Abstract
Corrosion driven delamination of coatings is crucially determined by the rate of the cathodic oxygen reduction reaction at the buried metal-organic coating interface. Quantitative measurement of this rate at such interfaces by conventional techniques is impeded due to the blocking of ion transport by the coating. A new approach where hydrogen permeation is used as a tool to measure the oxygen reduction rate underneath coatings has been recently introduced. This permeation based potentiometry approach measures the rate of the oxygen reduction reaction by correlating the open circuit potential established as a consequence of the dynamic electrochemical equilibrium between hydrogen oxidation and oxygen reduction reactions on the coated exit side with the hydrogen uptake rate on the entry side. In this work, the interfacial reaction kinetics of the oxygen reduction reaction causing delamination of the coating are investigated by this hydrogen permeation based potentiometric approach. Moreover, thus performed prolonged cathodic polarizations of the palladium/coating interface have been found to destroy the interface, just like it is the case in the cathodic delamination process. Initial results obtained on ultra-thin films of iron on palladium are also presented, showing that the technique is applicable also on technically more relevant metal surfaces.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
Corrosion driven degradation of the buried metal/organic coating interface is of utmost interest in the contemporary space of coatings science. Earlier studies have shown conclusively that the oxygen reduction reaction (ORR) plays a fundamental role in the destruction of this buried metal/organic coating interface. The role of the interface between organic coating and metal has been object of intense research over last decades. Leidheiser et al.1 pointed out the key role of the cathodic ORR for the degradation process and also showed that diffusion processes such as water and oxygen transport through the coating can influence this delamination rate. Stratmann and co-workers2–4 proposed a detailed delamination mechanism for a basic polymer coating on steel where galvanic coupling between the defect site of metal dissolution (local anode) and delamination site of electrochemical oxygen reduction (local cathode) supported by cation migration along the polymer/metal interface lead to the progressive deadhesion of the polymer from the underlying metal based on comprehensive analysis of the local electrode potential at the metal/polymer interface using primarily the Scanning Kelvin Probe (SKP) technique along with complementary Auger electron spectroscopy and mechanical deadhesion tests. Fürbeth and Stratmann5–7 extended their studies to coated zinc (or galvanized steel), which also involves anodic processes at the interface. Also other works by Brewis et al.8 and Dickie et al.9 show the importance of interfacial electrochemical reactions on metal/polymer adhesion. Up to now our knowledge about the reactions and how they are determined by the interface itself is too restricted to be able to make reliable prediction about interfacial degradation rates, although first attempts, albeit based on experimental input parameters, have been tried.10 An important question is how the electrochemical reactivity of such metal/polymer coated systems is determined by the interfacial chemistry of the metal oxide/coating interface. As an example, using XPS, Wielant et al.11 showed that a large fraction of hydroxyls present on the oxide surface aided bonding of amine/amide based model compounds through nitrogen protonation. This points to possibilities for improving the chemical bonding of coatings to metal surfaces. Although the exact importance of chemical bonds over physical ones is not fully clarified yet, there has been intriguing work indicating a significant direct influence of chemical bonds on delamination behavior.12
Electrochemical reactions at the metal/polymer interface play an important role on delamination kinetics. Hence, a quantitative knowledge of the reaction rate at this buried metal/coating interface is needed. Until now, there is only little information about the reaction rates at such buried interfaces since it is difficult to directly monitor the active processes taking place underneath the coating. The Scanning Kelvin Probe (SKP) provides direct information of the corrosion potential underneath coatings at buried interfaces, but, it does not provide information about interfacial reaction rates.2–4 Recently, inspired by earlier work on controlling the potential on one side of a palladium membrane by applying a potential on the other side to control the defined hydrogen activity across the sample,13–16 a new potentiometric approach using hydrogen permeation as a tool to measure interfacial oxygen reduction rates at this buried metal/organic coating interface has been developed.17 In this method, the classical Devanathan-Stachurski technique has been adapted, wherein hydrogen permeation from the back side of the sample is used to quantitatively measure the oxygen reduction rate at the metal/organic coating interface on the other side.
Briefly, the Devanathan-Stachurski18,19 permeation technique is widely applied for determining the diffusion constant of hydrogen and the hydrogen binding energies at trap sites in metals. The technique makes use of a double electrochemical cell to electroadsorb hydrogen from an aqueous solution on the back side (entry side) of the metal by suitable cathodic polarization. Atomic hydrogen then diffuses through the metal due to the chemical potential gradient maintained by anodic polarization on the other side (exit side), which keeps the hydrogen activity at the exit side close to zero by instantaneous oxidation of the emerging hydrogen. This exit current is a direct measure of the volume of hydrogen that has permeated through the metal.
In this new hydrogen permeation based potentiometry approach, the former technique has been modified by introducing oxygen in the exit cell for oxidizing the hydrogen that is permeating through, thereby creating the opportunity to monitor the oxygen reduction reaction kinetics. Fig. 1 describes the scheme of the various electrochemical processes taking place in this approach, on both faces of the metal membrane (Pd) coated with a model (Polyvinyl butyral (PVB)) organic coating on one side. Firstly, a defined amount of hydrogen is produced by cathodic polarization of an acidic electrolyte on the back side of the Pd membrane. Here, H is formed on the Pd surface and is forced to diffuse through the Pd membrane in the form of atomic H by the establishment of a steep chemical potential gradient. This steep gradient is achieved by using oxygen on the front side which nearly completely oxidizes the atomic H coming through to produce water as a net product and thus maintains [Hexit] close to zero. Thus, at the exit side, a dynamic equilibrium between hydrogen oxidation reaction (HOR) and oxygen reduction reaction (ORR) is established. It was proved17 that once this dynamic equilibrium is established, one is able to obtain direct access to the oxygen reduction rate at the exit side from the known hydrogen uptake on the entry side. Further, by monitoring the open circuit potential established as a result of this dynamic equilibrium between ORR and HOR at the exit side, the current-potential relationship (I(U) curve) can be quantitatively measured. By proving consistency17 between the I(U) curve for ORR from this approach and that obtained from standard three electrode measurements on bare Pd surface in acid, alkaline and buffer electrolyte medium, the assumption of [Hexit] ≈ 0 was validated. Further, the rate determining step (rds) for ORR has been proved by earlier reports20 to be the formation of the superoxide O−2, since H is not directly involved in this rds, i.e. the [Hexit] does not play a role on the derived I(U) curve for ORR kinetics. Underneath organic coatings, the technique was able to measure the true reaction rate by accessing the interfacial charge transfer process taking place between the oxygen molecules readily available at the exit metal side by fast diffusion through the organic coating and the atomic hydrogen exiting the buried interface. The I (U) relationship could be successfully obtained over a wide potential range. This technique clearly distinguished itself from current three electrode based electrochemical measurements which cannot provide reliable measurement of the ORR rate underneath coatings due to the blocking of ion transport (if there are no pinholes or defects in the coating) between the working and counter electrodes in the three-electrode setup by the organic coating, thus preventing polarization of the buried interface. In the new approach, supply of electrons and cations (protons) to the interface is simultaneously provided by the hydrogen emerging at the interface. This new potentiometric technique, free of cross-coating ion transport, allows the buried interface kinetics to be uniquely characterized.
