
Abstract
Carbon gel and carbon–nickel–palladium doped gels (C–Ni–Pd) were prepared by carbonising resorcinol–formaldehyde (RF) hydrogel and resorcinol–formaldehyde–nickel–palladium (RF–Ni–Pd) hydrogels at 900 °C in a nitrogen atmosphere. RF and RF–Ni–Pd hydrogels were synthesized through sol–gel polycondensation followed by ambient drying. The aim of this study was the determination of the effect of heat treatment in air at 450 °C on the properties of C–Ni–Pd gels prepared using different Pd salts. In the present work, Ni was added as acetate whereas Pd was added as acetate (CA–Ni–Pd) and as chloride (CB–Ni–Pd). Samples were examined by scanning electron microscopy and X-ray diffraction. Surface area was characterized by N2 adsorption at −195.5 °C. Thermogravimetric analysis was carried out in order to determine the thermal characteristics of carbon gel and nickel–palladium composites in air atmosphere. CA–Ni–Pd composite had a higher activity and two-phase reaction compared to the CB–Ni–Pd composite. Further improvement of the electrolyte diffusion into the particles of nickel and palladium was obtained by oxidative thermal treatment. During this process a structural modification of the material took place, consequently leading to changes in the electrochemical properties of the composites.
Similar content being viewed by others

1 Introduction
Organic aerogels are relatively new compounds, obtained first by Pekala [1] using resorcinol (R) and formaldehyde (F) as substrates. R and F are mixed with water in appropriate proportions, with the addition of a catalyst (usually Na2CO3), and then aqueous solution is allowed to polycondensation. The obtained resorcinol–formaldehyde (RF) gels are subjected to dehydration, most often by the exchange of solvents [1], drying with supercritical CO2 [1, 2], drying at ambient temperature [3–5], microwave drying [6] or by freeze drying [7, 8]. The products of drying are then subjected to thermal treatment to obtain carbon gels.
Recent efforts have focused on the modification of carbon gels through the use of different metal dopants [9–16]. It is worth mentioning that various metals can be easily incorporated into carbon gels to create carbon–metal composites. Such materials can be produced by the addition of soluble metal salts in the process of a sol–gel polymerization reaction. During this process the precipitation or crystallization of metal salts within the pore network occurs. The presence of metal salts can also play the role of catalyst, modifying the degree of polymerization or gelation, and can have an influence on the morphology and structure of pores of the organic gels. One area of significant interest is incorporation of metal species into the carbon framework with the goal of modifying the structure, conductivity, and catalytic and electrochemical activity of the carbon gels. Various metal-doped carbon gels have already been prepared and characterized from the point of view of physicochemical properties [17–21]. Metal-doped carbon materials are attractive for several applications, for example in electrochemistry [9, 14, 22–24] or catalysis [25–28], but the pores of carbon gels obtained by drying in ambient conditions are often closed, the chemical and electrochemical activity can not be fully utilized. Therefore, activation processes for the composites should be carried out, leading to the modification of the structural surface and resulting in an increase in the volume of open pores, allowing unconstrained diffusion of ions into their interior, and consequently, increasing their activity. The trials of the carbon gel activation were so far conduced only through activation in a CO2 atmosphere at different temperatures [29–31]. Carbon gels were also activated chemically with KOH and NaOH [32] or using high-energy ball milling [33].
The goal of the present paper is to prepare transition metal-containing composites using Ni and different Pd salts simultaneously and to determine differences depending on the kind of palladium salt used for the preparation of C–Ni–Pd composites and to determine the effect of heat treatment in an oxidizing atmosphere on the electrochemical activity of such prepared composites.
2 Experimental
2.1 Reagents
All chemicals were obtained commercially and were used without further purification: resorcinol (POCH, Poland), formaldehyde (37 wt%, stabilized 10 wt% methanol, Merck, Germany), sodium carbonate (POCH, Poland), nickel acetate (C4H6NiO4·4H2O) (Fluka, UK), palladium acetate (Sigma-Aldrich, USA), palladium chloride (POCH, Poland), acetone (POCH, Poland), potassium hydroxide (POCH, Poland). All reagents were of an analytical degree. Distilled water was used for preparing all solutions.
