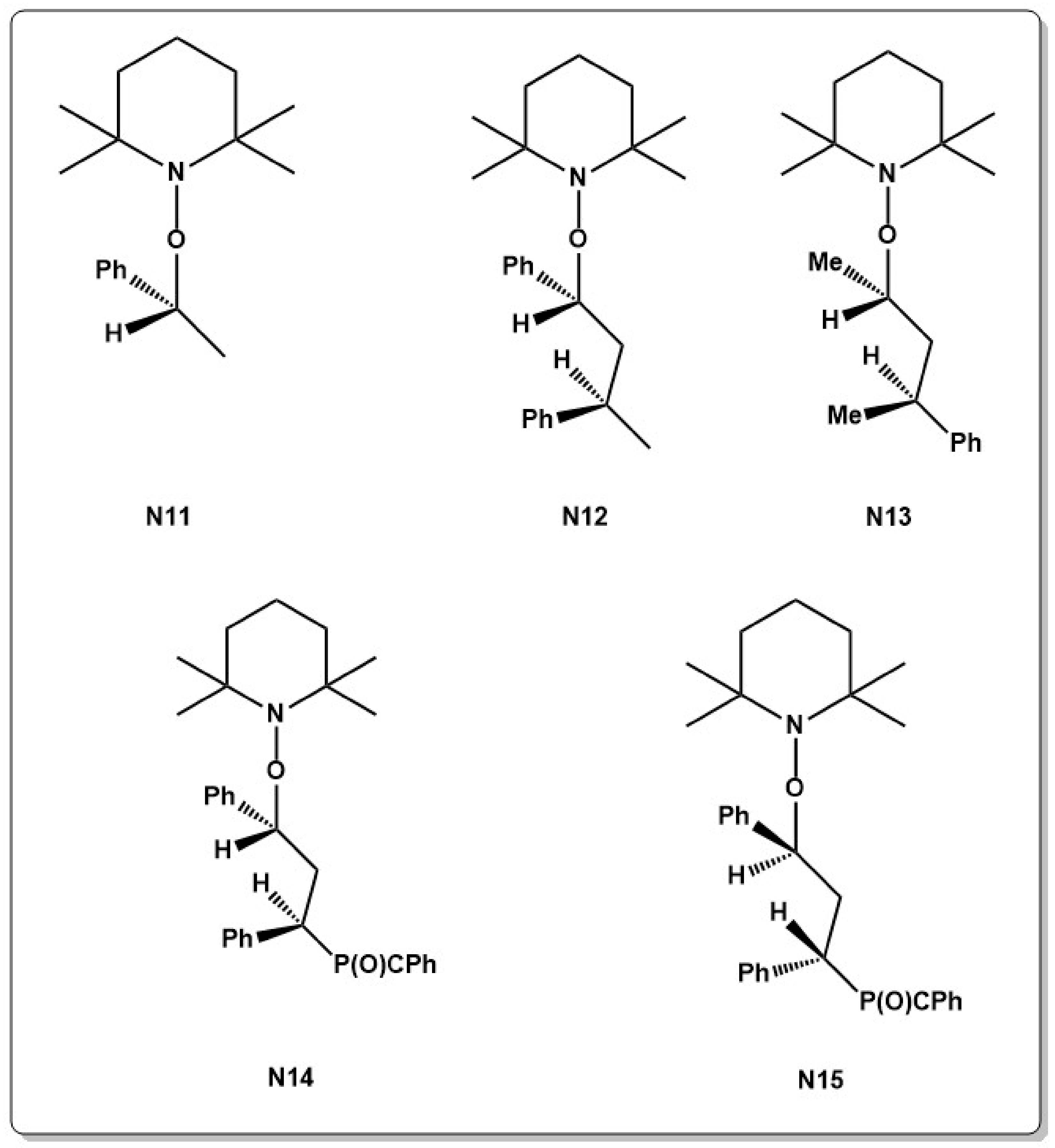
Diastereomeric effects on the homolysis of C–ON bond were observed in a theoretical study by Marque and co-workers in TEMPO alkoxyamine systems, bearing stereo centers on the alkoxy fragment (N11–N15 in Chart 3). The difference in the dissociation constant was identified as an entropic effect of the transition state of the bond dissociation. For the RS/SR pair the released alkyl radical showed the same conformation as in the alkoxyamine while for the RR and SS pairs, this effect was not observed. Thus, reorganization had to occur for the SS/RR pairs during bond cleavage, which increases the entropy term [84].
Challenges in the reaction control of NMRP concern typical side reactions such as bimolecular self-termination of transient polymeric radicals, competing N–OR bond homolysis, formation of mid-chain radicals, chain transfer to the solvent and disproportionation, alternatively named H-transfer or cross-termination. While most of the side reactions can be minimized by adjusting the reaction conditions and the choice of the alkoxyamine, cross-termination remains a bigger drawback, especially for monomers such as MA. In cross-termination, a proton is abstracted from the propagating chain end to give an olefin terminated polymer chain and a hydroxylamine. This results in the formation of dead chain ends and loss of control over the polymerization reaction with increased dispersity (D) [85,86,87]. Double bonds on the polymer chain end suffer from auto oxidation and decrease the service life and performance of the polymer [88]. The hydroxylamine can transfer the proton to another living polymer chain, thus giving a second dead end [89].
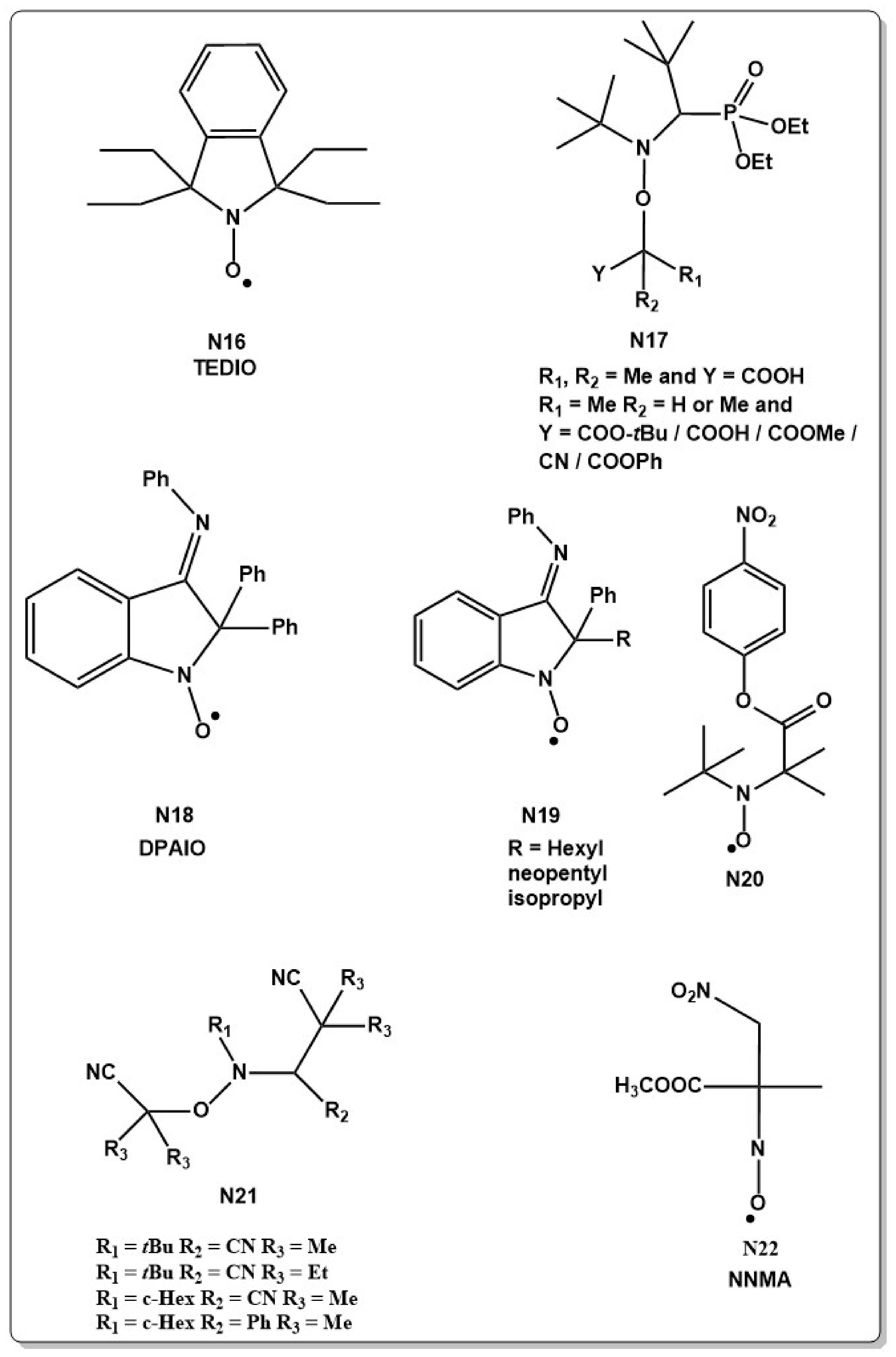
This disproportionation reaction becomes especially evident in the polymerization of methyl methacrylate (MMA) [86,90,91,92,93] which appeared to be an on-going challenge in NMRP. Since the first reports, NMRP was often criticized due to its inefficient polymerization of MMA which shows typically a low degree of control over polymerization. Poly(methyl methacrylate) (PMMA), however, is considered to be one of the most important industrial polymers. Thus, many studies were devoted to improving the controlled character. Different TEMPO and 1,1,3,3,-tetraethylisoindolin-2-oxyl (TEDIO, N16 in Chart 4) based nitroxides were tested by Rizzardo but conversion remained relatively low in the range of 30%–40% [90].
The development of SG1 and TIPNO nitroxides could partially solve this problem, however the reaction equilibrium was then shifted to the active radical and an increase in termination reactions and low conversion rates were observed [94,95]. Copolymerization of MMA with small amounts of St or other co-monomers shifts the equilibrium to the dormant species and allows reduction of termination reactions [96,97]. Presence of 4%–8% St at 90 °C for example allowed to polymerize MMA in a moderately controlled manner [96,98]. Different tertiary SG1 type alkoxyamines (N17 in Chart 4) were developed, which allowed to decrease the reaction temperature further to below 50 °C, making the polymerization reaction less prone to side reactions at lower temperatures [99] but the SG1 system did not show to be very suitable to polymerize MMA. A large dissociation constant and a small recombination constant indicates that the equilibrium constant becomes too large to effectively control the polymerization [94]. Although polymerization reactions with MMA at low temperatures were carried out successfully, a small amount of co-monomer was always necessary to prevent side reactions.
Besides St, acrylonitrile (AN) [100,101], 9-(4-vinylbenzyl)-9H-carbazole (4VBC) [102,103,104], or cyclic ketene acetals [105,106] were used as a co-monomer to control the polymerization of MA. However, copolymer formation is not always desired, even if the amount of co-monomer is small. For this reason, development of NMRP systems that allow the RDRP of MMA in the absence of co-monomer was long sought after. The requirements for successful NMRP of MMA are a low reactivity, which prevents side reactions such as disproportionation, slow homolysis and fast recombination ability [94,107,108]. Indol type nitroxides fulfill part of these conditions and 2,2-diphenyl-3-phenylimino-2,3-dihydroindol-1-yloxyl (DPAIO, N18 in Chart 4), was used to homopolymerize MMA for the first time with 80% conversion in a controlled way. The chain-end fidelity was investigated by chain extension experiments [108]. Based on this work, new DPAIO derivatives were developed for the polymerization of MMA at low temperatures (N19 in Chart 4) [109,110,111] but showed in general a high (), indicating that efficient control was not achieved. Furthermore, experiments to polymerize other monomers than MMA with this class of nitroxides were not successful. Similarly, 4-nitrophenyl-2-methylpropionat-2-yl nitroxide (N20 in Chart 4) showed control over MMA polymerization but failed to polymerize other monomers such as St [112]. Lately a new family of nitroxides (N21 in Chart 4) was reported to be able of polymerizing MMA as well as St under good control and the livingness was shown by chain extension experiments [92]. A new approach involving in-situ NMRP at 50 °C to polymerize MMA was recently reported, giving MMA with narrow molecular weight distribution. Methyl-2-methyl-3-nitro-2-nitrosopropionate (NMMA, N22 in Chart 4) was used as a mediator and the reaction was thermally initiated with the low temperature azo initiator 2,2′-azobis(4-methoxy-2,4-dimethyl valeronitrile) (V70) [113]. The polymerization showed a linear behavior in the time-conversion plot, which points to a RDRP. The observed relatively long induction time of approximately 2 h is considered to be typical for in-situ NMRP, as time is needed for the nitroxide to be formed. Furthermore, RDRP of benzyl methacrylate (BzMA) and trifluoroethyl methacrylate with the same nitroxide also appeared to be successful.
A relatively low polymerization rate independent of the employed monomer system is a further shortcoming of NMRP. Special accelerating agents such as organic acids [32,33,34], polar additives [35], reductants [36] or alkylating agents [37] could partially circumvent this problem. Lately, it was shown that also electron accepting monomers could improve polymerization rates. Copolymerization of St with acceptor monomers such as maleimide-N-phenylmaleimide (NPI), N-benzylmaleimide (BMI) and N-cyclohexylmaleimide (CMI) resulted in shortened induction periods and an increase of polymerization rates when compared to St homopolymerization [114]. Thereby, the polymerization rate increased by a factor of 5–8 in the sequence NPI > BMI > CMI.
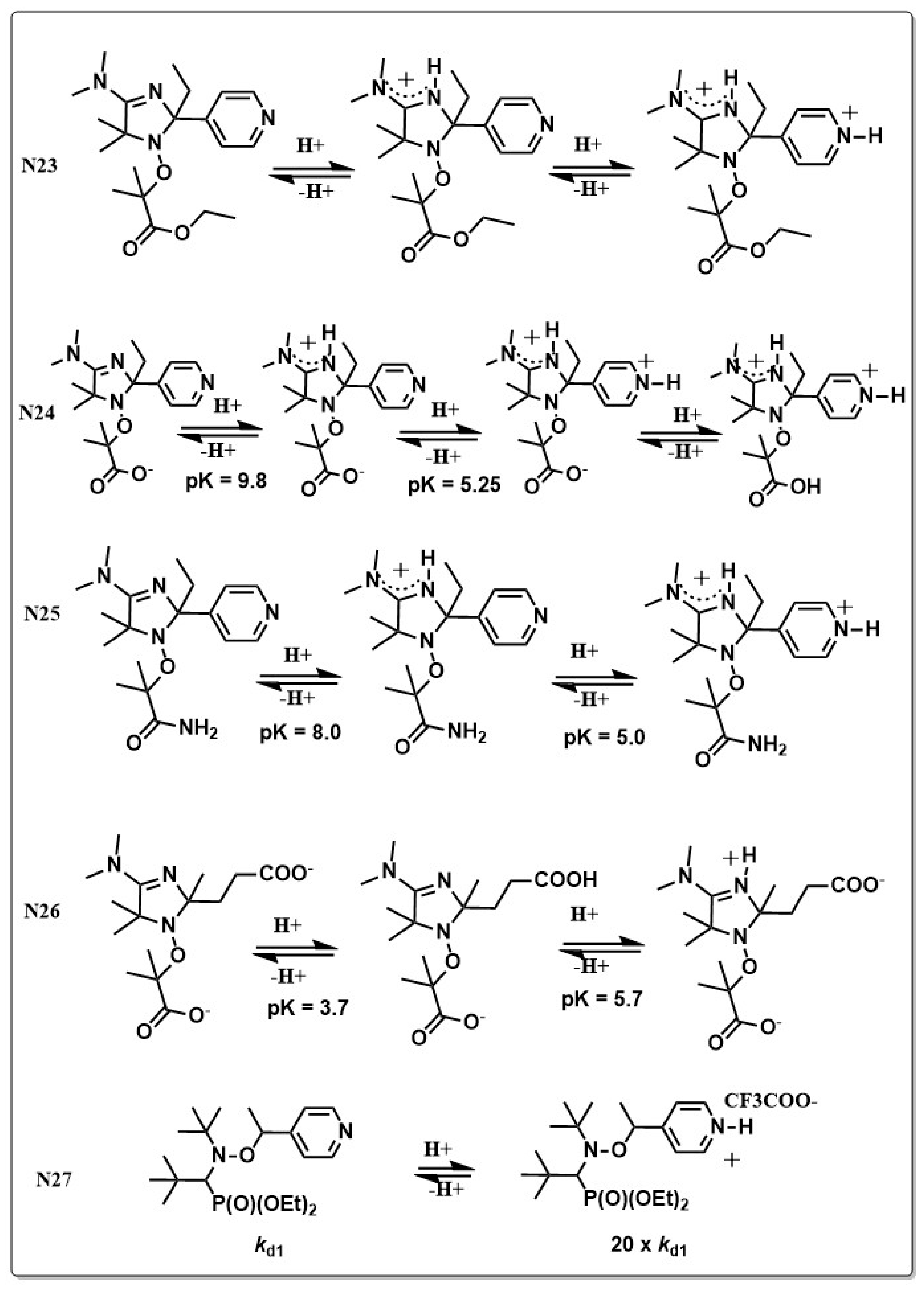
Most of the alkoxyamine initiator/mediator systems are based on thermal cleavage of the alkoxyamine. Lately much effort has been put into developing new initiating systems that can be either chemically or photochemically triggered. The first pH triggered C–ON bond homolysis and successful application in NMRP was reported in 2011 with imidazoline derivatized alkoxyamines [115]. Imidazole alkoxyamines (N23–N26 in Chart 5) bearing different protonable groups were prepared and the polymerization of St, acrylamide (AAm) and styrene sulfonate (SS) in organic and aqueous media showed successful polymerization. The degree of control, as well as the molecular weight of the formed polymer were dependent on the pH. Both protonation of the alkoxide moiety [116] or the nitroxide moiety [115] can be used to trigger chemically C–ON homolysis. Protonation of the alkoxyamine results in a drastic change of the dissociation constant, which can be increased up to 64-fold compared to the unprotonated form. The increase of the dissociation constant might stem from a higher partial negative charge on the carbon of the C–ON bond and a stabilization of the alkyl radical [117]. Furthermore, the choice of counter ion had a drastic effect as well [118]. The dissociation constant in (N27 in Chart 5) e.g., increased by 20 times when going from the unprotonated form to the protonated form with trifluorocarboxylate as counter anion. Changing the counter anion from triflate to camphorsulfonate showed an 80-fold increase when compared with the unprotonated form. This effect was explained by the solubility characteristics of the alkyl radical depending on the counter anion [118]. Furthermore, the counter anion of an activated species can help to stabilize the formed alkyl radical in solution after bond homolysis. Based on a lately published theoretical study, several TIPNO derivatives are able to control NMRP of St at room temperature when their acid groups are deprotonated, while the protonated form remains inactive and only controls polymerization at elevated temperatures. The reason was again found to be a stabilizing effect of the anion on the radical [119].
A completely different possibility to trigger NMRP was shown by photoacid generators. A tricomponent system composed of 4-methoxy TEMPO (MTEMPO) (N3 in Chart 1) and bis(alkylphenyl)iodoniumhexafluorophosphate (BAI) as the photo-acid generator and AIBN as initiator were effective in RDRP of MMA at room temperature [26]. In this approach, the photosensitizer initiates the disproportionation of the AIBN initiator and the nitroxide mediates the reaction. Further studies of this system concentrated on the influence of different initiators [120,121] and on the polymerization of different monomers [27,122,123,124,125], on the performance of alkoxyamines instead of nitroxides as mediators [126] and on the study of different photoacid generators [121,127]. Eight different azo-initiators in combination with different photoacids were examined and control over the polymerization reaction could be achieved in all cases. Expectedly, photoacids with higher excitation coefficients showed a higher control of the polymerization and provided higher initiator efficiency. The half-life of the azoinitiators on the other side had little to no effect on the control or the molecular weight of the polymers thus formed. A detailed mechanistic study, however, is lacking until now. Isopropyl thioxanthone (ITX), BPO and 2,2-dimethoxy-2-phenylacetophenone (DMPA) were used in combination with TEMPO and SG1 alkoxyamines bearing an α-hydrogen at the C–ON carbon. All experiments were carried out with Ebercyl 605 resin and gave conversions of approximately 40% in a reasonable time. A mechanism was proposed, in which the photo-initiator in the excited state abstracts a proton from the alkoxyamine, leading to a carbon centered C–ON radical which facilitates C–ON bond homolysis [128,129].
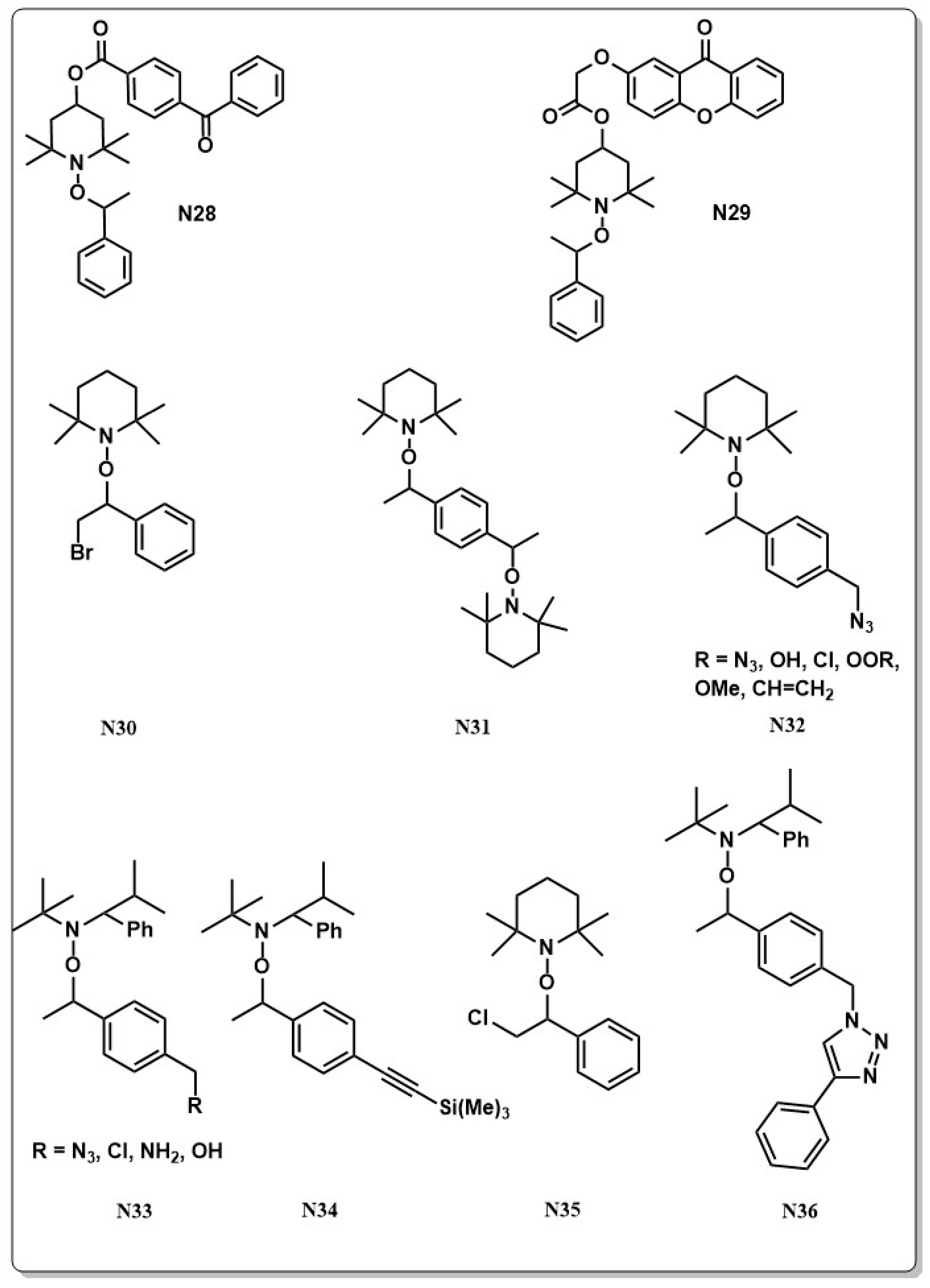
Direct attachment of chromophoric groups to the alkoxyamine guarantees a more efficient energy transfer and the design of so-called photoiniferter alkoxides (N28, N29 in Chart 6) showed controlled characteristics during the polymerization. The first report on RDRP NMRP with photoiniferter alkoxyamines was published by Lalevee and Gigmes and co-workers in 2010, who attached a chromophore group directly on an acyclic alkoxyamine to yield the (methyl 2-((4-benzoylphenyl) ((1-methoxy-2-methyl-1-oxopropan-2-yl) oxy)-amino)-2-methylpropanoate iniferter (N28 in Chart 6). Laser flash photolysis revealed that the major contribution to the C–ON bond homolysis stems from cleavage in the singlet state and polymerization of butyl acrylate (BA) with a partially living character was demonstrated [130]. Full control over the reaction was not achieved, mainly because of similar bond dissociation energies of the C–ON and C–NO bond. This problem, however, can be overcome by introducing a spacer between the chromophore and the nitroxide moiety [131]. A big advantage of photochemically triggered NMRP is the low reaction temperature at which the reaction can be performed. This not only minimizes the risk of side reactions but also allows for partial stereo-control which is not common in radical polymerization because of the planarity of the generated sp2 hybridized radicals. For this reason, polymerizations in the presence of additives and mediators or by using monomers with predetermined stereo specificity are commonly employed techniques to still gain control over stereospecifity [132,133,134]. Partial stereo-control, however, could also be achieved lately by photo NMRP where approximately 60% syndiotactic content was obtained [135].
An interesting feature of iniferter alkoxyamines is the possibility to fabricate macro photo-initiators. Chromophoric groups which are positioned on the nitroxide or alkoxy moiety can be transferred to the polymer and should be able to initiate a photopolymerization. In addition, this approach provides possibility to accomplish a dual initiation mechanism, where the NMRP is initiated thermally and the formed chromophore functionalized polymer is able to undergo photopolymerization in a second step [136]. The conceptually opposite way, which is the photochemical attachment of nitroxide moieties to a polymer backbone to yield a NMRP macroinitiator was demonstrated by our group. Benzophenone (BP) as Norrish Type II initiator in presence of TEMPO radicals was used to abstract hydrogen atoms of poly(ethylene oxide) (PEO) upon light irradiation. The TEMPO radicals can recombine with the formed radicals on the PEO backbone giving a TEMPO macroinitiator. Grafting from experiments demonstrated that TEMPO was attached successfully to the backbone and remains active for NMRP reactions [137]. The approach was further extended to visible light incorporation of TEMPO units to polyethylene (PE). PE-g-PSt copolymers were synthesized by combination of ring-opening metathesis polymerization (ROMP), Mn2(CO)10-Assisted TEMPO Substitution and NMRP [138].
One of the biggest advantages of NMRP is the accessibility of mono- or bi-functional polymers. This enables a facile approach to design complex polymer structures such as star polymers. A vast number of functional nitroxides are now reported in literature carrying e.g., chlorides, bromides, ketone, amine, alkoxy or ester groups on the alkoxide fragment (N30–N35 in Chart 6) [139,140,141,142,143]. These compounds as well as other alkoxyamines such as TEMPO, TIPNO or SG1 [28,144,145,146,147,148,149,150] were used for the preparation of functional polymers [151,152,153] or to investigate the chain end fidelity of the polymers [142,145,154,155,156,157,158,159,160,161,162].
2.3. Application of NMRP in Materials Synthesis
The development of click chemistry inspired the synthesis of alkyne and azide functional alkoxyamines (N30, N36 in Chart 6) [144,163,164] and synthesis of α-functionalized azide and alkyne polymers. Interestingly, polymerization with azide functional alkoxyamines appeared to be challenging and either click reaction had to be done before using the nitroxide as initiator (e.g., N36 in Chart 6) or chloro functional alkoxyamines (e.g., N32 in Chart 6) were used with subsequent substitution of the chloride to the azide group after polymerization. The first azide functional alkoxyamine capable of successfully initiating polymerization was reported by Braslau and co-workers. In this process, the azide functionality was conserved after NMRP. Specifically, the easiness to introduce groups capable of undergoing click reactions and the fact that NMRP is a transition metal free polymerization technique opened up new synthetic pathways in bio-conjugation applications. Besides alkyne-azide click reactions, thiol-ene Michael addition is a widely used technique for such applications. The design of a new alkene functionalized alkoxyamine based on the structure of the well-known SG1 nitroxide turned out to be a useful strategy for the synthesis of α-alkene and ω-nitroxide functionalized PSt. Interestingly, the alkene group on the alkoxy fragment was conserved during the polymerization reaction and was active in thiol-ene reactions afterwards [165].
Further examples where NMRP was employed in bio-conjugation made use of specially functionalized alkoxyamines which allow grafting to or grafting from reactions. Polymer peptide conjugates for example are an interesting class of biomaterials which show self-organizing properties [166,167,168] and the possible application as drug carriers or for molecular targeting [169,170,171]. They can be easily immobilized on surfaces with the polymer fragment to fabricate bioactive surfaces. Direct attachment of the peptide on the surface often causes structural changes of the peptide and can influence the activity of enzymes. Using a polymer spacer between the peptide and the surface is an easy way to overcome this problem [172].
A SG1 type N-hydroxysuccinimide (NHS) functionalized alkoxyamine was used to synthesize PMA comprising poly(ethylene glycol) (PEG) sidechains. The NHS functionalized polymer could then be completely or partially coupled to proteins such as lysozyme or a neuroprotective peptide [173]. The inverse sequence, which is first coupling the nitroxide to a peptide and the polymerization as grafting from the peptide afterwards, was elegantly demonstrated by Gigmes and co-workers. Thereby they could keep up the selectivity and obtained well defined conjugates [174,175]. In their approach a MMA–SG1 nitroxide attached to the glycine end-group of a preformed peptide via the carboxylate group was used as macroinitiator to obtain a St-protein conjugate [176]. Similarly, NMRP was used for both grafting to and grafting from approaches to essentially yield chitosan (CTS) modified polymers [177,178]. One of the first studies of CTS modified by NMRP reports the functionalization of N-phthaloylchitosan with 4-OH–TEMPO under γ-irradiation. The CTS-TEMPO macroinitiator was then used for grafting St [179] and SS [180]. Functionalization of the amino groups in PCTS with AAm or acrylate (Ac) fragments and radical addition of BlocBuilderTM gave a macroinitiator for grafting St, MMA and AN [181].
Very recently Marque and co-workers reported the preparation of a series of interesting spin labelled alkoxyamines. A Finland-trityl radical, which gives an easily interpretable pattern in ESR spectroscopy, was chosen as spin label of a TEMPO type alkoxyamine. No significant changes in the dissociation constants between the spin labelled and the parent alkoxyamines were observed regardless of the position of the spin label in the structure. Furthermore, the radical centered on the bulky marker did not participate in the polymerization of St, leading to spin labelled PS. This enables many possibilities in the preparation of polymers for theranostics or in vivo imaging. Proposed applications are e.g., tracking the distribution of polymer drugs or to transfer magnetic properties to organic materials [182].
Multivalent alkoxyamines possessing more than one nitroxide fragment in the molecular structure are interesting initiators for the synthesis of star polymers and complex polymer structures. Early works concentrated on the ESR investigation of TEMPO biradicals, which are separated by an alkyl chain. It was found that the coupling between the two radicals was transmitted through space rather than through the alkyl bond. The coupling coefficient turned out to be larger for longer alkyl spacers and at higher temperatures, thus at conditions where the bending of the chain is more likely, facilitating radicals to approach each other [183]. For example, the dissociation rate of a TIPNO bisalkoxyamine, where the two TIPNO fragments are separated by a (–CH2–)4 chain was twice as big as that of the mono-alkoxyamine. The proposed interaction of the radicals through space [184] was confirmed shortly later by theoretical studies [185]. Increasing the spacer length, however, has a drastic effect on the performance in NMRP. The mobility of TEMPO bisalkoxyamines with different spacer lengths with (–CH2–)1 unit to (–CH2–)4 units decreases with increasing spacer length and leads to a partially uncontrolled behavior. This was observed in the case of the (–CH2–)4 spacer, which gave PSt with a trimodal GPC trace. A better control could be achieved in other cases where (–CH2–)1–(–CH2–)3 spacers were used [186,187,188]. Reduced mobility of the nitroxide often comes along with loss of control and is also observed for smaller bi-functional nitroxide initiators. Typically, polymerization proceeds in a controlled fashion at the beginning of the reaction, which leads to a two-arm macromolecule. At higher conversions, often one arm breaks and forms a dead chain. This behavior can be explained by the bimolecular process to form the dormant species. Thereby the macromolecule needs to be captured on both sides by a free nitroxide radical in order to keep the control of the polymerization which bears steric difficulties as the chains grow. Typically, the GPC traces show a bimodal distribution, which corresponds to the formation of one and two arm fractions [189]. Although control in NMRP with bi-functional alkoxyamines suffers from steric and mobility shortcomings, they are proved to be useful in the synthesis of complex block copolymers via “inside-out” and “outside-in” polymerization. For example, a PSt-b-poly(t-butylstyrene)(PtBS)-b-PSt triblock copolymer was prepared with a TEMPO based dialkoxyamine in a facile two step “inside-out” protocol. First, PSt was polymerized, giving a bisnitroxide core from which two PSt chains were grown. Subsequent initiation of t-butylstyrene (tBS) polymerization allowed inserting a PtBS block between the PSt blocks and the nitroxide fragment. The GPC trace of the first polymerization step showed a bimodal behavior, thus some dead chains were formed during the first step. In the second step both, the triblock polymer and alkyl terminated chains were formed [190]. Furthermore, the opposite approach of an “outside-in” polymerization was applied successfully in the preparation of poly(t-butyl acrylate)PtBA-b-PSt-b-PtBA and PtBA-b-PBA-b-PtBA triblock polymers in a two-step protocol. GPC trace for the PSt block showed again a bimodal distribution, while a unimodal distribution for the PtBA block was observed [191]. This approach was also extended to tri- and tetra-functional SG1 based initiators for the preparation of three and four arm star PSt with controlled polymer weights [192] or even to six- and twelve fold functional TIPNO based alkoxyamines for the formation of star polymers with St, MA, N,N-dimethylacrylamide (NNDMAA) and isoprene (I) [193]. Increasing the number of functional groups in the nitroxide, however, comes along with a steady loss of control during the polymerization which is mainly reflected in multimodal distribution of the GPC traces.
NMRP was used excessively in the preparation of block and graft copolymers. Monomer sequence for the preparation of block copolymers in NMRP is in general very flexible. However, unfavorable kinetic parameter or side reactions should be considered by the choice of the sequence. PBA-b-PSt e.g., can be prepared by first polymerizing BA and using the macroinitiator for the initiation of the PSt block. Typically, well defined block copolymers with low are obtained. Changing the order, thus first polymerizing the PSt block and re-initiation of the BA block leads to a high in most cases [28,194,195]. However, adding free nitroxide can overcome this problem and in this way, PSt-b-BA could be obtained in a well-defined fashion [196]. Similarly, the sequence must be well considered when PMMA block copolymers are prepared.
3. Atom Transfer Radical Polymerization (ATRP)
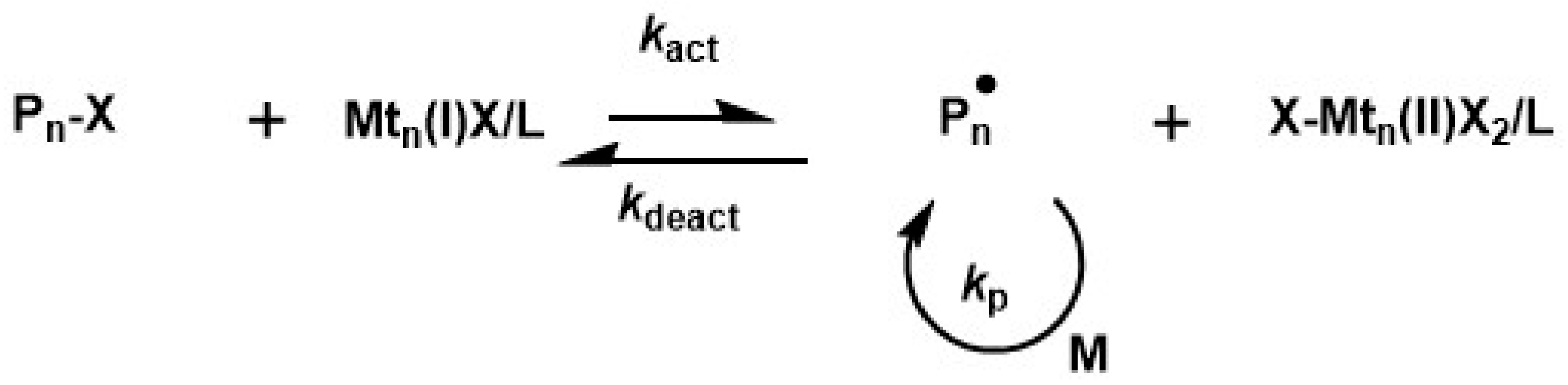
ATRP is currently the most widely studied RDRP technique. Its appeal stems from the possibility to polymerize a wide range of monomers as well as the commercial availability of reagents and catalysts. In its pristine form, ATRP is catalyzed by transition metal (Mtn) catalysts such as Cu complexes with multi-dentate ligands. Control is achieved by establishing an equilibrium, in which a dormant species R–X, bearing a halide (typically alkylhalide or halide terminated polymer), is reversely activated by a Mtn complex in its lower oxidation state. This activation step results in the formation of the Mtn complex in its higher oxidation state and a radical capable to initiate polymerization or to propagate (see Scheme 4).
3.1. Mechanism in ATRP
The radical concentration Pn∙ is determined by the ATRP equilibrium constant (KATRP) (see Equation (2)), the concentration of the alkylhalide and the ratio of the activator and deactivator concentration Cu(I)X/L and Cu(II)X2/L. Typical values of KATRP in Cu catalyzed systems [197,198] are below 10−4, thus the equilibrium is shifted to the left side and polymerization is typically operated at small radical concentrations to prevent termination.