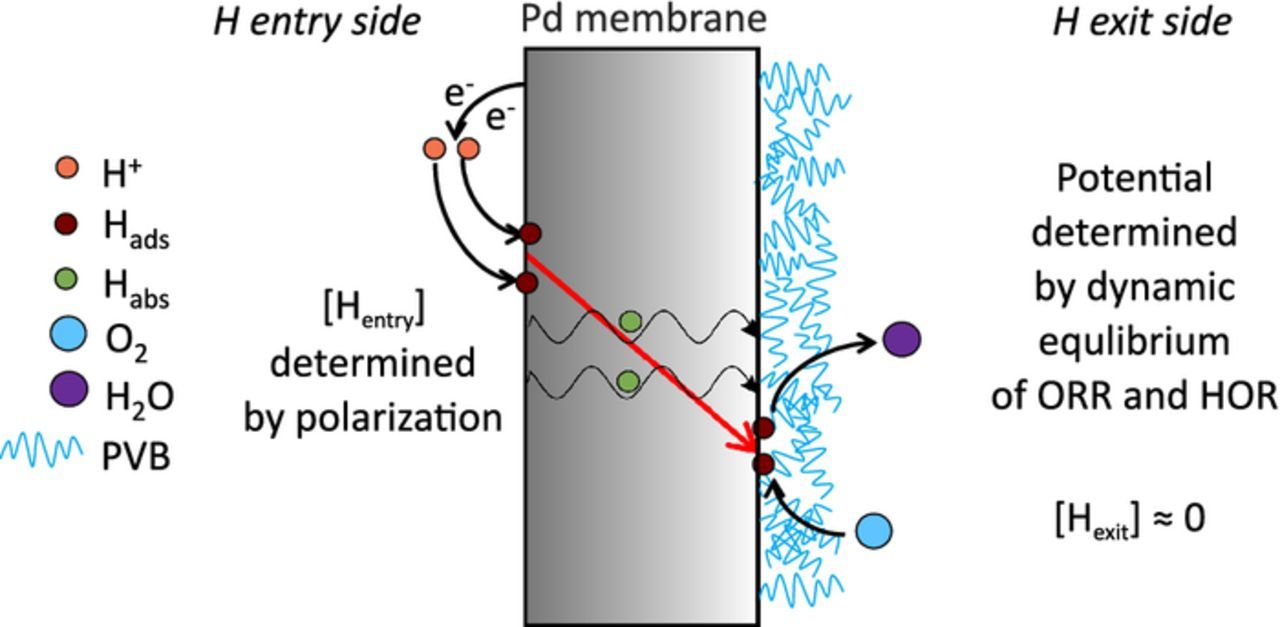
Figure 1. Schematic of the hydrogen permeation based potentiometry approach describing the various electrochemical reactions taking place on both sides of the Pd membrane with one side coated by model PVB coating. A steep [H] gradient in the Pd membrane is established by using oxygen on the exit side to nearly completely oxidize the [H] permeating through thereby maintaining [Hexit] ≈ 0.
The driving force for this work is to understand the basics of coating degradation. In this context, the kinetic reaction mechanisms of degradation at the palladium/PVB interface have been elucidated by this permeation based potentiometry approach. By performing measurements of oxygen reduction reaction with hydrogen oxidation as counter reaction at the buried interface, it is observed that the interface gradually degrades, most likely due to the same mechanisms as during cathodic delamination, i.e. by intermediate radicals of oxygen reduction. Further, due to significant production of water as the final product of the prolonged oxygen reduction and hydrogen oxidation reactions at the exit side, the pH at the buried interface approaches toward neutral values consequently shifting the measured I(U) curve of the ORR, depending on the initial pH prevailing at the interface. Results of delamination experiments at tailored Pd/PVB interfaces are found to correlate with the ORR rate at the corresponding interfaces obtained from the permeation based potentiometry approach. Preliminary results on the extension of this approach to relevant metals such as iron show promisingly good correlation between the I(U) curve for ORR derived from this approach on ultra-thin layers of iron coated on palladium and that obtained from standard three electrode electrochemistry. One important motivation behind the development of this approach is to be able in the future to provide input parameters for simulating delamination through finite element modeling in technically relevant metal/coating systems.
Experimental
Materials
As-rolled Pd foil (99.99+ %, light tight) of 25 μm thickness and 25 mm × 25 mm linear size and Au (99.99%) foil of 1 mm thickness and 15 mm × 15 mm linear size, were made use of as the working and counter electrodes respectively, and were both procured from Goodfellow Cambridge Limited. Electrolyte stock solution of 1 mol dm−3 H2SO4 (0.2% accuracy), NaClO4, C2H5OH (absolute, analytical grade) and HNO3 (69%) were purchased from VWR Chemicals BDH Prolabo, Belgium. KCl, H2O2 (30%), NH4OH (28–30%) and NaOH pellets were procured from Merck KGaA Darmstadt. H2SO4 (≥95%) was purchased from Bernd Kraft, Duisburg. Poly (vinyl butyral-co-vinyl alcohol-co-vinyl acetate) (PVB) composed of an average molecular weight 50,000 – 80,000 was purchased from Sigma Aldrich. ARMCO iron (99.85%) samples for SKP measurements were procured in-house. Electrolytic iron (99.9%) was purchased from Carboleg GmbH, Essen in the form of flakes and was used as the source for electron beam physical vapor deposition (EBPVD) of ultra-thin layers of Fe on Pd.