2.2 Preparation of carbon gel and carbon–nickel–palladium composites
Resorcinol–formaldehyde (RF) hydrogels were synthesized by the polycondensation of resorcinol (R) with formaldehyde (F), and sodium carbonate (C) as a base catalyst following a method already applied elsewhere [21]. Molar ratios R/F and R/C were 0.5 and 1000, respectively. The mass percentage of the reactants in water solution was set at RF = 40 %.
In order to obtain carbon–nickel–palladium composites (C–Ni–Pd) Na2CO3 catalyst was replaced with two pair of metal precursors: nickel acetate (C4H6NiO4·4H2O) and palladium acetate (C4H6O4Pd) or nickel acetate (C4H6NiO4·4H2O) and palladium chloride (PdCl2). The mass of nickel and palladium salts in the solution were adjusted so as to the theoretical metal mass in carbon formed after RF carbonization (carbonization conditions hereafter) was 5 wt% for each metal. The mass percentage of RF and metals in solution was set at RF/Ni/Pd = 40 %.
All the samples underwent the operation of water removal through the exchange with acetone. The exchange process was repeated three times using fresh acetone to remove residual water from the pores of wet gels. Subsequently the dried samples of gels were carbonized in a tube furnace under nitrogen flow at 900 °C for 3 h using a heating rate of 5 °C/min.
The obtained products were denoted as follows:
- CG:
-
carbon gel (without nickel and palladium),
- CA–Ni–Pd:
-
carbon gel doped with 5 wt% of nickel and 5 wt% of palladium (both as acetate),
- CB–Ni–Pd:
-
carbon gel doped with 5 wt% of nickel (as acetate) and 5 wt% of palladium (as chloride)
After carbonization all the gels were powdered in a high-energy ball mill (Pulverisette 6, Fritsch) in an argon atmosphere. Carbon gel and carbon–metal composites were milled twice at 350 rpm for 15 min, using a 10-min interruption between each milling. The mass ratio of stainless steel balls to milled material was 55:1.
2.3 Sample characterization
A scanning electron microscope (SEM) (Hitachi S-3400N) coupled with the energy dispersive spectrometer (EDS) (Thermo Electron Corp., model No. 4481B-1UES-SN with the NSS Spectral Imaging System software) was used for observing the morphology and surface distribution of active particles of samples as well as determining semi-quantitatively their chemical composition.
The X-ray analyses were performed with a diffractometer BRUKER D8 Advance, equipped with a computer control set. The measurements were done using a CuKα radiation (wavelength of 0.1542 nm) with nickel filtering.
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) of samples were carried out using Setaram device. Samples of about 20 mg were heated up to 1000 °C with a heating rate of 5 °C min−1 in order to determine the thermal characteristics of carbon gel and nickel–palladium composites in air atmosphere.
The BET specific surface areas (S BET) of samples were determined from the isotherms measured by N2 adsorption at −195.5 °C using a relative pressure ranging from 0.06 to 0.30 (ASAP2020 V3.01 H–Micromeritics porosimeter). The cumulative pore volume (V t) between 1.7 and 300 nm and mesopore size distributions were determined from the desorption branch of the N2 isotherm using the Barret–Joyner–Halenda (BJH) method.
2.4 Preparation of electrodes and evaluation of electrochemical properties
The working electrode was made by the insertion of 20 mg of powdered electrode material placed on a gold spiral, playing the role of current collector, mounted on the bottom of the vessel of measuring cell. Before the measurement electrode was immersed in 6 M KOH solution for 24 h to ensure the diffusion of aqueous electrolyte solution into the pores of carbon material.
Cyclic voltammetry (CV) was used to determine the electrochemical properties of the carbon gel and carbon–metal composite electrodes. A Hg/HgO/6 M KOH system (-0.098 V vs. normal hydrogen electrode) and a platinum spiral were used as the reference and counter electrode, respectively. The CV measurements were carried out in the potential range from −1.2 to 0.0 V with the scan rate 1 mV/s, according to the conditions described in detail in the earlier work [21].