It is important to highlight that the ATRP rate constant, as it is given by Equation (2), depends on the ratio of activator and deactivator complex concentration. This means that in theory the amount of Cu species can be arbitrarily decreased, making Cu based ATRP a catalytic process. Cu mediated ATRP follows the PRE, thus each chain termination results in accumulation of the oxidized Cu species which eventually will drastically decrease KATRP as the reaction proceeds with time [199,200]. Mechanistic aspects of the catalytic cycle were thoroughly investigated. There is currently a debate about whether halogen transfer from X–R to the activator complex Cu(I)X/L proceeds through an inner-sphere electron transfer (ISET) process with a halogen bridging transition state [201,202,203,204,205,206] or through outer-sphere electron transfer (OSET). OSET can proceed in a stepwise mechanism (OSET-SW) under formation of the RX∙ species or concerted (OSET-C) with electron transfer and bond cleavage happening in a single step. It was shown that electron transfer to an alkyl halide under ATRP conditions never gives RX∙ and that OSET-SW can therefore be ruled out [203,205]. OSET-C, however, still remains a possible pathway in ATRP. The presence of metal and other transition metal catalysts introduces Cu impurities in the final product which are problematic regarding biological and electronical applications. In the past, different new ATRP techniques such as activators regenerated by electron transfer (ARGET) ATRP [207], initiators for continuous activator regeneration (ICAR) ATRP [208,209], electrochemically mediated (e) ATRP [210,211], or zerovalent metals or sulphite in supplemental activators and reducing agents (SARA) ATRP [212,213,214,215] were developed to achieve well controlled polymerization even at low catalyst loading. Inspired by photo-induced Cu alkyne azide click reactions [216] light triggered ATRP showed to be promising in minimizing the metal catalyst loading. Thereby, light induced ATRP not only proceeds faster with higher control at lower catalyst loadings but also allows for temporal and spatial control of the reaction [217,218,219,220,221,222,223].
3.2. Recent Developments in ATRP
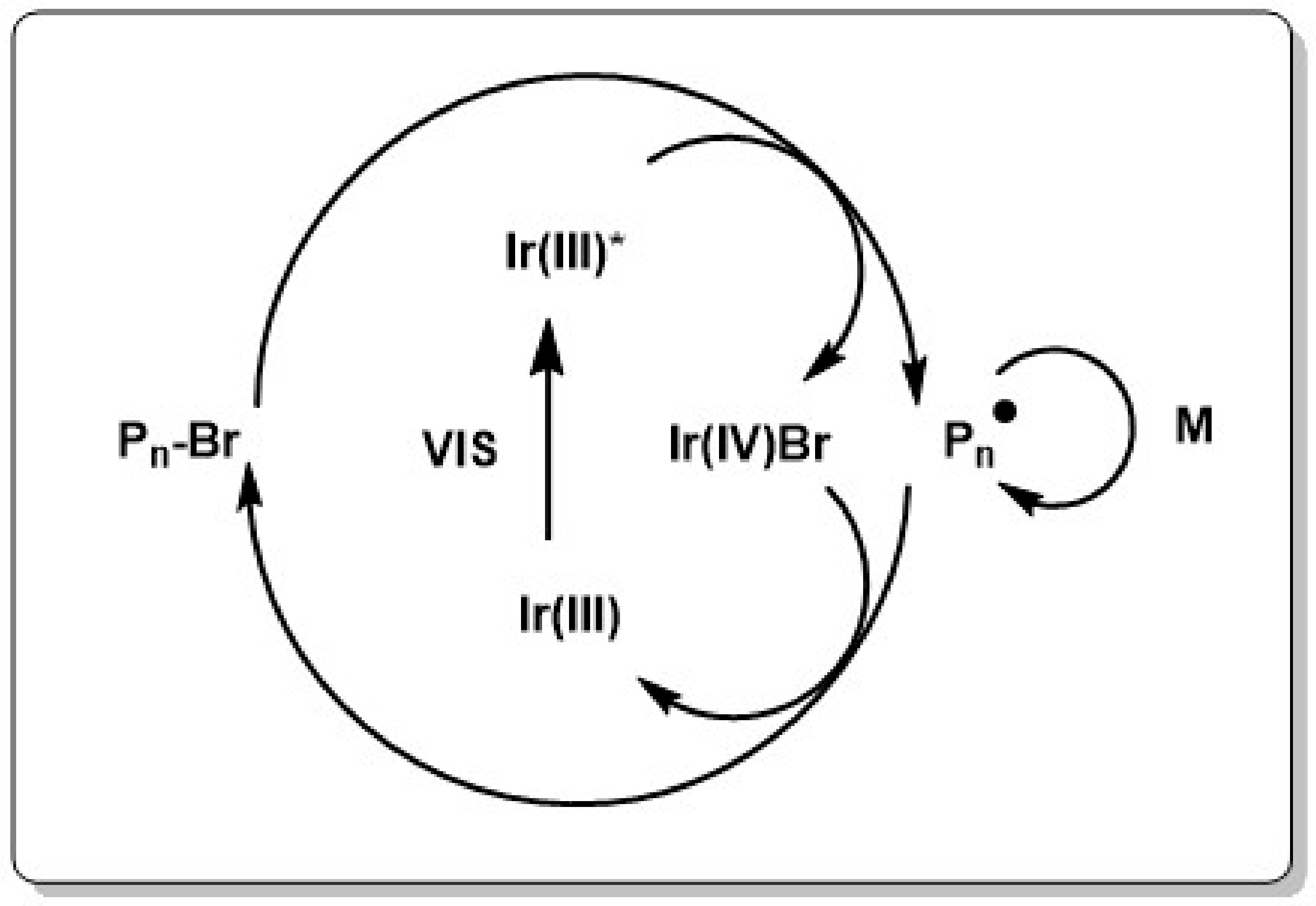
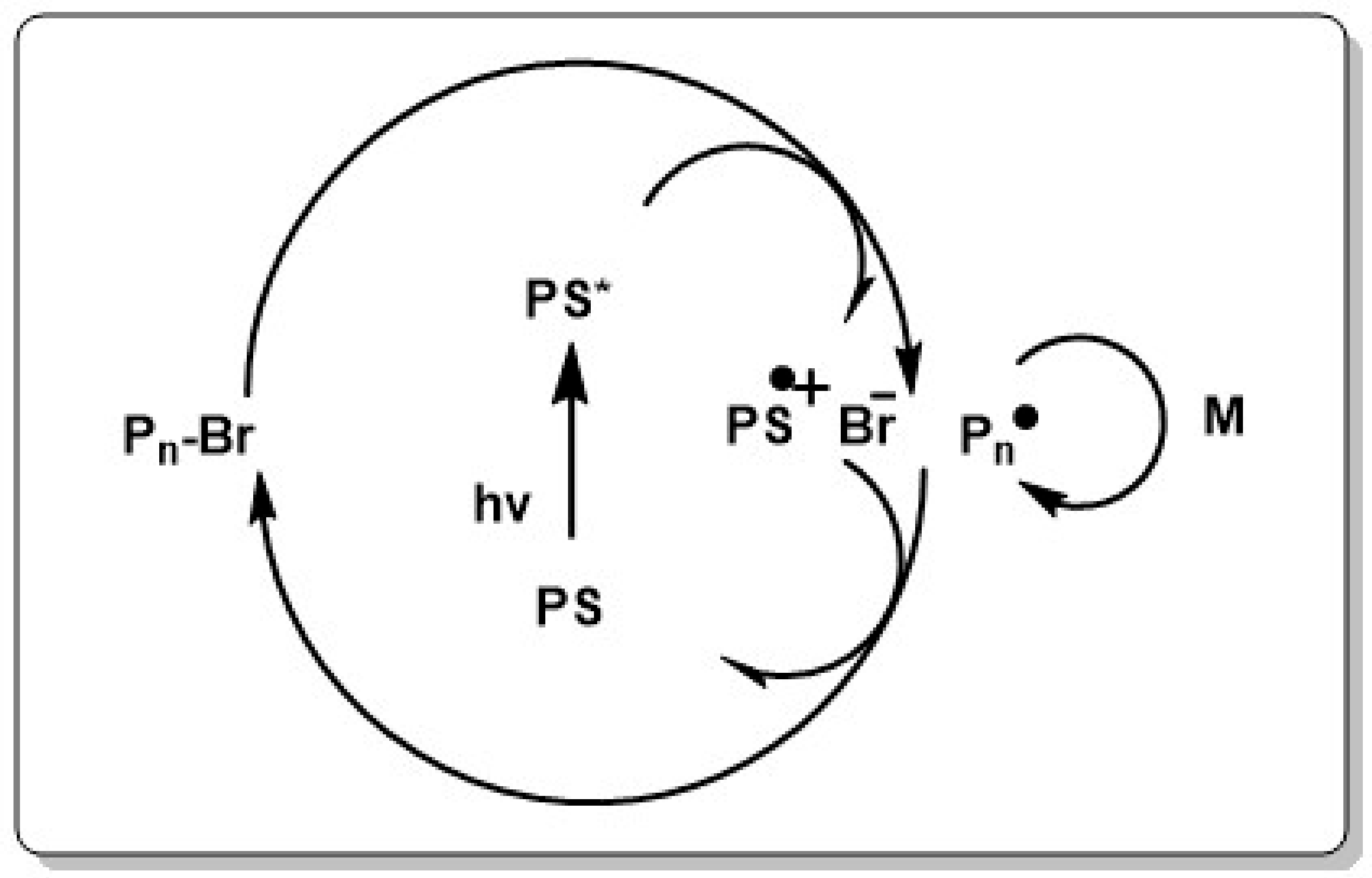
Mechanistically, photo ATRP can be activated through different light triggering reactions such as activation of a charge-transfer complex between the metal catalyst and the alkylhalide, activation of the inner sphere complex between catalyst and halide or generation of Cu(I)X/L from Cu(II)X2/L or vice versa. Recently our group could show that the spectral sensitivity can be extended to the visible spectrum by using commercially available photo-initiators [220,224,225], dyes [221], dimanganese decacarbonyl [226] or semiconductor photocatalysts [227,228], Lately in situ generation of Cu(I) from metallic Cu(0) without addition of photo-initiator was realized. Although this method showed dark polymerization, light strongly enhanced the polymerization rate. Additionally, using Cu(0) facilitates the handling of the catalyst and the recovery of the metal after polymerization [229]. An interesting case is the photo-induced ATRP polymerization with Ir(ppy)3 complex as catalyst. The fac-Ir(ppy)3 complex is able to reduce an alkylhalide upon irradiation to generate the Ir catalyst radical cation, bromine anion and an alkyl radical which is capable of initiating the polymerization. Deactivation and activation then proceeds as in conventional ATRP by switching between the Ir halide complex and the catalyst in its pristine form (see Scheme 5) [230]. ATRP reactions which follow this photocatalytic mechanism are commonly named as photo-induced electron transfer (PET) ATRP. Inspired by the photo-initiated oxidative quenching mechanism of Ir(ppy)3 it became soon clear that replacing the Ir catalyst with an organic catalyst, which is reducible in the excited state would lead to a stringent metal free system. To be able to replace the Ir catalyst, the organic catalyst must have a reduction potential in the excited state which is similar to the reduction potential of Ir(ppy)3 and must be able to form stable cation radicals which can coordinate reversibly a halide radical to act as deactivator throughout the polymerization (see Scheme 6).
Phenothiazine (PTH) has these characteristics and first attempts with 10-methylphenothiazine (Me–PTH) showed to be partially successful. While the experimental molecular weight agreed well with the theoretical molecular weight, the control over molecular weight distribution remained low. Tuning the nitrogen environment and replacing the methyl substituent with phenyl (10-phenylphenothiazine Ph–PTH) finally allowed for completely controlled polymerization, similar to conventional ATRP systems. It was assumed that change from the methylene to the phenyl substituent in the PTH reduces the catalyst decomposition, thus keeping the catalyst active for the deactivation step. This was later also supported by theoretical studies [231]. A detailed investigation of the metal free system showed that (i) the reaction is photo-catalyzed and no polymerization in absence of organocatalyst or light takes place and (ii) the reducing power of the excited state (Ered(Ph–PTH∙+/Ph–PTH* = −2.1 V vs. SCE) is the driving force of the reaction. Replacing Ph–PTH with dyes which are oxidizing in the excited state such as Eosin Y and Methylene Blue, did not result in polymerization. The highly reducing excited state of Ph–PTH also allowed for a wider functional group tolerance. 2-(dimethylamine)ethyl methacrylate (MAEMI) which failed to polymerize under Ir(ppy)3 catalysis, could be polymerized with Ph–PTH [232]. Soon after the first report on the metal free system, different other dyes were investigated as catalysts in an oxidative quenching mechanism and subsequently the range of organocatalysts including benzo[b]phenothiazines [233], N-aryl and N,N-diarylphenothiazines and perylene [234,235], dihydrophenazine [231] and N-arylphenothiazines [234] acting under UV and visible light irradiation were successfully applied. The mechanism of the oxidative quenching is depicted in Scheme 6.
Our group lately compared the performance of pyrene and anthracene as potential catalysts for metal free ATRP. Results showed that at both low and high concentrations of anthracene polymerizations resulted in lower yields. Furthermore, the turned out to be rather large and GPC traces showed a bimodal distribution when anthracene was used as catalyst. A detailed photochemical investigation revealed that excited anthracene can undergo either [4 + 4] cycloaddition with anthracene in the ground state, or reduces the alkylhalide to give alkyl radical, halide radical anion and the anthracene radical cation. The rate constant for the reduction step is rather high, which allows for efficient generation of radicals. However, the possibility of [4 + 4] cycloaddition is increased at higher concentrations, thus decreasing the polymer yield. Furthermore, NMR investigation revealed that polymer was grafted on the anthracene ring, which leads to the conclusion that anthracene radicals are not stable enough to prevent termination reactions via coupling.
In contrast, pyrene was much more tolerant towards the catalyst concentration. Initiation of the polymerization and the deactivation pathway followed the same way as anthracene but [4 + 4] cycloaddition did not occur. Excited pyrene, however, can form an exciplex with ground state pyrene molecules which can also undergo electron transfer with alkyl halides [236]. Very recently, we reported the successful photo-induced metal free ATRP using highly conjugated thienothiophene derivatives. Due to their high reducing nature these compounds favor an oxidative quenching mechanism [237]. Besides the oxidative quenching mechanism of excited state reducing dyes, the scope of metal free ATRP was lately extended to the reductive quenching mechanism (see Scheme 7) in presence of electron sacrificing compounds, such as amines.
In the proposed mechanism, the excited state dyes undergo electron transfer with electron donor amines. The formed radical anion dyes reduce the initiator alkyl halide to yield radicals which are responsible for the initiation. A back electron transfer from the halide anion to the amine radical cation concludes the formation of the dormant macroalkyl halide which returns to the polymerization cycle [238]. A detailed review highlighting the mechanistic aspects of organocatalyzed ATRP was recently published [239]. We have also demonstrated that reductive quenching with commercially available photo-initiators can be used to control polymerization in a metal free ATRP system. BP, camphorquinone (CQ), thioxanthone (TX) and ITX as long wavelength photo-initiators were used in the presence of N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as electron sacrificing compound. Experiments showed all features of a RDRP and chain extension experiments were performed successfully [240]. Further studies included Eosin Y and Erythrosin B as reducible dyes which absorb in the visible spectral range [238,241] or Fluorescein [242], all showing good performance as catalysts in the reductive quenching mechanism.
An initial problem in organocatalyzed photo ATRP was the necessity of a rather high catalyst loading which is typically in the range of 500–1000 ppm [231,232,233,234,235,236,237,238,239,240,241,242,243]. This not only introduces problems such as high cost but also runs against the demand for more sustainable “green” chemistry. Therefore, current research in metal free ATRP concentrates also on the development of highly efficient organocatalysts which are able to control polymerization even in the low ppm range. Very recently, the dicyanobenzene based donor-acceptor fluorophore 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) displayed good performance when applied with only 15 ppm concentration, yielding 90% monomer conversion within 3 h [244]. Increasing the concentration of the catalyst resulted in higher , which might stem from the tendency of 4CzIPN to independently initiate polymerization or the occurrence of side redox reactions during ATRP [245]. Another important parameter concerns light intensity as lower light intensity generates a lower amount of excited state catalyst and decreases the concentration of the catalytically effective species [246,247]. Although the oxidative quenching as well as the reductive quenching mechanism for dyes were investigated thoroughly and experimental results confirm the proposed mechanisms, the role of the excited state remained a matter of discussion. Regarding the activation and deactivation mechanism it was proposed that in metal free ATRP with Ph–PTHZ catalyst, the electron transfer activation step from the excited state of the catalyst to the alkylhalide most likely proceeds via OSET due to the strong negative reduction potential of the catalyst in the excited state. Regarding the deactivation step, it was proposed that the alkyl radical first oxidizes to a carbocation which subsequently traps a halide anion [248,249]. In polymerization of MMA and AN, the carbocation should be unstable and should lead to side reactions, which however, were not observed in the experiments. Recently, Matyjaszewski and co-workers reported a detailed mechanistic study on photomediated metal free ATRP using several PTH derivatives and other related compounds as photoredox catalysts [250]. The authors performed a detailed mechanistic study on metal free ATRP to identify the structure-reactivity relationship and to answer the question if electron transfer proceeds via ISET or OSET mechanism as well as to shed light on the deactivation mechanism. All employed catalysts possess a strong reduction potential in the excited singlet state, thus OSET might be the preferred pathway. Density functional theory (DFT) calculations revealed that electron transfer from the singlet excited state of the catalyst to the alkylhalide is a concerted dissociative electron transfer process (DET) which most likely involves a termolecular reaction of catalyst cation radical, alkyl radical and bromine anion. The most likely deactivation pathway was found to be an associative electron transfer from alkyl radical and halide anion to the catalyst radical cation to form the catalyst in its ground state and alkylhalide. In the presented calculations, it was assumed that the singlet excited state participates in the electron transfer process. This was, however, questioned lately, since also the triplet state possesses a reduction potential high enough to activate the reaction [251]. Experimental results from laser flash photolysis, fluorescence, phosphorescence and electron spin resonance spectroscopy revealed that PTHs in their triplet excited state can undergo electron transfer processes with alkyl halides in N,N-dimethylacetamide (DMA) solution. The development of photo-initiated ATRP covering the current status and future perspectives was recently summarized in an excellent review [252].
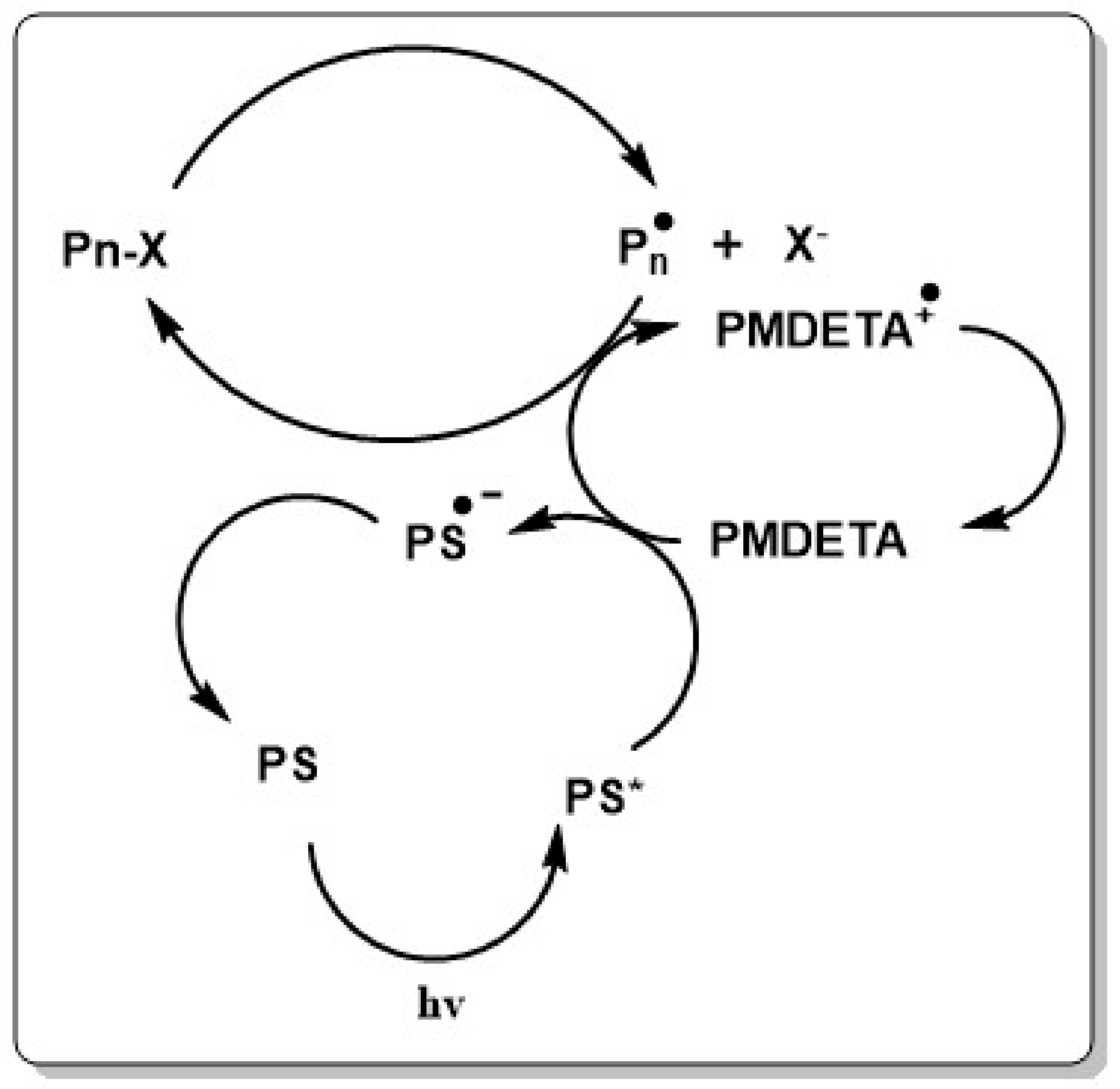
Besides using organic catalysts in metal free ATRP, a mechanistically surprisingly similar photo controlled technique called reversible complexation mediated polymerization (RCMP) evolved lately [253]. The development goes back to RCTP (see Scheme 1) which makes use of carbon centered radicals generated from alkyl iodides by organic catalysts [254,255,256,257]. Typically, the system consists of a conventional initiator, an alkyliodide as dormant species and O, Ge, Sn, P or N based catalysts [255]. Both techniques use iodine as capping agent and have the advantage of using nontoxic, cheap and easy to handle components. Both techniques, however, have important limitations with respect to monomer choice and molecular weight, thus limiting the system essentially to the polymerization of styrenics and methacrylates, leaving RDRP of acrylates unattainable [258]. In the proposed mechanism, the conventional initiator starts the polymerization reaction. The propagating polymer chain can then capture an iodide radical which stems from the iodine-catalyst complex (A–I) giving the dormant polymer chain P–I and A∙. The dormant species can then be activated subsequently by A to form the deactivator complex AI∙. The polymerization rate can be determined as
where Rp,0 is the polymerization rate without the deactivator catalyst AI. In RTCP, the polymerization rate decreases with increasing ration of (AI)/(P–X). While RCTP makes use of conventional initiators to start polymerization, RCMP works mechanistically different and resembles more the photo-induced metal free ATRP. The system is typically composed of an alkyliodide as initiator, an organic reducing amine such as tetrakis(dimethylamino)ethylene (TDAE) or triethylamine (TEA) as catalyst and the monomer. In the proposed mechanism, the amine abstracts an iodine radical from the dormant polymer chain to form an iodine-amine radical complex. This complex is unstable and reacts with a second iodine to form an iodine molecule amine complex. The propagating polymer chain can react with this complex to give the dormant species and the amine. In addition to TDAE and TEA, also tributylamine (TBA), N,N-dimethylethylenediamine (DMEDA), tetramethylethylenediamine (TMEDA), ethylenediamine (EDA) and NBS are used as amines, while 2-cyanopropyl iodide (CP–I) commonly serves as iodine source [259]. Lately RCMP could be extended to a wide range of different monomers such as BzMA, St, AN, 2-hydroxyethyl methacrylate (HEMA) and 2-(dimethylamino)ethyl methacrylate (DMAEMA).
3.3. ATRP in Materials Synthesis
The fast development of metal free ATRP led recently to first applications in surface polymerization from nanoparticles [260,261,262,263,264,265], development of metal free ATRP in continuous flow reactors [266] and application to polymerization of biomass based monomers [267].
The development of thermoplastic elastomers, which are derived from sustainable resources, gained recently attraction due to environmental issues which are raised by petroleum-based thermoplastic elastomers. Thermoplastic elastomers are typically ABA triblock copolymers in which A represents a glassy block and B a rubbery middle block. Polyacrylates, polymyrcene, polyesters or poly(menthide) are polymers which are synthesized from fatty acids or vegetable oil derived monomers. They show typically a low glass transition temperature (Tg) and are therefore used as replacement of the rubbery middle-block in ABA triblock polymers. Lactide and lactone derivatives or rosin derivatives on the other side can be used as sustainable monomers in the synthesis of glassy end-blocks. Tang and co-workers reported lately the synthesis of well controlled ABA triblock copolymers comprising a PBA middle block and rosin-derived end-blocks by ATRP. By keeping the mid-block size constant but varying the end-block size, thermoplastic elastomers with elongation at break similar to petroleum derived elastomers and with block size dependent order-disorder temperatures were obtained [268].
Further examples of ATRP polymerization of biomass derived monomers led to the development of materials with interesting properties, such as shape memory polymers [269] or cellulose rosin copolymers with a tuneable Tg [270,271].
Very recently also photo-induced metal free ATRP was used for the polymerisation of soybean oil methacrylate (SBMA), furfuryl methacrylate (FMA) and dehydroabietic ethyl methacrylate (DAEMA). SBMA, which is derived from soybean oil, has long alkyl chains and can decrease the Tg in copolymers. Tang and co-workers used metal free ATRP with PTH as the photocatalyst to obtain polymers with low and could show the synthesis of diblock copolymers with PSBMA as a soft block [268].
The development of organocatalyzed ATRP led also to application in biochemistry and biomedical sciences and in synthesis of polymer-protein conjugates. Lately, the immobilization of N-bromosuccinimide (NBS) initiator on proteins and ATRP polymerization with the protein initiator was reported [272]. A DNA synthesizer was used to prepare well-defined homopolymers, diblock copolymers and biohybrids by ATRP and MA as well as Ac polymers with excellent control were obtained. Grafting of hydrophilic and hydrophobic monomers from DNA was also reported [273]. The effective grafting under bio-relevant conditions of AAm, NNDMAA and N-vinylimidazole (NVI) from bovine serum albumin (BSA) as model protein via ICAR-ATRP was demonstrated lately. Both, homo and block copolymers could be grafted from the protein, thus livingness was retained during the polymerization [274].
4. Reversible Addition Fragmentation Chain Transfer Radical Polymerization (RAFT)
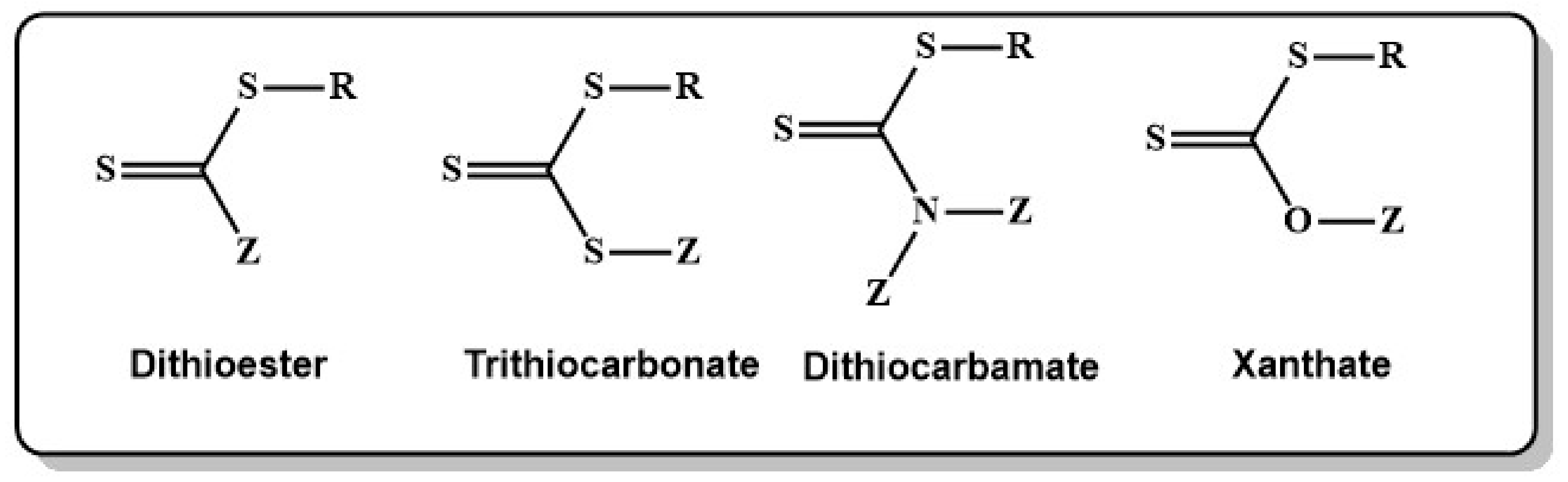
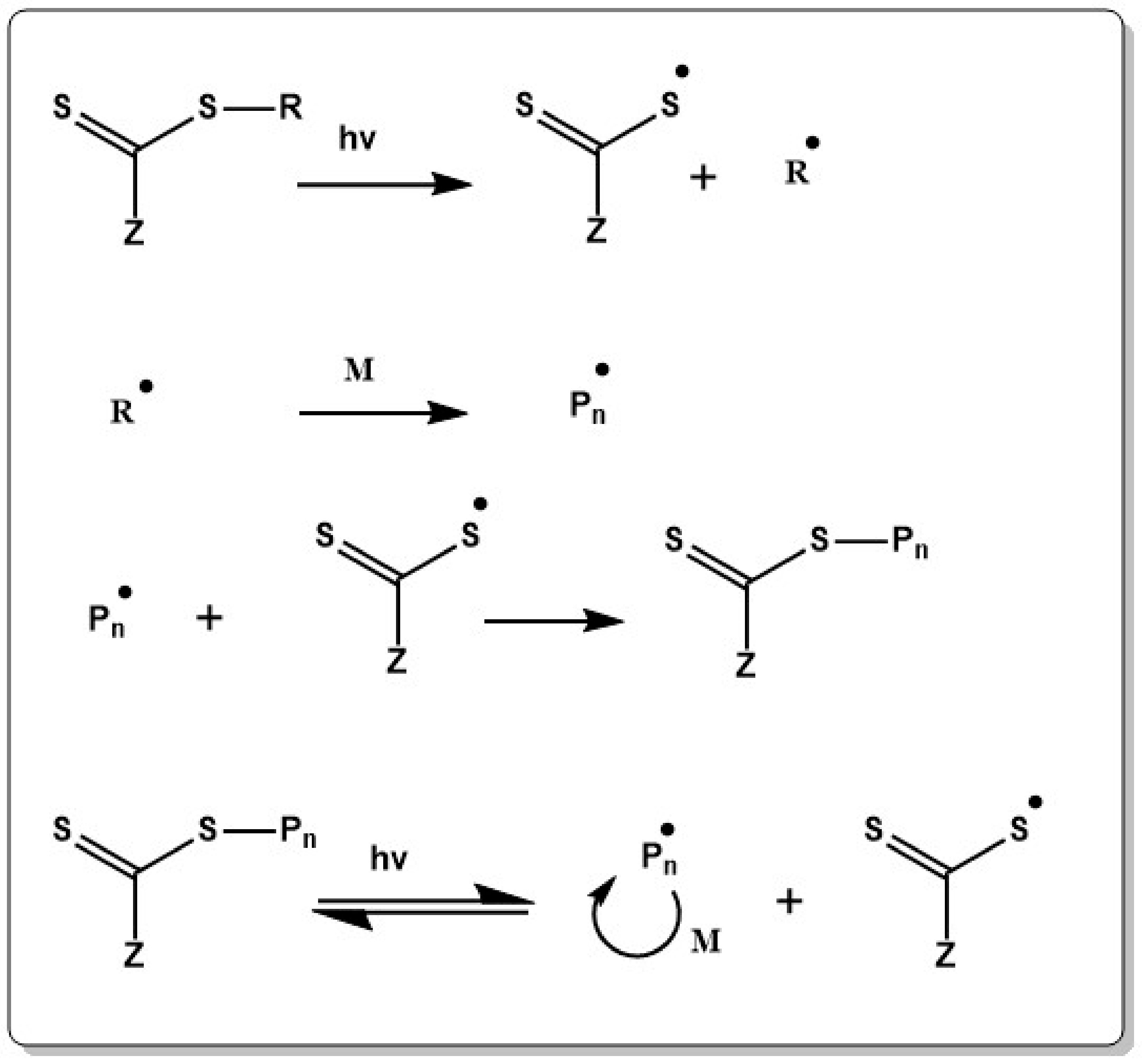
RAFT emerged as one of the most robust and versatile RDRP techniques since a first seminal publication by Rizzardo, Moad and Thang in 1998 [275]. Control of polymerization in RAFT is established by the addition of a chain transfer agent (CTA) or RAFT agent, typically a thiocarbonylthio compound (see Chart 1), which allows for reversible activation deactivation reactions.
Importantly, the equilibrium between activation and deactivation reaction is established by DT (see Scheme 1), a process which involves only exchange of the functionality. Therefore, a conventional external initiator radical (I∙) is required in the initiation step (Equation (1) in Scheme 8). The oligomeric radicals (Pn∙) formed at the first stage react with the CTA to give a radical intermediate. Fragmentation of the intermediate species releases a thiocarbonylthio group and a new radical (R∙). This newly formed radical can react with a thiocarbonyl group (back reaction in Equation (2) in Scheme 8) or initiate polymerization by reaction with monomer (Equation (3) in Scheme 8) and formation of a new propagating radical (Pm∙). After the initialization period (Equations (1)–(3) in Scheme 8), which is the time in which the CTA is fully consumed, the equilibrium between active and dormant species is established (Equation (4) in Scheme 8).
4.1. Mechanism of RAFT Polymerization
The transfer constant (ktr) for the chain transfer can be defined in terms of rate constant for the addition (kadd) and the fragmentation (kβ) reaction as given in Equation (4).
Transfer constants for different dithio compounds depend on Z, R (see Chart 7) and the type of monomer and can span more than five orders of magnitude, ranging from <0.01 to 1000. However, a transfer constant bigger than 2 is necessary to obtain small [276,277]. A good RAFT agent should therefore possess a reactive C=S double bond (high kadd), the intermediate radicals should have a high dissociation constant (high kβ) and fragment in favor of the products (kβ ≥ kadd) [278]. Bicciocchi et al. tried to decouple the R and Z group effects of the CTA and the monomer effect by polymerization of St with a benzylthiocarbonylthio RAFT agent with varying substitution of Z. Thus, changes in consumption of monomer and RAFT agent could be attributed to the influence of Z in the RAFT agent. The transfer constants decreased in the order Z = aryl > alkyl ~ alkylthio ~ pyrrole > aryloxy > amido > alkoxy > dialkylamino [279].
The mechanism proposed in Scheme 8 on the other side suggests that R must be a good homolytic leaving group relative to the propagating macromolecule as well as a radical capable of reinitiating polymerization. Importantly, the CTAs used as the RAFT agent act highly selective towards different types of monomers and must be carefully selected for each monomer system. More active monomers (MAM) such as MMA, St, MA and AN are polymerized under good control by dithioesters while the same CTA retard the polymerization of less activated monomers (LAM) such as vinylacetate (VAc), N-vinylpyrrolidone (NVP) and N-vinylcaprolactam (NVC). Latter ones, however, can be polymerized in a controlled way by xanthates (see Chart 7), N,N-dialkyl or N-alkyl-N-aryl dithiocarbamates. The equilibrium is typically rapidly established at the beginning of the polymerization, which guarantees, together with a fast initiation, a continuous growth of all polymer chains and yields polymers with narrow molecular weight distribution. Since conventional initiators are used in the initiation step, termination processes are unavoidable. The degree of termination reactions depends mainly on the initiator concentration and the dissociation rate of the initiator. Under typical RAFT conditions, the number of dormant thiocarbonylthio polymer chains is higher than the number of initiator derived chains and therefore most chains remain living. However, to maximize the chain fidelity, the initiator concentration can be minimized and a target molar mass, which is lower than that of polymer formed in absence of CTA (free radical polymerization condition), must be aimed for [278,280,281]. Additionally, the monomer conversion should be lower than 90% which makes isolation and purification of the polymer necessary before it can be used in chain extension reactions.
According to the mechanism, the number of propagating species and the polymerization rate should not be affected by the concentration of the RAFT agent. However, the rate of polymerization in RAFT with dithiobenzoates as CTAs appears retarded with increasing CTA concentration [281] The retardation of the polymerization rate for some specific RAFT agents was intensively studied and three possible mechanisms are reported:
In the slow fragmentation model, retardation takes place due to a slow fragmentation of the intermediate two arms adduct. This results in a small fragmentation constant (large equilibrium constant), which is in agreement with the experimental observations. However, the model predicts concentrations of propagating radicals and intermediate adduct radicals which differ from the experimental conditions [281,282].
In the intermediate radical termination (IRT) model, the retardation is assumed to be caused by cross termination reaction between adduct radicals and propagating radicals [283]. The intermediate radical termination with oligomers (IRTO) model finally proposes termination between adduct radicals and oligomeric propagating radicals. The equilibrium constants and concentration of radical species are close to the experimentally obtained values. However, retardation should occur over the full conversion range which is not observed experimentally [284,285].