Equipment
Commercial Red Rod reference electrodes (micro diameter model) from Radiometer Analytical SAS, France were made use of for both the electrochemical polarization and open circuit potential measurement experiments. These individual reference electrodes were measured to have a mean variation of 10 mV vs Ag/AgCl reference electrodes (viz. 240 mV vs standard hydrogen electrode (SHE)). The potentials expressed here are with respect to the Ag/AgCl reference electrode. An Ivium COMPACTSTAT electrochemical interface and a PalmSens EmStat potentiostat were utilized to carry out electrochemical permeation experiments. Electron beam physical vapor deposition was performed in PVD cluster from BESTEC GmbH under high vacuum conditions. Delamination experiments were performed using a commercial SKP with height regulation from KM Soft Control (Wicinski–Wicinski GbR, Wuppertal, Germany).21
Methods
0.1 mol dm−3 H2SO4 acid electrolyte solution (pH = 0.97–1.1) was prepared by dilution of 100 ml of the 1 mol dm−3 H2SO4 stock solution with 900 ml of deionized water. 0.1 mol dm−3 each of NaOH alkaline electrolyte solution (pH = 12.5–12.8) and NaClO4 near neutral electrolyte solution (pH = 5.15) was prepared by separately dissolving 4 g of NaOH pellets and 14.046 g of NaClO4 crystals in 1000 ml deionized water respectively. Palladium foils were cleaned thoroughly by ultrasonification in ethanol, rinsing in deionized water, followed by treatment in a base piranha solution (5:1:1:: H2O:NH4OH:H2O2) at 70°C for 30 minutes and a final cleaning step in concentrated HNO3 for 3 minutes. To prepare model physisorbed coatings on Pd, two stock solutions of 5 wt% and 10 wt% PVB in ethanol were made use of. Three different types of PVB coating on Pd were prepared by spin coating: (i) 5 wt% PVB solution was twice spin coated on Pd at ω = 2000 rpm for 20 seconds (30 second interval in between) and annealed at 70°C for 10 minutes (in the following referred to as PVB_thin) (ii) Next, the first step of spin coating twice with 5 wt% PVB was kept constant to prepare identical interfaces, followed by baking in the oven at 70°C for 10 minutes, and subsequent spin coating twice with a 10 wt% PVB solution (30 second interval in between). This coating was then similarly dried (referred to as PVB_thick) (iii) a final so called compact coating was prepared by spin coating twice only with a 10 wt% PVB solution (30s interval) and dried similarly (referred to as PVB_compact). The aim was to check if identical interfaces (PVB_thin vs PVB_thick) exhibited similar ORR characteristics and different interfaces (PVB_thin or PVB_thick vs PVB_compact) displayed contrasting ORR behavior. The thicknesses of the three coatings PVB_thin, PVB_thick and PVB_compact were measured to be around 1 μm, 3 μm and 2 μm respectively using ellipsometry. For measurements on iron coated palladium, ultra-thin Fe layers of thicknesses 100 nm and 200 nm were prepared by electron beam deposition (i = 87 mA) at 1 Å/s with the sample stage spinning at ω = 30 rpm.
The Devanathan-Stachurski permeation setup used in this work has been described extensively in the previous publication.17 Briefly, a double electrochemical cell with the one-side PVB coated Pd as the working electrode is sandwiched in-between the two individual cells (working electrodearea = 0.3286 cm²). Each independent cell consisted of a Luggin capillary that accommodated the Red Rod reference electrode and only the entry cell contained the Au foil as a counter electrode inside a glass shield. Stepwise cathodic polarization was applied to the entry side (bare Pd side) filled with 0.1 mol dm−3 H2SO4 to produce hydrogen under potentiostatic conditions in argon atmosphere. The associated current is basically the hydrogen entry current or the hydrogen permeation current "I". The hydrogen exit side (PVB coated Pd) was filled with 0.1 mol dm−3 either of H2SO4, NaOH or NaClO4 and oxygen was purged here to oxidize the permeating atomic hydrogen depending on the purpose of the investigation being carried out. For PVB/Pd interfacial degradation experiments, 0.1 mol dm−3 H2SO4 was always used on the hydrogen exit side, while for verifying the dilution effect all the three different electrolytes were explored. For measurements with ultra-thin layers of iron on palladium, the hydrogen exit side was filled with 0.1 mol dm−3 NaOH, since iron layers would corrode in acidic and near neutral conditions. The open circuit potential established by the dynamic equilibrium between the hydrogen oxidation and oxygen reduction reactions on the exit side, "U" was measured as a function of the hydrogen uptake current "I" to obtain the I(U) curve, thereby describing the ORR. The current and potential values were allowed to reach steady state for around 30 minutes before noting down measurements.
Delamination experiments at different tailored Pd/PVB interfaces were performed, using iron as an artificial defect, at 23°C in humid air with relative humidity of about 95%. Glass samples with precut 15 mm × 15 mm size dimensions were first cleaned with acid piranha solution (3:1::H2O2:H2SO4), rinsed with deionized water, followed by ultrasonification in ethanol and final cleaning with deionized water before physical vapor depositing a 200 nm palladium film with a 10 nm chromium adhesion layer in-between (to improve the poor adhesion between Pd and glass). With the aim to reproduce identical PVB coated interfaces such as those tested in the permeation based potentiometry approach using the Devanathan-Stachurski cell, a similar spin coating procedure as described above was carried out on Pd coated glass to prepare glass/Pd/PVB_thin and glass/Pd/PVB_thick samples respectively. This composite sample was then glued using a two-component epoxy on to iron samples of 20 mm × 25 mm size dimensions, which were previously mechanically ground using P1000 grinding paper and cleaned in ethanol and distilled water.22,23 A reservoir was thus made at the junction between the composite sample and iron as shown in Fig. 2 where 1 mol dm−3 KCl was provided as an electrolyte medium for corrosion initiation on iron and PVB coating delamination. Iron corrodes spontaneously and polarizes the PVB/Pd interface providing the necessary driving force for PVB delamination and therefore serves as an artificial defect. An SKP system from KM Soft Control was used to monitor the progress of the cathodic delamination front by performing surface scan measurements on an area of 5000 μm × 100 μm on the composite PVB/Pd/glass/iron sample at a distance of 1 mm away from the edge of the defect with a step size of 50 μm (x-axis) and 100 μm (y-axis). SKP tip was made of Ni/Cr (80/20 wt%) alloy with a diameter of 100 μm and its calibration was done with a Cu/CuSO4 (saturated) solution.