All electrochemical measurements were carried out at room temperature using AUTOLAB potentiostat–galvanostat (model PGSTAT 30).
3 Results and discussion
3.1 Physico-chemical characterization
The samples of CG and nickel–palladium doped carbon gels were examined using thermal analysis in order to determine their thermal stability against air. Thermal scanning was done at the temperature range from 20 to 1000 °C.
All the TG curves indicate a common behavior (Fig. 1). Weight loss for the gel CG (Fig. 1a) as well as for both C–Ni–Pd composites (Fig. 1b, c) began at approximately 500 °C. All samples were completely burned-out at around 700 °C.

TG and DSC curves recorded in air atmosphere (heating rate: 5 °C/min) for sample CG (a), CA–Ni–Pd (b) and CB–Ni–Pd (c)
Also DSC curves obtained for the composites upon heating are similar in shapes (Fig. 1b, c). For sample CG a peak with the maxima at 550 °C is noted (Fig. 1a) whereas for samples C–Ni–Pd double exothermic peaks are recorded. The first peaks with their maxima at 550 °C are related to the oxidation reaction of carbon and the second ones (maxima at 700 °C) can be assigned to an oxidation of nickel and palladium particles.
These data gave the information that the process of heat treatment of composites should be carried out at temperatures below 500 °C to avoid thermal decomposition of samples. Because of this some parts of the composites were activated in air at 450 °C. For this purpose the samples were heated up to 450 °C at a heating rate of 5 °C/min and then left at this temperature for 30 min.
The influence of the addition of the nickel–palladium catalyst to the RF gel and the gels’ subsequent conversion to metallic nickel and palladium due to carbonization can be evaluated by the comparison of the SEM images shown in Fig. 2 with those presented in Fig. 3.

SEM images of carbon gel CG

SEM images of composites before heat treatment CA–Ni–Pd (a, b) and CB–Ni–Pd (c, d)
One can observe the changes in morphology of carbon gel and C–Ni–Pd composites caused by the addition of different Pd catalysts. It is known that the presence of metal precursor in the RF solution changes the mechanism of the polymerization process [24, 34, 35]. The particles’ dimensions of sample CG are from 5 to 10 μm in diameter (Fig. 2) and they have different shapes.
The evidence for the catalytic effect of transition metals on the polymerization process can be observed on the SEM images for samples of C–Ni–Pd composites (Figs. 3, 4), where spherical particles are in contrast to differently shaped particles visible for the sample free of metal dopants (Fig. 2). The particles’ dimensions of both composites (1–5 μm in diameter) are smaller than for those of gel CG.

SEM images of composites after heat treatment CA–Ni–Pd (a) and CB–Ni–Pd (b)
One can see on the carbon surface of C–Ni–Pd composites nickel and palladium inclusions which are observed as light spots (Figs. 3, 4). Their size is different and varies from 0.01 to 0.3 μm for composite CA–Ni–Pd and from 0.01 to 1 μm for composite CB–Ni–Pd.
Through the comparison of Fig. 3 with Fig. 4 it is apparent that the morphology of both composites is not altered by the process of heat treatment in air at 450 °C.
The EDS data are shown in Table 1. One can see that chemical composition of composites CA–Ni–Pd and CB–Ni–Pd changed after thermal treatment. The main change is the increase in oxygen content which can be assigned to both the formation of oxygen functional groups on the carbon surface of the composites and the formation of metal oxides on the metal surface.
Surface properties of carbon gel and nickel–palladium doped carbon gels were characterized using the N2 adsorption–desorption isotherms and are given in Table 2.
The comparison of the specific surface area (S BET) for the carbon gel free of metals (sample CG) and the nickel–palladium doped carbon gels showed the decrease of the specific surface area. The specific surface area is smaller, about 63 and 49 m2/g for sample CA–Ni–Pd and CB–Ni–Pd, respectively. The porous structure of these gels is significantly changed by nickel and palladium particles incorporated in the carbon matrix. As seen from Table 2 mesopores disappeared in composite CA–Ni–Pd and CB–Ni–Pd and, as a consequence a significant reduction of the average width of the pores down to 2.40–2.24 nm compared to that of the original sample CG of d BJH = 15.25 nm. The measurements also showed that carbon gel without metals had a total pore volume (Vt) at p/po = 0.99 about two times larger than those for carbon–metal composites. One can see from the structural data presented in this Table that the conversion of nickel and palladium salts to metallic nickel and palladium due to carbonization lead to significant changes. The effect of these reactions was a more than six-fold decrease in the average pore diameter of mesopores and, finally, was responsible for the decrease in their cumulative volume of pores.