The partially contradictory models and the fact that a mechanism which holds in all cases could not be established, led to the formation of a IUPAC task group in 2005 with the aim to understand in detail the mechanism of RAFT polymerization. The group lately summarized the different models which partially explain the retardation in RAFT and an extended discussion in a report [286].
RAFT is the RDRP which comes closest to the conditions of conventional free radical polymerization. This is especially an advantage, since the strength of free radical polymerization, such as large choice of monomers, large range of solvents to be used [287,288,289], the possibility to carry out polymerization at a wide range of temperatures [290,291,292] and a high functional group tolerance [293,294] can be directly transferred to RAFT. RAFT essentially does not require high temperatures as for example NMRP or metal catalysts as in ATRP. This underpins its strength for biological applications [293]. Furthermore, RAFT appeared suitable for controlled polymerization of cyanoacrylates (CA) when macromolecular RAFT agents were used [295] while ATRP or NMRP are unsuitable for the polymerization of CA mainly due to the basic character of the reagents, such as amine ligands in ATRP. RAFT, however, also has some drawbacks, such as slow polymerization time, oxygen intolerance and the necessity to use external initiators, which increase the probability of termination reactions.
4.2. Recent Developments in RAFT
As outlined before, the choice of the right RAFT agent is crucial for successful polymerization. The efficiency highly depends on the interplay between the monomer and the properties of the leaving group R and the activity of the group Z [296]. The main families of RAFT agents are dithioesters [297], dithiocarbamates [298], trithiocarbonates [299] and dithiocarbonates (or xanthates) [298] (see Chart 7) with the common structure representing a Z group linked by a carbon, a nitrogen, a sulphur or an oxygen atom. Different phosphorus containing RAFT agents which bear the phosphorus atom in the R group, such as phosphine oxide-containing (R1R2P(O)–) R groups were developed and found to be useful for anchoring RAFT functionalities on the surface of inorganic quantum dots [300,301].
Furthermore, RAFT agents with phosphine and phosphine boron complex groups were applied for the preparation of end-group functionalized polymers as intermediates for the fabrication of polymer-peptide conjugates or biocompatible polymers [302]. First studies with agents bearing the phosphorus atom in the Z group were undertaken with phosphoryl and (thiophosphoryl)dithioformates of the formula R–S–(C=S)–P(X)(OR)2 (X = O or S) [303,304]. However, polymerization of St resulted in the formation of polymers with uncontrolled molecular weight, most probably due to a too slow rate of radical formation. Lately, Barner-Kowollik and co-workers were able polymerize 2-hydroxyethylacrylate (HEA) mediated by 2-cyano-prop-2-yl-(diethoxyphosphoryl)dithioformate [305] yielding polymers with small . Furthermore, low molecular PSt could be obtained in a controlled polymerization by benzyl(diethoxyphosphoryl)dithioformate as the RAFT agent [306,307,308]. Dithiocarbamates can adopt zwitterionic forms which localize a positive charge on the nitrogen and a negative charge on sulphur atom. This reduces the double bond character of the thiocarbonyl unit and results in a lower reactivity towards radical addition and in smaller transfer constants [309,310]. As a consequence, they show reduced efficiency to polymerize MAMs [309]. The inhibition effect of dithioestes and trithiocarbonates on the polymerization of LAMs, on the other side, stems from the poor radical leaving group ability, which depends on the nature of R [311]. This selectivity effects hamper successful synthesis of block copolymers with MAMs and LAMs, such as the synthesis of poly(MAM)-b-poly(LAM) copolymers. To overcome this problem, stimuli-responsive RAFT agents which can polymerize both LAMs and MAMs were developed. N-(4-Pyridinyl)-N-methyldithiocarbamates for example can effectively polymerize LAMs such as VAc, NVP or NVC. In the presence of a strong acid, however, the protonated form can control successfully the polymerization of MAMs such as MA, BA and MMA [311,312,313]. Thus, switchable RAFT agents allow to synthesize block copolymers which are composed of more activated and less activated blocks, which would otherwise not be possible with typical RAFT agents [314]. For example PDMAA-b-PNVC block copolymer was prepared recently by the sequential polymerization of the respective monomers [315]. However, reactivity with respect to the different monomer classes is often compromised. Introducing electron-withdrawing groups to Z or R significantly enhances the transfer constant of the RAFT agents. This was lately exploited by the design of highly effective switchable RAFT agents with a pyrazole ring as Z group and substitution of the pyrazole in 4-position. The new agents appeared to be highly active in controlling the polymerization of MAMs and LAMs and PMAM-b-PLAM block copolymer formation [316].
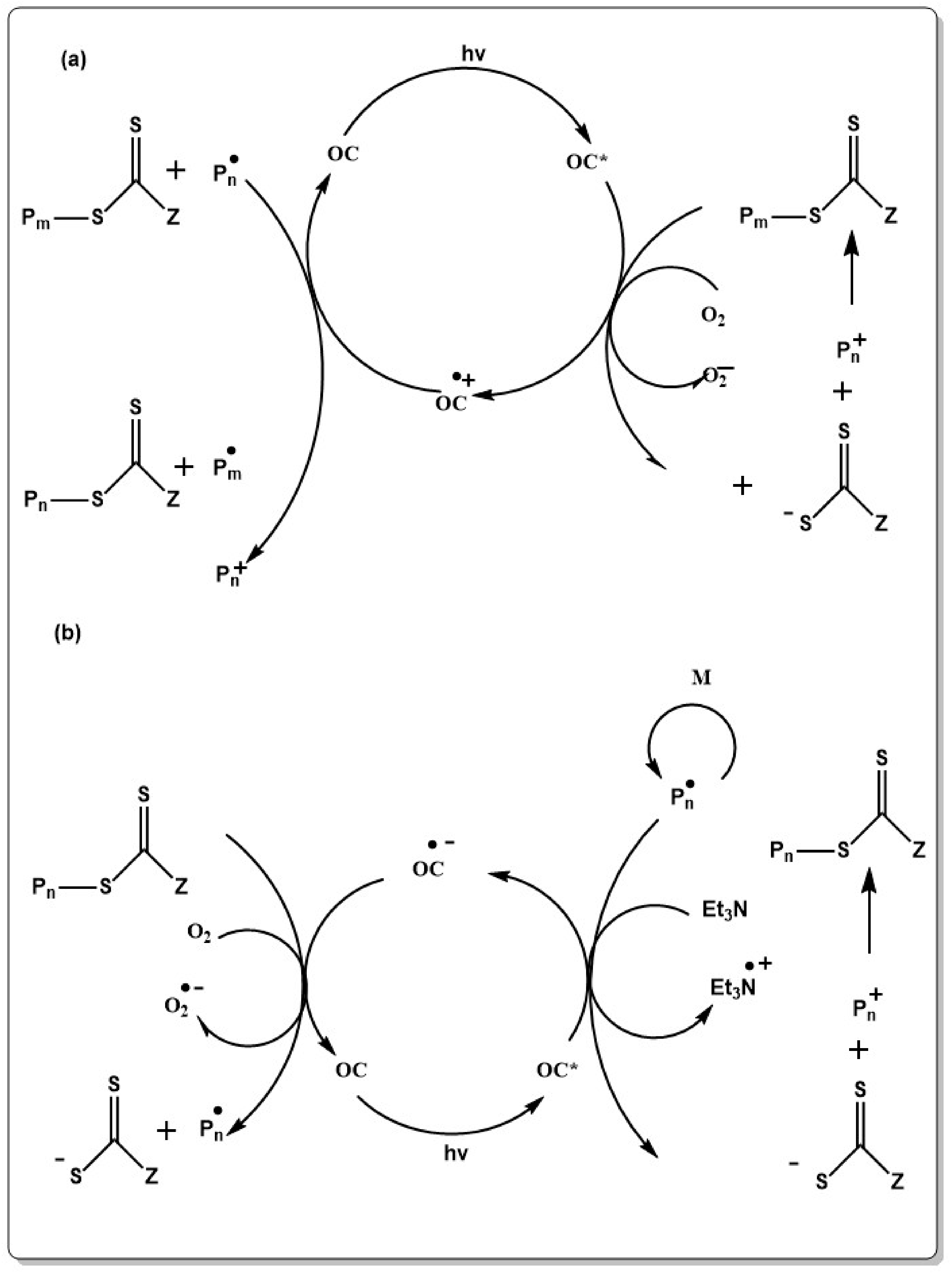
Generally, thermal initiators are used in RAFT polymerization and the reaction is carried out at elevated temperatures. This causes problems associated with self-termination or irreversible chain transfer which lowers the degree of living chains [317,318,319]. Thus, extensive effort was put in finding alternative initiation methods, which allow performing RAFT under initiator free conditions or at lower temperatures. Obviously, initiation mechanisms which use light or high energy radiation appeared to be useful in developing RAFT polymerization free of external initiators. In the first report on irradiation initiated RAFT polymerization, MA, St and MMA was polymerized in the presence of dibenzyl trithiocarbonate under γ-irradiation resulting polymers with small [320]. In the proposed mechanism, the S-benzyl linkage is sequentially cleaved under γ-irradiation liberating the benzyl radical as initiating species and a thiocarbonylthiyl radical. However, it soon became evident that the high energy radiation enables side reactions such as branching. Thus, initiating systems which respond to longer wavelength irradiation were developed. In the initial studies, dithiocarbamates termed as iniferters were used [321]. The reaction mechanism under UV light irradiation, however, most probably proceeds via a photochemical cleavage of the dithio compounds rather than through DT as depicted in Scheme 9 [322,323].
Polymerization of MMA and St under UV irradiation showed controlled behaviour and a narrow molecular weight distribution (MWD) at low monomer conversions. A significant broadening of the MWD was observed at higher monomer conversions, which was traced back to the decomposition of transfer sites at the end of the polymer chain and which eventually led to deactivation of the RAFT process [324]. The limitation in terms of conversion, however, can be overcome by using long wavelength photo initiators for radical generation and CTAs which do not absorb in the spectral regime of the radiation source. This was shown by Cai and co-workers who used S-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate (DDMAT) and the long wavelength photo initiator (2,4,6-trimethylbenzoyl)diphenylphosphine oxide (TPO) as a bi-component system. Upon irradiation, the photo-initiator generates radicals which start the polymerization reaction and the RAFT polymerization proceeds according to the mechanism described in Scheme 8. Importantly, the wavelength of the light source must match the spectral response of the photo initiator but must not induce cleavage of the CTA. MMA could be polymerized in a controlled way with up to 85% conversion [325,326]. Furthermore, the choice of the photo initiator allows polymerization at long wavelengths, specifically at the visible range or under sunlight [327]. Very recently this approach was extended to RAFT polymerization of AN under blue LED light. 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene was used as initiator in presence of 2-cyanoprop-2-yl-1-dithionaphthalate as transfer agent and gave PAN with narrow molecular weight distribution in a RDRP [328]. Inspired by a vast variety of phenacyl based photo initiators our group lately developed a phenacyl morpholine-4-dithiocarbamate (PMDC) RAFT agent for UV irradiated RAFT polymerization [329]. The phenacyl unit as R compound in PMDC appeared to be an easy leaving group, which is necessary for fast initiation. The polymerization of St proceeded in a controlled fashion and the morpholino thiocarbamate moiety was attached to the chain end with high end-group fidelity. Similar to the other reports on thiocarbonyl initiators [324,330] PMDC acted as RAFT agent in the process. Thus, light intensity as well as wavelength must be carefully selected to maintain high end-group fidelity in photo RAFT polymerization.
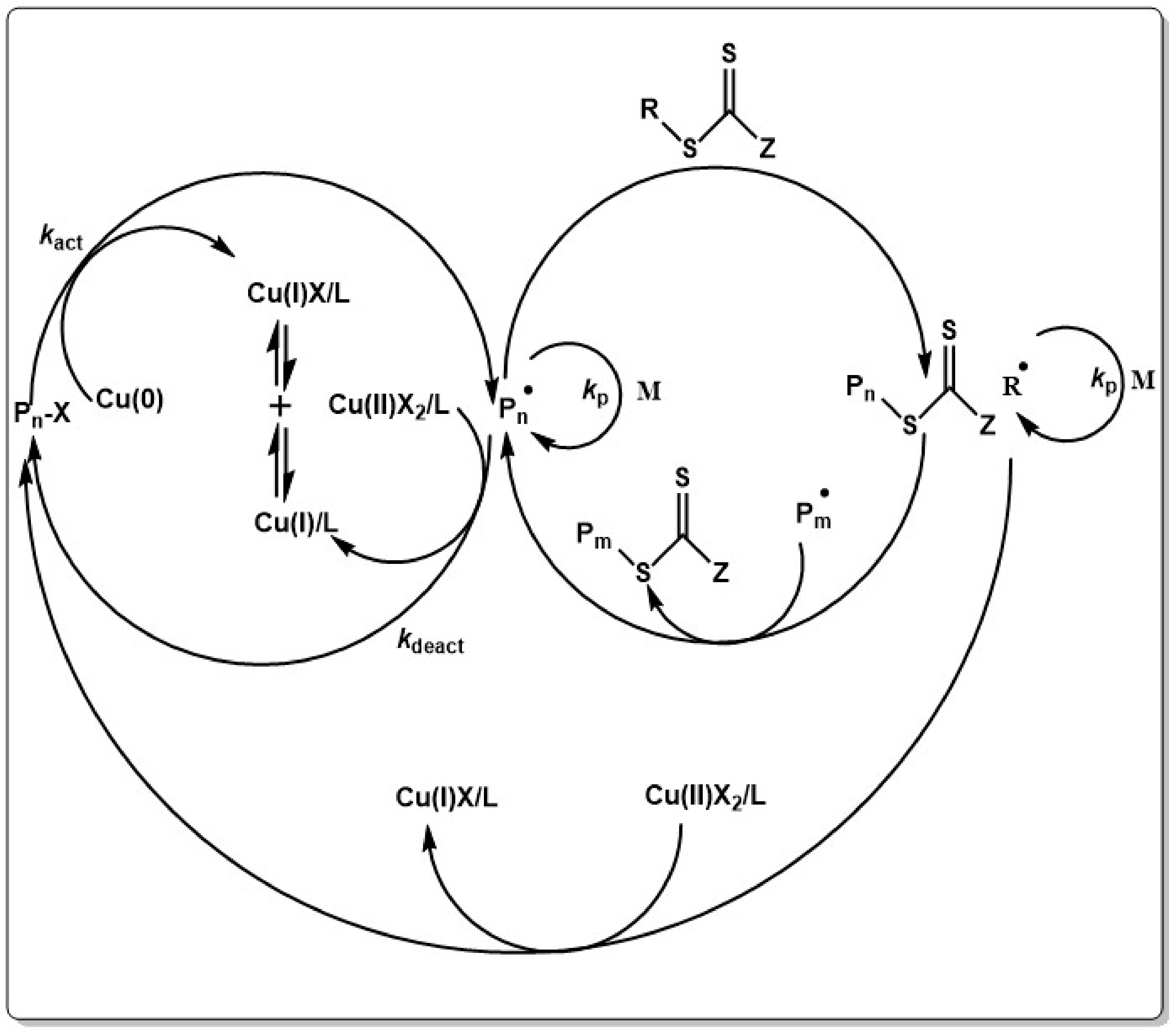
Sunlight activated RAFT polymerization by using a bifunctional thiocarbonylthio compound was realized, offering great promise for industrial applications. The reaction could be temporally controlled by light on/off experiments. Typically, thiocarbonylthio compounds show a distinct absorption in the visible region at 400–550 which stems from a n → π* transition [331]. By selective excitation with blue light at 460 nm, this transition allows for activation of a range of thiocarbonylthio compounds which can polymerize a vast amount of monomers [332]. A further step forward was the development of single electron transfer RAFT (SET-RAFT) which combines single electron transfer living radical polymerization (SET-LRP) via RAFT process. Typically, an alkyl halide oxidizes Cu(0) to yield an alkyl radical and Cu(I)X. Cu(I)X instantaneously disproportionates into Cu(0) and Cu(II)X2. The formed alkyl radical can react with the addition of monomer, or can be deactivated by the RAFT agent, releasing the R group of the RAFT agent as a radical along with the dormant species (see Scheme 10) [317,318,333,334].
Cu(0) species used in the original reports of SET-RAFT are sensitive to oxidizing environment and therefore, the reactions had to be carried out in a strictly inert atmosphere. Addition of reducing agents, such as hydrazine [335,336,337,338,339,340], strong acids [341] or ascorbic acid [342] allows to use insensitive CuO as catalyst and to carry out the reaction under ambient atmosphere. Application of SET-RAFT was extended to other transition metal catalyzed systems, such as iridium or ruthenium based catalysts [203,343,344], however, similar as in transition metal catalyzed ATRP traces of metals can potentially impede SET-RAFT for the application in biomedical or medical fields.
Lately a transition metal free SET-RAFT process was developed in which sodium sulfites replace the conventional catalytic system. Inorganic sulfites were reported to activate dormant polymer chains during SARA-ATRP. Thus, application of these reducing agents in presence of 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid as RAFT agent resulted in the polymerization of MMA. In the proposed mechanism, a reductive cleavage of the RAFT agent by one electron injection from the sulfite results in the formation of stable radical anions from the RAFT agent, which can then fragment in the initiating radical R. and the Ph–C(=S)–S anion. In accordance with the reaction involved in the process, the method is called dissociative electron transfer RAFT (DET–RAFT) [345]. The mechanism developed for PET–ATRP [230] was soon transferred to RAFT polymerization (PET–RAFT) [346]. The iridium complex fac-[Ir(ppy)3] was employed as photo catalyst and, upon irradiation, thiocarbonylthio compound was reduced via photo induced electron transfer. In the process, the thiocarbonylthio compound acts as initiator as well as CTA. The generated radical can participate in the RAFT reaction or regenerate the catalyst by reduction of the Ir(IV) species to Ir(III). The reaction can be performed at room temperature with catalyst loadings in the ppm range. This strategy was further extended to other catalytic systems, such as Ru(bpy)3Cl2 [346,347,348,349,350,351] and metalloporphyrins [352]. The aim to replace transition metal catalysts in RDRP led to the development of purely organic catalyzed photo RAFT systems. Boyer and co-workers investigated a series of organic dyes, namely Eosin-Y, Methylene Blue, Fluorescein, Nile Red and Rhodamine 6G as organocatalysts in the presence and absence of amine and with cyanopentanoic acid dithiobenzoate (CPADB) as CTA. Experiments revealed that the reactivity decreased in the order Eosin Y >> Fluorescein >> Nil Red, Rhodamine 6G and Methylene Blue. In the proposed mechanism, (see Scheme 11a) the organocatalyst in its excited state can transfer an electron to the RAFT agent. The RAFT agent acts as initiator and also as CTA to mediate chain growth in a living manner. Light on/off experiments with MMA showed that the photo polymerization remains dormant with no polymerization taking place in the dark state.
Tertiary amines are known to be excellent reducing agents and capable to act as a sacrificial agent to provide an electron for the reduction of organocatalysts. Addition of TEA to Eosin Y catalyzed PET–RAFT resulted in doubling the polymerization rate with shorter induction periods. In presence of electron donors, a reductive mechanism (see Scheme 11b) is preferred over an oxidative pathway. In the reductive pathway, an electron transfer from the donor to the organocatalyst occurs. Under aerobic conditions part of the built catalyst radical anion will reduce the oxygen, while the other part will transfer an electron to the RAFT agent. Because of the mild conditions and oxygen tolerance of the reaction, PET–RAFT using organocatalysts was then further extended to aqueous systems which showed successful polymerization even in the presence of oxygen [353]. The process was also applied to biochemical systems, such as grafting from proteins [354].
Very recently the range of initiating systems was extended to enzymatic reactions. Considering the big amount of enzymatic catalytic systems in traditional radical polymerization and the mild reaction conditions used in RAFT, an enzyme catalyzed RAFT polymerization should be feasible. First attempts to use horseradish peroxidase (HRP) in RDRP, however, failed. Polymerization of poly(ethylene glycol) acrylate (PEGA) for example gave polymers with larger than 1.5 [355] and control was completely lost upon polymerization of poly(ethylene glycol) methacrylate (PEGMA). Lately An and co-workers reported successfully a study on HRP initiated RAFT polymerization which showed control in polymerization of a wide range of MAMs and LAMs under different conditions, such as solution polymerization, dispersion polymerization and in biological environment [356]. The authors used the HRP/H2O2/Acetylacetone (AcAc) system, originally developed for enzyme catalyzed radical polymerization [357]. In the system, the enzyme undergoes a two electron oxidation which is driven by H2O2 to reduce two AcAc molecules. The AcAc radical species then initiates the polymerization. After the initiation step, the mechanism proceeds same as in a typical RAFT polymerization. Thus, enzymatic action is limited to the initiation step and does not participate in RAFT specific reaction steps. The reaction could be performed in a pH range from 4 to 7 and reached almost quantitative yield for NNDMAA with high chain end fidelity and narrow molecular weight distribution in short reaction times. The distinct pH dependence of the conversion was explained with the optimum reaction conditions of the enzyme as well as with the enol form of AcAc being the initiating species. Furthermore, an enzymatic cascade reaction using glucoseoxidase (GOx) and HRP could be demonstrated successfully. GOx catalyzes the oxidation of glucose by oxygen to H2O2 which then fuels the enzymatic reaction of HRP. Enzyme catalyzed RAFT was further extended to the synthesis of protein-polymer conjugates [358].
4.3. RAFT in Materials Synthesis
Since the development of RAFT, many multifunctional RAFT agents were reported in the literature and used for the preparation of star polymers for drug delivery [359,360,361]. Typically, two approaches are widely used in RAFT star polymerization. In the R-Star approach, the RAFT agent is attached via a reinitiating R group. In this method, however, linear by-products are commonly formed [362,363,364,365]. In the Z-Star RAFT polymerization the RAFT agent is attached via a stabilizing Z-group [366]. Homogeneous star polymers, however, can be best prepared by using a star RAFT agent that contains a stabilizing Z-group at the core [363,367,368,369]. Star polymer which are joint together by reinitiating R groups were employed in the fabrication of polymer gold nanocomposites. By joining the arms together though the R group, sulfur containing RAFT groups are displayed on the outer surface. After reduction, the sulfur groups can then attach to gold particles or surfaces [370,371,372,373]. This principle was employed by Rossner and co-workers to synthesize satellite nanostructures based on star polymers. The facile control of RAFT polymerization on the chain length thereby allows for tuning of the distance between the core in the middle and the satellites [374].
Since the RAFT mechanism proceeds via insertion of monomer into the C-S bond, end-functional polymers are obtained by using functional CTAs [311,375,376,377]. Incorporation of carboxylates [375], peptides [165], lipids [378], proteins [379] and many other groups [377,380,381,382,383] in the R group of the CTA for example led to end-functionalized polymers serving for specific applications. Obviously, the thiocarbonyl group is present on the ω-end of the polymer as a consequence of the polymerization mechanism.
One of the prerequisites for successful RAFT is a weak C–S bond, which allows establishing the equilibrium during polymerization. However, the weak C–S bond can become a problem for practical applications. The thiocarbonyl fragment is easily released from the polymer and is responsible for colour appearance and odour. Thus, much effort was put in developing various methods for removing the thiocarbonyl end-group [380]. Furthermore, the thiocarbonyl group can be seen as protected thiol, alkene or dienophil which enables post polymerization reactions.
Sequence control during polymerization is becoming increasingly important in polymer chemistry. The first approach to achieve sequence control in RAFT goes back to Zard and co-workers, who reported RAFT single unit monomer insertion (SUMI) with xanthates as CTA for LAMs [384,385]. However, the reaction suffered from low yield and polymerization of activated monomers failed. Later on Moad [386,387] and Chen [388] could show that RAFT SUMI is achievable for MAMs when trithiocarbonate or dithiobenzoate was used as the RAFT agent. In general, a high transfer constant for the initial RAFT agent and a low rate constant for the propagation steps are beneficial. However, side product formation which makes purification by GPC necessary hampers the applicability is necessary. Initiator derived side reactions are one source of by-product formation. This problem could be solved by using light induced RAFT, where the RAFT agent is cleaved under UV light and initiates the polymerization as outlined before [389,390,391].
The robustness of RAFT and the possibility to carry out reaction in aqueous environment makes RAFT a suitable method for the synthesis of glycopolymers. Lately glycosylated core-shell colloidal nanoparticles were realised through surfactant-free emulsion copolymerization of sugars and St by RAFT. The reaction allowed obtaining core-shell particles with a narrow size distribution in a one-pot fashion. The size of the particles could be tuned by adjusting the sugar content with respect to the St content [392]. Grafting a glycopolymer from silica nanoparticle surfaces was realized lately by RAFT and binding of proteins to the glycopolymer was shown [393].
RAFT was also used intensively in the synthesis of block copolymers for drug delivery. Synthetic and neutral polymers showed great potential to improve bioavailability, shelf-life and therapeutic efficacy of drugs for oral administration. Spray drying is conveniently used to disperse active pharmaceutical compounds in polymers. Thereby polymer-drug interaction plays an important role in the performance of drug-polymer spray dried dispersions. Lately Reineke and co-workers investigated the effect of block chemistry and length towards polymer-drug spray dried formulations and solubility in oral delivery conditions. They used RAFT polymerization to synthesize five well defined block copolymers, namely poly(ethylene-alt-propylene), poly(N-isopropylacrylaminde) (PNIPAm) and poly(N,N-diethylaminoethyl methacrylate) (PDEAEMA), each copolymerized with a second block of a gradient copolymer of NNDMAA and 2-methacrylamidotrehalose. Thereby PNIPAm exhibits both hydrophilic and hydrophobic character depending on the temperature, while PDEAEMA being a hydrophilic and pH responsive polymer. They could show that the solubility of the polymer matrices in the dissolution media and an increase in hydrogen bonding sites in the polymer matrix are critical to decrease the drug crystallization, increase the drug solubility and achieve supersaturation concentration of hydrophobic drugs in the dissolution media [394,395].
Block polymers also find use as toughening agents, thermoplastic elastomers and pressure sensitive adhesives. For example, traditionally PSt-PI-PSt triblock copolymers are used as pressure sensitive adhesives. Lately, however, it was shown that acrylic block copolymers showed certain advances over St based polymers mainly in terms of UV light and oxidation stability. Furthermore, the functional versatility of acrylates allows forming block polymers with a broad range of polarity and Tg values. Additional the combination of biobased resources with petroleum based feedstocks became a vital aspect to improve the sustainability of pressure sensitive adhesives. Isosorbide, a bicyclic sugar derivative gained much attention as biobased feedstock lately because it imparts desireable qualities such as high Tg and thermal stability. Recently polymerization of isosorbide monomers was extended to RDRP because of the good control over the polymerization reaction and narrow [396,397,398]. Furthermore, glucose based triblock copolymers were prepared by RAFT, where Glucose-6-acrylate-1,2,3,4-tetraacetate was used as the glassy component in thermoplastic elastomers [399].
5. Conclusions and Outlook
Significant advances in RDRP provided the basis to control the composition of polymers on a microscale and provides now the ability to fabricate materials which were not accessible a decade ago. Since the first reports on living anionic polymerization until the transfer of the living polymerization concept to radical polymerization, the synthetic techniques to precisely determine the structure of polymers were steadily improved. Today, we have a wide choice of methods to synthesize living polymers in a controlled fashion out of almost an arbitrary choice of monomers and terminated by a wide range of chemical functions. This enables to build up more and more complex structures and to develop tailored polymer materials for very specialized applications.
Research work during the last decade provided deep insight in the mechanisms governing control in radical polymerization. This allowed for developing new and highly efficient initiation or mediating systems which are able to provide a high degree of control during the reaction. Development of photo-induced RDRP in NMRP allowed to reduce reaction temperatures and to decrease the amount of side reactions. Photo controlled catalytic systems also allowed to minimize the amount of transition metal catalysts in ATRP or even to develop purely organic and metal free ATRP systems. Switchable RAFT agents allow for polymerization of more or less activated polymers, whose limitation was one of the big drawbacks in RAFT for long time.