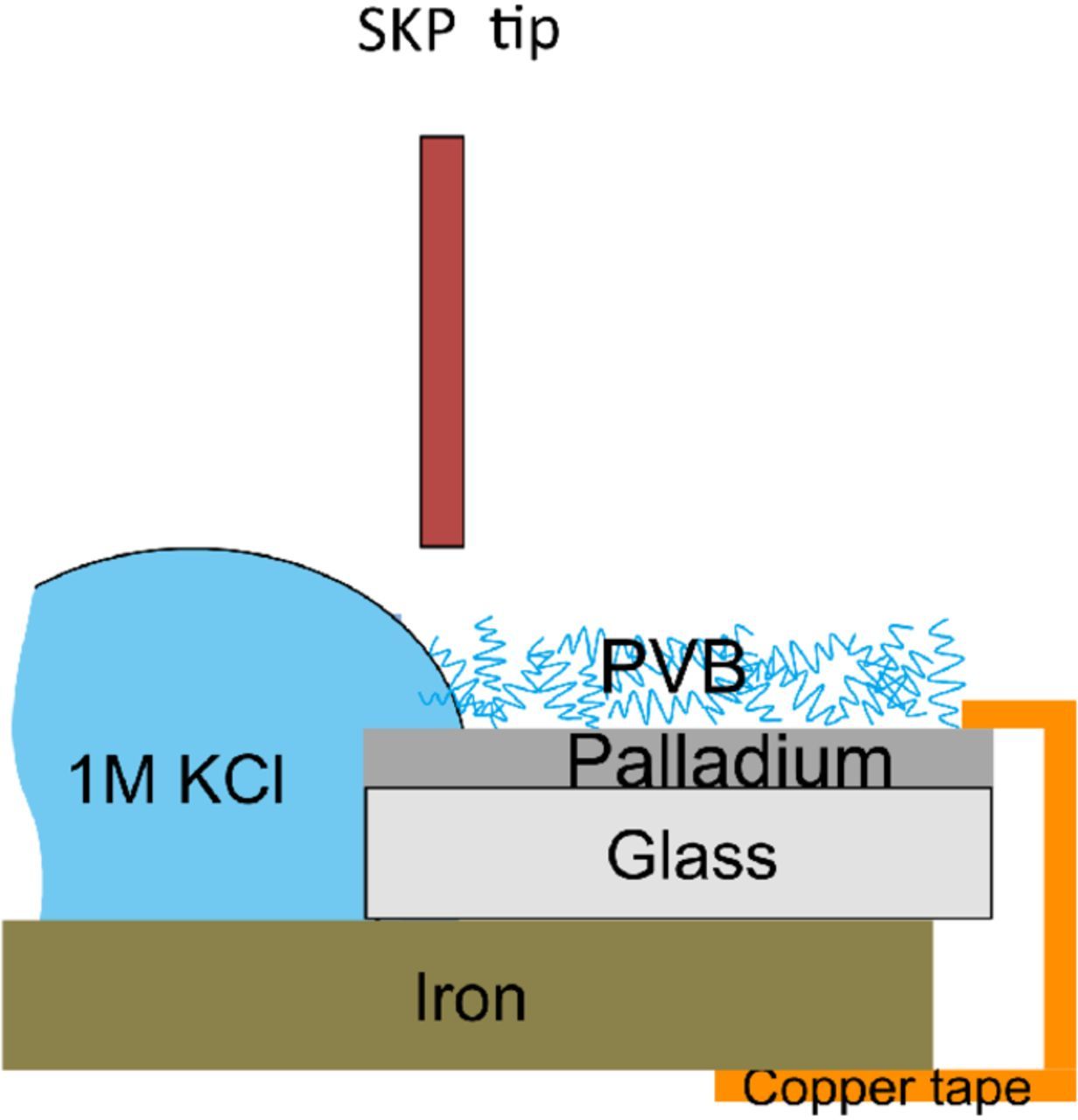
Figure 2. Schematic for measuring progress of cathodic delamination front at PVB/Pd interface using iron as an artificial defect.
Results and Discussion
Construction of the I(U) curve by the permeation based potentiometry approach has been described in detail previously.17 Briefly, the I(U) curve is obtained by measuring the open circuit potential established by the dynamic equilibrium of the hydrogen oxidation and oxygen reduction reactions at the exit side and the corresponding hydrogen entry current density. For a given quantity of hydrogen entering the Pd membrane and being completely oxidized, the same amount of oxygen needs to get reduced in order to establish the open circuit potential on the exit side. By performing stepwise cathodic polarization on the entry side, increasing amounts of defined hydrogen current is allowed to permeate through the Pd membrane, enabling increased cathodic oxygen reduction current on the exit side, thereby necessitating the measured open circuit potential on the exit side to shift cathodically. Monitoring the evolution of the steady state open circuit potential on the exit side "Vexit" with the corresponding entry hydrogen permeation current "I", the I(U) curve is derived. Fig. 3 shows for three types of coatings PVB_thin, PVB_thick and PVB_compact, the current-potential (I(U)) curve for two successive runs derived from the permeation based potentiometry approach. Here, "successive runs" refers to performing a linear cathodic permeation sweep on a fresh Pd membrane, followed by switching off the cathodic polarization to allow the system to return to open circuit conditions overnight and subsequently repeating the linear cathodic permeation sweep on the same used Pd membrane with fresh electrolyte. From Fig. 3, for all the three coatings, the measured ORR current density "I" increases with successive runs. For instance; around an exit potential of +200 mV a five-fold increase in "I" for PVB_thin and a two-fold increase for PVB_thick and PVB_compact between runs 1 and 2 can be clearly seen. This is due to the degradation of the PVB/Pd interface by the aggressive intermediate radicals of the ORR. Here degradation implies a local deadhesion of the interface due to the oxygen reduction, most likely due to reactive intermediates.2 This degradation leads to a more reactive interface, with the presence of underfilm Pd-water interfaces (presumably in the form of nanoscopic pockets, this is subject of current research) supporting higher ORR currents after successive runs as compared to the original intact adherent PVB-Pd interface. Further, the preparation procedure outlined in this work involving the use of 5 and 10 wt% PVB solutions for spin coating as well as an intermediate annealing step results in different coatings with different interfaces which slightly changes the derived I(U) curves as compared to those reported in our earlier work.17