The adsorption studies also provided information about the changes in the structural properties of composites C–Ni–Pd due to oxidative thermal treatment. As seen from the comparison of data gathered in Table 2, such a treatment gave rise to an increase in the specific surface area. The surface became about 13 % higher than that for untreated composites. A further effect of the heat treatment of composites was an increase in pore diameter to 2.80 and 2.53 nm for samples CA–Ni–Pd and CB–Ni–Pd, respectively.
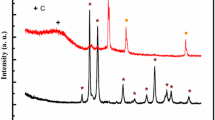
The XRD spectra for carbon gel CG is shown in Fig. 5. The X-ray pattern exhibit broad reflections at 2θ = 23.6° and 44.1° which correspond to the (002) and (101) diffractions of the graphitic framework respectively, buried in the carbon matrix. On all XRD patterns recorded for composite CA–Ni–Pd and CB–Ni–Pd (Fig. 6) a broad diffraction peak at about 2θ = 24°, point to the presence of turbostratic carbon is also presented. For both untreated composites (Fig. 6a,c) three sharp peaks positioned at 2θ = 41°, 47° and 69° are visible. These peaks are assigned to crystalline palladium. Two other peaks at 2θ = 44°, 52°, corresponding to crystalline nickel can also be observed. On the XRD pattern recorded for carbon gel CB–Ni–Pd one additional peak at about 42° is presented, corresponding to palladium oxide. Such a peak is not visible for sample CA–Ni–Pd.

XRD pattern of carbon gel CG

XRD patterns for composites: CA–Ni–Pd before (a) and after (b) heat treatment and CB–Ni–Pd before (c) and after (d) heat treatment. Peak positions: (asterisk) graphite, (filled square) Pd, (plus symbol) PdO, (circle) Ni, (multiply symbol) NiO
From the Fig. 6b, d it can be observed that intensities of peaks for both composites increase after heat treatment. This is particularly noticeable for peaks arising from crystalline palladium. These peaks are also a trifle broader. From the literature it is known that the peak broadening has often been correlated with electrochemical activity [36–38]. The diffraction peak located at about 52° shows the existence of nickel, disappeared on the XRD patterns for both heat treated composites. Three additional peaks for sample CA–Ni–Pd are noted. The first of them centered at 37.3°, corresponding to the presence of nickel oxide is also recorded for sample CB–Ni–Pd. The second one at 2θ about 34° indicating palladium oxide [32, 34] is also noted for both composites. The third of them at 2θ = 42° proves the presence of palladium oxide appeared only on the XRD pattern for composite CA–Ni–Pd.
3.2 Electrochemical properties
To examine the electrochemical properties of the carbon gel and composites, CV tests have been conducted in 6 M KOH solution from −1.2 V to 0 V. Cyclic voltammograms obtained for carbon gel and metal-doped gels are depicted in Figs. 7, 8 and 9, respectively. The character of CV curves recorded for C-Ni–Pd composites is distinctly different from that of the carbon gel free of metals. The presence of metals noticeably influenced both the shape and the current charges of the CV curves. One can also see differences in the recorded reactions depending on the kind of palladium salt used.

Cyclic voltammograms recorded for carbon gel CG. Potential range from −1.2 V to 0.0 V. Scan rate 1 mV/s

Cyclic voltammograms recorded during the 10th and 25th cycle for carbon gel CA–Ni–Pd (a) and CB–Ni–Pd (b). Potential range from −1.2 V to 0.0 V. Scan rate 1 mV/s

Cyclic voltammograms recorded during the 10th and 25th cycle for carbon gel CA–Ni–Pd (a) and CB–Ni–Pd (b) after heat treatment in air. Potential range from −1.2 V to 0.0 V. Scan rate 1 mV/s
In Fig. 7 it can be observed that for carbon gel CG there are no apparent redox peaks connected with the reduction/oxidation of the carbon matrix. One can only see capacitive effects related to the process of charging/discharging of the electrical double layer and faradaic reactions [24, 32, 39].