The possibility to carry out RDRP under very mild conditions resulted in increased research effort in using these techniques for the synthesis of biocomposites, polymer-protein conjugates, glycopolymers or polymers for biomedical applications. Furthermore, RDRP finds more and more application in developing high end materials from renewable biopolymers. The full potential of the technique in these emerging fields, however, will just be explored in the future.
Acknowledgments
Johannes Kreutzer gratefully acknowledges TÜBITAK for financial support in the framework of the project 116C038.
Author Contributions
The manuscript was written through contributions of both authors.
Conflicts of Interest
The authors declare no competing financial interest.
Abbreviations
| 4CzIPN | 2,4,5,6-tetra(9H-carbazol-9-yl) isophthalonitril |
| 4VBC | 9-(4-vinyl-benzyl)-9H-carbazole |
| Ac | Acrylate |
| AAm | Acrylamide |
| AcAc | Acetylacetone |
| AIBN | 2,2′-azobis(2-methylpropionitril) |
| AN | Acrylonitrile |
| ARGET ATRP | Activator Regenerated by Electron Transfer ATRP |
| ATRP | Atom Transfer Radical Polymerization |
| BA | Butyl acrylate |
| tBA | t-butyl acrylate |
| BAI | Bis-(alkylphenyl)iodonium hexafluorophosphate |
| BMI | N-benzylmaleimide |
| BP | Benzophenon |
| BPO | Benzoylperoxide |
| tBS | t-butylstyrene |
| BSA | Bovine Serum A |
| BzMA | Benzyl methacrylate |
| CA | Cyanoacrylate |
| CMI | N-cyclohexylmaleimide |
| CPADB | Cyanopentanoic acid dithiobenzoate |
| CP–I | 2-cyanopropyl iodide |
| CQ | Camphorquinone |
| CTA | Chain Transfer Agent |
| CTS | Chitosan |
| Dispersity | |
| DAEMA | Dehydroabietic ethyl methacrylate |
| DBN | Di-t-butyl nitroxide |
| DC | Dissociation-Combination |
| DCF | Dead Chain Fraction |
| DDMAT | S-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate |
| DET | Dissociative Electron Transfer |
| DET-RAFT | Dissociative Electron Transfer RAFT |
| DFT | Density Functional Theory |
| DMA | N,N-dimethylacetamide |
| DMAEMA | 2-(dimethylamino)ethyl methacrylate |
| DMEDA | N,N-dimethylethylenediamine |
| DMPA | 2,2-dimethoxy-2-phenylacetophenone |
| DPAIO | 2,2-diphenyl-3-phenylimino-2,3-dihydroindol-1-yloxyl |
| DT | Degenerative Exchange Chain Transfer |
| e-ATRP | Electro ATRP |
| EDA | Ethylenediamine |
| ESR | Electron Spin Resonance |
| FMA | Furfuryl methacrylate |
| GOx | Glucose Oxidase |
| HEA | 2-hydroxyethyl acrylate |
| HEMA | 2-hydroxyethyl methacrylate |
| HRP | Horseraddish Peroxidase |
| I | Isoprene |
| ICAR ATRP | Initiators for Continuous Activator Regeneration ATRP |
| IRT | Intermediate Radical Termination |
| IRTO | Intermediate Radical Termination with Oligomers |
| ISET | Inner Sphere Electron Transfer |
| ITX | Isopropyl thioxanthone |
| LAM | Less Activated Monomers |
| MA | Methacrylate |
| MAEMI | 2-(dimethylamine)ethyl methacrylate |
| MAM | More Activated Monomers |
| Me-PTH | 10-methyl phenothiazine |
| MMA | Methyl methacrylate |
| MTEMPO | 4-methoxy TEMPO |
| Mtn | Transition Metal |
| MWD | Molecular Weight Distribution |
| NBS | N-bromosuccinimide |
| NHS | N-hydroxysuccinimide |
| NMMA | Methyl-2-methyl-3-nitro-2-nitrosopropionate |
| NMRP | Nitroxide Mediated Radical Polymerization |
| NNDMAA | N,N-dimethylacrylamide |
| NPI | Maleimide-N-phenylmaleimide |
| MMA | N-vinylcaprolactam |
| NVI | N-vinylimidazole |
| NVP | N-vinylpyrrolidone |
| OSET | Outer Sphere Electron Transfer |
| OSET-C | Continuous Outer Sphere Electron Transfer |
| OSET-SW | Stepwise Outer Sphere Electron Transfer |
| PDEAEMA | poly(N,N-diethylaminoethyl methacrylate) |
| PEG | Poly(ethylene glycol) |
| PEGA | Poly(ethylene glycol) methyl ether acrylate |
| PEGMA | Poly(ethylene glycol) methyl ether methacrylate |
| PEO | Poly(ethylene oxide) |
| PET-ATRP | Photo-induced Electron Transfer ATRP |
| PET-RAFT | Photo-induced Electron Transfer RAFT |
| Ph-PTH | 10-phenyl phenothiazine |
| PMDC | Morpholine-4-dithiocarbamate |
| PMDETA | N,N,N′,N″,N″-pentamethyldiethylenetriamine |
| PNIPAm | Poly(N-isopropylacrylamide) |
| PRE | Persistent Radical Effect |
| PTH | Phenothiazine |
| RAFT | Reversible Addition Fragmentation Chain Transfer Polymerization |
| RCMP | Reversible Complexation Mediated Polymerization |
| RCT | Reversible Chain Transfer |
| RCTP | Reversible Chain Transfer Polymerization |
| RDRP | Reversible-Deactivation Radical Polymerization |
| ROMP | Ring-Opening Metathesis Polymerization |
| SARA-ATRP | Supplementary Activator and Reducing Agent ATRP |
| SBMA | Soybean oil methacrylate |
| SET-LRP | Single Electron Transfer Living Radical Polymerization |
| SET-RAFT | Single Electron Transfer RAFT |
| SG1 | N-t-butyl-1-diethoxyphosphoryl-N-oxidanyl-2,2-dimethylpropan-1-amine |
| SS | Styrene sulfonate |
| St | Styrene |
| SUMI | Single Unit Monomer Insertion |
| TBA | Tributylamine |
| TDAE | Tetrakis (dimethylamino) ethylene |
| TEA | Triethylamine |
| TEDIO | 1,1,3,3,-tetraethylisoindolin-2-oxyl |
| TEMPO | 2,2,6,6,-tetramethyl-2-piperidin-1-oxyl |
| TIPNO | 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide |
| TMEDA | Tetramethylethylenediamine |
| TPO | 2,4,6-trimethylbenzoyl-diphenylphosphine oxide |
| TX | Thioxanthone |
| V70 | 2,2′-azobis(4-methoxy-2,4-dimethyl valeronitrile) |
| Vac | Vinyl acetate |
References
- Szwarc, M. “Living” Polymers. Nature 1956, 176, 1168–1169. [Google Scholar] [CrossRef]
- Szwarc, M.; Levy, M.; Milkovich, R. Polymerization Initiated by Electron Transfer Monomer. A New Method of Formation of Block Copolymers. J. Am. Chem. Soc. 1956, 78, 2656–2657. [Google Scholar] [CrossRef]
- Pan, X.; Tasdelen, M.A.; Laun, J.; Junkers, T.; Yagci, Y.; Matyjaszewski, K. Photomediated controlled radical polymerization. Prog. Polym. Sci. 2016, 62, 73–125. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Kahveci, M.U.; Yagci, Y. Telechelic polymers by living and controlled/living polymerization methods. Prog. Polym. Sci. 2011, 36, 455–567. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E. The History of Nitroxide-mediated Polymerization. In Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science, 19th ed.; Gigmes, D., Ed.; RSC Polymer Series; RSC: London, UK, 2016; Volume 19, pp. 1–44. [Google Scholar]
- Jansen, G. Spin Trapping. Acc. Chem. Res. 1971, 4, 31–40. [Google Scholar] [CrossRef]
- Chalfont, G.R.; Perkins, M.J.; Horsfield, A. Probe for homolytic reactions in solution. II. Polymerization of styrene. J. Am. Chem. Soc. 1968, 90, 7141–7142. [Google Scholar] [CrossRef]
- Kunitake, T.; Murakami, S. Study of Radical Polymerizations by Spin Trapping I. Trapping of Initiating and Propagating Radicals. Polym. J. 1972, 3, 249–251. [Google Scholar] [CrossRef]
- Kunitake, T.; Murakami, S. An application of spin trapping to radical polymerization. J. Polym. Sci. Chem. Ed. 1974, 12, 67–81. [Google Scholar] [CrossRef]
- Sato, T.; Otsu, T. Evaluation of relativities of vinyl monomers towards t-butoxy radical by means of spin trapping technique. Polymer 1975, 16, 389–391. [Google Scholar] [CrossRef]
- Sato, T.; Otsu, T. Application of spin trapping technique to radical polymerization, 14. Initiation reaction of vinyl monomers with tert-butoxyl radical and evaluation of monomer reactivities. Macromol. Chem. 1977, 178, 1941–1950. [Google Scholar] [CrossRef]
- Roszantsev, E.G.; Sholle, V.D. Synthesis and Reactions of Stable Nitroxyl Radicals II. Reactions. Synthesis 1971, 8, 401–414. [Google Scholar] [CrossRef]
- Berdnarek, D.; Moad, G.; Rizzardo, E.; Solomon, D.H. End groups of poly(methyl methacrylate-co-styrene) prepared with tert-butoxy, methyl, and/or phenyl radical initiation: Effects on solvent, monomer composition, and conversion. Macromolecules 1988, 21, 1522–1528. [Google Scholar] [CrossRef]
- Bizilj, S.; Kelly, D.; Serelis, A.; Solomon, D.; White, K. The Self-Reactions of 1-Methoxycarbonyl-1-Methylethyl and Higher Ester Radicals: Combination vs. Disproportionation and Oligomeric Products from Secondary Reactions. Aust. J. Chem. 1985, 38, 1657–1673. [Google Scholar] [CrossRef]
- Cutberthson, M.J.; Rizzardo, E.; Solomon, D.H. The reactions of t-butoxyl with unsaturated hydrocarbons: Structure and reactivity of allylic radicals. Aust. J. Chem. 1983, 36, 1957–1973. [Google Scholar] [CrossRef]
- Rizzardo, E.; Serelis, A.K.; Solomon, D.H. Initiation mechanisms in radical polymerizations: Reaction of cumyloxy radicals with methyl methacrylate and styrene. Aust. J. Chem. 1982, 35, 2013–2024. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Solomon, D.H. The Reaction of Benzoyloxy Radicals with Styrene-Implications Concerning the Structure of Polystyrene. J. Macromol. Sci. 1982, A17, 51–59. [Google Scholar] [CrossRef]
- Rizzardo, E.; Solomon, D.H. On the Origins of Nitroxide Mediated Polymerization (NMP) and Reversible Addition-Fragmentation Chain Transfer (RAFT). Aust. J. Chem. 2012, 65, 945–969. [Google Scholar] [CrossRef]
- Solomon, D.H.; Rizzardo, E.; Cacioli, P. Polymerization Processes and Polymers Produced Thereby. U.S. Patent 4,581,429, 8 April 1986. [Google Scholar]
- Georges, M.K.; Veregin, R.P.N.; Kazmaier, P.M.; Harmer, G.K. Narrow molecular weight resins by a free-radical polymerization process. Macromolecules 1993, 26, 2987–2988. [Google Scholar] [CrossRef]
- Hawker, C.J.; Barklay, G.C.; Orellana, A.; Dao, J.; Devonport, W. Initiating systems for Nitroxide-Mediated “Living” Free Radical Polymerization: Synthesis and Evaluation. Macromolecules 1996, 29, 5245–5254. [Google Scholar] [CrossRef]
- Hawker, C.J. Molecular Weight Control by a “Living” Free-Radical Polymerization Process. J. Am. Chem. Soc. 1994, 116, 11185–11186. [Google Scholar] [CrossRef]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerization. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef] [PubMed]
- Hawker, C.J. “Living” Free Radical Polymerization: A Unique Technique for the Preparation of COntrolled Macromolecular Architectures. Acc. Chem. Res. 1997, 30, 373–382. [Google Scholar] [CrossRef]
- Hawker, C.J.; Barklay, G.C.; Dao, J.L. Radical Crossover in Nitroxide Mediated “Living” Free Radical Polymerization. J. Am. Chem. Soc. 1996, 118, 11467–11471. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by a nitroxide mediator. Colloid Polym. Sci. 2008, 286, 1663–1666. [Google Scholar] [CrossRef]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of vinyl acetate. Colloid Polym. Sci. 2010, 288, 73–78. [Google Scholar] [CrossRef]
- Benoit, D.; Chaplinski, V.; Braslau, R.; Hawker, C.J. Development of a Universal Alkoxyamine for “Living” Free Radical Polymerization. J. Am. Chem. Soc. 1999, 121, 3904–3920. [Google Scholar] [CrossRef]
- Grimaldi, S.; Finet, J.-P.; Moigne, F.L.; Zheghdaoui, A.; Tordo, P.; Benoit, D.; Fontanille, M.; Gnanou, Y. Acyclic β-Phosphonylated Nitroxides: A New Series of Counter-Radicals for “Living”/Controlled Free Radical Polymerization. Macromolecules 2000, 33, 1141–1147. [Google Scholar] [CrossRef]
- Sciannamea, V.; Jerome, R.; Detrembleur, C. In-Situ Nitroxide-Mediated Radical Polymerization (NMP) Processes: Their Understanding and Optimization. Chem. Rev. 2008, 108, 1104–1126. [Google Scholar] [CrossRef] [PubMed]
- Green, A.C.; Grubs, R.B. Current Methods for N-Alkoxyamine Synthesis. In Controlled/Living Radical Polymerization: Progress in RAFT, DT, NMP & OMRP; Matyjaszewski, K., Ed.; ACS: Washington, DC, USA, 2009; Chapter 6; pp. 81–93. [Google Scholar]
- Georges, M.K.; Veregin, R.P.N.; Katzmaier, P.M.; Hamer, G.K.; Saban, M. Narrow Polydispersity Polystyrene by a Free-Radical Polymerization Process-Rate Enhancement. Macromolecules 1994, 27, 7228–7229. [Google Scholar] [CrossRef]
- Veregin, R.P.N.; Odell, P.G.; Michalka, L.M.; Georges, M.K. Mechanism of Rate Enhancement Using Organic Acids in Nitroxide-Mediated Living Free-Radical Polymerization. Macromolecules 1996, 29, 4161–4163. [Google Scholar] [CrossRef]
- Baldovi, M.V.; Mohtat, N.; Scaiano, J.C. Influence of Acids on Reaction Rates of Free Radical Scavanging by TEMPO. Relevance to “Living” Free Radical Polymerization. Macromolecules 1996, 29, 5497–5499. [Google Scholar] [CrossRef]
- Odell, P.G.; Veregin, R.P.N.; Michalka, L.M.; Georges, M.K. Characteristics of the Stable Free Radical Polymerization of Styrene in the Presence of 2-Fluoro-1-methylpyridinium p-Toluenesulfonate. Macromolecules 1997, 30, 2232–2237. [Google Scholar] [CrossRef]
- Keoshkerian, B.; Georges, M.; Quinlan, M.; Veregin, R.; Goodbrand, B. Polyacrylates and Polydienes to High Conversion by a Stable Free Radical Polymerization Process: Use of Reducing Agents. Macromolecules 1998, 31, 7559–7561. [Google Scholar] [CrossRef]
- Malmstroem, E.E.; Miller, R.D.; Hawker, C.J. Development of a new class of rate-accelerating additives for nitroxide-mediated ‘living’ free radical polymerization. Tetrahedron 1997, 53, 15225–15236. [Google Scholar] [CrossRef]
- Greszta, D.; Matyjaszewski, K. TEMPO-mediated polymerization of styrene: Rate enhancement with dicumyl peroxide. J. Polym. Sci. A Polym. Chem. 1997, 35, 1857–1861. [Google Scholar] [CrossRef]
- Ohno, K.; Tsujii, Y.; Fukuda, T. Mechanism and Kinetics of Nitroxide-Controlled Free Radical Polymerization. Thermal Decomposition of 2,2,6,6,-Tetramethyl-1-polystyroxypiperidines. Macromolecules 1997, 30, 2503–2506. [Google Scholar] [CrossRef]
- Baethge, H.; Butz, S.; Schmidt-Naake, G. Rate enhancement of the N-oxyl-controlled free radical copolymerization of styrene with N-vinylcarbazole. Angew. Makromol. Chem. 1999, 267, 52–56. [Google Scholar] [CrossRef]
- Gerard, P.; Couvreur, L.; Magnet, S.; Ness, J.; Schmidt, S. Controlled Architecture Polymers at Arkema: Synthesis, Morphology and Properties of All-Acyrylic Block Copolymers. In Controlled/Living Radical Polymerization: Progress in RAFT, DT, NMP & OMRP; Matyjaszewski, K., Ed.; ACS: Washington, DC, USA, 2009; Chapter 24; pp. 361–376. [Google Scholar]
- Lessard, B.; Tervno, C.; Maric, M. High-Molecular-Weight Poly(tert-butyl acrylate) by Nitroxide-Mediated Polymerization: Effect of Chain Transfer to Solvent. Macromol. React. Eng. 2009, 3, 245–256. [Google Scholar] [CrossRef]
- Sundararman, A.; Stephan, T.; Grubbs, R.B. Reversible Restructuring of Aqueous Block Copolymer Assemblies through Stimulus-induced Changes in Amphiphilicity. J. Am. Chem. Soc. 2008, 130, 12264–12265. [Google Scholar] [CrossRef] [PubMed]
- Junkers, T.; Wong, E.H.H.; Stenzel, M.H.; Barner-Kowollik, C. Formation Efficiency of ABA Blockcopolymers via Enhanced Spin Capturing Polymerization (ESCP): Locating the Alkoxyamine Function. Macromolecules 2009, 42, 5027–5035. [Google Scholar] [CrossRef]
- Theato, P.; Killinger, D. Synthesis of Photoreactive Block Copolymer Based on 1-Iminopyridinium Ylides. Aust. J. Chem. 2010, 63, 1164–1168. [Google Scholar] [CrossRef]
- Gamsy, C.G.; Beyou, E.; Bourgeat-Lami, E. Micellar behaviour of well-defined polystyrene-based block copolymers with triethoxysilyl reactive groups and their hydrolysis-condensation. J. Polym. Sci. A Polym. Chem. 2010, 48, 784–793. [Google Scholar] [CrossRef]
- Zhang, W.; Yan, Y.; Zhou, N.; Cheng, Z.; Zhou, J.; Xia, C.; Zhu, X. Controlled synthesis and fluorescence properties of poly(9-(4-vinylbenzyl)-9H-carbazole) via nitroxide-mediated living free-radical polymerization. Eur. Polym. J. 2008, 44, 3300–3305. [Google Scholar] [CrossRef]
- Quinn, J.D.; Register, R.A. Nitroxide-mediated radical polymerization of N-ethyl-2-vinylcarbazole. Polym. Adv. Technol. 2008, 19, 556–559. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Sakaguchi, K.; Kawakami, A.; Taka, H.; Kita, H.; Shimomura, T.; Ogino, K. Charge transporting block copolymer for morphology control in light emitting device based on polymer blends. Synth. Mat. 2010, 160, 1679–1982. [Google Scholar] [CrossRef]
- Pan, T.N.T.; Bertin, D. Synthesis of Water-Soluable Homopolymers and Block Copolymers in Homogeneous Aqueous Solution via Nitroxide-Mediated polymerization. Macromolecules 2008, 41, 1886–1895. [Google Scholar]
- Enright, T.E.; Cunningham, M.F.; Keoshkerian, B. Nitroxide-Mediated Bulk and Miniemulsion Polymerization in a Continuous Tubular Reactor: Synthesis of Homo-, Di- and Triblock Copolymers. Macromol. React. Eng. 2010, 4, 186–196. [Google Scholar] [CrossRef]
- Ramirez-Wong, D.G.; Posada-Valez, C.A.; Saldivar-Guerra, E.; Luna-Barcenas, J.G.; Ott, C.; Schubert, U.S. Silicon-based and fluorinated polymeric surfactants for nitroxide mediated dispersion polymerization in supercritical carbon dioxide. Macromol. Symp. 2009, 283–284, 120–129. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Men, X.; Xu, X.; Zhu, X. Reversible conversion of water-droplet mobility from rollable to pinned on a superhydrophobic functionalized carbon nanotube film. J. Colloid Interface Sci. 2010, 346, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Guillet, P.; Fustin, C.-A.; Mugemana, C.; Ott, C.; Schubert, U.S.; Gohy, J.-F. Tuning block copolymer micelles by metal-ligand interactions. Soft Matter 2008, 4, 2278–2282. [Google Scholar] [CrossRef]
- Datsuyuk, V.; Billon, L.; Guerret-Piecourt, C.; Dagreou, S.; Passade-Boupatt, N.; Bourrigaud, S.; Guerret, O.; Couvreur, L. In situ nitroxide-mediated polymerized poly(acrylic acid) as a stabilizer/compatibilizer carbon nanotube/polymer composites. J. Nanomater. 2007, 2007, 74769. [Google Scholar] [CrossRef]
- Fleischmann, S.; Komber, H.; Voit, B. Diblock Copolymer as Scaffold for Efficient Functionalization via Click Chemistry. Macromolecules 2008, 41, 5255–5264. [Google Scholar] [CrossRef]
- Maeda, R.; Hayakawa, T.; Tokita, M.; Kakimoto, M.-A.; Urushibata, H. Double Ordered Layers within Microphase-Stucture of Double Liquid Crystalline Side-chain Type Block Copolymer. Chem. Lett. 2008, 37, 1174–1175. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E. Alkoxyamine-Initiated Living Radical Polymerization: Factors Affecting Alkoxyamine Homolysis Rate. Macromolecules 1995, 28, 8722–8728. [Google Scholar] [CrossRef]
- Puts, R.D.; Sogah, D.Y. Control of Living Free-Radical Polymerization by a New Chiral Nitroxide and Implcation for the Polymerization Mechanism. Macromolecules 1996, 29, 3323–3325. [Google Scholar] [CrossRef]
- Veregin, R.P.N.; Georges, M.K.; Hamer, G.K.; Kazmaier, P.M. Mechanism of Living Free Radical Polymerization with Narrow Polydispasity: Electro Spin Resonance and Kinetic Studies. Macromolecules 1995, 28, 4391–4398. [Google Scholar] [CrossRef]
- Grimaldi, S.; Finet, J.P.; Zeghadaoui, A.; Tordo, P.; Benoit, D.; Gnanou, Y.; Fontaille, M.; Nicol, P.; Piersond, J.F. Synthesis and Application to “Living” Free Radical Polymerization of a New Class of Nitroxyl Radicals. ACS Dev. Polym. Chem. 1997, 38, 651–652. [Google Scholar]
- Benoit, D.; Grimaldi, S.; Robin, S.; Finet, J.-P.; Tordo, P.; Gnanou, Y. Kinetics and Mechanism of Controlled Free-Radical Polymerization of Styrene and n-Butyl Acrylate in the Presence of an Acyclic β-Phosphonylated Nitroxide. J. Am. Chem. Soc. 2000, 122, 5929–5939. [Google Scholar] [CrossRef]
- Listigovers, N.A.; Georges, M.K.; Odell, P.G.; Keoshkerian, B. Narrow Polydispersity Diblock and Triblock Copolymers of Alkyl Acrylates by a “Living” Stable Free Radical Polymerization. Macomolecules 1996, 29, 8992–8993. [Google Scholar] [CrossRef]
- Ciriano, M.V.; Korth, H.G.; Scheppinger, W.B.V.; Mulder, P. Thermal stability of 2,2,6,6,-Tetramethylpipiridine-1-oxyl (TEMPO) and Related N-Alkoxyamines. J. Am. Chem. Soc. 1999, 121, 6375–6381. [Google Scholar] [CrossRef]
- Skene, W.G.; Belt, S.T.; Connoly, T.J.; Hahn, P.; Scaiano, J.C. Decomposition Kinetics, Arrhenius Parameters, and Bond Dissociation Energies for Alkoxyamines of Relevance in “Living” Free Radical Polymerization. Macromolecules 1998, 31, 9103–9105. [Google Scholar] [CrossRef]
- Bon, S.A.F.; Chambard, G.; German, A.L. Nitroxide-Mediated Living Polymerization: Determination of the Rate Coefficient for Alkoxyamine C-O Bond Homolysis by Quantitative ESR. Macromolecules 1999, 32, 8269–8276. [Google Scholar] [CrossRef]
- Kothe, T.; Marque, S.; Martscheke, R.; Popov, M.; Fischer, H. Radical reaction kinetics during homolysis of N-alkoxyamines: Verification of the persistent radical effect. J. Chem. Soc. Perkin Trans. 1998, 2, 1553–1560. [Google Scholar] [CrossRef]
- Engel, P.S.; Duan, S.; Arhancet, G.B. Thermolysis of a Tertiary Alkoxyamine. Recombination and Disproportionation of α-Phenylethyl/Diethyl Nitroxyl Radical Pairs. J. Org. Chem. 1997, 62, 3537–3541. [Google Scholar] [CrossRef]
- Li, I.; Howell, B.A.; Matyjaszewski, K.; Shigemoto, T.; Smith, P.B.; Priddy, D.B. Kinetics and decomposition of 2,2,6,6,-tetramethyl-1-(1-phenylethoxy)piperidine and its implication on nitroxyl-mediated styrene polymerization. Macromolecules 1995, 28, 6692–6693. [Google Scholar] [CrossRef]
- Grattan, D.W.; Carlson, D.J.; Howard, J.A.; Wiles, D.M. The thermal decomposition of 1-(2′-cyano-2′-propoxy)-4-oxo-2,2,6,6,-tetramethylpiperidine. Can. J. Chem. 1979, 57, 2834–2842. [Google Scholar] [CrossRef]
- Mercier, C.L.; Gaudel, A.; Siri, D.; Tordo, P.; Marque, S.; Martschke, R.; Fischer, H. Characteristics of phosphonylated nitroxides and alkoxyamines used in controlled/”living” radical polymerization. Polym. Prepr. Am. Chem. Soc. Div. Polym. Chem. 1999, 313–314. [Google Scholar]
- Katzmaier, P.M.; Moffat, K.A.; Georges, M.K.; Veregin, R.P.N.; Hamer, G.K. Free-Radical Polymerization for Narrow-Polydispersity Resins. Semiempirical Molecular Orbital Calculations as a Criterion for Selecting Stable Free Radical Reversible Terminators. Macromolecules 1995, 28, 1841–1846. [Google Scholar] [CrossRef]
- Marque, S.R.A.; Mercier, C.L.; Tordo, P.; Fischer, H. Factors Influenceing the C-O-Bond Homolysis of Trialkylhydroxylamines. Macromolecules 2000, 33, 4403–4410. [Google Scholar] [CrossRef]
- Mercier, C.L.; Acerbis, S.; Bertin, D.; Chauvin, F.; Gigmes, D.; Guerret, O.; Lansalot, M.; Marque, S.; Moigne, F.L.; Fischer, H.; Tordo, P. Design and use of β-phosphorus nitroxides and alkoxyamines in controlled/”living” free radical polymerization. Macromol. Symp. 2002, 182, 225–247. [Google Scholar] [CrossRef]
- Braslau, R.; Naik, N.; Zipse, H. Stereoselective Coupling of Prochiral Radicals with a Chrial C2-Symmetric Nitroxide. J. Am. Chem. Soc. 2000, 122, 8421–8434. [Google Scholar] [CrossRef]
- Cuatepotoz-Diaz, R.; Albores-Valesco, M.; Saldivar-Guerra, E.; Jimenez, F.B. Nitroxide mediated polymerization using diphenyl azabutane N-oxides. A study of electron effects and of the [nitroxide]/[initiator] ratio on the polymerization control. Polymer 2004, 45, 815–824. [Google Scholar] [CrossRef]
- Gaudel-Siri, A.; Siri, D.; Tordo, P. Homolysis of N-alkoxyamine: A Computational Study. Chem. Phys. Chem. 2006, 7, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Nakamura, N.; Taniguchi, J. Low-Temperature “Living” Radical Polymerization of Styrene in the Presence of Nitroxides with Spiro Structures. Macromolecules 2001, 34, 447–455. [Google Scholar] [CrossRef]
- Miura, Y.; Nakamura, N.; Taniguchi, J. Radical polymerization of butyl acrylate and random copolymerization of styrene and butyl acrylate and styrene and methyl methacrylate mediated by monospiro- and dispiropiperidinyl-N-oxyl radicals. Polymer 2003, 44, 3461–3467. [Google Scholar] [CrossRef]
- Jing, Y.; Mardyukov, A.; Bergander, K.; Daniliuc, C.G.; Struder, A. Synthesis of Bulky Nitroxides, Characteriyation, and Their Application in Controlled Radical Polymerization. Macromolecules 2014, 47, 3595–3602. [Google Scholar] [CrossRef]
- Jing, Y.; Tesch, M.; Wang, L.; Daniliuc, C.G.; Struder, A. Synthesis of bulky nitroxides and its application in the nitroxide-mediated radical polymerization. Tetrahedron 2016, 72, 7665–7671. [Google Scholar] [CrossRef]
- Siegenthaler, K.O.; Studer, A. Nitroxide-Mediated Radical Polymerization—Increase of Steric Demand in Nitroxides. How Much is Too Much? Macromolecules 2006, 39, 1347–1352. [Google Scholar] [CrossRef]
- Hodgson, J.L.; Roskop, L.B.; Gordon, M.S.; Lin, C.Y.; Coote, L. Side Reaactions of Nitroxide-Mediated Polymerization: N- versus O-C Cleavage of Alkoxyamines. J. Phys. Chem. A 2010, 114, 10458–10466. [Google Scholar] [CrossRef] [PubMed]
- Blachon, A.; Marque, S.R.A.; Roubaud, V.; Siri, D. Diastereomer Effect on the Homolysis of the C-ON Bond in Alkoxyamines: A DFT investigation of 1,3-Diphenylbutyl-TEMPO. Polymers 2010, 2, 353–363. [Google Scholar] [CrossRef]
- Cunningham, M.F. Recent progress in nitroxide-mediated polymerization in miniemulsion. C. R. Chim. 2003, 6, 1351–1374. [Google Scholar] [CrossRef]
- Dire, C.; Belleney, J.; Nicolas, J.; Bertin, D.; Magnet, S.; Charleux, B.J. β-Hydrogen transfer from poly(methyl methacrylate) propagating radicals to the nitroxide SG1: Analysis of the chain-end and determination of the rate constant. J. Polym. Sci. A: Polym. Chem. 2008, 46, 6333–6345. [Google Scholar] [CrossRef]