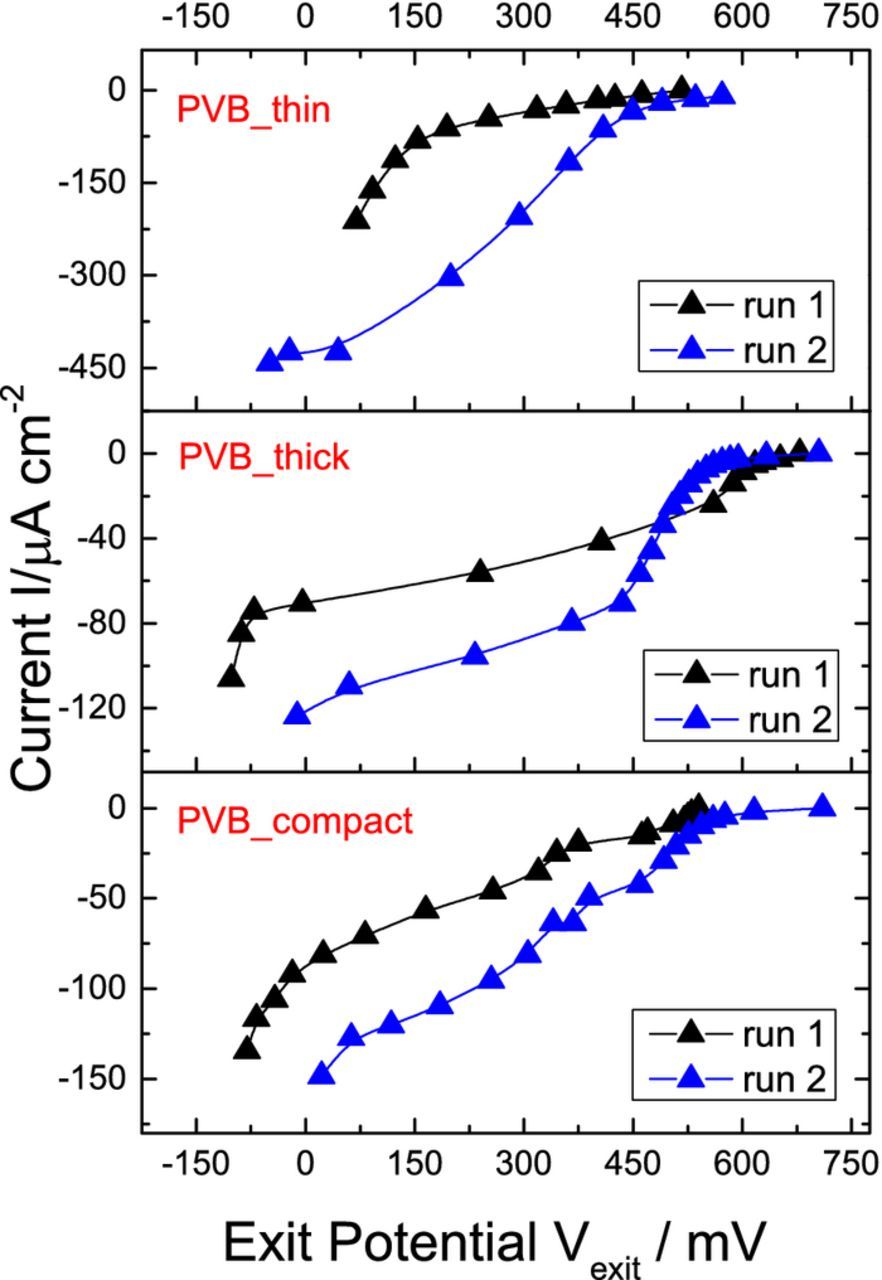
Figure 3. I(U) curves measured by the permeation based potentiometry approach17 for PVB_thin, PVB_thick and PVB_compact for two successive runs. 0.1 mol dm−3 H2SO4 in both entry and exit cells, with Ar being purged in entry cell and O2 in exit cell.
Fig. 4 shows the I(U) curve of the forward and backward runs for PVB_thick and PVB_compact in acidic, near neutral and alkaline environments at the exit side. Here, the forward run is a linear cathodic permeation sweep starting from the open circuit potential of the Pd membrane on the entry side while the backward run refers to a linear anodic permeation sweep continuing from the end entry potential of the forward sweep back to the open circuit potential of Pd on the entry side. From Fig. 4, we see a cathodic shift of the exit potential between the backward and forward I(U) curve for PVB_thick with acidic medium (pH = 1.1) at the exit side. For instance, at a ORR current density of ∼ − 55 μAcm−2, the backward I(U) curve is shifted cathodically by around ∼230 mV with respect to the forward one. This is proposed to be due to a pH increase at the interface by dilution by produced water.
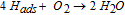
Since water is the main product of the hydrogen oxidation and oxygen reduction reactions, dilution at the interface might cause significant increase in pH (the initial pH at the coating/metal interface is determined by the pH in the electrolyte) approaching neutral pH when starting from an acidic pH which directly shifts the I(U) curves cathodically with respect to potential according to the Nernst equation.
In contrast, when starting from an alkaline medium (pH = 12.5) at the exit side, pH decreases to neutral pH by dilution of the interface by water subsequently shifting the I(U) curve anodically with respect to potential between the backward and forward runs. For instance, at a ORR current density of ∼ − 40 μAcm−2, the backward I(U) curve is shifted anodically by around ∼ 80 mV with respect to the forward one. In the case of near neutral medium (pH = 5.15) at the exit side, there is no shift in the I(U) curve between the forward and backward runs as there is not much change in pH as dilution does not affect the already near neutral exit side. Fig. 4, also shows a very similar behavior for PVB_compact with a cathodic, anodic and no shift in exit potential between the backward and forward I(U) curves for acidic, alkaline and near neutral exit side environments, respectively, providing support to the dilution effect hypothesis.