The comparison of cyclic voltammograms recorded for samples CA–Ni–Pd and CB–Ni–Pd before heat treatment during the 10th and 25th cycle is presented in Fig. 8. We can observe the differences originating from the kind of palladium salt used. The created changes concern the processes taking place during both cathodic and anodic polarization. The differences consist in both the decrease in the anodic and cathodic charge and the shapes of registered curves. It is proved that the carbon gels with pore diameter between 3 and 13 nm have good voltammetry characteristics and high capacitance values [32].
No effects arising from the redox reactions of nickel and palladium during the first 10 cycles of scanning (CV curves not shown here) are observed for sample CB–Ni–Pd, whereas a couple of current peaks appeared for sample CA–Ni–Pd. In contrast to the CV curves recorded for sample CA–Ni–Pd, also during further cyclization for sample CB–Ni–Pd only one small and flat anodic peak at about −0.84 V is observed. This peak is connected with the oxidation of sorbed hydrogen. Also cathodic peak noted for sample CA–Ni–Pd at about −0.9 V is flat and small. This peak can be related with both the reaction of hydrogen adsorption/absorption and reduction of NiOOH. After the polarization direction is changed the oxidation of sorbed hydrogen and nickel and/or palladium takes places within the broad anodic peak noted at about −0.8 V. Both peaks become more and more profound upon cyclization.
The higher activity of CA–Ni–Pd composite is in agreement with studies that the carbon atoms with rounded shapes are the most reactive [40, 41]. Sample CA–Ni–Pd has a slightly higher amount of particles with such shapes which suggests a better access of the electrolyte.
The comparison of cyclic voltammograms recorded during the 10th and 25th cycle for samples CA–Ni–Pd and CB–Ni–Pd after heat treatment is presented in Fig. 9. A detailed consideration of these CV curves reveals further changes in the kinetics and mechanism of the reactions depending on the kind of palladium salt used for the preparation of C–Ni–Pd composites. It is clear that peaks measured for heat treated samples are more acute and the current noted for them is around three- and four-times higher, respectively for samples CB–Ni–Pd and CA–Ni–Pd. The release of Ni–Pd particles could be a reason for their effects as a result of the oxidation of the carbon surface and subsequently their exposure to the electrolyte and their improvement of access to the electrolyte. The higher electrochemical activity of C–Ni–Pd composites after heat treatment in the reactions of hydrogen sorption/desorption is also correlated with data derived from an XRD analysis (more intensive peaks arising from crystalline palladium).
In contrast to the double and sharp anodic/cathodic peaks of high intensity noted for composite CA–Ni–Pd, only one small cathodic peak at −0.95 V associated with the reactions of hydrogen sorption and the anodic peak at −0.84 V related to hydrogen release were recorded for sample CB–Ni–Pd. An interesting feature of the anodic peak observed for composite CA–Ni–Pd after thermal treatment is its splitting (Fig. 8). This may indicate changes in the reaction mechanism. The formation of a double peak might be related to transformation from the α- to β-(Pd–Ni)/H phase within the carbon matrix [42] associated with the Ni ↔ Ni(OH)2 reaction [43]. One can also see the improvement of the electrochemical activity in the alkaline solution for CA–Ni–Pd composite during further cyclization.
As an elucidation of the higher activity of sample CA–Ni–Pd it is reasonable to assume that the use of Ni and Pd acetate leads to Ni–Pd particles of a higher activity due to their smaller dimensions (Figs. 3, 4). The higher activity of this composite compared to the composite prepared using Ni acetate and Pd chloride, lends credence to such a hypothesis. The presence of nickel and palladium acetates in the carbon matrix also contributed to the increase of pore diameter as compared to composite CB–Ni–Pd. The further increase in electrochemical activity of the composite CA–Ni–Pd due to heat treatment and two-phase reaction, might be ascribed to the formation of the porous structure ensuring a better transport of ions to Ni–Pd particles. The reason probably lies in the more activating interaction of CO2, being liberated due to thermal decomposition of the acetate, with the carbon skeleton.