- Souaille, M.; Fischer, H. Living Free Radical Polymerization Mediated by the Reverseible Combination of Transient Propagating and Persistent Nitroxide Radicals. The Role of Hydroxylamine and Alkene Formation. Macromolecules 2001, 34, 2830–2838. [Google Scholar] [CrossRef]
- Gryn’ova, G.; Hodgson, J.L.; Coote, M.L. Revising the mechanism of polymer autooxidation. Org. Biomol. Chem. 2011, 9, 480–490. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, L.; Yang, Y. Effect of Hydrogen Transfer Reaction on Kinetics of Nitroxide-Mediated Free-Radical Polymerization. Macromolecules 2000, 33, 2286–2289. [Google Scholar] [CrossRef]
- Moad, G.; Anderson, A.G.; Ercole, F.; Johnson, C.H.J.; Kristina, J.; Moad, C.L.; Rizzardo, E.; Spurling, T.H.; Thang, S.H. Controlled-Growth Free-Radical Polymerization of Methylacrylate Esters: Reversible Chain Transfer versus Reversible Termination. ACS Symp. Ser. 1998, 685, 332–360. [Google Scholar]
- Burguiere, C.; Dourges, M.; Charleux, B.; Varion, J. Synthesis and Characterization of ω-Unsaturated Poly(styrene-b-n-butylmethacrylate) Block Copolymer Using TEMPO-Mediated Controlled Radical Polymerization. Macromolecules 1999, 32, 3883–3890. [Google Scholar] [CrossRef]
- Ballard, N.; Aguirre, M.; Simula, A.; Agirre, A.; Leiza, J.R.; Asua, J.M.; Es, S.V. New Class of Alkoxyamines for Efficient Controlled Homopolymerization of Methacrylates. ACS Macro Lett. 2016, 5, 1019–1022. [Google Scholar] [CrossRef]
- Ananchenko, G.S.; Fischer, H. Decomposition of model alkoxyamines in simple and polymerizing systems. 1,2,2,6,6-tetramethylpiperidinyl-N-oxyl-based compounds. J. Polym. Sci. A Polym. Chem. 2001, 39, 3604–3621. [Google Scholar] [CrossRef]
- Guillaneuf, Y.; Gigmes, D.; Marque, S.R.A.; Tordo, P.; Bertin, D. Nitroxide-Mediated Polymerization of Methyl Methacrylate Using a SG1-Based Alkoxyamine: How the Penultimate Effect Could Lead to Uncontrolled and Unliving Polymerization. Macromol. Chem. Phys. 2006, 207, 1278–1288. [Google Scholar] [CrossRef]
- McHalle, R.; Aldabbagh, F.; Zetterlund, P.B. The role of excess nitroxide in the SG1 (N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide)-mediated polymerization of methyl methacrylate. Polym. Sci. A Polym. Chem. 2007, 45, 2194–2203. [Google Scholar] [CrossRef]
- Charleux, B.; Nicholas, J.; Guerret, O. Theoretical Expression of the Average Activation-Deactivation Equilibrium Constant in Controlled/Living Free-Radical Copolymerization Operating via Reversible Termination. Application to a Strongly Improved Control in Nitroxide-Mediated Polymerization of Methyl Methacrylate. Macromolecules 2005, 38, 5485–5492. [Google Scholar]
- Ananchenko, G.S.; Souaille, M.; Fischer, M.H.; Mercier, C.L.; Tordo, P. Decomposition of model alkoxyamines in simple and polymerizing systems. II Diastereomeric N-(2-methylpropyl)-N-(1-diethyl-phosphono-2,2-dimethylpropyl)-aminoxyl-based compounds. J. Polym. Sci. A Polym. Chem. 2002, 40, 3264–3283. [Google Scholar] [CrossRef]
- Nicolas, J.; Dire, P.; Mueller, L.; Belleney, J.; Charleux, B.; Marque, S.R.A.; Bertin, D.; Magnet, S.; Couvreur, L. Living Character of Polymer Chains Prepared via Nitroxide-Mediated Controlled Free-Radical Polymerization of Methyl Methacrylate in the Presence of a Small Amount of Styrene at Low Temperature. Macromolecules 2006, 39, 8274–8282. [Google Scholar] [CrossRef]
- Chauvin, F.; Couturier, J.-L.; Dulfis, P.E.; Gerard, P.; Gigmes, D.; Guerret, O.; Guillaneuf, Y.; Marque, S.R.A.; Bertin, D.; Tordo, P. Crowded Phosphonylated Alkoxyamines with Low Dissociation Temperatures: A Milestone in Nitroxide-Mediated Polymerization. ACS Symp. Ser. 2006, 944, 326–341. [Google Scholar]
- Nicolas, J.; Brusseau, S.; Charleux, B. A minimal amount of acrylonitrile turns the nitoxide-mediated polymerization of methyl methacrylate into an almost ideal controlled/living system. J. Polym. Sci. A Polym. Chem. 2010, 48, 34–47. [Google Scholar] [CrossRef]
- Chenal, M.; Mura, S.; Marchal, C.; Gigmes, D.; Charleux, B.; Fattal, E.; Couvreur, P.; Nicolas, J. Facile Synthesis of Innocuous Comb-Shaped Polymethacrylates with PEG Side Chains by Nitroxide-Mediated Radical Polymerization in Hydroalcoholic Solutions. Macromolecules 2010, 43, 9291–9303. [Google Scholar] [CrossRef]
- Lessard, B.; Ling, E.J.; Morin, M.S.T.; Maric, M. Nitroxide-mediated radical copolymerization of methyl methacrylate controlled with a minimal amount of 9-(4-vinylbenzyl)-9H-carbazole. J. Polym. Sci. A Polym. Chem. 2011, 49, 1033–1045. [Google Scholar] [CrossRef]
- Lessard, B.H.; Ling, E.J.Y.; Maric, M. Fluorescent, thermoresponsive Oligo(ethylene glycol) Methacrylate/9-(4-Vinylbenzyl)-9H-carbazole Copolymers Designed with Multiple LCSTs via Nitroxide Mediated Controlled Radical Polymerization. Macromolecules 2012, 45, 1879–1891. [Google Scholar] [CrossRef]
- Lessard, B.H.; Guillaneuf, Y.; Mathew, M.; Liang, K.; Clement, J.-L.; Gigmes, D.; Hutchinson, R.A.; Maric, M. Understanding the Controlled Polymerization of Methyl Methacrylate with Low Concentration of 9-(4-Vinylbenzyl)-9H-carbazole Comonomer by Nitroxide-Mediated Polymerization: The Pivotal Role of Reactivity Ratios. Macomolecules 2013, 46, 805–813. [Google Scholar] [CrossRef]
- Delplace, V.; Guegian, E.; Harrisson, S.; Gigmes, D.; Guillaneuf, Y.; Nicolas, J. A ring to rule them all: A cyclic kenete acetal comonomer controls the nitroxide-mediated polymerization of methacrylates and confers tunable degradability. Chem. Commun. 2015, 51, 12847–12850. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Guegian, E.; Ibraim, N.; Harrisson, S.; Nicolas, J. Efficient synthesis of 2-methyl-4-phenyl-1,3-dioxolane, a cyclic ketene acetal for controlling the NMP of methyl methacrylate and confering tunable degradability. Polym. Chem. 2016, 7, 4427–4435. [Google Scholar] [CrossRef]
- Chauvin, F.; Dulfis, P.E.; Gigmes, D.; Guillaneuf, Y.; Marque, S.R.A.; Tordo, P.; Bertin, D. Nitroxide-Mediated Polymerization: The Pivotal Role of the kd Vlaue of the Initiating Alkoxyamine and the Importance of the Experimental Conditions. Macromolecules 2006, 39, 5238–5250. [Google Scholar] [CrossRef]
- Guillaneuf, Y.; Gigmes, D.; Marque, S.R.A.; Astolfi, P.; Greci, L.; Tordo, P.; Bertin, D. First Effective Nitroxide-Mediated Polymerization of Methyl Methacrylate. Macromolecules 2007, 40, 3108–3144. [Google Scholar] [CrossRef]
- Astolfi, P.; Greci, L.; Stipa, P.; Rizzoli, C.; Ysacco, C.; Rollet, M.; Autissier, L.; Tardy, A.; Guillaneuf, Y.; Gigmes, D. Indolinic nitroxides: Evaluation of their potential as universal control agents for nitroxide mediated polymerization. Polym. Chem. 2013, 4, 3694–3906. [Google Scholar] [CrossRef]
- Greene, A.C.; Grubbs, R.B. Synthesis and Evaluation of N-Phenylalkoxyamines for Nitroxide-Mediated Polymerization. Macromolecules 2009, 42, 4388–4390. [Google Scholar] [CrossRef]
- Fukuyama, T.; Kajihara, Y.; Ryu, I.; Studer, A. Nitroxide-Mediated Polymerization of Styrene, Butyl Acrylate, or Methyl Methacrylate by Microflow Reactor Technology. Synthesis 2012, 44, 2555–2559. [Google Scholar]
- Greene, A.C.; Grubbs, R.B. Nitroxide-Mediated Polymerization of Methyl Methacrylate and Styrene with New Alkoxyamines from 4-Nitrophenyl 2-Methylpropionat-2-yl Radicals. Macromolecules 2010, 43, 10320–10325. [Google Scholar] [CrossRef]
- Detrembleur, C.; Jerome, C.; Winter, J.D.; Gerbaux, P.; Clement, J.-L.; Guillaneuf, Y.; Gigmes, D. Nitroxide mediated polymerization of methacrylates at moderate temperatures. Polym. Chem. 2014, 5, 335–340. [Google Scholar] [CrossRef]
- Butz, S.; Baethge, H.; Schmidt-Naake, G. N-oxyl mediated free radical donor-acceptor co- and terpolymerization of styrene, cyclic maleimide monomers and n-butyl methacrylate. Macromol. Chem. Phys. 2000, 201, 2143–2151. [Google Scholar] [CrossRef]
- Edeleva, M.V.; Kirilyuk, I.A.; Zhurko, I.F.; Parkhomenko, D.A.; Tsentalovich, Y.P.; Bagryanskaya, E.G. pH-Sensitive C-NO Bond Homolysis of Alkoxyamines of Imidazoline Series with multiple Ionizable Groups as an Approach for Control of Nitroxide Mediated Polymerization. J. Org. Chem. 2011, 76, 5558–5573. [Google Scholar] [CrossRef] [PubMed]
- Bremond, P.; Marque, S.R.A. First proton triggered C-ON bond homolysis in alkoxyamines. Chem. Commun. 2011, 47, 4291–4293. [Google Scholar] [CrossRef] [PubMed]
- Bagryanskaya, E.; Bremond, P.; Edeleva, M.; Marque, S.R.A.; Parkhomenko, D.; Roubaud, V.; Siri, D. Chemically Triggered C-ON Bond Homolysis in Alkoxyamines. Part 2: DFT Investigation and Application of the pH Effect on NMP. Macromol. Rapid Commun. 2012, 33, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Audran, G.; Bremond, P.; Marque, S.R.A.; Obame, G. Chemically Triggered C-ON Bond Homolysis in Alkoxyamines. 6. Effect of the Counteranion. J. Org. Chem. 2013, 78, 7754–7757. [Google Scholar] [CrossRef] [PubMed]
- Gryn’ova, G.; Smith, L.M.; Coote, M.L. Computational design of pH-switchable control agents for nitroxide mediated polymerization. Phys. Chem. Chem. Phys. 2017, 19, 22678–22683. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by 2,2,6,6,-tetramethylpiperidine-1-oxyl in the presence of a photo-acid generator. Colloid Polym. Sci. 2009, 287, 767–772. [Google Scholar] [CrossRef]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of methyl methacrylate using (4-tert-butylphenyl)dipphenyl-sulfonium triflate as a photo-acid generator. Colloid Polym. Sci. 2010, 288, 239–243. [Google Scholar] [CrossRef]
- Yoshida, E. Synthesis of poly(methyl methacrylate)-block-poly(tetrahydrofuran) by photo-living radical polymerization using a 2,2,6,6,-tetramethylpiperidine-1-oxyl macromediator. Colloid Polym. Sci. 2009, 287, 1417–1424. [Google Scholar] [CrossRef]
- Yoshida, E. Stability of growing polymer chain ends for nitroxide-mediated photo-living radical polymerization. Colloid Polym. Sci. 2010, 288, 1027–1130. [Google Scholar] [CrossRef]
- Yoshida, E. Graft copolymerization of methyl methacrylate on polystyrene backbone through nitroxide-mediated photo-living radical polymerization. Colloid Polym. Sci. 2011, 289, 837–841. [Google Scholar] [CrossRef]
- Yoshida, E. Selective Controlled/Living Photoradical Polymerization of Glycidyl Methacrylate, Using a Nitroxide Mediator in the presence of a Photosensitive Triarylsulfonium Salt. Polymers 2012, 4, 1580–1589. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate using alkoxyamine as an initiator. Colloid Polym. Sci. 2010, 288, 7–13. [Google Scholar] [CrossRef]
- Yoshida, E. Effects of initiators and photo-acid generators on nitroxide-mediated photo-living radical polymerization of methyl methacrylate. Colloid Polym. Sci. 2010, 288, 901–905. [Google Scholar] [CrossRef]
- Versace, D.L.; Lalevee, J.; Fouassier, J.-P.; Gigmes, D.; Guillaneuf, Y.; Bertin, D. Photosensitized alkoxyamines as bicomponent radical photoinitiators. J. Polym. Sci. A Polym. Chem. 2010, 48, 2910–2915. [Google Scholar] [CrossRef]
- Su, J.; Liu, X.; Hu, J.; You, Q.; Cui, Y.; Chen, Y. Photo-induced controlled radical polymerization of methyl methacrylate mediated by photosensitive nitroxides. Polym. Int. 2015, 64, 867–874. [Google Scholar] [CrossRef]
- Guillaneuf, Y.; Bertin, D.; Gigmes, D.; Versace, D.-L.; Lalevee, J.; Fouassier, J.-P. Toward Nitroxide-Mediated Photopolymerization. Macromolecules 2010, 43, 2204–2212. [Google Scholar] [CrossRef]
- Guillaneuf, Y.; Versace, D.L.; Bertin, D.; Lalevee, J.; Gigmes, D.; Fouassieer, J.-P. Inportance of the Position of the Chromophore Group on the Dissociation Process of Light Sensitive Alkoxyamines. Macromol. Rapid Commun. 2010, 31, 1909–1913. [Google Scholar] [CrossRef] [PubMed]
- Farina, M. Stereochemical control by inclusion polymerization. Macromol. Chem. Phys. 1981, 4, 21–35. [Google Scholar] [CrossRef]
- Isobe, Y.; Yamada, K.; Nakano, T.; Okamoto, Y. Stereocontrol in the free-radical polymerization of methacrylates with fluoroalcohols. J. Polym. Sci. A Polym. Chem. 2000, 38, 4693–4703. [Google Scholar] [CrossRef]
- Habaue, S.; Okamoto, Y. Stereocontrol in radical polymerization. Chem. Rec. 2001, 1, 46–52. [Google Scholar] [CrossRef]
- Su, J.; Liu, X.; Li, M.; Zhang, T.; Cui, Y. Stereocontrol of Methyl Methacrylate during Photoinduced Nitroxide-Mediated Polymerization in the Presence of Photosensitive Alkoxyamine. Int. J. Polym. Sci. 2016, 1, 1–8. [Google Scholar] [CrossRef]
- Morris, J.; Telitel, S.; Fairfull-Smith, K.E.; Bottle, S.E.; Lalevee, J.; Clement, J.-L.; Guillaneuf, Y.; Gigmes, D. Novel polymer synthesis methodologies using combinations of thermally- and photochemically-induced nitroxide mediated polymerization. Polym. Chem. 2015, 6, 754–763. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Moszner, N.; Yagci, Y. The use of poly(ethylene oxide) as hydrogen donor in type II photoinitiated free radical polymerization. Poylm. Bull. 2009, 63, 173–183. [Google Scholar] [CrossRef]
- Arslan, M.; Ciftci, M.; Buchmeiser, M.; Yagci, Y. Polyethylene-g-Polystyrene Copolymer by Combination of ROMP, Mn2(CO)10-Assisted TEMPO Substitution and NMRP. ACS Macro Lett. 2016, 5, 946–949. [Google Scholar]
- Kobatake, S.; Harwood, H.J.; Quick, R.P.; Priddy, D.B. Nitroxide-mediated styrene polymerization initiated by an oxoamonium chloride. J. Polym. Sci. A Polym. Chem. 1998, 36, 2555–2561. [Google Scholar] [CrossRef]
- Dao, J.; Benoit, D.; Hawker, C.J. A versatile and efficient synthesis of alkoxyamine LFR via manganese based asymmetric epoxidation catalysts. J. Polym. Sci. A Polym. Chem. 1998, 36, 2161–2167. [Google Scholar] [CrossRef]
- O’Brian, G.; Nielsen, A.; Braslau, R. Ketone Functionalized Nitroxides: Synthesis, Evaluation of N-Alkoxyamine Initiators, and Derivatization of Polymer Termini. Macromolecules 2007, 40, 7848–7854. [Google Scholar]
- Rodler, M.; Harth, E.; Rees, I.; Hawker, C.J. End-group fidelity in nitroxide-mediated living-free radical polymerizations. J. Polym. Sci. A Polym. Chem. 2000, 38, 4749–4763. [Google Scholar] [CrossRef]
- Ruehl, J.; Morimoto, C.; Stevens, D.J.; Braslau, R. Carboxylic acid- and hydroxyl-functionalized alkoxyamine initiators for nitroxide mediated radical polymerization. React. Funct. Polym. 2008, 68, 1563–1577. [Google Scholar] [CrossRef]
- Nil, N.L.; Braslau, R. Synthesis of arylethyl-functionalized N-alkoxyamine initiators and use in nitroxide-mediated radical polymerization. J. Polym. Sci. A Polym. Chem. 2007, 45, 2341–2349. [Google Scholar]
- Hawker, C.J.; Hendrik, J.L. Accurate Control of Chian End by a Novel “Living” Free-Radical Polymerization Process. Macromolecules 1995, 28, 2993–2995. [Google Scholar] [CrossRef]
- Braslau, R.; Tsimelzon, A.; Gewandter, J. Novel Methodology of the Synthesis of N-Alkoxyamines. Org. Lett. 2004, 6, 2233–2235. [Google Scholar] [CrossRef] [PubMed]
- Bosman, A.W.; Vestberg, R.; Heumann, A.; Frechter, J.M.J.; Hawker, C.J. A Modular Approach toward Functionalized Three-Dimensional Macromolecules: From Synthetic Concepts to Practical Applications. J. Am. Chem. Soc. 2003, 125, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Acerbis, S.; Bertin, D.; Boutevin, B.; Gigmes, D.; Lacroix-Desmanez, P.; Mercier, C.L.; Lutz, J.-F.; Marque, S.R.A.; Siri, D.; Tordovo, P. Intramolecular Hydrogen Bonding: The Case of β-Phosphorylated Nitroxide (=Aminoxyl) Radical. Helv. Chim. Acta 2006, 89, 2119–2132. [Google Scholar] [CrossRef]
- Nicolay, R.; Marx, L.; Hemery, P.; Matyjaszewski, K. Synthesis and Evaluation of a Functional, Water- and Organo-Soluble Nitroxide for “Living” Free Radical Polymerization. Macromolecules 2007, 40, 6067–6075. [Google Scholar] [CrossRef]
- Bertin, D.; Gigmes, D.; Marque, S.R.A.; Tordo, P. Polar, Steric, and Stabilization Effects in Alkoxyamines C-ON Bond Homolysis: A Multiparameter Analysis. Macromolecules 2005, 38, 2638–2650. [Google Scholar] [CrossRef]
- Bowden, N.B.; Willets, K.A.; Moerner, W.E.; Waymouth, R.M. Synthesis of Fluorescentyl Labeled Polymers and Their Use in Single-Molecule Imaging. Macromolecules 2002, 35, 8122–8125. [Google Scholar] [CrossRef]
- Gavranovic, G.T.; Csihoni, S.; Bowden, N.B.; Hawker, C.J.; Waymouth, R.M.; Moerner, W.E.; Fuller, G.G. Well-Controlled Living Polymerization of Perylene-Labeled Polyisoprenes and Their Use in Single-Molecule Imaging. Macromolecules 2006, 39, 8121–8127. [Google Scholar] [CrossRef]
- Contrella, N.D.; Tillman, E.S. Synthesis and characterization of fluorene end-labeled polymers prepared by nitroxide-mediated polymerization. Polymer 2008, 49, 4076–4079. [Google Scholar] [CrossRef]
- Ballesteros, O.G.; Maretti, L.; Sastre, R.; Scaiano, J.C. Kinetics of Cap Separation in Nitroxide-Regulated “Living” Free Radical Polymerization: Application of a Novel Methodology Involving a Perfluorescent Nitroxide Switch. Macromolecules 2001, 34, 6184–6187. [Google Scholar] [CrossRef]
- Aspee, A.; Garcia, O.; Maretti, L.; Sastre, R.; Scaiano, J.C. Free Radical Reactions in Poly(methyl methacrylate) Films Monitored Using a Perfluorescent Quinoline-TEMPO Sensor. Macromolecules 2003, 36, 3550–3556. [Google Scholar] [CrossRef]
- Scott, M.E.; Parent, J.S.; Hennigar, S.L.; Whitney, R.A.; Cunningham, M.F. Determination of Alkoxyamine Concentrations in Nitroxyl-Mediated Styrene Polymerization Products. Macromolecules 2002, 35, 7628–7633. [Google Scholar] [CrossRef]
- Beija, M.; Charreyre, M.T.; Martinho, J.M.G. Dye-labelled polymer chains at specific sites: Synthesis by living/controlled polymerization. Prog. Polym. Sci. 2011, 36, 568–602. [Google Scholar] [CrossRef]
- Zhu, Y.; Howell, B.A.; Priddy, D.B. Nitroxide Initiated/Mediated Polymerization of Styrene: Analysis of End-Groups. Am. Chem. Soc. Polym. Prepr. Dev. Polym. Chem. 1997, 38, 97–98. [Google Scholar]
- Chmela, S.; Hrckova, L. Nitroxide mediated styrene radical polymerization using a fluorescent marked mediator. Eur. Polym. J. 2009, 45, 2580–2586. [Google Scholar] [CrossRef]
- Bucisova, L.; Yin, M.; Chmela, S.; Habicher, W.D. Nitroxide-mediated Living Radical Polymerization of Styrene with Fluorescent Initiator. J. Macromol. Sci. Part A Pure Appl. Chem. 2008, 45, 761–768. [Google Scholar] [CrossRef]
- Greene, A.C.; Grubss, R.B. Synthesis and evaluation of an ester-functional alkoxyamine for nitroxide-mediated polymerization. J. Polym. Sci. A Polym. Chem. 2009, 47, 6342–6352. [Google Scholar] [CrossRef]
- Chmela, S.; Kollar, J.; Hrckova, L. Fluorescent dye-labeled TIPNO type regulator for nitroxide mediated reversible-deactivation radical polymerization. J. Photochem. Photobiol. A Chem. 2015, 307–308, 123–130. [Google Scholar] [CrossRef]
- Binder, W.H.; Gloger, D.; Weinstabl, H.; Allmaier, G.; Pittenauer, E. Telechelic Poly(N-iospropylacrylamides) via Nitroxide-Mediated Controlled Polymerization and “Click” Chemistry: Livingness and “Grafting-from” Methodology. Macromolecules 2007, 40, 3097–3107. [Google Scholar] [CrossRef]
- Fleischmann, S.; Kronber, H.; Appelhans, D.; Voit, B.I. Diastereotopic Styrene Arrangement in the Heterosequence of Random Styrene-Ethylene Copolymer. Macromol. Chem. Phys. 2007, 208, 1050–1060. [Google Scholar] [CrossRef]
- Bernhardt, C.; Stoffelbach, F.; Charleux, B. Synthesis and use of a new alkene-functionalized SG1-based alkoxyamine. Polym. Chem. 2011, 2, 229–235. [Google Scholar] [CrossRef]
- Hentschel, J.; Bleck, K.; Ernst, O.; Lutz, J.F.; Börner, H.G. Easy Access to Bioactive Peptide-Polymer Conjugates via RAFT. Macromolecules 2008, 41, 1073–1075. [Google Scholar] [CrossRef]
- Smeek, J.M.; Otten, M.B.J.; Thies, J.; Tirell, D.A.; Stunnenberg, H.G.; Hest, J.C.M.V. Controlled Assembly of Macromolecular β-Sheet Fibrils. Angew. Chem. 2005, 117, 2004–2007. [Google Scholar] [CrossRef]
- Boerner, G.; Smarsly, B.M.; Rank, A.; Schubert, R.; Geng, Y.; Discher, D.E.; Hellweg, T.; Brandt, A. Organization of Self-Assembled Peptide-Polymer Nanofibers in Solution. Macromolecules 2008, 41, 1430–1437. [Google Scholar] [CrossRef]
- Kessel, S.; Thomas, A.; Börner, H.G. Mimicking Biosilification: Programmed Coassembly of Peptide-Polymer Nanotapes and Silica. Angew. Chem. Int. Ed. 2007, 46, 9023–9026. [Google Scholar] [CrossRef] [PubMed]
- Paira, T.K.; Banerjee, S.; Raula, M.; Kotal, A.; Si, S.; Mandal, T.K. Peptide-Polymer Bioconjugates via Atom Transfer Radical Polymerization and Their Solution Aggregation into Hyrid/Nanospheres for Dye Uptake. Macromolecules 2010, 43, 4050–4061. [Google Scholar] [CrossRef]
- Wester, H.-J.; Kessler, H. Molecular Targeting with Peptides or Peptide-Polymer Conjugates: Just a Question of Size? J. Nucl. Med. 2005, 46, 1940–1945. [Google Scholar] [PubMed]
- Kühnle, H.; Börner, H.G. Biotransformation an Polymer-Peptid-Konjugaten—Ein universelles Werkzeug zur Mikrostrukturkontrolle. Angew. Chem. 2009, 121, 6552–6556. [Google Scholar] [CrossRef]
- Chenal, M.; Boursier, C.; Guillaneuf, Y.; Taverana, M.; Couvreur, P.; Nicolas, J. First peptide/protein PEGylation with functional polymers designed by nitroxide-mediated polymerization. Polym. Chem. 2011, 2, 1523–1530. [Google Scholar] [CrossRef]
- Molawi, K.; Studer, A. Reversible switching of substrate activity of poly-N-isopropylacrylamide peptide conjugates. Chem. Commun. 2007, 48, 5173–5175. [Google Scholar] [CrossRef] [PubMed]
- Moller, M.; Hentschel, C.; Chi, X.; Studer, A. Aggregation behaviour of peptide-polymer conjugates containing linear peptide backbones and multiple polymer side chains prepared by nitroxide-mediated radical polymerization. Org. Biomol. Chem. 2011, 9, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Trimaille, T.; Mabrouk, K.; Monnier, V.; Charles, L.; Bertin, D.; Gigmes, D. SG1-Functionalized Peptides as Precursors for Polymer-Peptide Conjugates: A Straightforward Approach. Macromolecules 2010, 43, 4864–4870. [Google Scholar] [CrossRef]
- Garcia-Valdez, O.; George, S.; Champagne-Hertley, R.; Saldivar-Guerra, E.; Champagne, P.; Cunningham, M.F. Chitosan modification via nitroxide-mediated polymerization and grafting to approach in homogeneous media. Polymer 2015, 67, 139–147. [Google Scholar] [CrossRef]
- Garcia-Valdez, O.; Champagne-Hertley, R.; Saldivar-Guerra, E.; Champagne, P.; Cunningham, M.F. Modification of chitosan with polystyrene and poly(n-butyl acrylate) via nitroxide-mediated polymerization and grafting from approach in homogeneous media. Polym. Chem. 2015, 6, 2827–2836. [Google Scholar] [CrossRef]
- Hua, D.; Deng, W.; Tang, J.; Cheng, J.; Zhu, X. A new method of controlled grafting modification of chitosan via nitroxide-mediated polymerization using chitosan-TEMPO macroinitiator. Int. J. Biol. Macromol. 2008, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hua, D.; Tang, J.; Zhu, X.L. Synthesis and propety of poly(sodium 4-styrenesulfonate) grafted chitosan by nitroxide-mediated polymerization with chitosan-TEMPO macroinitiator. Carbohydr. Polym. 2010, 81, 358–364. [Google Scholar] [CrossRef]
- Lefay, C.; Guillaneuf, Y.; Morreira, G.; Thevarajah, Y.Y.; Castignoles, P.; Ziarelli, F. Heterogeneous modification via nitroxide-mediated polymerization. Polym. Chem. 2013, 4, 322–328. [Google Scholar] [CrossRef]
- Audran, G.; Bagryanskaya, E.G.; Bremond, P.; Edeleva, M.V.; Marque, S.R.A.; Parkhomenko, D.A.; Rogozhnikova, O.Y.; Tormyshev, V.M.; Tretyakov, E.V.; Trukhin, D.V.; et al. Trityl-based alkoxyamines as NMP controllers and spin-labels. Polym. Chem. 2016, 7, 6490–6499. [Google Scholar] [CrossRef] [PubMed]
- Szydlowska, J.; Pietrasik, K.; Glaz, L.; Kaim, A. An ESR study of biradicals formed from two 4-amino-TEMPO linked by -(CH2)n-, (n = 2,3,4,6). Chem. Phys. Lett. 2008, 460, 245–252. [Google Scholar] [CrossRef]
- Ruehl, J.; Hill, N.L.; Walter, E.D.; Millhauser, G.; Braslau, R. A Proximal Bisnitroxide Initiator: Studies in Low-Temperature Nitroxide-Mediated Polymerizations. Macromolecules 2008, 41, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Marque, S.R.A.; Siri, D. Is Experimental Evidence Sufficient Enough To Account for the Stabilization Effect of Bisnitroxide on the Fate of NMP Experiments? Macromolecules 2009, 42, 1404–1406. [Google Scholar] [CrossRef]
- Kaim, A.; Szydlowska, J.; Pietrasik, K. The effect of the spacer length on binitroxide radical polymerization of styrene. Macromol. Res. 2011, 19, 1041–1047. [Google Scholar] [CrossRef]
- Pietrasik, K.; Swiatkowska, O.; Kaim, A. Direct synthetic route for water-dispersible polythiophene nanoparticles via surfactant-free oxidative polymerization. Polymer 2010, 55, 812–816. [Google Scholar]
- Kaim, A.; Pietrasik, K.; Stoklosa, T. N,N′-Diaminoethane linked bis-TEMPO-mediated free radical polymerization of styrene. Eur. Polym. J. 2010, 46, 519–527. [Google Scholar] [CrossRef]
- Huang, W.; Chiarelli, R.; Charleux, B.; Rassat, A.; Vairon, J.-P. Unique Behaviour of Nitroxide Biradicals in the Controlled Radical Polymerization of Styrene. Macromolecules 2002, 35, 2305–2317. [Google Scholar] [CrossRef]