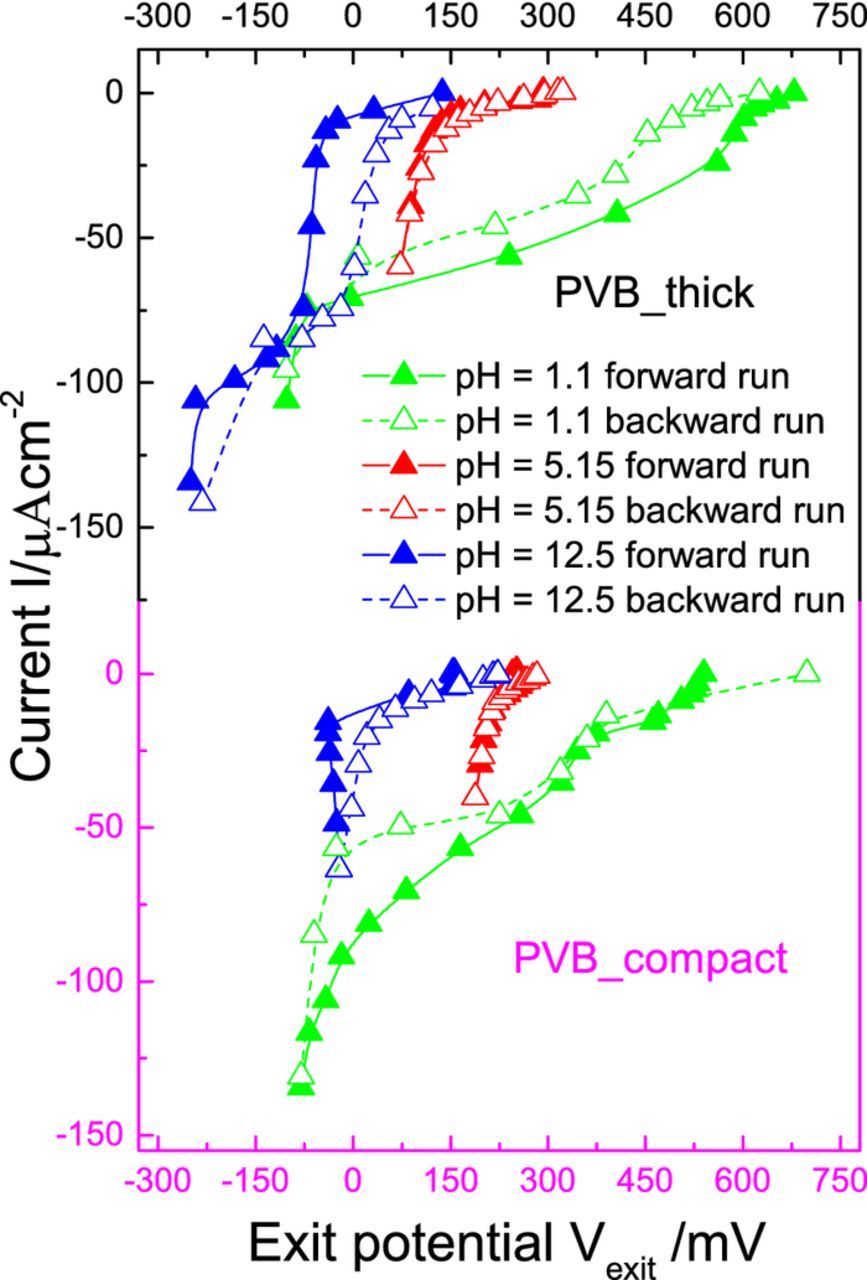
Figure 4. I(U) curves of the forward and backward runs measured by the permeation based potentiometry approach17 for PVB_thick and PVB_compact at acidic (0.1 moxl dm−3 H2SO4, pH = 1.1), alkaline (0.1 mol dm−3 NaOH, pH = 12.5) and near neutral (0.1 mol dm−3 NaClO4, pH = 5.15) exit cell environment. 0.1 mol dm−3 H2SO4 was present in the entry cell for all the runs, with Ar purged in entry cell and O2 in exit cell.
Figs. 5a, 5b show the correlation between delamination experiments performed with the SKP using iron as an artificial defect and ORR current densities obtained from permeation based potentiometry approach for PVB_thin and PVB_thick with near neutral (pH = 5.15) electrolyte environment in the exit side. The aim is to see if there is a correlation between the onset of ORR at the still intact coating from the permeation based potentiometry approach and the delamination rate calculated using the SKP for tailored interfaces. From Fig. 5a, PVB_thick is seen to have an earlier onset for ORR as compared to PVB_thin indicating that the former has a weaker interface. For instance, an ORR current I ∼ −1.3 μA cm−2 is measured at a higher potential of around ∼ 291 mV for PVB_thick as compared to ∼161 mV for PVB_thin. Fig. 5b shows corresponding relationship of measured delamination distance "x" with time "t" for PVB_thin and PVB_thick. The variation of x vs t for PVB_thin shows a more linear dependence while PVB_thick tends more toward a √t behavior indicating that the interfacial oxygen reduction reaction is the rate determining step (rds) for PVB_thin delamination while cation migration along the PVB_thick/Pd interface could also play a role in the rds for PVB_thick. Higher delamination rate of ∼1645 μm h−1 was extracted by a linear regression fit of x vs t for PVB_thick as compared to a lower rate ∼ 706 μm h−1 for PVB_thin, indicating that PVB_thick is the weaker interface possibly due to interfacial defect formation during the intermediate annealing step between the two spin coatings for PVB_thick. This agrees well with the results from the permeation based potentiometry approach that PVB_thick is the weaker interface. On the other hand, there are differences between the two cases: While in the delamination experiment there will be an increase in pH as the final product of ORR is OH− at the interface and also the concentration of cations compensating this negative charge will increase, whereas neither of these two occur for the experiment providing the I(U) curve. However, we assume that the crucial step for delamination where the interfacial reaction is rate determining will be the initial degradation step. At this stage pH shift and also increase in cation concentration will be still very low. This is the subject of further research.