4 Conclusions
Nickel–palladium doped carbon gels prepared by the carbonization of organic gels containing nickel and palladium acetate (CA–Ni–Pd) and nickel acetate and palladium chloride (CB–Ni–Pd) appeared to be electrochemically active in 6 M KOH aqueous solution.
For composites CA–Ni–Pd a higher electrochemical activity was obtained. The increased activity of these composites and the biphasic reaction can already be observed before the oxidation heat treatment. The reason probably lies in a more activating interaction of CO2, which is being liberated due to thermal decomposition of the acetate, with the carbon skeleton.
The improvement of the electrolyte diffusion into the particles of nickel and palladium was obtained by thermal treatment in air at 450 °C. The electrochemical treatment carried out in the potential range from −1.2 to 0.0 V resulted in the changes in the electrochemical properties of thermally treated composites C–Ni–Pd. As a result of such treatment the transport of electrolyte ions improved, allowing contact with particles of nickel and palladium present in the coal matrix. In the case of thermally treated composites one can notice an increase in the registered current of hydrogen sorption/desorption.
References
R.W. Pekala, J. Mater. Sci. 24, 3221 (1989)
H. Pröbstle, M. Wiener, J. Fricke, J. Porous Mater. 10, 213 (2003)
D. Wu, R. Fu, S. Zhang, M.S. Dresselhaus, G. Dresselhaus, Carbon 42, 2033 (2004)
H.H. Jung, S.W. Hwang, S.H. Hyun, K.H. Lee, G.T. Kim, Desalination 216, 377 (2007)
S.J. Kim, S.W. Hwang, S.H. Hyun, J. Mater. Sci. 40, 725 (2005)
L. Zubizarreta, A. Arenillas, J.A. Menédez, J.J. Pis, J.-P. Pirard, N. Job, J. Non-Cryst. Solids 354, 4024 (2008)
H. Tamon, H. Ishizaka, T. Yamamoto, T. Suzuki, Carbon 38, 1099 (2000)
T. Yamamoto, A. Endo, T. Ohmori, M. Nakaiwa, S.R. Mukai, H. Tamon, Drying Technol. 23, 2119 (2005)
N. Yoshizawa, H. Hatori, Y. Soneda, Y. Hanzawa, K. Kaneko, M.S. Dresselhaus, J. Non-Cryst. Solids 330, 99 (2003)
N. Job, R. Pirard, B. Vertruyen, J.-F. Colomer, J. Marien, J.-P. Pirard, J. Non-Cryst. Solids 353, 2333 (2008)
F.J. Maldonado-Hodar, C. Moreno-Castilla, J. Rivera-Utrilla, Y. Hanzawa, Y. Yamada, Langmuir 16, 4367 (2000)
T.F. Baumann, G.A. Fox, J.H. Sachter, N. Yosizawa, R. Fu, M.S. Dresselhaus, Langmuir 18, 7073 (2002)
L.C. Cotet, M. Baia, I.C. Popescu, V. Cosoveanu, E. Indrea, J. Popp, V. Danciu, J. Alloys Compd. 434–435, 854 (2007)
N. Job, F. Maillard, J. Marie, S. Berthon-Fabry, J.-P. Pirard, M. Chatenet, J. Mater. Sci. 44, 6591 (2009)
J. Aguado-Serrano, M.L. Rojas-Cervantes, R.M. Martín-Aranda, A.J. López-Peinado, V. Gómez-Serrano, Appl. Surf. Sci. 252, 6075 (2006)
Z. Zapata-Benabithe, F. Carrasco-Marín, C. Moreno-Castilla, Mater. Chem. Phys. 138, 6075 (2013)