- Lizotte, J.R.; Anderson, S.G.; Long, T.E. Novel dinitroxide mediating agent for stable free-radical polymerization. J. Polym. Sci. A: Polym. Chem. 2004, 42, 1547–1556. [Google Scholar] [CrossRef]
- Hill, N.L.; Breslau, R. Synthesis and Characterization of a Novel Bisnitroxide Initiator for Effecting “Outside-In” Polymerization. Macromolecules 2005, 38, 9066–9074. [Google Scholar] [CrossRef]
- Dulfis, P.-E.; Chagneux, N.; Gigmes, D.; Trimaille, T.; Marque, S.R.A.; Bertin, D.; Tordo, P. Intermolecular radical addition of alkoxyamines onto olefins: An easy access to advanced macromolecular architectures precursors. Polymer 2007, 48, 5219–5225. [Google Scholar]
- Miura, Y.; Dairoku, M. Synthesis and characterization of 6- and 12-arm star polymers by nitroxide-mediated radical polymerization of St and MA from dendritic TIPNO-based hexafunctional and dodecafunctional macroinitiator. J. Polym. Sci. A Polym. Chem. 2007, 45, 4364–4376. [Google Scholar] [CrossRef]
- Tortosa, K.; Smith, J.A.; Cunningham, M.F. Synthesis of Polystyrene-block-poly(butyl acrylate) Copolymers Using Nitroxide-Mediated Living Radical Polymerization in Miniemulsion. Macromol. Rapid Commun. 2001, 22, 957–961. [Google Scholar] [CrossRef]
- Nicolas, J.; Charleux, B.; Guerret, O.; Magnet, S. Nitroxide-Mediated Controlled Free-Radical Emulsion Polymerization of Styrene and n-Butyl Acrylate with a Water-Soluble Alkoxyamine as Initiator. Angew. Chem. Int. Ed. 2004, 43, 6186–6189. [Google Scholar] [CrossRef] [PubMed]
- Marx, L.; Henrey, P. Synthesis and evaluation of a new polar, TIPNO type nitroxide for “living” free radical polymerization. Polymer 2009, 50, 2752–2761. [Google Scholar] [CrossRef]
- Tan, W.; Tsarevski, N.V.; Matjyaszewski, K. Determination of equilibrium constants for atom transfer radical polymerization. J. Am. Chem. Soc. 2006, 128, 1598–1604. [Google Scholar]
- Bubak, M.; Morik, J. Equilibrium constants and activation rate coefficients for atom tranfer radical polymerization at pressures up to 2500 bar. Macromol. Chem. Phys. 2010, 211, 2154–2161. [Google Scholar] [CrossRef]
- Matjyaszewski, K. Atom transfer radical polymerization (ATRP): Current status and future perspectives. Macromolecules 2002, 45, 4015–4039. [Google Scholar] [CrossRef]
- Fischer, H. The persistent radical effect, a principle for selective radical reactions and living radical polymerizations. Chem. Rev. 2001, 101, 3581–3610. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.D.; Isse, A.A.; Bartolomei, N.; Gennaro, A. New Insights into the mechanism of activation of atom transfer radical polymerization by Cu(I) complexes. Chem. Commun. 2011, 47, 3580–3582. [Google Scholar] [CrossRef] [PubMed]
- Braunecker, W.A.; Matjyaszewski, K. Recent mechanistic developments in atom transfer radical polymerization. J. Mol. Catal. A Chem. 2006, 254, 155–164. [Google Scholar] [CrossRef]
- Lin, C.Y.; Coote, M.L.; Gennaro, A.; Matjyaszewski, K. Ab Initio Evaluation of the Thermodynamic and Electrochemical Properties of Alkyl Halides and Radicals and Polymerization. J. Am. Chem. Soc. 2008, 12, 12762–12774. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, F.; Matjyaszewski, K. Temperure effect on activation rate constants in ATRP: New mechanistic insights into the activation process. Macromolecules 2009, 42, 6050–6055. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A.; Lin, C.Y.; Hodgson, J.L.; Coote, M.L.; Guliashivili, T. Mechanism of carbon-halogen bond reductive cleavage in activated alkyl halide initiators relevant to living radical polymerizatio: Theoretical and experimental study. J. Am. Chem. Soc. 2011, 133, 6254–6264. [Google Scholar] [CrossRef] [PubMed]
- Abdirisak, A.I.; Bortolamei, N.; Paoli, P.D.; Gennaro, A. On the mechanism of activator of copper-catalyzed atom transfer radical polymerization. Electrochim. Acta 2013, 110, 655–662. [Google Scholar]
- Jakubowski, W.; Matjyaszewski, K. Activators regenerated by electron transfer for atom transfer radical polymerization of (meth)acylates and related block copolymers. Angew. Chem. Int. Ed. 2006, 45, 4482–4486. [Google Scholar] [CrossRef] [PubMed]
- Matjyaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.H.; Braunecker, W.A.; Tsarevsky, N.V. Diminishing Catlyst concentration in atom transfer radical polymerization with reducing agents. Proc. Nat. Acad. Sci. USA 2006, 103, 15309–15314. [Google Scholar] [CrossRef] [PubMed]
- Konkolewicz, K.; Magneau, A.J.D.; Averik, S.E.; Simokava, A.; He, H.K.; Matjyaszewski, K. ICAR ATRP with ppm Cu catlyst in water. Macromolecules 2012, 45, 4461–4468. [Google Scholar] [CrossRef]
- Magneau, A.J.D.; Strandwitz, N.C.; Gennaro, A.; Matjyaszewski, K. Electrochemically mediated atom transfer radical polymerization. Science 2011, 332, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Bortolamei, N.; Isse, A.A.; Magneau, A.J.D.; Gennaro, A.; Matjyaszewski, K. Controlled Aqueous atom transfer radical polymerization with electrochemical generation of the active catalyst. Angew. Chem. Int. Ed. 2011, 50, 11391–11394. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.R.; Mendonca, P.V.; Serra, A.C.; Popov, A.V.; Matjyaszewski, K.; Guliashvili, T.; Coelho, J.F.J. Inorganic sulfites: Efficient reducing agents and supplemental aactivators for atom transfer radical polymerization. ACS Macro Lett. 2012, 1, 1308–1311. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Wang, Y.; Zhong, M.; Krys, P.; Isse, A.A.; Gennaro, A.; Matjyaszewski, K. Reversible-deactivation radical polymerization in the presence of metallic copper. A critical assessment of the SARA ATRP and SET-LRP mechanisms. Macromolecules 2013, 46, 8749–8772. [Google Scholar] [CrossRef]
- Guliashvili, T.; Mendonca, P.V.; Serra, A.C.; Popov, A.V.; Coehlo, J.F.J. Copper mediated controlled living radical polymerization in polar solvents: Insights into some relevant mechanistic aspects. Chem. Eur. J. 2012, 18, 4607–4612. [Google Scholar] [CrossRef] [PubMed]
- Perce, V.; Guliashvili, T.; Ladislaw, J.S.; Wistrand, A.; Stjerndahl, A.; Sienkowska, M.J.; Monteiro, M.J.; Sahoo, S. Ultrafast Synthesis of ultralight molar mass polymers by metal-catalyzed living radical polymerization of acrylates, methacrylates and vinyl chloride mediated by SET at 25 °C. J. Am. Chem. Soc. 2006, 128, 14156–14165. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Iskin, B.; Yagci, Y. Photo-induced Copper(I)-Catalyzed Click Chemistry by Electron transfer Process Using Polynuclear Aromatic Compounds. Macromol. Chem. Phys. 2014, 215, 662–668. [Google Scholar] [CrossRef]
- Guan, Z.; Smart, B.; Remarkable, A. Remarkable Visible Light Effect on Atom-Transfer Radcial Polymerization. Macromolecules. 2000, 33, 6904–6906. [Google Scholar] [CrossRef]
- Pan, X.; Malhotra, M.; Simakova, A.; Wang, Z.; Konkolewicz, D.; Matjyaszewski, K. Photoinduced Atom Transfer Radical Polymerization with ppm-Level Cu Catalyst by Visible Light in Aqueous Media. J. Am. Chem. Soc. 2015, 137, 15430–15433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Z.; Parker, B.; Matjyaszewski, K. ATRP of MMA with ppm Levels of Iron Catalyst. Macromolecules 2011, 44, 4022–4025. [Google Scholar] [CrossRef]
- Taskin, O.S.; Yilmaz, G.; Tasdelen, M.A.; Yagci, Y. Photo-induced reverse atom transfer radical polymerization of methylmethacrylate using camphorquinone/benzhodryl system. Polym. Int. 2014, 63, 902–907. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Ciftci, M.; Yagci, Y. Visible Light-Induced Atom Transfer Radical Polymerization. Macromol. Chem. Phys. 2012, 213, 1391–1396. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Schroeder, K.; Buback, J.; Matjyaszewski, K. Visible Light and Sunlight Photo-induced ATRP with ppm of Cu Catalyst. ACS Macro Lett. 2012, 1, 1219–1223. [Google Scholar] [CrossRef]
- Discekici, E.; Anastasaki, A.; Kaminker, R.; Willenbacher, J.; Truong, N.P.; Fleischmann, C.; Oschmann, B.; Lunn, D.J.; Alaniz, J.R.D.; Davis, T.P.; et al. Light Mediated Atom Transfer Radical Polymerization of Semi-Fluorinated (Meth)acrylates: Facile Access to functional Materials. J. Am. Chem. Soc. 2017, 139, 5939–5945. [Google Scholar] [CrossRef] [PubMed]
- Tasdelen, M.A.; Uygun, M.; Yagci, Y. Studies on Photoinduced ATRP in the presence of Photoinitiator. Macromol. Chem. Phys. 2011, 212, 2036–2042. [Google Scholar] [CrossRef]
- Yagci, Y.; Tasdelen, M.A.; Jokusch, S. Recduction of Cu(II) by photochemically generated phosphonyl radicals to generate Cu(I) as catalyst for atom transfer radical polymerization and azide-alkyne cycloaddition click reactions. Polymer 2014, 55, 3468–3474. [Google Scholar] [CrossRef]
- Ciftci, M.; Tasdelen, M.A.; Yagci, Y. Sunlight induced atom transfer radical polymerization by using dimanganese decacarbonyl. Polym. Chem. 2014, 5, 600–606. [Google Scholar] [CrossRef]
- Dadashi-Silab, S.; Tasdelen, M.A.; Kiskan, B.; Wang, X.; Antonietti, M.; Yagci, Y. Photochemically Mediated Atom Transfer Radical Polymerization Using Polymeric Semiconductor Mesoporous Graphitic Carbon Nitride. Macromol. Chem. Phys. 2014, 215, 675–681. [Google Scholar] [CrossRef]
- Dadashi-Silab, S.; Tasdelen, M.A.; Asiri, A.M.; Khan, S.B.; Yagci, Y. Photoinduced Atom Transfer Radical Polymeriztion Using Semiconductor Nanoparticles. Macromol. Rapid Commun. 2014, 35, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Kork, S.; Ciftci, M.; Tasdelen, M.A.; Yagci, Y. Photoinduced Cu(0)-Mediated Atom Transfer Radical Polymerization. Macromol. Chem. Phys. 2016, 217, 812–817. [Google Scholar] [CrossRef]
- Fors, B.P.; Hawker, C.J. Control of a Living Radical Polymerization of Methacrylates by Light. Angew. Chem. Int. Ed. 2012, 51, 8850–8853. [Google Scholar] [CrossRef] [PubMed]
- Theriot, J.C.; Lim, C.-H.; Yang, H.; Ryan, M.D.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed atom transfer radical polymerization driven by visible light. Science 2016, 27, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Treat, N.J.; Sprafke, H.; Kramer, J.W.; Clark, P.G.; Berton, B.E.; Alaniz, J.R.D.; Fors, B.P.; Hawker, C.J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [Google Scholar] [CrossRef] [PubMed]
- Dadashi-Silab, S.; Pan, X.; Matjyaszewski, K. Phenyl Benzo[b]phenothiazine as a Visible Light Photoredox Catalyst for Metal-Free Atom Transfer Radical Polymerization. Chem. Eur. J. 2017, 23, 5972–5977. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Lim, C.-H.; McCarthy, B.G.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc. 2016, 138, 11399–11407. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-H.; Ryan, M.D.; McCarthy, B.G.; Thierot, J.C.; Sartor, S.M.; Damrauer, N.H.; Musgrave, C.B.; Miyake, G.M. Intramolecular Charge Transfer and Ion Pairing in N,N-Diaryl Dihydrophenazine Photoredox Catalysts for Efficient Organocatalyzed Atom Transfer Polymerization. J. Am. Chem. Soc. 2017, 139, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Allushi, A.; Jokusch, S.; Yilmaz, G.; Yagci, Y. Photo-initiated Metal-Free Controlled/Living Radical Polymerization Using Polynuclear Aromatic Hydrocarbons. Macromolecules 2016, 49, 7785–7792. [Google Scholar] [CrossRef]
- Kutahya, C.; Allushi, A.; Isci, R.; Kreutzer, J.; Öztürk, T.; Yilmaz, G.; Yagci, Y. Photoinduced Metal-Free Atom Transfer Radical Polymerization Using Highly Conjugated Thiophene Derivatives. Macromolecules 2017, 50, 6903–6910. [Google Scholar] [CrossRef]
- Kutahya, C.; Aykac, F.; Yilmaz, G.; Yagci, Y. Using reducible dyes in the presence of amines. Polym. Chem. 2016, 7, 6094–6098. [Google Scholar] [CrossRef]
- Theriot, J.C.; McCarthy, B.G.; Lim, C.-H.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization: Perspectives on Catalyst Design and Performance. Macromol. Rapid Commun. 2017, 38, 1700040. [Google Scholar] [CrossRef] [PubMed]
- Allushi, A.; Kütahya, C.; Aydogan, C.; Kreutzer, J.; Yilmaz, G.; Yagci, Y. Conventional Type II photo-initiators as activators for photo-induced metal-free atom transfer radical polymerization. Polym. Chem. 2017, 8, 1972–1977. [Google Scholar] [CrossRef]
- Niu, T.; Jiang, J.; Li, S.; Ni, B.; Liu, X.; Chen, M. Well-Defined High-Molecular-Weight Polyacrylontrile Formation via Visible-Light-Induced Metal-Free Radical Polymerization. Macromol. Chem. Phys. 2017, 218, 1700169. [Google Scholar] [CrossRef]
- Liu, X.D.; Zhang, L.F.; Cheng, Z.P.; Zhu, X.L. Metal-free photo-induced electron transfer-atom transfer radical polymerization (PET-ATRP) via a visible light organic photocatalyst. Polym. Chem. 2016, 7, 689–700. [Google Scholar] [CrossRef]
- Miyake, G.M.; Thierot, J.C. Perylene as an Organic Photocatalyst for the Radical Polymerization of Functionalized Vinyl Monomers through Oxidative Quenching with Alkyl Bromides and Visible Light. Macromolecules 2014, 47, 8255–8261. [Google Scholar] [CrossRef]
- Huang, Z.; Gu, Y.; Liu, X.; Zhang, L.; Cheng, Z.; Zhu, X. Metal-Free Atom Transfer Radical Polymerization of Methyl Methacrylate with ppm Level of Organic Photocatalyst. Macromol. Rapid Commun. 2017, 38, 1600461–1600469. [Google Scholar] [CrossRef] [PubMed]
- Tsaversky, N.V.; Braunecker, W.A.; Matjyaszewski, K. Electron transfer reaction relevant to atom transfer radical polymerization. J. Organomet. Chem. 2007, 692, 3212–3222. [Google Scholar]
- Ryan, M.D.; Pearson, R.M.; French, T.A.; Miyake, G.M. Impact of Light on Control in Photoinduced Organocatalyzed Atom Transfer Radical Polymerization. Macromolecules 2017, 50, 4616–4662. [Google Scholar] [CrossRef]
- Borska, K.; Moravcikova, D.; Mosnacek, J. Photochemically Induced ATRP of (Meth)Acrylates in the Presence of Air: The Effect of Light Intensity, Ligand, and Oxygen Concentration. Macromol. Rapid Commun. 2017, 38, 1600639. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, C.-J.; Nguyen, J.D.; Finkbeiner, P.; Stephenson, C.R. Visible Light-Mediated Atom Tranfer Radical Addition via Oxidative and Reductive Quenching of Photocatalyst. J. Am. Chem. Soc. 2012, 134, 8875–8884. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.D.; Tucker, J.W.; Konieczynska, M.D.; Stephenson, C.R. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fang, C.; Fantini, M.; Malhotra, N.; So, W.Y.; Peteanu, L.A.; Isse, A.A.; Gennaro, A.; Liu, P.; Matjyaszewski, K. Mechanism of Photo-induced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J. Am. Chem. Soc. 2016, 138, 2411–2425. [Google Scholar] [CrossRef] [PubMed]
- Jokusch, S.; Yagci, Y. The Active Role of Excited States of Phenothiazines in Photo-induced Metal Free Atom Transfer Polymerization: Singlet or Triplet States? Polym. Chem. 2016, 7, 6039–6043. [Google Scholar] [CrossRef]
- Dadashi-Silab, S.; Tasdelen, M.A.; Yagci, Y. Photoinitiated atom transfer radical polymerization: Current status and future perspectives. J. Polym. Sci. A Polym. Chem. 2014, 52, 2878–2888. [Google Scholar] [CrossRef]
- Goto, A.; Tsujii, Y.; Kaji, H. Reversible Complexation Mediated Polymerization (RCMP) of Methyl Methacrylate. In Progress in Controlled Radical Polymerization: Mechanism and Techniques; ACS: Washington, DC, USA, 2012; Volume 20, pp. 305–315. [Google Scholar]
- Goto, A.; Hirai, N.; Nagasawa, K.; Tsujii, Y.; Fukuda, T.; Kaji, H. Phenols and Carbon Compounds as Efficient Organic Catalyst for Reversible Chain Transfer Catalyzed Living Radical Polymerization (RTCP). Macromolecules 2010, 43, 7971–7978. [Google Scholar] [CrossRef]
- Goto, A.; Nagasawa, K.; Shinjo, A.; Tsuji, Y.; Fukuda, T. Reversible Chain Transfer Catalyzed Polymerization of Methyl Methacrylate with In-Situ Formed Alkyl Iodide Initiator. Aust. J. Chem. 2009, 62, 1492–1495. [Google Scholar] [CrossRef]
- Goto, A.; Zushi, H.; Hirai, N.; Wakada, T.; Tsuji, Y.; Fukuda, T. Living Radical Polymerization with Germanium, Tin and Phosphorus Catalysts - Reversible Chain Transfer Catalyzed Polymerization (RTCPs) J. Am. Chem. Soc. 2007, 129, 13347–13354. [Google Scholar] [CrossRef] [PubMed]
- Yorizane, M.; Nagasuga, T.; Kitayama, Y.; Tanaka, A.; Minami, H.; Goto, A.; Fukuda, T.; Okubo, M. Reversible Chain Transfer Catalyzed Polymerization (RTCP) of Methyl Methacrylate with Nitrogen Catalyst in an Aqueous Microsuspension System. Macromolecules 2010, 43, 8703–8705. [Google Scholar] [CrossRef]
- Goto, A.; Ohtsuki, A.; Ohfuji, H.; Tanishima, M.; Kaji, H. Reversible Generation of a Carbon-Centered Radical from Alkyl Iodide Using Organic Salts and Their Application as Organic Catalysts in Living Radical Polymerization. J. Am. Chem. Soc. 2013, 135, 11131–11139. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Suzuki, T.; Ohfuju, H.; Tanishima, M.; Fukuda, T.; Tsujii, Y.; Kaji, H. Reversible Complexation Mediated Living Radical Polymerization (RCMP) Using Organic Catalysts. Macromolecules 2011, 44, 8709–8715. [Google Scholar] [CrossRef]
- Discekici, E.H.; Pester, C.W.; Treat, N.J.; Lawrence, J.; Mattson, K.M.; Narupai, B.; Toumayan, E.P.; Luo, Y.; McGrath, A.J.; Clark, P.G.; et al. Simple Benchtop Approach to Polymer Brush Nanostructures Using Visible-Light-Mediated Metal-Free Atom Transfer Radical Polymerization. ACS Macro Lett. 2016, 5, 258–262. [Google Scholar] [CrossRef]
- Ma, L.; Li, N.; Zhu, J.; Chen, X. Visible Light-Induced Metal Free Surface Initiated Atom Transfer Radical Polymerization of Methyl Methacrylate on SBA-15. Polymers 2017, 9, 58. [Google Scholar] [CrossRef]
- Yan, J.; Pan, X.; Schmitt, M.; Wang, Z.; Bockstaller, M.R.; Matyjaszewski, K. Enhancing Initiation Efficiency in Metal-Free Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP). ACS Macro Lett. 2016, 5, 661–665. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Ye, G.; Zhu, S.; Wang, Z.; Huo, X.; Matyjaszewski, K.; Lu, Y.; Chen, J. Metal-Free Photo-induced Electron Transfer-Atom Transfer Radical Polymerization Integrated with Bioinspired Polydopamine Chemistry as a Green Strategy for Surface Engineering of Magnetic Nanoparticles. Appl. Mater. Interfaces 2017, 9, 13637–13646. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Liu, M.; Heng, C.; Huang, Q.; Mao, L.; Huang, H.; Hui, J.; Deng, F.; Zhang, X.; Wei, Y. Applied Surface Science Surface polyPEGylation of Eu3+ doped luminescent hydroxyapatite nanorods through the combination of ligand exchange and metal free surface initiated atom transfer radical polymerization. Appl. Surf. Sci. 2017, 399, 499–505. [Google Scholar] [CrossRef]
- Zeng, G.; Liu, M.; Shi, K.; Heng, C.; Mao, L.; Wang, Q.; Huang, H.; Deng, F.; Zhang, X.; Wei, Y. Surface modification of nanodiamond through metal free atom transfer radical polymerization. Appl. Surf. Sci. 2016, 390, 710–717. [Google Scholar] [CrossRef]
- Ramsey, B.L.; Pearson, R.M.; Beck, L.R.; Miyake, G.M. Photo-induced Organocatalyzed Atom Transfer Radical Polymerization Using Continuous Flow. Macromolecules 2017, 50, 2668–2674. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yuan, L.; Wang, Z.; Rahman, M.A.; Huang, Y.; Zhu, T.; Wang, R.; Cheng, J.; Wang, C.; Chu, F.; et al. Photo-induced Metal-Free Atom Transfer Radical Polymerization of Biomass-Based Monomers. Macromolecules 2016, 49, 7709–7717. [Google Scholar] [CrossRef]
- Ding, W.; Wang, S.; Yao, K.; Ganewatta, M.S.; Tang, C.; Robertson, M.S. Physical Behavior of Triblock Copolymer Thermoplastic Elastomers Containing Sustainable Rosin-Derived Polymethacrylate End Blocks. ACS Sustain. Chem. Eng. 2017, 5, 11470–11480. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Yuan, L.; Hayat, J.; Trenor, N.M.; Lamm, M.E.; Vlaminck, L.; Bilet, S.; Du Prez, F.E.; Wang, Z.; et al. Biomass Approach toward Robust, Sustainable, Multi-Shape-Memory Materials. ACS Macro Lett. 2016, 5, 602–606. [Google Scholar] [CrossRef]
- Yu, J.; Liu, X.; Wang, C.; Wang, J.; Chu, F.; Tang, C. Integration of renewable cellulose and rosin towards sustainable copolymers by “grafting from” ATRP. Green Chem. 2014, 16, 1854–1864. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, K.; Chen, X.; Wang, J.; Wang, Z.; Ploehn, H.J.; Wang, C.; Chu, F.; Tang, C. Sustainable thermoplastic elastomers derived from renewable cellulose, rosin and fatty acids. Polym. Chem. 2014, 5, 3170–3181. [Google Scholar] [CrossRef]
- Caramli, S.; Murata, H.; Amemiya, E.; Matyjaszewski, K.; Russel, A.J. Tertiary Structure-Based Predicition of How ATRP Initiators react with Proteins. ACS Biomater. Sci. Eng. 2017, 3, 2086–2087. [Google Scholar] [CrossRef]
- Pan, X.; Lathwal, S.; Mack, S.; Yan, J.; Das, S.R.; Matyjaszewski, K. Automated Synthesis of Well-Defined Polymers and Biohybrids by Atom Transfer Radical Polymerization Using a DNA Synthesizer. Angew. Chem. Int. Ed. 2017, 56, 2740–2743. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Karni, D.; Kovaliov, M.; Ramelot, T.; Konkolewicz, D.; Graner, S.; Averick, S. Grafting challenging monomers from proteins using aqueous ICAR ATRP under bio-relevant conditions. Polym. Chem. 2017, 8, 3992–3998. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.C.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.; Mayadunne, R.T.; Moad, G.F.; Moad, C.L. Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Müller, A.H.E.; Zhuang, R.; Yan, D.; Litvenko, G. Kinetic Analysis of “Living” Polymerization Processes Exhibiting Slow Equilibria. 1. Degenerative Transfer (Direct Activity Exchange between Active and “Dormant” Species). Application to Group Transfer. Macromolecules 1995, 28, 4326–4333. [Google Scholar] [CrossRef]
- Litvenko, G.; Müller, A.H.E. General Kinetic Analysis and Comparison of Molecular Weight Distributions for Various Mechanisms of Activity Exchange in Living Polymerization. Macromolecules 1997, 30, 1253–1266. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Radical addition-fragmentation chemistry in polymer synthesis. Polymer 2008, 49, 1079–1131. [Google Scholar] [CrossRef]
- Bicciocchi, E.; Chong, Y.K.; Giorgini, L.; Moad, G.; Rizzardo, E.; Thang, S.H. Substitution Effects on RAFT Polymerization with Benzyl Aryl Trithiocarbonates. Macromol. Chem. Phys. 2010, 211, 529–538. [Google Scholar] [CrossRef]
- Moad, G.; Chiefari, J.; Moad, C.L.; Postma, A.; Mayadunne, R.T.A.; Rizzardo, E.; Thang, S.H. Initiating free radical polymerization. Macromol. Symp. 2002, 182, 65–80. [Google Scholar] [CrossRef]
- Moad, G.; Chiefari, J.; Chong, B.Y.K.; Krstina, J.; Mayudanne, R.T.A.; Postma, A.; Rizzardo, E.; Thang, S.H. Living free radical polymerization with reversible addition—Fragmentation chain transfer (the life of RAFT) Polym. Int. 2000, 49, 993–1001. [Google Scholar]
- Barner-Kowollik, C.; Quinn, J.F.; Nguyen, T.L.U.; Heuts, J.P.A.; Davis, T.P. Kinetic Investigations of Reversible Addition Fragmentation Chain Transfer Polymerization: Cumyl Phenyldithioacetate Mediated Homopolymerizations of Styrene and Methyl Methacrylate. Macromolecules 2001, 34, 7849–7857. [Google Scholar] [CrossRef]
- Monteiro, M.J.; Brouwer, H.D. Intermediate Radiacal Termination as the Mechanism for Retardation in Reversible Addition-Fragmentation Chain Transfer Polymerization. Macromolecules 2001, 34, 349–352. [Google Scholar] [CrossRef]
- Ting, S.R.; Davis, T.P.; Zetterlund, P.B. Retardation in RAFT Polymerization: Does Cross-Termination Occur with Short Radicals Only? Macromolecules 2011, 44, 4187–4193. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Hawkett, B.S.; Gray-Weale, A.; Perrier, S. RAFT polymerization kinetics: How long are the cross-terminatig oligomers? J. Polym. Sci. A Polym. Chem. 2009, 47, 3455–3466. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Buback, M.; Charleux, B.; Coote, M.L.; Drache, M.; Fukuda, T.; Goto, A.; Klumperman, B.; Lowe, A.B.; McLeary, J.B.; et al. Mechanism and kinetics of dithiobenzoate-mediated RAFT polymerization. I. The current situation. J. Polym. Sci. A Polym. Chem. 2006, 44, 5809–5831. [Google Scholar] [CrossRef]
- Li, M.; Li, H.; De, P.; Summerlin, B.S. Thermoresponsive Block Copolymer-Protein Conjugates Prepared by Graftng-from via RAFT Polymerization. Macromol. Rapid Commun. 2011, 32, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, M.; Yu, X.; Bapat, A.P.; Sumerlin, B.S. Block copolymer conjugates prepared by sequentially grafting from proteins via RAFT. Polym. Chem. 2011, 2, 1531–1535. [Google Scholar] [CrossRef]
- Sumerlin, B.S. Proteins as Initiators of Controlled Radical Polymerization: Grafting-from via ATRP and RAFT. ACS Macro Lett. 2012, 1, 141–145. [Google Scholar] [CrossRef]
- Su, X.-L.; He, W.-D.; Li, J.; Li, L.-Y.; Zhang, B.-Y.; Pan, T.-T. RAFT cryopolymerization of N,N-dimethylacrylamide and N-isopropylacrylamide in moderatly frozen aqueous solution. J. Polym. Sci. A Polym. Chem. 2009, 47, 6863–6872. [Google Scholar]
- Su, X.-L.; He, W.-D.; Pan, T.-T.; Ding, Z.-L.; Zhang, Y.-J. RAFT cryopolymerization of acrylamides and acrylates in dioxane at −5 °C. Polymer 2010, 51, 110–114. [Google Scholar]
- Paulus, R.M.; Beer, C.R.; Hoogenboom, R.; Schubert, U.S. High Temperature Initiator-Free RAFT Polymerization of Methyl Methacrylates in a Microwave Reactor. Aust. J. Chem. 2009, 62, 254–259. [Google Scholar] [CrossRef]