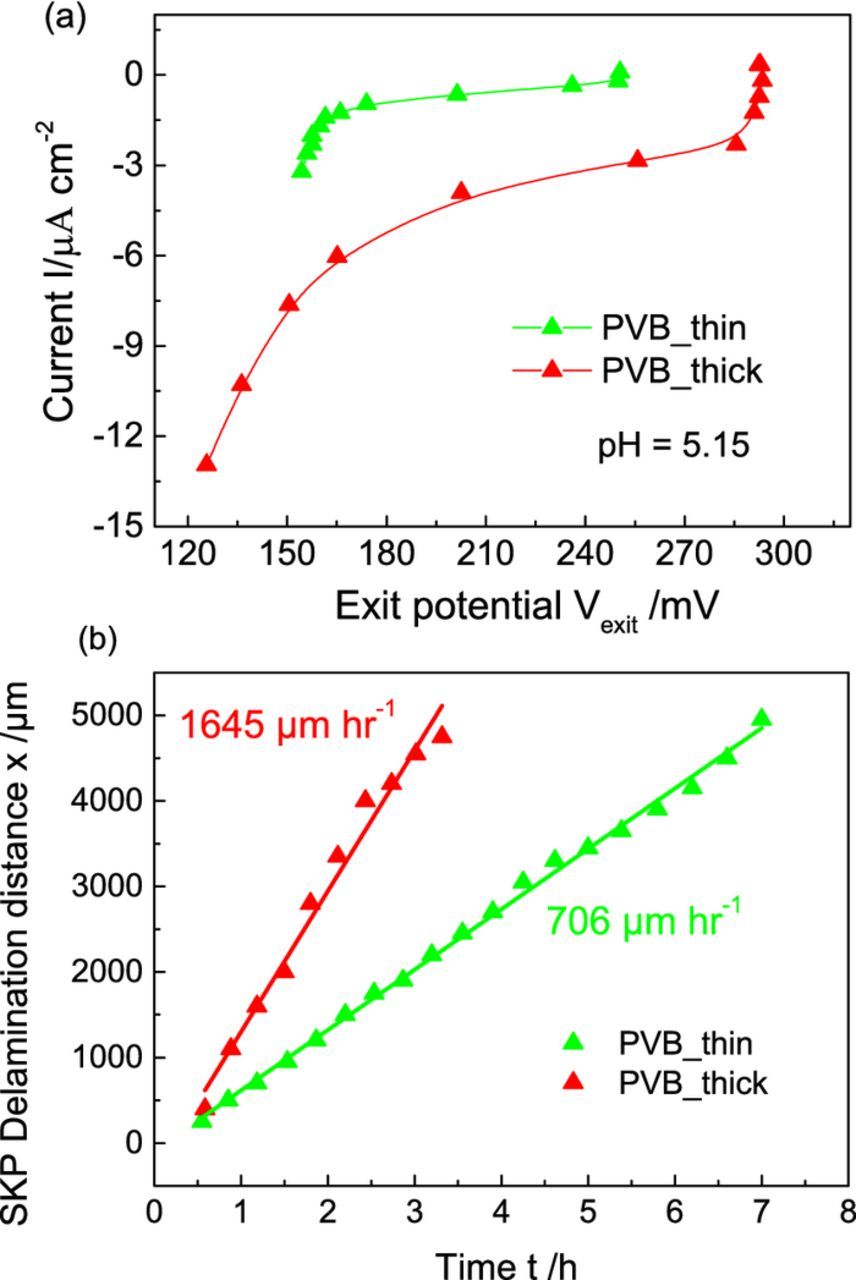
Figure 5. (a) I(U) curves for PVB_thin and PVB_thick measured using the permeation based potentiometry approach17 in near neutral (0.1 mol dm−3 NaClO4, pH = 5.15) exit cell environment and 0.1 mol dm−3 H2SO4 in the entry cell. Entry and exit cell were purged with Ar and O2 respectively (b) Delamination distance 'x' vs time 't' measured using the Scanning Kelvin Probe (SKP) for PVB_thin and PVB_thick using iron as an artificial defect in humid air atmosphere.
Finally, the viability of extending the permeation based potentiometry approach to more relevant metals was explored by depositing ultra-thin layers of iron of thicknesses 100 nm and 200 nm on one side of the Pd membrane using physical vapor deposition technique. Such ultra-thin iron films should not significantly affect the hydrogen permeation and hence the uptake at the entry side of the Pd membrane and provide the possibility to investigate the initial stages of ORR at more relevant interfaces. Fig. 6 shows the I(U) curves for ORR obtained from the permeation based potentiometry approach for both thicknesses along with the I(U) characteristics for ORR measured using a standard three electrode electrochemical setup in 0.1 mol dm−3 NaOH (pH = 12.5). An alkaline electrolyte medium was chosen at the exit side for this study as the iron layers would readily corrode in acidic medium. Firstly, the I(U) curves of ORR for both 100 nm and 200 nm of iron look very similar in the ORR kinetic region of interest, i.e., in the potential window of 50 mV to −400 mV. This implies that diffusion of hydrogen through the ultra-thin iron films is not determining at least up to 200 nm Fe. Further, there is a good fit of the I(U) curve with the I(U) characteristics for ORR in this potential window (identical curvature and similar magnitudes) measured in a standard three electrode setup for both the thicknesses of Fe, implying that the hydrogen uptake on the entry side is not limited by the ultra-thin Fe layers. The potential change caused by hydrogen permeation through iron oxide is usually interpreted as being due to reduction of FeIII sites to FeII (while oxygen oxidizes the FeII sites to FeIII).24,13 This is also in accordance with earlier work by Stratmann et al. who postulated that the potential on iron oxide surfaces is determined by the  ratio.25 Indeed, as the measurement of the I(U) curve is performed on point by point basis, it can be safely assumed that the according equilibria are established, ie. the dynamic equilibrium between ORR and HOR will determine also an according
ratio.25 Indeed, as the measurement of the I(U) curve is performed on point by point basis, it can be safely assumed that the according equilibria are established, ie. the dynamic equilibrium between ORR and HOR will determine also an according  ratio in the native oxide layer present on the ultrathin Fe films. Of course, there is a cross-dependence between the two because the oxygen reduction depends very much on the FeII concentration on the surface as these are the actual catalytic sites as previously shown by Vago et al.,25 Krieg et al.26 But this is also the case if the potential is changed potentiostatically (as the case for the three electrode measurement also presented shows). Only below - 400 mV the I(U) characteristics obtained from standard electrochemical setup deviate from the I(U) curve measured by this technique as ORR rate is no longer being measured here and the entry hydrogen current is obviously not identical anymore to the ORR rate at the exit side with the hydrogen recombination reaction dominant on the entry side due to excess hydrogen evolution. This proves that the permeation based potentiometry approach can be extended to such ultra-thin metal films for studies in the relevant potential range and hold promise to future simulation studies in real corrosion systems.
ratio in the native oxide layer present on the ultrathin Fe films. Of course, there is a cross-dependence between the two because the oxygen reduction depends very much on the FeII concentration on the surface as these are the actual catalytic sites as previously shown by Vago et al.,25 Krieg et al.26 But this is also the case if the potential is changed potentiostatically (as the case for the three electrode measurement also presented shows). Only below - 400 mV the I(U) characteristics obtained from standard electrochemical setup deviate from the I(U) curve measured by this technique as ORR rate is no longer being measured here and the entry hydrogen current is obviously not identical anymore to the ORR rate at the exit side with the hydrogen recombination reaction dominant on the entry side due to excess hydrogen evolution. This proves that the permeation based potentiometry approach can be extended to such ultra-thin metal films for studies in the relevant potential range and hold promise to future simulation studies in real corrosion systems.