V.M. Gun’ko, V.M. Bogatyrov, O.I. Oranska, I.V. Urubkov, R. Leboda, B. Charmas, J. Skubiszewska-Zięba, Appl. Surf. Sci. 30, 263 (2014)
Z. Zapata-Benabithe, F. Carrasco-Marín, C. Moreno-Castilla, Mater. Chem. Phys. 138, 870 (2013)
S. Chandra, S. Bag, R. Bhar, P. Pramanik, Microporous Mesoporous Mater. 138, 149 (2011)
A.F. Pérez-Cadenas, C. Ros, S. Morales-Torres, M. Pérez-Cadenas, P.J. Kooyman, C. Moreno-Castilla, F. Kapteijn, Carbon 56, 324 (2013)
J.M. Skowroński, M. Osińska, J. Sol-Gel. Sci. Technol. 71, 109 (2014)
Y.J. Lee, J.C. Jung, S. Park, J.G. Seo, S.-H. Baeck, J.R. Yoon, J. Yi, I.K. Song, Curr. Appl. Phys. 10, 947 (2010)
Y.J. Lee, J.C. Jung, S. Park, J.G. Seo, S.-H. Baeck, J.R. Yoon, J. Yi, I.K. Song, Curr. Appl. Phys. 11, 1 (2011)
J.M. Skowroński, M. Osińska, Curr. Appl. Phys. 12, 911 (2012)
N. Job, J. Marie, S. Lambert, S. Berthon-Fabry, P. Achard, Energy Convers. Manag. 49, 2461 (2008)
W. Kiciński, J. Lasota, J. Mater. Sci. Technol. 28, 249 (2012)
M. Sánchez-Polo, J. Rivera-Utrilla, U. von Gunten, Water Res. 40, 3375 (2006)
C.I. Fort, L.C. Cotet, V. Danciu, G.L. Turdean, I.C. Popescu, Mater. Chem. Phys. 138, 893 (2013)
E. Guilminot, F. Fisher, M. Chatenet, A. Rigacci, S. Berthon-Fabry, P. Achard, E. Chainet, J. Power Sources 166, 104 (2007)
C. Robertson, R. Mokaya, Microporous Mesoporous Mater. 179, 151 (2013)
G. Rasines, C. Macias, M. Haro, J. Jagiello, C.O. Ania, Microporous Mesoporous Mater. 209, 18 (2015)
S.W. Hwang, S.H. Hyun, J. Non-Cryst. Solids 347, 238 (2004)
M. Osińska, Microporous Mesoporous Mater. 214, 95 (2015)
J.M. Skowroński, M. Osińska, Przem. Chem. 91, 1407 (2012)
C. Moreno-Castilla, F.J. Maldonado-Hódar, A.F. Pérez-Cadenas, Langmuir 19, 5650 (2003)
H. Chen, J.M. Wang, T. Pan, H.M. Xiao, J.Q. Zhang, C.N. Cao, Int. J. Hydrogen Energy 28, 119 (2003)
M.C. Bernard, R. Cortes, M. Keddam, H. Takenouti, P. Bernard, S. Senyarich, J. Power Sources 63, 247 (1996)
D. Claude, T. Cecile, J. Mater. Chem. 7, 1439 (1997)
L.L. Zhang, X.S. Zhao, Chem. Soc. Rev. 38, 2520 (2009)
T.S. Ong, H. Yang, Carbon 38, 2077 (2000)
T.D. Shen, W.Q. Ge, K.Y. Wang, M.X. Quan, J.T. Wang, W.D. Wei, C.C. Koch, Nanostruct. Mater. 7, 393 (1996)
J.M. Skowroński, A. Czerwiński, T. Rozmanowski, Z. Rogulski, P. Krawczyk, Electrochim. Acta 52, 5677 (2007)
J.M. Skowroński, A. Ważny, J. New Mater. Electrochem. Syst. 9, 345 (2006)
Acknowledgments
This research was supported by the Grant No. 03/31/DSPB/0274.
Author information
Authors and Affiliations
Corresponding author
Additional information
In memory of Professor Jan M. Skowroński
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Osińska, M. The effect of catalyst and heat treatment on the properties of carbon–metal composites. J Porous Mater 23, 365–373 (2016). https://doi.org/10.1007/s10934-015-0089-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-015-0089-4