- Boyer, C.; Bulmus, V.; Davis, T.P.; Ladmiral, V.; Liu, J.; Perrier, S. Bioapplicatins of RAFT Polymerization. Chem. Rev. 2009, 109, 5402–5436. [Google Scholar] [CrossRef] [PubMed]
- Semsarilar, M.; Perrier, S. ‘Green’ reversible addition-fragmentation chain-transfer (RAFT). Nat. Chem. 2010, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Duffy, C.; Phelan, M.; Zwitterlund, P.; Aldabbagh, F. Reversible addition-fragmentation chain transfer polymerization of alkyl-2-cyanoacrylates: An assessment of livingness. J. Polym. Sci. A Polym. Chem. 2017, 55, 1397–1408. [Google Scholar] [CrossRef]
- Mazieres, S.; Kulai, I.; Geagea, R.; Ladeira, S.; Destarac, M. Mechanistic Understanding of the Divergent Cylcization of 0-Alkynylbenzaldehyde Acetals and Thioacetals Catalyzed by Metal Halides. Chem. Eur. J. 2015, 21, 1726–1734. [Google Scholar] [PubMed]
- Destarac, M.; Gauthier-Gillaizeau, I.; Vuong, C.-T.; Zard, S.Z. Vinylogous Thionothio Compounds for RAFT Polymerization. Macromolecules 2006, 39, 912–914. [Google Scholar] [CrossRef]
- Destarac, M.; Charmot, D.; Franck, X.; Zard, S.Z. Dithiocarbonates as universal reversible addition-fragmentation chain transfer. Macromol. Rapid Commun. 2000, 39, 1035–1039. [Google Scholar] [CrossRef]
- Thang, S.H.; Chong, Y.H.; Mayadunne, R.T.A.; Moad, G.; Rizzardo, E. A novel synthesis of functional diethioesters, diethiocarbamates, xanthates and trithiocarbonates. Tetrahedron Lett. 1999, 40, 2435–2438. [Google Scholar] [CrossRef]
- Esteves, A.C.C.; Hodge, P.; Trindade, T.; Barros-Timmons, A.M. Preparation of nanocomposites by reverssible addition-fragmentation chain transfer polymerization from the surface of quantum dots in mini emulsion. J. Polym. Sci. A Polym. Chem. 2009, 47, 5367–5377. [Google Scholar] [CrossRef]
- Skaff, H.; Emrick, T. Reversible Addition Fragmentation Chain Transfer (RAFT) Polymerization from Unprotected Cadmium Selenide Nanoparticles. Angew. Chem. Int. Ed. 2004, 43, 5383–5386. [Google Scholar] [CrossRef] [PubMed]
- Poetzsch, R.; Fleischmann, S.; Tock, C.; Komber, H.; Voit, B.I. Combining RAFT and Staudinger Ligation: A Potentially New Synthetic Tool for Bioconjugation Formation. Macromolecules 2011, 44, 3260–3269. [Google Scholar] [CrossRef]
- Laus, M.; Papa, R.; Sparnacci, K.; Alberti, A.; Benaglia, M.; Macciantelli, D. Controlled Radical Polymerization of Styrene with Phosphoryl- and (Thiophosphoryl)dithioformates as RAFT Agents. Macromolecules 2001, 34, 7269–7275. [Google Scholar] [CrossRef]
- Alberti, A.; Benaglia, M.; Laus, M.; Macciantelli, D.; Sparnacci, K. Direct ESR Detection of Free Radicals in the RAFT Polymerization of Styrene. Macromolecules 2003, 36, 736–740. [Google Scholar] [CrossRef]
- Glassner, M.; Delaitre, G.; Kaupp, M.; Blico, J.P.; Barner-Kowollik, C. (Ultra)-Fast Catalyst-Free Macromolecular Conjugation in Aqueous Environment at Ambient Temperature. J. Am. Chem. Soc. 2012, 132, 7274–7277. [Google Scholar] [CrossRef] [PubMed]
- Sinnwell, S.; Inglis, A.J.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. An Atom-Efficient Conjugation Approach to Well-Defined Block Copolymer Using RAFT Chemistry and Hetero Diels-Alder Cycoaddition. Chem. Commun. 2008, 2052–2054. [Google Scholar] [CrossRef] [PubMed]
- Inglis, A.J.; Sinnwell, S.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, N.H. Reversible Addition Fragmentation Chain Transfer (RAFT) and Hetero-Diels-Alder Chemistry as Convenient Conjugation Tool for Access to Complex Molecular Design. Macromolecules 2008, 41, 4120–4126. [Google Scholar] [CrossRef]
- Inglis, A.J.; Sinnwell, S.; Stenzel, M.H.; Barner-Kowollik, C. Ultrafast Click Conjugation of Macromolecular Building Blocks at Ambient Temperature. Angew. Chem. Int. Ed. 2009, 48, 2411–2414. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, J.; Mayadunne, R.T.A.; Moad, C.L.; Moad, G.; Rizzardo, E.; Postama, A.; Skidmore, M.A.; Thang, S.H. Thiocarbonylthio Compounds (S = C(Z)S-R) in Free Radical Polymerization with Reversible Addition-Fragmentation Chain Transfer (RAFT Polymerization). Effect of the Activating Group Z. Macromolecules 2003, 36, 2273–2283. [Google Scholar] [CrossRef]
- Mayadunne, R.T.A.; Rizzardo, E.; Chiefari, J.; Chong, Y.K.; Moad, G.; Thang, S.H. Living Radcial Polymerization with Reversible Addition-Fragmentation Chain Transfer (RAFT Polymerization) Using Dithiocarbamates as Chain Transfer Agents. Macromolecules 1999, 32, 6977–6980. [Google Scholar] [CrossRef]
- Benaglia, M.; Cheifari, J.; Chong, Y.K.; Moad, G.; Rizzardo, E.; Thang, S.H. Universal (Switchable) RAFT Agents. J. Am. Chem. Soc. 2009, 131, 6914–6915. [Google Scholar] [CrossRef] [PubMed]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Rizzardo, E.; Chiefari, J.; Mayadunne, R.T.A.; Moad, G.; Thang, S.H. Synthesis of Defined Polymers by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process. ACS Symp. Ser. 2000, 768, 278–296. [Google Scholar]
- Moad, G.; Keddie, D.; Guerro-Sanchez, C.; Rizzardo, E.; Thang, S.H. Advances in Switchable RAFT Polymerization. Macromol. Symp. 2015, 350, 34–42. [Google Scholar] [CrossRef]
- Keddie, D.; Guerro-Sanchez, C.; Moad, G.; Rizzardo, E.; Thang, S.H. Switchable Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization in Aqueous Solution, N,N-Dimethylacrylamide. Macromolecules 2011, 44, 6738–6745. [Google Scholar] [CrossRef]
- Gardiner, J.; Martinez-Botella, I.; Kohl, T.M.; Krstina, J.; Moad, G.; Tyrell, J.H.; Coote, M.L.; Tsanaktsidis, J. 4-Halogeno-3,5-dimethyl-1H-pyrazole-1-carbodithioates: Versatile reversible addition fragmentation chain transfer agents with broad applicability. Polym. Int. 2017, 55, 1438–1447. [Google Scholar] [CrossRef]
- Subramanian, S.H.; Babu, R.P.; Dhamodharan, R. Ambient Temperature Polymerization of Styrene by Single Electron Transfer Initiation, Followed by Reversible Addition Fragmentation Chain Transfer Control. Macromolecules 2008, 41, 262–265. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, W.; Xia, H.; Zhu, J.; Zhang, W.; Zhu, X. Single-Electron Transfer Living Radical Polymerization (SET-LRP) of Methyl Methacrylate (MMA) with a Typical RAFT Agent as an Initiator. Macromolecules 2009, 42, 7360–7366. [Google Scholar] [CrossRef]
- Marenich, A.V.; Ho, J.; Coote, M.L.; Cramer, C.J.; Truhlar, D. Computational electrochemistry: Prediction of liquid-phase reduction potentials. Phys. Chem. Chem. Phys. 2014, 16, 15068–15106. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.K.; You, Y.Z.; Pan, C.Y. 60Co-γ-Irradiation-Initiated “Living” Free-Radical Polymerization in the Presence of Dibenzyl Trithiocarbonate. Macromol. Rapid Commun. 2001, 22, 315–319. [Google Scholar] [CrossRef]
- Otsu, T. Iniferter concept and living radical polymrization. J. Polym. Sci. A Polym. Chem. 2000, 38, 2121–2136. [Google Scholar] [CrossRef]
- Hong, C.; You, Y.Z.; Bai, R.K.; Bai, C.Y.; Borjihan, G.J. Controlled polymerization of acrylic acid under 60Co irradiation in the presence of dibenzyl trithiocarbonate. Polym. Sci. A Polym. Chem. 2001, 39, 3934–3939. [Google Scholar] [CrossRef]
- You, Y.Z.; Hong, C.H.; Bai, R.K.; Pan, C.Y.; Wang, J. Photo-Initiated Living Free Radical Polymerization in the Presence of Dibenzyl Trithiocarbonate. Macromol. Chem. Phys. 2002, 203, 477–483. [Google Scholar] [CrossRef]
- Quinn, J.F.; Barner, L.; Barner-Kowollik, C.; Rizzardo, E.; Davis, T.P. Reversible Addition-Fragmentation Chain Transfer Polymerization Initiated with Ultraviolet radiation. Macromolecules 2002, 35, 7620–7627. [Google Scholar] [CrossRef]
- Lu, L.; Yang, N.; Cai, Y. Well-controlled reversible addition-fragmentation chain trasfer radical polymerization under ultraviolet radiation at ambient temperature. Chem. Commun. 2005, 5287–5288. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, H.; Yang, N.; Cai, Y. Toward Rapid and Well-Controlled Ambient Temperature RAFT Polymerization under UV-Vis Radiation: Effect of Radiation Wave Range. Macromolecules 2006, 39, 3770–3776. [Google Scholar] [CrossRef]
- Jiang, W.; Lu, L.; Cai, Y. Highly Efficient and Well-Controlled Ambient Temperature RAFT Polymerization under Solar Radiation. Macromol. Rapid Commun. 2007, 28, 725–728. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, L.; Cheng, Z.; Zhu, X. Reversible Addition-Fragmentation Chain Transfer Polymerization of Acrylonitrile under Irradiation of Blue LED Light. Polymers 2017, 9, 4. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Durmaz, Y.Y.; Karagöz, B.; Bicak, N.; Yagci, Y. A new photoiniferter/RAFT agent for ambient temperature rapid and well-controlled radical polymerization. J. Polym. Sci. A Polym. Chem. 2008, 46, 3387–3395. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Pan, E.H.; Stenzel, M.H.; Barner-Kowollik, C.; Davis, T.P.; Lewis, D.; Barner, L. Ambient Temperature RAFT Polymerization of Acrylic Acid Initiated with Ultraviolet Radiation in Aqueous Solution. Macromolecules 2007, 40, 2978–2980. [Google Scholar] [CrossRef]
- Coyle, J.D. The photochemistry of thiocarbonyl compounds. Tetrahedron 1985, 41, 5393–5425. [Google Scholar] [CrossRef]
- McKenzie, T.G.; Fu, Q.; Wong, E.H.H.; Dunstan, D.E.; Qiao, G.G. Visible Light Mediated Controlled Radical Polymerization in the Absence of Exogeneous Radical Sources or Catalysts. Macromolecules 2015, 48, 3864–3872. [Google Scholar] [CrossRef]
- Hadriharan, N.; Ponnusamy, K.; Dhamodharan, R. Controlled polymerization of methacrylate at ambient temperature using trithiocarbonate chain transfer agents via SET-RAFT-cyclohexyl methacrylate: A model study. J. Polym. Sci. A Polym. Chem. 2010, 48, 5329–5338. [Google Scholar] [CrossRef]
- Hadriharan, N.; Dhamodharan, R. Controlled polymerization of carbazole-based vinyl and methacrylate monomers at ambient temperature: A comparative study through ATRP, SET, and SET-RAFT polymerization. J. Polym. Sci. A Polym. Chem. 2011, 49, 1021–1032. [Google Scholar] [CrossRef]
- Littrell, D.M.; Bowers, D.H.; Tatarchuk, B.J. Hydrazine reduction of transition-metal oxides. J. Chem. Soc. Faraday Trans. 1 1987, 83, 3271–3280. [Google Scholar] [CrossRef]
- Fleischmann, S.; Rosen, B.M.; Perec, V. SET-LRP of acrylate in air. J. Polym. Sci. A Polym. Chem. 2010, 48, 1190–1196. [Google Scholar] [CrossRef]
- Fleischmann, S.; Rosen, B.M.; Perec, V. Dramatic acceleration of SET-LRP of methyl acrylate during catalysis with activated Cu(0) wire. J. Polym. Sci. A Polym. Chem. 2010, 48, 5109–5119. [Google Scholar]
- Fleischmann, S.; Rosen, B.M.; Perec, V. SET-LRP of methyl methacrylate initiated with CCl4 in the presence and absence of air. J. Polym. Sci. A Polym. Chem. 2010, 48, 2243–2250. [Google Scholar] [CrossRef]
- Fleischmann, S.; Rosen, B.M.; Perec, V. Disproportionating versus nondisproportionatig solvent effect in the SET-LRP of methyl acrylate during catalysis with nonactivated and activated Cu(0) wire. J. Polym. Sci. A Polym. Chem. 2011, 49, 4227–4240. [Google Scholar]
- Fleischmann, S.; Rosen, B.M.; Perec, V. SET-LRP of methyl acrylate catalyzed with activated Cu(0) wire in methanol in the presence of air. J. Polym. Sci. A Polym. Chem. 2011, 49, 4756–4765. [Google Scholar]
- Nguyen, N.H.; Perec, V. Acid dissolution of copper oxides as a method for the activation of Cu(0) wire catalyst for Set-LRP. J. Polym. Sci. A Polym. Chem. 2011, 49, 4241–4252. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Wang, W.; Zhu, J.; Cheng, Z.; Zhou, N.; Zhang, W.; Zhu, X. SET-RAFT of MMA mediated by ascorbic acid-activated copper oxide. J. Polym. Sci. A Polym. Chem. 2012, 50, 1424–1426. [Google Scholar] [CrossRef]
- Hodgson, J.L.; Namazian, M.; Bottle, S.E.; Coote, M.L. One-Electron Oxidation and Reduction Potentials of Nitroxide Antioxidants: A Theoretical Study. J. Phys. Chem. A 2007, 111, 13595–13605. [Google Scholar] [CrossRef] [PubMed]
- Zare, H.R.; Eslami, M.; Namazian, M.; Coote, M.L. Experimental and Theoretical Studies of Redox Reactions of o-Chloranil in Aqueous Solution. J. Phys. Chem. B 2009, 113, 8080–8085. [Google Scholar] [CrossRef] [PubMed]
- Maximiano, P.; Mendonca, P.V.; Costa, J.R.C.; Haworth, N.L.; Serra, A.C.; Guliashvili, T.; Coote, M.L.; Coehlo, J.F.J. Ambient Temperature Transition-Metal-Free Dissociative Electron Trnasfer Reverible Addition-Fragmentation Chain Transfer Polymerization (DET-RAFT) of Methacrylates, Acrylates, and Styrenes. Macromolecules 2016, 49, 1597–1604. [Google Scholar] [CrossRef]
- Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. A Robust and Versatile Photoinduced Living Polymerization of Conjugated and Unconjugated Monomers and Its Oxygen Tolerance. J. Am. Chem. Soc. 2014, 136, 5508–5519. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jung, K.; Boyer, C. Oxygen Tolerance Study of Photoinduced Electron Transfer-Reversible Addition-Fragmentation Chain Transfer (PET-RAFT) Polymerization Mediated by Ru(bpy)3Cl2. Macromolecules 2014, 47, 4217–4299. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, J.; Boyer, C. Photoinduced Electron Transfer-Reversible Addition-Fragmentation Chain Transfer (PET-RAFT) Polymerization of Vinyl Acetate and N-Vinylpyrrolidone: Kinetic and Oxygen Tolerance Study. Macromolecules 2014, 47, 4930–4942. [Google Scholar] [CrossRef]
- Xu, J.; Jung, K.; Corrigan, N.A.; Boyer, C. Aqueous photoinduced living/controlled polymerization: Tailoring for bioconjugation. Chem. Sci. 2014, 5, 3568–3575. [Google Scholar] [CrossRef]
- Xu, J.; Shanmugam, S.; Duong, H.; Boyer, C. Organo-photocatalysts for photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization. Polym. Chem. 2015, 6, 5615–5624. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, J.; Boyer, C. Utilizing the electron transfer mechanism of chlorophyll a under light for controlled radical polymerization. Chem. Sci. 2015, 6, 1341–1349. [Google Scholar] [CrossRef]
- Shanugam, S.; Xu, J.; Boyer, C. Exploiting Metalloporphorins for Selective Living Radical Polymerization. J. Am. Chem. Soc. 2015, 137, 9174–9185. [Google Scholar] [CrossRef] [PubMed]
- Yeow, J.; Chapman, R.; Xu, J.; Boyer, C. Oxygen tolerant photopolymerization for ultralow volumes. Polym. Chem. 2017, 8, 5012–5022. [Google Scholar] [CrossRef]
- Tucker, B.S.; Coughlin, M.L.; Figg, C.A.; Sumerlin, B.S. Grafting-From Proteins Using Metal-Free PET-RAFT Polymerizations under Mild Visible-Light Irradiation. ACS Macro Lett. 2017, 6, 452–457. [Google Scholar] [CrossRef]
- Ng, Y.-H.; Lena, F.D.; Chai, C.L.L. Metalloenzymatic radical polymerization using alkyl halides as initiators. Polym. Chem. 2011, 2, 589–594. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, X.; Zhu, A.; Ma, K.; Lv, Y.; Wang, X.; An, Z. Enzym-Initiated Reversible Addition-Fragmentation Chain Transfer Polymerization. Macromolecules 2015, 48, 7792–7802. [Google Scholar] [CrossRef]
- Emery, O.; Lalot, T.; Brigodiot, M.; Marechal, E. Free-radical polymerization of acrylamide by horseradish peroxidase-mediated initiation. J. Polym. Sci. A Polym. Chem. 1997, 35, 3331–3333. [Google Scholar] [CrossRef]
- Danielason, A.P.; Kuren, D.B.-V.; Lucius, M.E.; Makaroff, K.; Williams, C.; Page, R.C.; Berberich, J.A.; Konkolewicz, D. Well-Defined Macromolecules Using Horseradish Peroxidase as a RAFT Initiase. Macromol. Rapid Commun. 2016, 37, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.; Stenzel, M.H.; Davis, T.P. Photoresponsive nanocarriers based on PAMAM dendrimers with o-nitrobenzyl shell. J. Polym. Sci. A Polym. Chem. 2010, 49, 551–557. [Google Scholar] [CrossRef]
- Harth-Smith, G.; Chaffery-Millar, H.; Barner-Kowollik, C. Living Star Polymer Formation: Detailed Assessment of Poly(acrylate) Radical Reaction Pathways via ESI-MS. Macromolecules 2008, 41, 3023–3041. [Google Scholar] [CrossRef]
- Liu, J.; Duaong, H.; Whittaker, M.R.; Davis, T.P.; Boyer, C. Synthesis of Functional Core, Star Polymers via RAFT Polymerization for Drug Delivery Applications. Macromol. Rapid Commun. 2012, 33, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Mayadunne, R.T.A.; Jeffery, J.; Moad, G.; Rizzardo, E. Living Free Radical Polymerization with Reversible Addition-Fragmentation Chain Transfer (RAFT Polymerization): Approaches to Star Polymers. Macromolecules 2003, 36, 1505–1513. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Davis, T.P. Star polymer synthesis using trithiocarbonate functional β-cyckodextrin cores (reversible addition-fragmentation chain-transfer polymerization). J. Polym. Sci. A Polym. Chem. 2002, 40, 4498–4512. [Google Scholar] [CrossRef]
- Chaffey-Millar, H.; Stenzel, M.H.; Davis, T.P.; Coote, M.L.; Barner-Kowollik, C. Design Criteria for Star Polymer Formation Processes via Living Free Radical Polymerization. Macromolecules 2006, 39, 6406–6419. [Google Scholar] [CrossRef]
- Stenzel-Rosenbaum, M.; Davis, T.P.; Chen, V.; Fane, A.G. Star-polymer synthesis via radical reversible addition-fragmentation chain-transfer polymerization. J. Polym. Sci. A Polym. Chem. 2001, 39, 2777–2783. [Google Scholar] [CrossRef]
- Johnston-Hall, G.; Monteiro, M.J. Bimolecular radical termination: New perspectives and insights. J. Polym. Sci. A Polym. Chem. 2008, 46, 3155–3173. [Google Scholar] [CrossRef]
- Boschmann, D.; Vana, P. Poly(vinyl acetate) and Poly(vinyl propionate) Star Polymers via Reversible Addition Fragmentation Chain Transfer (RAFT) Polymerization. Polym. Bull. 2005, 53, 231–242. [Google Scholar] [CrossRef]
- Boschmann, D.; Vana, P. Z-RAFT Star Polymerization of Acrylates: Star Coupling via Intermolecular Chain Transfer to Polymer. Macromolecules 2007, 40, 2683–2693. [Google Scholar] [CrossRef]
- Darcos, V.; Dureault, A.; Taton, D.; Gnanou, Y.; Marchand, P.; Caminade, A.-M.; Majoral, J.-P.; Destarac, M.; Leising, F. Synthesis of hybrid dendrimer-star polymers by the RAFT process. Chem. Commun. 2004, 40, 2110–2111. [Google Scholar] [CrossRef] [PubMed]
- Duwez, A.-S.; Guilliet, P.; Colard, C.; Gohy, J.-F.; Fustin, C.A. Dithioester and Trithiocarbonates as Anchoring Groups for the “Grafting-To” Approach. Macromolecules 2006, 39, 2729–2731. [Google Scholar] [CrossRef]
- Fustin, C.A.; Colard, C.; Filali, M.; Guillet, P.; Duwez, A.-S.; Meier, M.A.R.; Schubert, U.S.; Gohy, J.-F. Tuning the Hydrophilicity of Gold Nanoparticles Templated in Star Block Copolymers. Langumir 2006, 22, 6690–6695. [Google Scholar] [CrossRef] [PubMed]
- Ebling, B.; Vana, P. RAFT-Polymers with Single and Multiple Trithiocarbonate Groups as Uniform Gold-Nanoparticle Coatings. Macromolecules 2013, 46, 4862–4871. [Google Scholar] [CrossRef]
- Rossner, C.; Ebling, B.; Vana, P. Spherical Gold-Nanoparticle Assemblies with Tunable Interparticle Distance Mediated by Multifunctional RAFT Polymers. ACS Macro Lett. 2013, 2, 1073–1076. [Google Scholar] [CrossRef]
- Rossner, C.; Vana, P. Planet-Satellite Nanostructures Made to Order by RAFT Star Polymers. Angew. Chem. Int. Ed. 2014, 53, 12639–12642. [Google Scholar] [CrossRef] [PubMed]
- Skey, J.; O’Reil, R.K. Facile one pot synthesis of a range of reversible addition-fragmentation chain transfer (RAFT) agents. Chem. Commun. 2008, 4183–4185. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.R.; Dunclaff, D.J.; Rannard, S.P.; Perrier, S. Selective One-Pot Synthesis of Trithiocarbonates, Xanthates, and Dithiocarbamates for Use in RAFT/MADIX Living Radical Polymerizations. Org. Lett. 2006, 8, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Coady, D.J.; Norris, B.C.; Lynch, V.M.; Bielawski, C.W. Ionic Dithioester-Based RAFT Agents Derived from N-Heterocyclic Carbenes. Macromolecules 2008, 41, 3775–3778. [Google Scholar] [CrossRef]
- Bathfield, M.; Daviot, D.; D’Agosto, F.; Spitz, R.; Ladaviere, C.; Charreyre, M.T.; Delair, T. Synthesis of Lipd-α-End-Functionalized Chains by RAFT Polymerization. Stabilization of Lipid/Polymer Particle Assemblies. Macromolecules 2008, 41, 8346–8353. [Google Scholar] [CrossRef]
- Liu, J.; Bulmus, V.; Herlambang, D.L.; Barner-Kowollik, C.; Stenzel, M.H.; Davis, T.P. In Situ Formation of Protein-Polymer Conjugates through Reversible Addition Fragmentation Chain Transfer Polymerization. Angew. Chem. Int. Ed. 2007, 46, 3099–3103. [Google Scholar] [CrossRef] [PubMed]
- Moad, G.; Chong, Y.K.; Postma, A.; Rizzardo, E.; Thang, S.H. Advances in RAFT polymerization: The synthesis of polymers with defined end-groups. Polymer 2005, 46, 8458–8468. [Google Scholar] [CrossRef]
- Zhou, G.; Harrunaa, I.I. Synthesis and Characterization of Bis(2,2′:6′,2″-terpyridine)ruthenium(II)—Connected Diblock Polymers via RAFT Polymerization. Macromolecules 2005, 38, 4114–4123. [Google Scholar] [CrossRef]
- Samakande, A.; Chaghi, R.; Derrien, G.; Charnay, C.; Hartmann, P.C. Aqueous behaviour of cationic surfactants containing a cleavable group. J. Colloid Int. 2008, 320, 315–320. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Li, M.; Gondi, S.R.; Sumerlin, B.S. Temperature-Regulated Activity of Responsive Polymer-Protein Conjugates by Grafting-from via RAFT Polymerization. J. Am. Chem. Soc. 2008, 130, 11288–11289. [Google Scholar] [CrossRef] [PubMed]
- Delduc, P.; Tailhna, C.; Zard, S.Z. A tetrameric copper(I) phosphide and its conversion into a bis(phosphido)cuprate and a bis(phosphine)-complexed tetrameric coper(I) thiolate. Chem. Soc. Chem. Commun. 1988, 308–310. [Google Scholar] [CrossRef]
- Quiclet-Sire, B.; Revol, G.; Zard, S.Z. A convergent, modular access to complex amines. Tetrahedron 2010, 66, 6656–6666. [Google Scholar] [CrossRef]
- Chen, M.; Haeussler, M.; Moad, G.; Rizzardo, E. Block copolymer containing organic semiconductor segments by RAFT polymerization. Org. Biomol. Chem. 2011, 9, 6111–6119. [Google Scholar] [CrossRef] [PubMed]
- Houshayr, S.; Keddie, D.J.; Moad, G.; Mulder, R.J.; Saubern, S.; Tsanaktsidis, J. The scope for synthesis of macro-RAFT agents by sequential insertion of single monomer units. Polym. Chem. 2012, 3, 1879–1888. [Google Scholar] [CrossRef]
- Chen, M.; Ghiggino, K.P.; Mau, A.W.H.; Rizzardo, E.; Sasse, W.H.F.; Thang, S.H.; Wilson, G.J. Synthesis of Functionalized RAFT Agents for Light Harvesting Macromolecules. Macromolecules 2004, 37, 5479–5481. [Google Scholar] [CrossRef]
- Xu, J.; Shanmugam, S.; Fu, C.; Aguey-Zinsou, K.-F.; Boyer, C. Selective Photoactivation: From a Single Unit Monomer Insertion Reaction to Controlled Polymer Architectures. J. Am. Chem. Soc. 2016, 138, 3094–3106. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fu, C.; Shanmugam, S.; Hawker, C.J.; Moad, G.; Boyer, C. Synthesis of Discrete Oligomers by Sequential PET-RAFT Single-Unit Monomer Insertion. Angew. Chem. Int. Ed. 2017, 129, 8376–8383. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Huang, Z.; Hawker, C.J.; Moad, G.; Xu, J.; Boyer, C. RAFT-mediated, visible light-initiated single unit monomer insertion and its application in the synthesis of sequence-defined polymers. Polym. Chem. 2017, 8, 4637–4643. [Google Scholar] [CrossRef]
- Kutcherlapati, S.N.R.; Yeole, N.; Gadi, M.R.; Perali, R.S.; Jana, T. RAFT mediated one-pot synthesis of glycopolymer particles with tunable core-shell morphology. Polym. Chem. 2017, 8, 1371–1380. [Google Scholar] [CrossRef]
- Kutcherlapati, S.N.R.; Koyilapu, R.; Boddu, U.M.R.; Datta, D.; Perali, R.S.; Swamy, M.J.; Jana, T. Glycopolymer-Grafted Nanoparticles: Synthesis Using RAFT Polymerization and Binding Study with Lectin. Macromolecules 2017, 50, 7309–7320. [Google Scholar] [CrossRef]
- Ting, J.M.; Tale, S.; Purchel, A.A.; Jones, S.D.; Widanapathirana, L.; Tolstyka, Z.P.; Guo, L.; Guillaudeau, S.J.; Bates, F.S.; Reineke, T.M. High-Throughput Excipient Discovery Enables Oral Delivery of Poorly Soluable Pharmaceuticals. ACS Cent. Sci. 2016, 2, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Li, Z.; LaBelle, A.J.; Bates, F.S.; Lodge, T.P.; Hillmeyer, M.A. Impact of Polymer Excipient Molar Mass and End Groups on Hydrophobic Drug Solubility Enhancement. Macromolecules 2017, 50, 1102–1112. [Google Scholar] [CrossRef]
- Gallagher, J.J.; Hillmyer, M.A.; Reineke, T.M. Isosorbide-based Polymethacrylates. ACS Sustain. Chem. Eng. 2015, 3, 662–667. [Google Scholar] [CrossRef]
- Beghdadi, S.; Miladi, I.A.; Romdhane, B.; Bernard, J.H.; Drockenmuller, E. RAFT polymerization of Bio-Based 1-Vinyl-4-Dianhydrohexitol-1,2,3-Triazole Stereoisomers obtained via Click Chemistry. Biomacromolecules 2012, 13, 4138–4145. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.J.; Hillmyer, M.A.; Reineke, T.M. Acrylic Triblock Copolymers Incorporating Isosorbide for Pressure Sensitive Adhesives. ACS Sustain. Chem. Eng. 2016, 4, 3379–3387. [Google Scholar] [CrossRef]
- Nasin, M.; Reineke, T.M. Sustainable glucose-based block copolymer exhibit elastomeric and adhesive behavior. Polym. Chem. 2016, 7, 5233–5240. [Google Scholar]
Scheme 1.
The most common mechanisms for reversible activation in polymerization reactions.