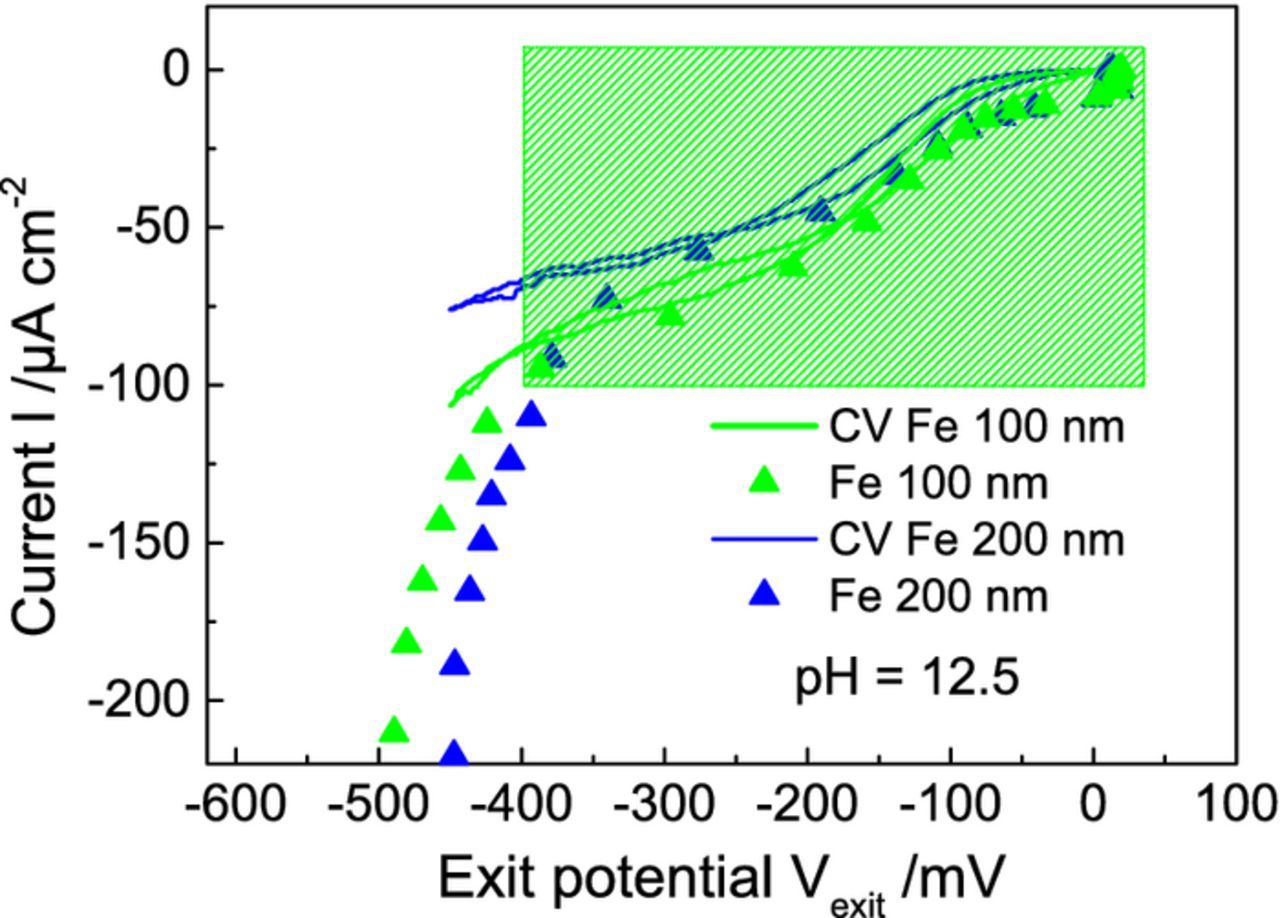
Figure 6. I(U) curves for 100 nm and 200 nm ultra-thin layers of iron coated on exit side of the palladium membrane measured in alkaline (0.1 mol dm−3 NaOH, pH = 12.5) exit cell environment (triangles). 0.1 mol dm−3 H2SO4 was present in the entry cell purged with Ar while the iron coated side was exposed to O2 in the exit cell. I(U) curves measured using the standard three electrode electrochemical setup for oxygen reduction reaction are also shown for the corresponding thicknesses of iron layers (lines).
By virtue of such fully immersed conditions, this hydrogen permeation based potentiometry approach gives one the ability to systematically change the pH at the metal/polymer interface by using a specific exit cell electrolyte environment (acidic, neutral, alkaline) and thereby to characterize the interfacial reactivity by obtaining pure current-potential relationship (I(U) curves) at controlled pH. This is an advantage over uncontrolled systems, for instance where the pH at the interface is unknown. Although one might argue, that in the real scenario, the pH at the interface is shifted to alkaline values (while it stays more neutral with the approach presented here), the capacity to control the pH at the exit side provides one the option to study degradation at any defined pH, including neutral conditions. Further, the crucial part for delamination is believed to be the initial stages where the pH is still neutral. The pH shift by dilution effect can be reconciled by making use of a correction factor, because in principle one can measure this shift. More fundamental information such as diffusion of water can also be obtained. Finally, studies on probing the interfacial reactivity using the Kelvin Probe on the exit side for coating/metal interfaces not under immersed conditions but exposed to atmosphere at different humidity, allowing examination under more typical delamination conditions as well as investigation of electrochemistry at "dry electrodes" are currently underway.
Conclusions
The ion-transport free hydrogen permeation based potentiometry approach has been established as a platform to measure interfacial reaction kinetic mechanisms at buried interfaces. Continuous loss in coating adhesion by aggressive attack of the ORR radical intermediates similar to cathodic delamination by ORR in real corrosion systems is evidenced through degradation experiments with this approach. Shift in exit potential between backward and forward I(U) permeation sweeps due to dilution by end reaction product water reflects the sensitivity of the technique in capturing key interfacial reaction mechanisms. Delamination experiments using the Kelvin Probe serve as supporting information for the feasibility of this technique to unravel information about the reactivity of the still intact interface during the initial stages of degradation. A good match in derived ORR rate for ultra-thin iron layers on palladium with I(U) characteristics obtained from standard electrochemical setup serve as a platform for future simulation of real corrosion systems.
Acknowledgments
The authors thank the Max-Planck-Society for financial support.