Scheme 2.
General scheme of the reaction mechanism in NMRP.

Scheme 3.
Initiation mechanism of uni- and bimolecular initiators in NMRP.

Chart 1.
TEMPO derivatives used as nitroxides in NMRP.

Chart 2.
Alkoxyamines and nitroxide derivatives showing improved bond hydrolysis. Ring enlargement in TEMPO derivatives (N6 and N7) and acyclic nitroxides (N8–N10).
Chart 2.
Alkoxyamines and nitroxide derivatives showing improved bond hydrolysis. Ring enlargement in TEMPO derivatives (N6 and N7) and acyclic nitroxides (N8–N10).

Chart 3.
Alkoxyamines bearing stereo centers used in NMRP.

Chart 4.
Nitroxides and alkoxyamines used for the RDRP of MMA in NMRP.

Chart 5.
Alkoxyamines for pH triggered C–ON bond homolysis used in NMRP.

Chart 6.
Functional alkoxyamines used in NMRP.

Scheme 4.
Activation and deactivation equilibrium reaction in ATRP.

Scheme 5.
Schematic depiction of light catalyzed ATRP with Ir(ppy)3 as catalyst.

Scheme 6.
Schematic depiction of the oxidative quenching mechanism in metal free ATRP.

Scheme 7.
Reductive quenching mechanism with N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as an example for electron sacrificing compound in metal free ATRP.
Scheme 7.
Reductive quenching mechanism with N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as an example for electron sacrificing compound in metal free ATRP.

Scheme 8.
Mechanism of RAFT polymerization.

Chart 7.
Typical chain transfer agents used in RAFT.

Scheme 9.
Reaction mechanism for photo-RAFT involving iniferter process.

Scheme 10.
Reaction mechanism for PET-RAFT.

Scheme 11.
Organo catalyzed (OC) PET-RAFT. (a) Oxidative pathway; (b) reductive pathway.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kreutzer, J.; Yagci, Y. Metal Free Reversible-Deactivation Radical Polymerizations: Advances, Challenges, and Opportunities. Polymers 2018, 10, 35. https://doi.org/10.3390/polym10010035
AMA Style
Kreutzer J, Yagci Y. Metal Free Reversible-Deactivation Radical Polymerizations: Advances, Challenges, and Opportunities. Polymers. 2018; 10(1):35. https://doi.org/10.3390/polym10010035
Chicago/Turabian StyleKreutzer, Johannes, and Yusuf Yagci. 2018. "Metal Free Reversible-Deactivation Radical Polymerizations: Advances, Challenges, and Opportunities" Polymers 10, no. 1: 35. https://doi.org/10.3390/polym10010035
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.